
Development of functionalized peptides for efficient inhibition of myostatin by selective photooxygenation†
Hideyuki Okamoto,
Atsuhiko Taniguchi *,
Shoya Usami,
Masahiro Katsuyama,
Sho Konno,
Akihiro Taguchi,
Kentaro Takayama‡
and
Yoshio Hayashi*
*,
Shoya Usami,
Masahiro Katsuyama,
Sho Konno,
Akihiro Taguchi,
Kentaro Takayama‡
and
Yoshio Hayashi*
Department of Medicinal Chemistry, School of Pharmacy, Tokyo University of Pharmacy and Life Sciences, Hachioji, Tokyo 192-0392, Japan. E-mail: atani@toyaku.ac.jp; yhayashi@toykau.ac.jp
First published on 3rd November 2020
Abstract
For the inhibition of myostatin, which is an attractive strategy for the treatment of muscle atrophic disorders including muscular dystrophy, myostatin-binding peptides were synthesized with an on/off-switchable photooxygenation catalyst at different positions on the peptide chain. These functionalized peptides oxygenated and inactivated myostatin upon irradiation with near-infrared light. Among the peptides tested, a peptide (5) with the catalyst moiety at the 16 position induced myostatin-selective photooxygenation, and efficiently inhibited myostatin. These peptides exhibited low phototoxicity. Such functionalized peptides would provide a precedented strategy for myostatin-targeting therapy, in which myostatin is irreversibly and catalytically inactivated by photooxygenation.
Introduction
Myostatin, a member of the transforming growth factor-β (TGF-β) family, is a major negative regulator of skeletal muscle growth and plays a key role in homeostasis of skeletal muscle.1 Myostatin is composed of two 109-mer peptide chains which dimerize by forming disulfide bonds. Similar to other members of the TGF-β family, myostatin is biosynthesized as promyostatin, a precursor protein which consists of an N-terminal prodomain and C-terminal mature domain.2 These domains are subsequently converted to a prodomain protein and myostatin, respectively, and these proteins bind noncovalently to form an inactive latent complex.3 Upon proteolysis of the prodomain protein, the active mature myostatin is released from the complex. It then forms heteromeric complex with activin type-II receptor and type-I receptor on the muscle cells.4 This initiates Smad signaling which suppresses the growth of muscle cells.A genetic defect of myostatin causes muscle hypertrophy,1 and overexpressed myostatin induces cachexia which is associated with muscle atrophy.5 Thus, the inhibition of myostatin is a possible strategy for treatment of various muscle atrophic disorders including muscular dystrophy, cancer cachexia, sarcopenia, sporadic inclusion body myositis, and disuse muscle atrophy. Some research groups have reported protein inhibitors such as anti-myostatin antibodies and a soluble decoy receptor,6–10 but no reported myostatin inhibitors have yet progressed into clinical use.
For the development of a smaller inhibitor, we focused the N-terminal peptide sequence of the prodomain protein which interacts with mature myostatin in the higher order structure of the latent myostatin complex.11 A 23-mer myostatin-binding peptide (1) from the mouse prodomain sequence has been reported (Fig. 1a).12 This peptide (1) exhibits inhibitory activity against myostatin with IC50 = 3.5 ± 0.3 μM.13 Although a structure–activity relationship (SAR) study of 1 afforded some stronger inhibitors,13–17 their inhibitory activities are lower than that of the prodomain protein, a 243-mer protein which is an endogenous inhibitor with IC50 = 2.5 ± 0.3 nM.§
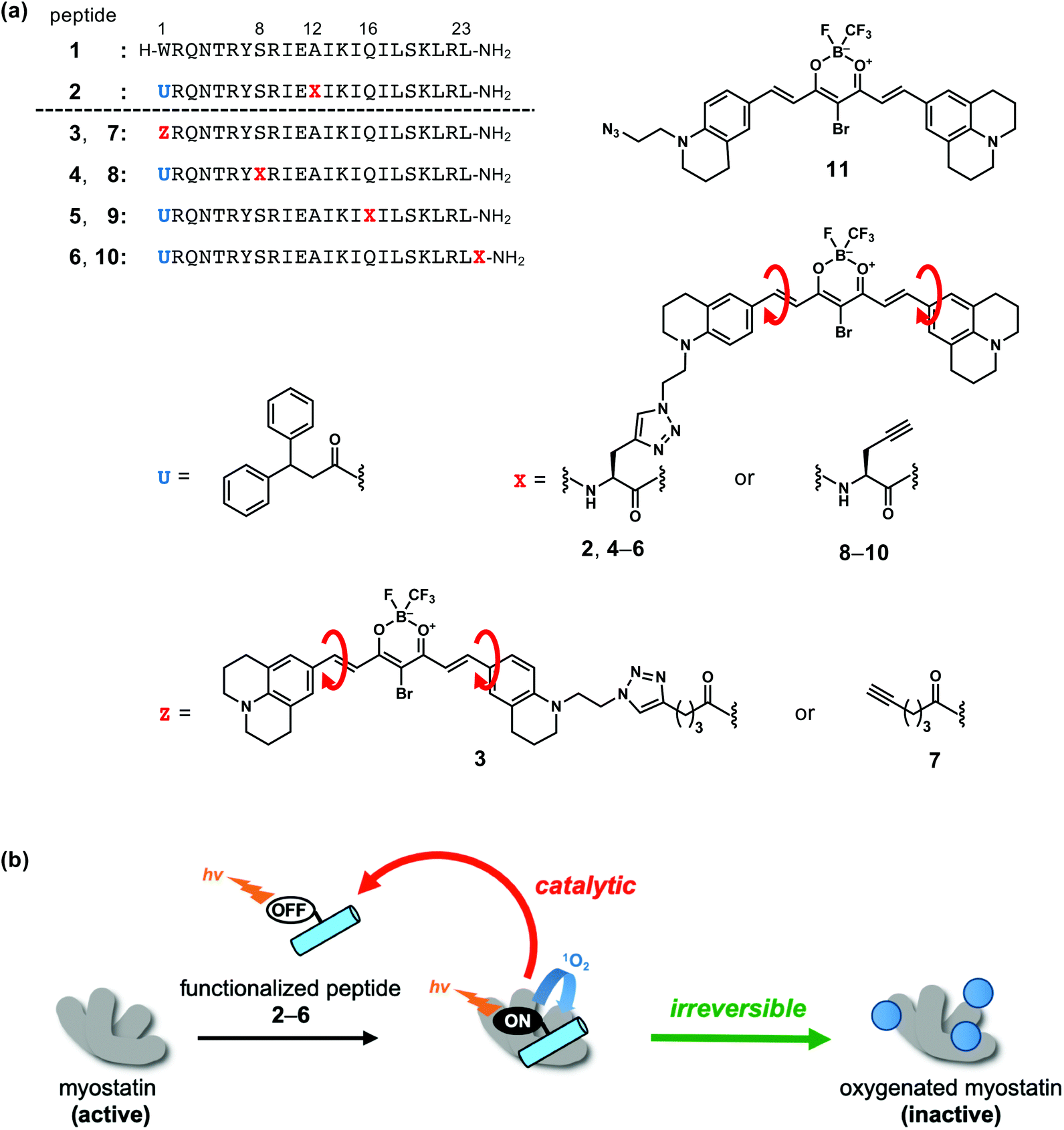 | ||
| Fig. 1 Functionalized peptides which induce photooxygenation of myostatin. (a) Structures of myostatin-binding peptide 1, functionalized peptides 2–6, peptide segments 7–10 and photocatalyst segment 11. (b) Inactivation of myostatin by photooxygenation using peptides 2–6. | ||
To enhance the inhibitory effect of the myostatin-binding peptide (1) while maintaining the molecular size, we functionalized 1 by attaching a photooxygenation catalyst at Ala12 to afford peptide 2 (Fig. 1a).18 Upon light irradiation, 2 successfully inactivates myostatin by a catalytic and irreversible photooxygenation reaction (Fig. 1b).
For the selective photooxygenation, we applied an on/off switchable photooxygenation catalyst19 to the functionalized peptide (2) (Fig. 1a). When 2 fails to bind with myostatin, its photoexcited catalyst moiety promptly relaxes through an intramolecular bond rotation (red arrows in Fig. 1a). When the peptide segment of 2 binds with myostatin, the bond rotation in the catalyst moiety is restricted by interactions with myostatin. As a result, the photoexcited catalyst moiety induces photooxygenation via a relaxation pathway. Thus, peptide 2 can selectively oxygenate myostatin (Fig. 1b).
In this strategy, while the binding mode between myostatin and the functionalized peptide is unclear, the position of the photooxygenation catalyst on the peptide chain should have an influence on the photooxygenation activity. Accordingly, in an effort to enhance the myostatin inhibitory effect, we synthesized a series of functionalized peptides (2–6) which differ in the catalyst position on the peptide chain. These peptides were compared in terms of the photooxygenation and inactivation of myostatin. We found that a peptide (5) possessing the catalyst at position 16 showed the efficient inhibition of myostatin.
Results and discussion
Design
Selection of the sites that could be modified to tether the photooxygenation catalyst on the peptide chain was based on our previous SAR study of original myostatin-binding peptide (1). Ser8 and Gln16 were first selected, because our previous Ala scan study of 1 revealed that these side chains were not essential to the myostatin affinity.14 Ser8 and Gln16 are located four amino acid residues away from Ala12 which is the catalyst site in functionalized peptide 2. Another site was the position 1. In our detailed SAR study of the position 1 of peptide 1, we found that this residue tolerated a carboxylic acid bearing an aromatic or aliphatic cyclic group at the γ-position, for example, 4-(indol-3-yl)butanoic acid, 4-phenylbutanoic acid or 4-cyclohexylbutanoic acid.13 In this study, the triazole ring, which is formed by a conjugation reaction of an azide with an alkyne,20,21 is such a cyclic group. In a fifth case, the catalyst was attached at the C-terminus. Thus, we prepared five functionalized peptides 2–6 which have the photooxygenation catalyst at the positions 1 (3), 8 (4), 12 (2), 16 (5) and the C-terminus (6) (Fig. 1a).Synthesis
The syntheses of novel functionalized peptides 3–6 were achieved in accordance with our reported synthesis of peptide 2.18 Peptides 3–6 were obtained from alkyne-containing peptide segments (7–10) and an azide-containing photooxygenation catalyst segment (11) (Fig. 1a and Scheme S1†). Peptide segments 7–10 were synthesized by a conventional Fmoc-based solid-phase peptide synthesis (SPPS). For the synthesis of peptide segment 7 (Scheme S1a†), hex-5-ynoic acid was coupled at position 1 to introduce an alkyne. For peptide segments 8–10 (Scheme S1b†), propargyl glycine was condensed at the corresponding position to introduce an alkyne moiety into the peptide chains. 3,3-Diphenyl propionic acid was used at the position 1 in peptides 8–10 corresponding to 4–6 as 2, because Trp1 of peptide 1 is very sensitive to photooxygenation.13 To the alkyne moiety in peptide segments 7–10, the azide-containing catalyst segment (11), which was prepared by a published method,18 was conjugated by a copper-catalysed alkyne–azide cycloaddition (CuAAC).20,21 As a result, functionalized peptides 3–6 were obtained in yields of 10–21% and purity of >95%.Absorbance
The absorption spectra of the functionalized peptides (2–6) were recorded (Fig. S1†). All peptides have absorption maxima between 600 and 650 nm, and a small peak around 750 nm. These results indicated that absorption property of the photooxygenation catalyst part was unaffected by the conjugation site, and the catalyst part on the peptide chain can be excited with 650–800 nm of near-infrared (NIR) light. This type of irradiation is biocompatible and is used in clinical practice, e.g. in photodynamic therapy (PDT), because NIR light exhibits high permeability and low phototoxicity in tissues.22Photooxygenation
To evaluate the photooxygenation capability, each functionalized peptide (2–6)¶ was irradiated with a light-emitting diode (LED, ∼730 nm) in the presence of myostatin. The treated myostatin was reduced by 1,4-dithiothreitol (DTT) for cleavage of native disulfide bonds, digested by endoproteinase Lys-C, and then the resulting peptide fragments were analysed by matrix-assisted laser desorption/ionization (MALDI)-time-of-flight (TOF) mass spectrometry (MS). As a representative result, that from the functionalized peptide (5) is shown in Fig. 2a. Myostatin fragments 40–54, 55–78, and 79–90 were clearly detected in the MS analysis. After irradiation, the MS peaks derived from one or two-oxygen atom adducts were increased in fragments 79–90 (left charts) and 55–78 (right charts), compared with a control study (upper charts). In contrast, no peak derived from an oxygen adduct was observed in fragment 40–54 (center charts). These results are consistent with our previous study using a functionalized peptide (2).18 Met residues at the positions 79 and/or 84 could be oxygenated in the fragment 79–90, and His residues at the positions 57, 59 and/or 62 could be oxygenated in the fragment 55–78.18 Other functionalized peptides 3, 4 and 6 gave results similar to those from peptide 5 (Fig. S2†). A surrogate oxygenation ratio was calculated from MS intensities of fragment 79–90 since this fragment in particular was clearly detected in the MS analysis.18 In the comparison among functionalized peptides 2–6, there is no significant difference (Fig. 2b, left), although conjugate 5 tended to induce the oxygenation more. From the overall results, a clear difference in oxygenation site and activity was not observed among peptides 2–6.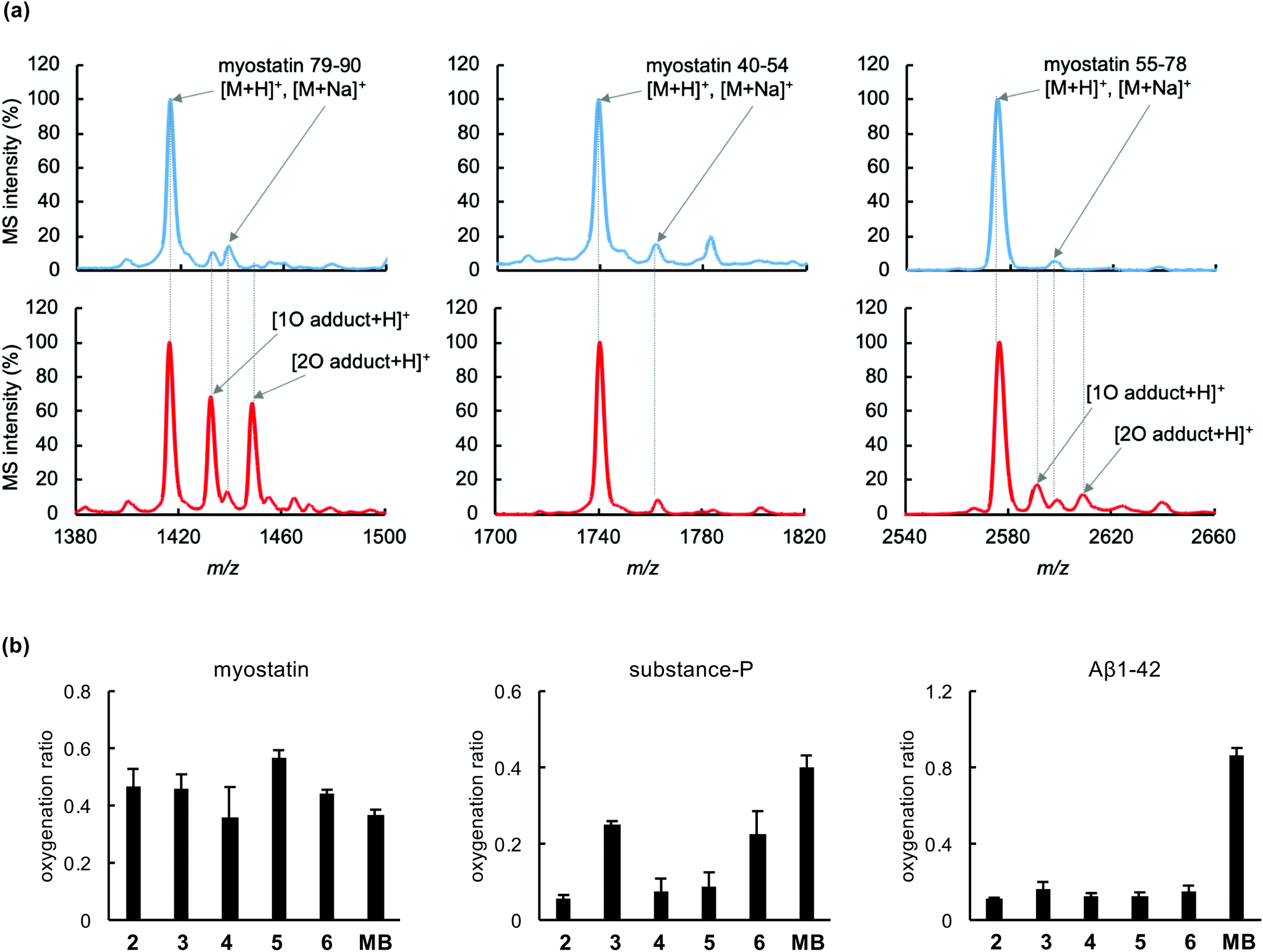 | ||
| Fig. 2 Photooxygenation of myostatin using functionalized peptides 2–6. Phosphate-buffered solution (10 mM, pH 7.4) containing myostatin (1 μM) and 2–6 (4 μM) was irradiated (λ = 730 nm) for 15 min, and analyzed by MALDI-TOF MS following fragmentation of the myostatin. (a) MS spectra of myostatin fragments 79–90, 40–54, and 55–78 which were obtained from myostatin before (blue) and after (red) oxygenation using 5. (b) Oxygenation ratio of myostatin, substance-P, and amyloid-β 1–42 (Aβ1–42) using peptide 2–6 or methylene blue (MB). The ratio was calculated from MS intensities of myostatin 79–90, substance-P, and Aβ1–42: oxygenation ratio = (sum of MS intensities of oxygenated species)/[(MS intensity of native species) + (sum of MS intensities of oxygenated species)]. | ||
The myostatin-selective oxygenation was evaluated, using substance-P and amyloid-β (Aβ) 1–42 as off-target models. Functionalized peptides (2–6) induced photooxygenation of myostatin as much as methylene blue, which was used as a non-selective control (Fig. 2b, left), while all peptides induced much less oxygenation of Aβ1–42 (Fig. 2b, right) than methylene blue. This selectivity could be attributed to myostatin affinity of the peptide part and on/off-switchable function of the catalyst part. In the case of substance-P, peptides 2, 4 and 5 induced less oxygenation than peptides 3 and 6, although the oxygenation ratios by all peptides were lower than that of methylene blue (Fig. 2b, center). The reason for this may be that peptides 2, 4, and 5 possess the photooxygenation catalyst in the middle range of the peptide chain (2: position 12, 4: position 8, and 5: position 16), preventing substance-P from approaching the catalyst section, while peptides 3 and 6 tether the catalyst in the N and C-terminus, respectively. In view of these results, peptides 2, 4, and 5 are promising candidates in terms of selectivity.
On/off-switch function
We attempted to compare the on/off-switch function of the catalyst moiety in peptides 2–6. In our previous study of peptide 2, we confirmed the switch function using the fluorescence-functionalized peptide 2′ (Fig. 3a).18 Comparing the structure of 2′ with 2, the bromine atom is absent, a fluorine is added to the B atom in place of the CF3 group, and an N,N-dimethylaniline moiety is substituted for the julolidine moiety (Fig. 3avs.1a). This derivative part of 2′ produced fluorescence when 2′ is bound to myostatin, resulting from the suppression of the bond rotation in the fluorescence part, while the catalyst part of 2 induces photooxygenation. We used newly prepared fluorescence-functionalized peptides (3′–6′) (Fig. 3a and Scheme S2†) corresponding to the photooxygenation-functionalized peptides (3–6).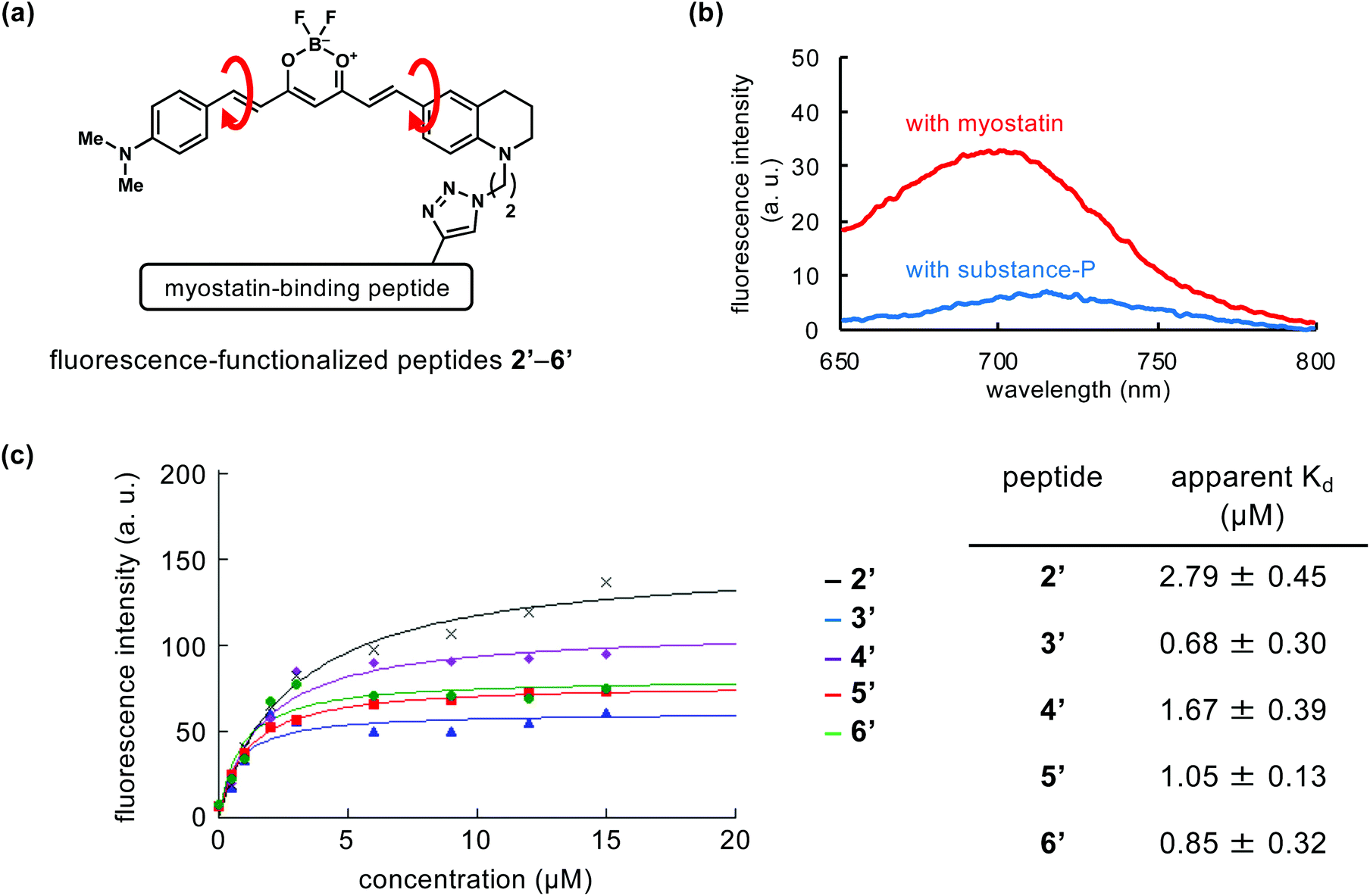 | ||
| Fig. 3 On/off-switch of fluorescence-functionalized peptides (2′–6′). (a) Structures of the fluorescent part in 2′–6′. (b) Fluorescence spectra of 5′ with myostatin (red) or substance-P (blue). Phosphate-buffered solution (10 mM, pH 7.4) containing 5′ (2 μM) with myostatin or substance-P (0.5 μM each) was used for the measurement (ex: 626 nm). (c) Concentration-dependency of fluorescence intensity of 2′–6′ (ex: 626 nm, em: 680 nm). The fitting curves and apparent Kd values were generated using KaleidaGraph 4.5 (Synergy Software, Reading, PA). | ||
Peptides 2′–6′ were added to the solution containing myostatin or substance-P, and the fluorescence spectra were measured. As a representative result, that in the use of peptide 5′ is shown in Fig. 3b. When 5′ exists with myostatin, fluorescence was observed around 700 nm. This indicates that peptide 5′ binds with myostatin and the bond rotation in the fluorescence section is restricted. In contrast, when peptide 5′ exists with substance-P as an off-target model, little fluorescence was observed. Peptides 2′–4′ and 6′ gave similar results (Fig. S3†). These indicate the on/off-switching functions in fluorescence-functionalized peptides (2′–6′). Considering from this, a similar switch system could behave in photooxygenation-functionalized peptides (2–6), and contribute to the myostatin-selective photooxygenation. On the other hand, in the photooxygenation study, peptides 3 and 6, which have the catalyst at the N and C-terminus, respectively, induced the photooxygenation of substance-P (Fig. 2b, center), while the corresponding peptides (3′ and 6′) did not produce fluorescence (Fig. S3†). Some level of switch function could work in 3 and 6, but the function might be not enough to suppress the photooxygenation of substance-P. Because the photooxygenation is irreversible, the changes in the photooxygenation study may be more clearly detected than those in the fluorescence study.
The fluorescence intensity was plotted as a function of the concentration of each peptide (2′–6′), and apparent Kd values of peptides 2′–6′ were calculated based on the fitting curves (Fig. 3c). The Kd values of peptides 3′ and 6′ (Kd = 0.68 ± 0.30 and 0.85 ± 0.32 μM, respectively) were lower than those of the other peptides (2′, 4′, and 5′). Because peptides 3′ and 6′ possess the fluorescence moiety at each terminus of the peptide chain (3′: position 1, 6′: C-terminus), the myostatin affinity of the original peptide might be unaffected by the additional moiety. Among the other peptides (2′, 4′, 5′) with fluorescence moiety in the middle range of peptide chain, 5′ (position 16) showed the lowest Kd value (1.05 ± 0.13 μM).
Inactivation
We compared inactivation of myostatin among photooxygenation-functionalized peptides 2–6. The biological activity of myostatin was evaluated by the previously established luciferase reporter assay using HEK293 cells.12–18 In this assay, the luciferase activity is correlated with the activity of myostatin. When myostatin was treated with peptide 2–6 (1 nM) and light, the luciferase activity was significantly lower than that in the absence of peptide (Fig. 4a, lane a vs. lanes b–f). This indicates that all peptides inhibit myostatin under irradiation. Peptide 5 with the catalyst at position 16 showed strong inhibitory activity (lane e), compared to the other peptides. This is consistent with the result that 5 shows slightly higher photooxygenation activity (Fig. 2b, left).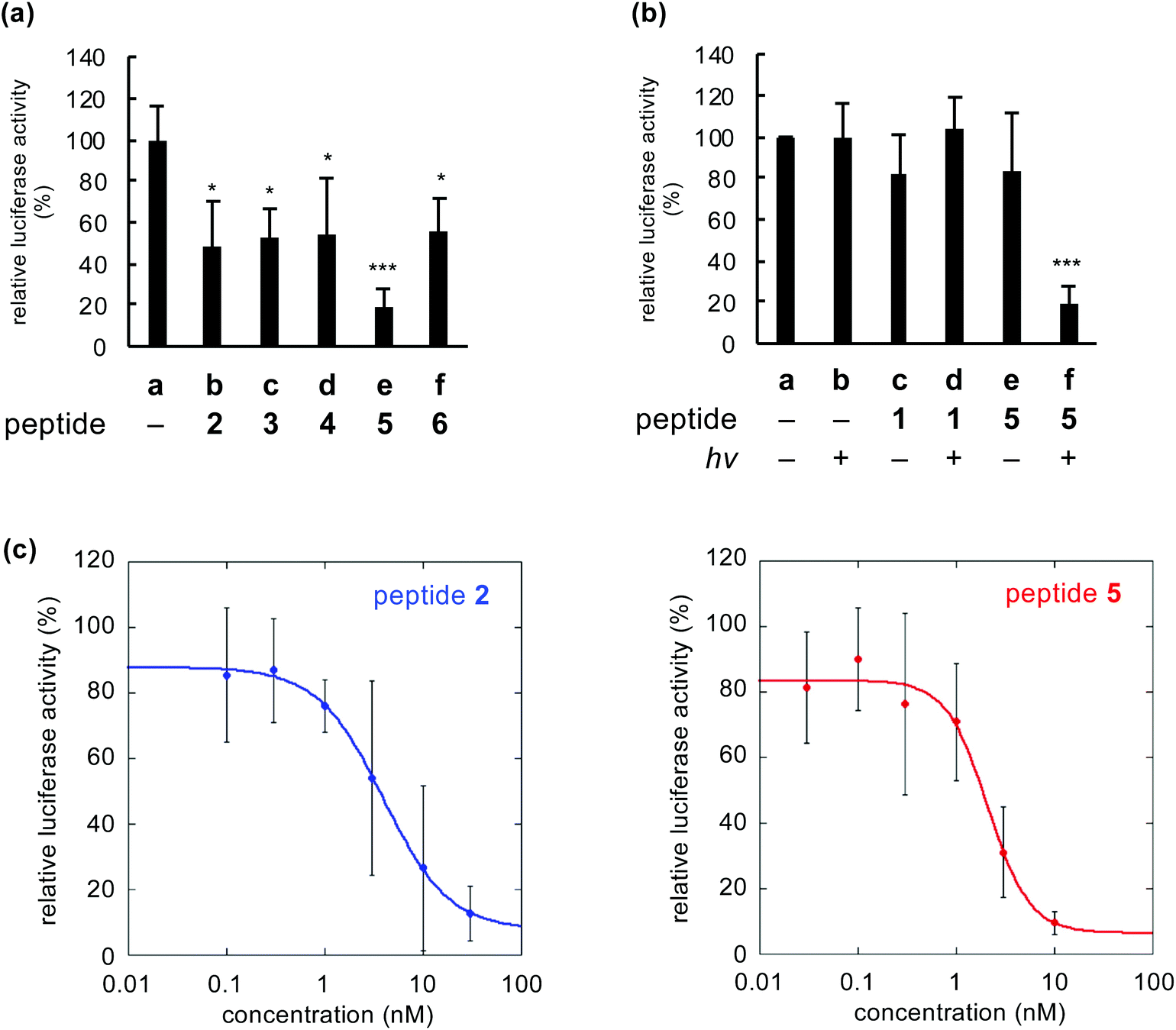 | ||
| Fig. 4 Inhibition of myostatin by photooxygenation using functionalized peptides 2–6. (a) Comparison of activities of myostatin treated with peptides 2–6 and light. (b) Activities of myostatin treated with peptide 1 or 5. (c) Dose–response curve of the inhibition using peptide 2 (left) or 5 (right). DMEM containing peptides 1–6 (1 nM) and myostatin (0.6 nM) was irradiated (λ = 730 nm) for 30 min, and added to the cell culture for the luciferase reporter assay (n = 3, mean ± s.d.; *p < 0.05, ***p < 0.001 vs. lane a by Tukey's test). The fitting curves were generated using KaleidaGraph 4.5. | ||
When myostatin was treated with peptide 5 and irradiated, the activity of myostatin was decreased to about 20% (Fig. 4b, lane f). In contrast, when myostatin was treated with 5 (lane e) or light (lane b), the activity was retained. These results indicated that myostatin was inactivated by photooxygenation using both peptide 5 and light. Meanwhile, the original myostatin-binding peptide (1) showed no inhibitory activity under same conditions (lanes c and d). The peptide concentration (1 nM) in the assay was too low, because IC50 value of 1 is 3.5 ± 0.3 μM.13 On the other hand, the IC50 value of peptide 5 with light was 2.1 ± 0.5 nM based on the dose–response curve (Fig. 4c, right). This suggests that the inhibitory effect of 5 was more than 1500-fold greater than that of the original peptide (1) due to the irreversible and catalytic inactivation. Moreover, the IC50 value of 5 was lower than that of previously reported 2 (IC50 = 3.9 ± 0.4 nM: Fig. 4c, left). Thus, peptide 5 is the promising functionalized peptide for the inhibition of myostatin by photooxygenation. Additionally, the IC50 value of 5 was equivalent to that of the prodomain protein which is an endogenous protein inhibitor (a 243-mer dimer, IC50 = 2.5 ± 0.3 nM)†. This suggests that the even mid-sized peptide 5 (a 23-mer) could realize the comparable inhibitory efficacy to such a large natural protein inhibitor by the photooxygenation strategy.
Phototoxicity
The phototoxicity of peptides 2–6 was evaluated. HEK293 cells were treated with peptide 2–6 or methylene blue as a control (3 μM each) under irradiation. After 2 days, the cell viability was measured using WST-1. In the absence of irradiation, the cell viabilities did not decrease in all cases, indicating that these specific compounds were not toxic under non-irradiated conditions (Fig. 5, black bars). The cell viability in irradiated methylene blue was decreased to 0.4, while the viabilities in peptides 2–6 were maintained even under irradiation (Fig. 5, white bars). These results demonstrated that peptides 2–6 were not subject to phototoxicity. Probably, this can be attributed to the target-selectivity resulting from both the myostatin-binding peptide and the on/off-switchable catalyst.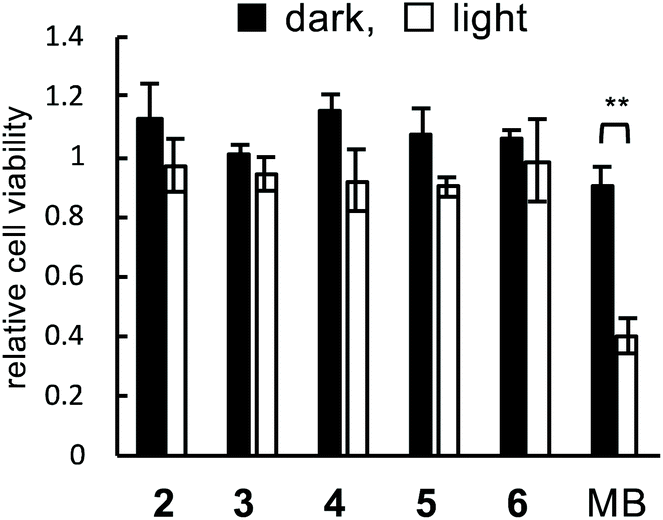 | ||
| Fig. 5 Phototoxicity of functionalized peptides 2–6. The cells treated with 2–6 or methylene blue (MB) (3 μM each) under irradiated (λ = 730 nm, 15 min: white bars) or non-irradiated (black bars) conditions (n = 3, mean ± s.d.; **p < 0.01 in the indicated pair by Student's t-test). | ||
Experimental
General procedures
Myostatin was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). Substance-P and Aβ1–42 isopeptide were purchased from Peptide Institute, Inc. (Osaka, Japan). Aβ1–42 isopeptide, which contains an ester structure between the Gly25–Ser26 sequence, converts to intact Aβ1–42 in situ at neutral pH through an O-to-N intramolecular rapid acyl migration (t1/2 ∼ 10 s).23,24 Methylene blue was purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Other reagents and solvents were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan), Nacalai Tesque, Inc., Peptide Institute, Inc., Sigma-Aldrich Co. LLC (St Louis, MO, USA), Tokyo Chemical Industries Co., Ltd (Tokyo, Japan), Wako Pure Chemical Industries, Ltd (Osaka, Japan), and Watanabe Chemical Industries, Ltd (Hiroshima, Japan). All chemicals were used as received.Fmoc-based SPPS was performed following the protocol of a peptide synthesizer Prelude (Gyros Protein Technologies AB, Uppsala, Sweden) on an Fmoc-NH SAL resin (0.37 mmol g−1). Analytical HPLC was carried out on a reverse-phase column (Nacalai Tesque COSMOSIL Protein-R or 5C18-AR-II, 4.6ID × 150 mm) using a linear gradient of CH3CN (5–85%, 40 min) in 0.1% aqueous trifluoroacetic acid (TFA) with a flow rate of 1.0 mL min−1, and detected at UV 230 or 200–800 nm. Preparative HPLC was carried out on a reverse-phase column (Waters SunFire Prep C18 OBD, 19ID × 150 mm) using a linear gradient of 0.1% TFA/CH3CN in 0.1% aqueous TFA with a flow rate of 5.0 mL min−1, and detected by absorbance at 230, 620, or 700 nm. MALDI-TOF MS spectra were recorded on Shimadzu Biotech AXIMA Assurance using α-cyano-4-hydroxy cinnamic acid as a matrix. HRMS spectra were recorded on Waters MICRO MASS LCT premier.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 (11 mM, 60 μL, 1.1 eq.) and L-ascorbic acid (120 mM, 120 μL, 24 eq.) in DMF/MeOH (1:1 v:v, 600 μL), tetrakis(acetonitrile)copper(I) hexafluorophosphate (4.09 mg, excess) was added. The solution was stirred at room temperature (RT) for 30 min then purified by preparative RP-HPLC in a 0.1% aqueous TFA–CH3CN system to obtain peptide 3 as a TFA salt (dark blue amorphus, 0.5 mg, 18%).
18 (11 mM, 60 μL, 1.1 eq.) and L-ascorbic acid (120 mM, 120 μL, 24 eq.) in DMF/MeOH (1:1 v:v, 600 μL), tetrakis(acetonitrile)copper(I) hexafluorophosphate (4.09 mg, excess) was added. The solution was stirred at room temperature (RT) for 30 min then purified by preparative RP-HPLC in a 0.1% aqueous TFA–CH3CN system to obtain peptide 3 as a TFA salt (dark blue amorphus, 0.5 mg, 18%).HRMS m/z [M + H]+ found 3463.8345 (calcd. for C156H250N45O34BrBF4 3463.8349); purity >95% (HPLC, tR = 21.4 min).
HRMS m/z [M + H]+ found 3585.8945 (calcd. for C167H256N45O33BrBF4 3585.8950); purity >95% (HPLC, tR = 20.1 min).
HRMS m/z [M + H]+ found 3544.8679 (calcd. for C165H253N44O33BrBF4 3544.8684); purity >95% (HPLC, tR = 20.7 min).
HRMS m/z [M + H]+ found 3672.9241 (calcd. for C170H261N46O35BrBF4 3672.9270); purity >95% (HPLC, tR = 20.4 min).
:5:2.5 v:v:v) for 90 min, filtered, concentrated, precipitated with diethyl ether, and purified by preparative RP-HPLC in a 0.1% aqueous TFA–CH3CN system to obtain peptide 7 as a TFA salt (white amorphous, 14 mg, 10%).LRMS(MALDI) m/z [M + H]+ found 2791.5 (calcd. for C125H218N40O32 2791.7); purity >95% (HPLC, tR = 17.0 min).
LRMS(MALDI) m/z [M + H]+ found 2912.7 (calcd for C136H224N40O31 2913.7); purity >95% (HPLC, tR = 21.5 min).
LRMS(MALDI) m/z [M + H]+ found 2872.7 (calcd. for C134H221N39O31 2872.7); purity >95% (HPLC, tR = 18.5 min).
LRMS(MALDI) m/z [M + H]+ found 3001.2 (calcd. for C139H229N41O33 3000.8); purity >95% (HPLC, tR = 18.2 min).
18 (11 mM, 30 μL, 1.1 eq.) afforded 3′ as a dark violet amorphous TFA salt (0.2 mg, 16%).HRMS m/z [M + H]+ found 3283.9072 (calcd. for C151H247N45O34BF2 3283.9043); purity >95% (HPLC, tR = 23.0 min).
HRMS m/z [M + H]+ found 3405.9568 (calcd. for C162H253N45O33BF2 3405.9564); purity >95% (HPLC, tR = 23.9 min).
HRMS m/z [M + H]+ found 3364.9285 (calcd. for C160H250N44O33BF2 3364.9298); purity >95% (HPLC, tR = 24.0 min).
HRMS m/z [M + H]+ found 3492.9895 (calcd. for C165H258N46O35BF2 3492.9884); purity >95% (HPLC, tR = 24.6 min).
Absorbance and fluorescence measurements
Absorbance measurements were performed using a Shimadzu UV-1700 UV-visible spectrophotometer with a rectangular quartz cell (10 mm path-length). Fluorescence measurement was performed using a Hitachi F-7100 fluorescence spectrophotometer with a rectangular quartz cell (5 mm path-length). The background was subtracted from each fluorescence spectrum.Photooxygenation
The photooxygenation study was similar to that described by Okamoto et al.18 A stock solution of myostatin (8 μM in 1 mM aqueous HCl) was diluted with 1 mM aqueous HCl and 20 mM phosphate buffer (pH 7.4) to produce a 1 μM solution of myostatin (10 mM phosphate, pH 7.4). A stock solution of substance-P (100 μM in water) and Aβ1–42 isopeptide (250 μM in 0.1% aqueous TFA, ultracentrifuged)24 were diluted with water and 20 mM phosphate buffer (pH 7.4) to 20 μM of each peptide (10 mM phosphate, pH 7.4). To each solution, functionalized peptide 2–6 or methylene blue (50 μM in CH3CN/DMSO = 9/1) was added to the final concentration (4 or 12 μM, respectively) for the oxygenation studies. The mixture was irradiated with LED ISLM-150 × 150-FF (λ = 730 nm, 14 mW: CCS Inc., Kyoto, Japan) at RT or 37 °C at approximately 3–5 cm from samples. The reaction mixtures were reduced with DTT at a final concentration of 50 mM at 37 °C for 0.5 h, digested with endoproteinase Lys-C (1/20 of protein by weight) at 37 °C for 2.5 h, de-salted with ZipTip U-C18 (Millipore Co.), and analysed using MALDI-TOF MS. For the comparison of oxygenation ratios based on the MS intensities, a comparative sample set was analysed at one time, and the analytical conditions were standardized within the comparative sample set, for example, matrix substance, matrix/sample concentrations, solvent, laser power and laser-shot number. For cell-based assay, the 10 mM phosphate-buffered solution containing myostation (1 μM) and peptide 2–6 was diluted with serum-free DMEM (Dulbecco's modified Eagle's medium) to 0.6 nM myostatin, irradiated (730 nm, 30 min), and then added to the cell culture.Luciferase-reporter assay
The cell-culture and luciferase-reporter assay were performed as described by Okamoto et al.18 HEK293 cells were cultured in DMEM (10% FBS, non-essential amino acids) at 37 °C under 5% CO2. The cells were seeded at 2.0 × 104 cells per well in the 96-well plates. After 24 h, transfection of reporter (pGL4.48[luc2P/SBE/Hygro], Promega) and control (pGL4.74[hRluc/TK], Promega) vectors was carried out using FuGENE HD (Promega) for 24 h. After the culture medium was exchanged to serum-free DMEM, the cells were incubated for 8 h. After the medium was exchanged to each sample solution (0.6 nM myostatin), the cells were incubated for 4 h then washed with PBS. The preparation of cell lysates and the measurement of luciferase reporter activities were conducted according to manufacturer's protocol of a Dual-Luciferase Reporter Assay System (Promega). Each experiment was carried out in triplicate. Values represent means ± s.d. (n = 3).Phototoxicity assay
HEK293 cells, which were cultured in DMEM (10% FBS, non-essential amino acids) at 37 °C under 5% CO2, were seeded at 2.0 × 104 cells per well in the 96-well plates. After 48 h, the culture medium was exchanged with serum-free DMEM containing peptide 2–6 or methylene blue (3 μM each). The cells were irradiated with LED (λ = 730 nm, 15 min), and then incubated for 24 h. Cell viability was determined using WST-1 (Roche Ltd).Conclusions
Based on the conjugation of myostatin-binding peptide (1) and on/off-switchable photooxygenation catalyst, a series of photooxygenation-functionalized peptides (2–6) were prepared, attached the catalyst moiety at different positions on the peptide chain. These peptides oxygenated and inactivated myostatin under irradiation. After comparison among them, peptide 5 with the catalyst part at position 16 in the peptide chain showed preferable properties in myostatin-selectivity and inactivation efficacy. Moreover, the peptides are excited by NIR light and exhibit low phototoxicity. Therefore, these functionalized peptides can be biocompatible inhibitors, which very efficiently inactivate myostatin through irreversible and catalytic photooxygenation. These peptide inhibitors would provide a precedented strategy for myostatin-targeting therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (C) 19K07001 (A. T.) and (B) 19H03356 (Y. H.), The Noguchi Institute (A. T.), The Takeda Science Foundation (A. T), The Uehara Memorial Foundation (A. T.), and Intramural Research Grant (2-5) for Neurological and Psychiatric Disorders of NCNP.Notes and references
- A. C. McPherron, A. M. Lawler and S.-J. Lee, Nature, 1997, 387, 83–90 CrossRef CAS.
- A. C. McPherron and S. J. Lee, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12457–12461 CrossRef CAS.
- N. M. Wolfman, A. C. McPherron, W. N. Pappano, M. V. Davies, K. Song, K. N. Tomkinson, J. F. Wright, L. Zhao, S. M. Sebald, D. S. Greenspan and S.-J. Lee, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15842–15846 CrossRef CAS.
- S. J. Lee and A. C. McPherron, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9306–9311 CrossRef CAS.
- T. A. Zimmers, M. V. Davies, L. G. Koniaris, P. Haynes, A. F. Esquela, K. N. Tomkinson, A. C. McPherron, N. M. Wolfman and S.-J. Lee, Science, 2002, 296, 1486–1488 CrossRef CAS.
- S. Bogdanovich, T. O. Krag, E. R. Barton, L. D. Morris, L. A. Whittemore, R. S. Ahima and T. S. Khurana, Nature, 2002, 420, 418–421 CrossRef CAS.
- X. Zhou, J. L. Wang, J. Lu, Y. Song, K. S. Kwak, Q. Jiao, R. Rosenfeld, Q. Chen, T. Boone, W. S. Simonet, D. L. Lacey, A. L. Goldberg and H. Q. Han, Cell, 2010, 142, 531–543 CrossRef CAS.
- E. Latres, J. Pangilinan, L. Miloscio, R. Bauerlein, E. Na, T. B. Potocky, Y. Huang, M. Eckersdorff, A. Rafique, J. Mastaitis, C. Lin, A. J. Murphy, G. D. Yancopoulos, J. Gromada and T. Stitt, Skeletal Muscle, 2015, 5, 34 CrossRef.
- C. Becker, S. R. Lord, S. A. Studenski, S. J. Warden, R. A. Fielding, C. P. Recknor, M. C. Hochberg, S. L. Ferrari, H. Blain, E. F. Binder, Y. Rolland, S. Poiraudeau, C. T. Benson, S. L. Myers, L. Hu, Q. I. Ahmad, K. R. Pacuch, E. V. Gomez, O. Benichou and STEADY Group, Lancet Diabetes Endocrinol., 2015, 3, 948–957 CrossRef CAS.
- C. Campbell, H. J. McMillan, J. K. Mah, M. Tarnopolsky, K. Selby, T. McClure, D. M. Wilson, M. L. Sherman, D. Escolar and K. M. Attie, Muscle Nerve, 2017, 55, 458–464 CrossRef CAS.
- Y. Ohsawa, K. Takayama, S. Nishimatsu, T. Okada, M. Fujino, Y. Fukai, T. Murakami, H. Hagiwara, F. Itoh, K. Tsuchida, Y. Hayashi and Y. Sunada, PLoS One, 2015, 10, e0133713 CrossRef.
- K. Takayama, Y. Noguchi, S. Aoki, S. Takayama, M. Yoshida, T. Asari, F. Yakushiji, S. Nishimatsu, Y. Ohsawa, F. Itoh, Y. Negishi, Y. Sunada and Y. Hayashi, J. Med. Chem., 2015, 58, 1544–1549 CrossRef CAS.
- K. Takayama, A. Nakamura, C. Rentier, Y. Mino, T. Asari, Y. Saga, A. Taguchi, F. Yakushiji and Y. Hayashi, ChemMedChem, 2016, 11, 845–849 CrossRef CAS.
- T. Asari, K. Takayama, A. Nakamura, T. Shimada, A. Taguchi and Y. Hayashi, ACS Med. Chem. Lett., 2017, 8, 113–117 CrossRef CAS.
- K. Takayama, C. Rentier, T. Asari, A. Nakamura, Y. Saga, T. Shimada, K. Nirasawa, E. Sasaki, K. Muguruma, A. Taguchi, A. Taniguchi, Y. Negishi and Y. Hayashi, ACS Med. Chem. Lett., 2017, 8, 751–756 CrossRef CAS.
- C. Rentier, K. Takayama, M. Saitoh, A. Nakamura, H. Ikeyama, A. Taguchi, A. Taniguchi and Y. Hayashi, Bioorg. Med. Chem., 2019, 27, 1437–1443 CrossRef CAS.
- K. Takayama, T. Asari, M. Saitoh, K. Nirasawa, E. Sasaki, Y. Roppongi, A. Nakamura, Y. Saga, T. Shimada, H. Ikeyama, A. Taguchi, A. Taniguchi, Y. Negishi and Y. Hayashi, ACS Med. Chem. Lett., 2019, 10, 985–990 CrossRef CAS.
- H. Okamoto, A. Taniguchi, S. Usami, A. Taguchi, K. Takayama and Y. Hayashi, Chem. Commun., 2019, 55, 9108–9111 RSC.
- J. Ni, A. Taniguchi, S. Ozawa, Y. Hori, Y. Kuninobu, T. Saito, T. C. Saido, T. Tomita, Y. Sohma and M. Kanai, Chem, 2018, 4, 807–820 CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS.
- Y. Sohma, M. Sasaki, Y. Hayashi, T. Kimura and Y. Kiso, Chem. Commun., 2004, 124–125 RSC.
- A. Taniguchi, Y. Sohma, Y. Hirayama, H. Mukai, T. Kimura, Y. Hayashi, K. Matsuzaki and Y. Kiso, ChemBioChem, 2009, 10, 710–715 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob02042g |
| ‡ Present address: Department of Environmental Biochemistry, Kyoto Pharmaceutical University, 5 Misasaginakauchi-cho, Yamashina, Kyoto 607-8414, Japan. |
| § The value was calculated based on the dose–response curve obtained in our group with the reported protocol.13 |
| ¶ Four equivalents of peptides 2–6 were used in the photooxygenation of myostatin to enhance the reaction rate. A catalytic amount of the peptide induced the photooxygenation with long-time irradiation in our previous study.18 |
| This journal is © The Royal Society of Chemistry 2021 |