
Using experimental and computational approaches to probe an unusual carbon–carbon bond cleavage observed in the synthesis of benzimidazole N-oxides†
Fabrizio
Politano
 ab,
Arturo León
Sandoval
a,
Jorge G.
Uranga
b,
Elba I.
Buján
b and
Nicholas E.
Leadbeater
*a
ab,
Arturo León
Sandoval
a,
Jorge G.
Uranga
b,
Elba I.
Buján
b and
Nicholas E.
Leadbeater
*a
aDepartment of Chemistry, University of Connecticut, 55 North Eagleville Road, Storrs, Connecticut 06269, USA. E-mail: nicholas.leadbeater@uconn.edu
bInstituto de Investigaciones en Físico Química de Córdoba (INFIQC)-CONICET, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, X5000HUA, Córdoba, Argentina
First published on 6th November 2020
Abstract
Experimental and computational studies have been performed in order to investigate an unusual carbon–carbon bond cleavage that occurs in the preparation of certain benzimidazole N-oxides from anilines. The key factor determining the outcome of the reaction was found to be the substituents on the amine functionality of the aniline.
Introduction
The N-oxide and N-hydroxy derivatives of imidazole and benzimidazole are interesting families of compounds due their potential biological activity. They can act as herbicides, insecticides, and nematicides, as well as possessing bacteriostatic, anthelmintic, and coccidiostatic properties.1 Benzimidazole N-oxides stand out in particular as they show broad versatility in catalysis, synthesis, and drug development for human and veterinary medicine.2 They are potential agents against tuberculosis, malaria and some neglected tropical diseases, such as Chagas disease and leishmaniasis.3The potential application of benzimidazole N-oxides has been a motivating factor in our research group for the development of efficient routes for their preparation and we have developed both batch microwave-heated5 and conventionally-heated continuous-flow4 protocols (Scheme 1). These methodologies have several advantages from a green chemistry perspective compared with previously-reported methods.6–8 They comprise of a one-pot two-step process: a nucleophilic aromatic substitution reaction (SNAr) of different amines on 2-nitrochlorobenzene derivatives, followed by a cyclization reaction in basic media. The intermediate is not isolated; simple precipitation or liquid–liquid extraction is employed as work-up, and ethanol and water are used as solvents.
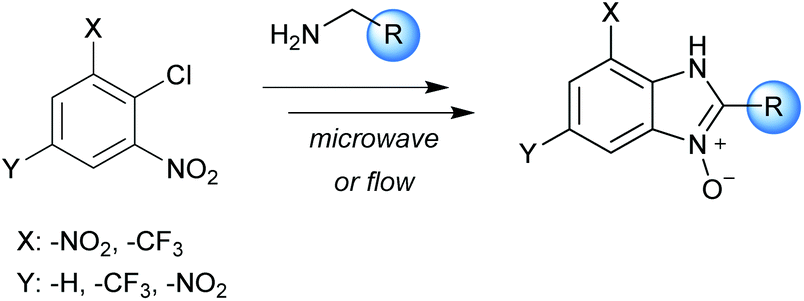 | ||
| Scheme 1 Preparation of benzimidazole N-oxides. | ||
Using our methodologies, we have prepared a library of over 40 benzimidazole-N-oxides. While most of the desired compounds were generated as expected, there were a few noteworthy exceptions. Thin-layer chromatography monitoring in these cases showed similar results to all the others, but 1H-NMR spectroscopic analysis showed an interesting anomaly. Fig. 1(I) shows the typical peak pattern over the aromatic region of the 1H-NMR spectrum of the benzimidazole N-oxide products (with -R being aliphatic), and Fig. 1(II) shows the 1H-NMR spectrum of the unexpected product, with a new signal in the aromatic region (peak d). This was observed in the reaction of 2-chloro-1,3-dinitrobenzene (1) with ethanolamine and with ethylenediamine. Instead of the expected products 4e and 4f, 7-nitro-1H-benzimidazole 3-oxide (5) was formed (Scheme 2), suggesting that a carbon–carbon bond cleavage occurs. While the reaction mechanism for the formation of benzimidazole N-oxides has been broadly studied,9–11 the only reported carbon–carbon bond cleavage reported for this class of reaction was due to a decarboxylation process.7,9 To our knowledge, there have not been any reports of the cleavage observed in our case. This motivated us to probe our observation in more detail.
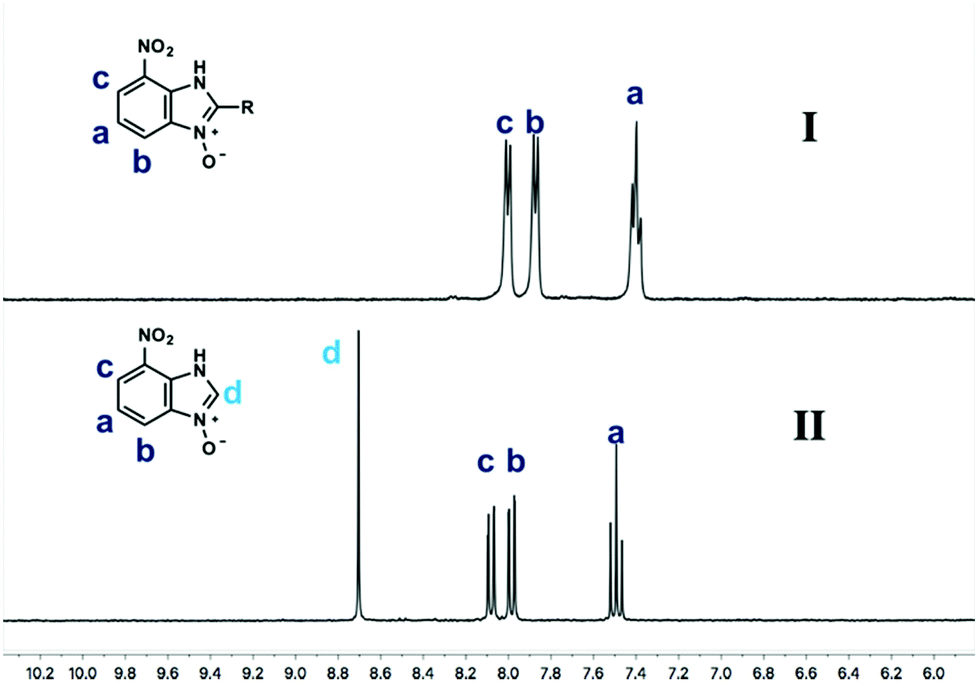 | ||
| Fig. 1 Peak pattern in the aromatic region of the 1H-NMR spectrum of benzimidazole-N-oxides: (I) typically; (II) in the case of the reaction of 2-chloro-1,3-dinitrobenzene (1) with ethanolamine and with ethylenediamine. | ||
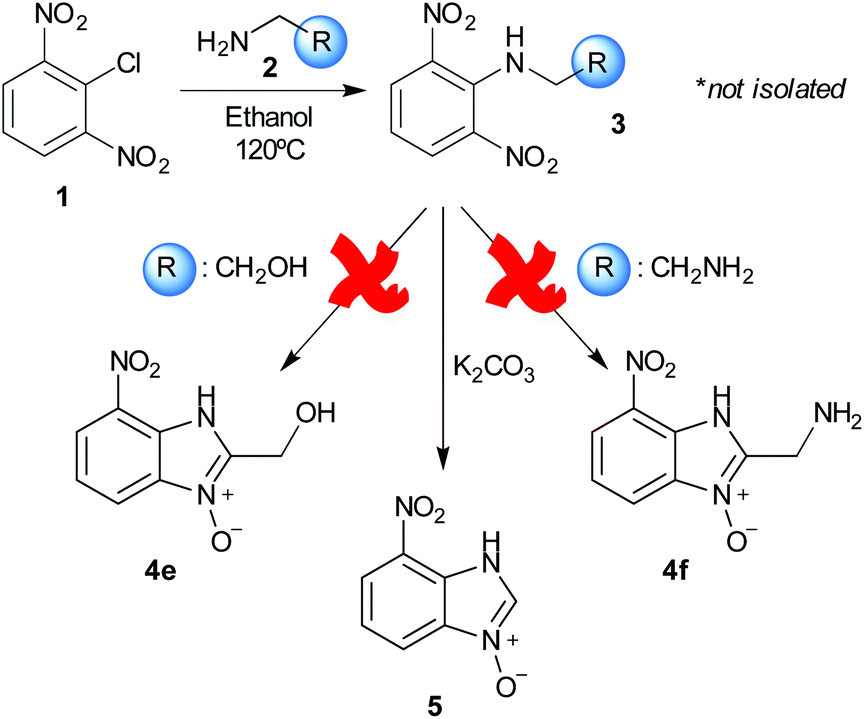 | ||
| Scheme 2 Products observed in the reaction of 1 with ethanolamine and ethylenediamine. | ||
Computational chemistry plays a very important role in the study of reaction mechanisms. The development of new computational methods, together with advances in computational power allows for the reduction of calculation times to a reasonable length.12,13 In this regard, implementing a computational approach to help us explain the divergent reaction pathway we observed seemed prudent. However, as in many other examples in the literature, product selectivity cannot be simply reduced to a choice between pathways with different activation energies.14–16 For instance, competitive reactions that contain the same or similar transition state (TS) could be governed by post-transition-state bifurcation.17 Solutions to these complex chemical problems are the focus of much current research and are resource-intensive. To this end, a simplified approach for the prediction of reaction selectivity would be highly attractive.
Here, we present results of our computational and experimental study of the anomalous C–C bond-cleavage observed. We bring insight to the reaction mechanism and establish the structural features that govern the divergent path leading to carbon–carbon bond cleavage.
Results and discussion
Experimental studies
We started our experimental study by revisiting our two methodologies for the preparation of benzimidazole-N-oxides. We adapted the reaction of 1 with a series of aliphatic amines (2) that we had initially performed under continuous-flow conditions, to the “one-pot two-step” batch approach we recently reported when using benzylamines as the amine component.4 The reactions were performed employing 1 equiv. of 2-chloro-1,3-dinitrobenzene (1) and 2 equiv. of the desired amine (2) in 3 mL of ethanol, heating at 120 °C for 20 min. Following this step, 2 mL of a 0.5 M aqueous solution of K2CO3 was added, and the heating step repeated. The products obtained are shown in Table 1.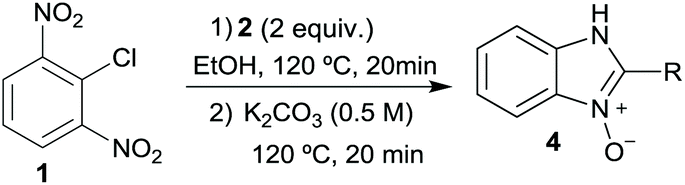
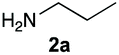
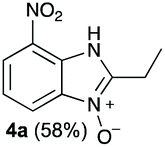

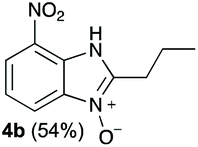
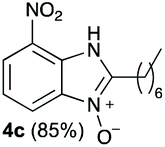
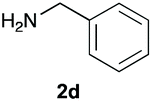
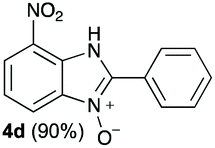
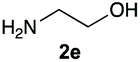
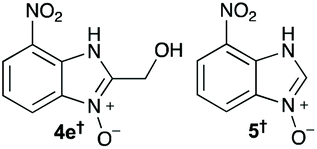
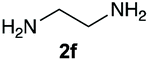
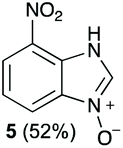
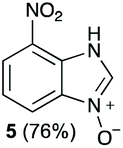
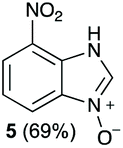
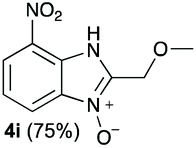

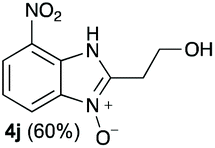
The reaction of 1 with propylamine (2a) and with butylamine (2b) led to the expected benzimidazole-N-oxide products, 4a and 4b, with retention of the alkyl chain (Table 1, entries 1 and 2). The reaction with an amine bearing a longer alkyl chain (octylamine, 2c) and with an aromatic ring (benzylamine, 2d) behaved similarly (entries 3 and 4). However, when the reaction was performed with ethanolamine (2e) and ethylenediamine (2f), the amines that motivated this study, we saw the divergence in product outcome that was first observed in our continuous-flow approach (entries 5 and 6). In the case of the reaction of 1 with 2e, we observed a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 mixture of the expected product with the alkyl chain intact (4e) and that with loss of the alkyl chain (5) and, when using 2f, product 5 was formed exclusively.
:60 mixture of the expected product with the alkyl chain intact (4e) and that with loss of the alkyl chain (5) and, when using 2f, product 5 was formed exclusively.
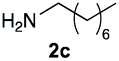
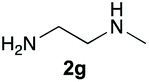
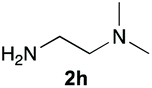
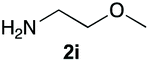
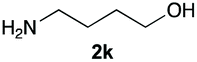
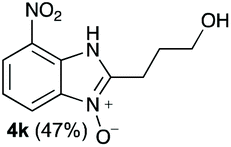
Upon considering the molecular structures of 2e and 2f, we posited that they could participate in hydrogen-bonding interactions and that this could be tied into the mechanism of alkyl group cleavage. To test this hypothesis, we performed the reaction of 1 with the corresponding N- and O-methylated amine analogs, N-methylethylenediamine (2g), N,N-dimethylethylenediamine (2h), and 2-methoxyethylamine (2i). With both 2g and 2h, the dealkylated product 5 was formed, but when using 2i the alkyl group remained intact (entries 7–9). This suggests that hydrogen-bonding is not a key factor. Next we considered the distance of the heteroatom with respect to the nitrogen atom involved in the cyclization reaction, to see if that may play a role in the dealkylation process. We screened 3-amino-1-propanol (2j, entry 10) and 4-amino-1-butanol (2k, entry 11) as amine substrates. In each case the product with retention of the alkyl group was observed. Thus, when the heteroatom is further away from the benzimidazole ring, the dealkylation mechanism is not operational, suggesting that a possible intramolecular process could be occurring.
Computational studies
Alongside our experimental work, we carried out a series of computational studies in order to probe further the origin of product selectivity in the reaction of 1 with amines. We performed DFT calculations at the B3LYP/6-31+G* level using Gaussian 09.18 Each structure was found by a manual conformational search based on chemical interactions considering all the possible orientations of the R group. In the case of the bicarbonate anion, different intermolecular interactions were considered and the most stable ones were selected for analysis. Stationary points were characterized as a minimum in the potential energy surface through normal mode analysis. Transition states were characterized as first-order saddle points and their connection with reagent and product were confirmed through IRC calculations. Solvent effects were considered in implicit form using the integral equation formalism variant (IEFPCM) method19 and ethanol (ε = 24.852) was used as a continuum solvent model.We used as our starting point the previously reported mechanism for the base-mediated cyclization of nitroanilines to yield benzimidazole N-oxides (Scheme 3).10 The first step involves the generation of intermediate II from aniline I. The second step is the reduction of a nitro group to a nitroso functionality,20,21 followed by the cyclization of the o-nitrosoimine (III) to give intermediate IV.9,22 A similar cyclization mechanism has also been reported recently for the formation of benzoxazoles.23 Intermediate IV bears an sp3 carbon in the nascent imidazole ring, bonded to a hydrogen and the substituent originating from the parent amine. We focused attention on this intermediate because we posited that it was at this stage of the mechanism that the divergence in pathway would occur; the next step being either C–H bond-cleavage to give V (path A) or C–C cleavage giving VI (path B).
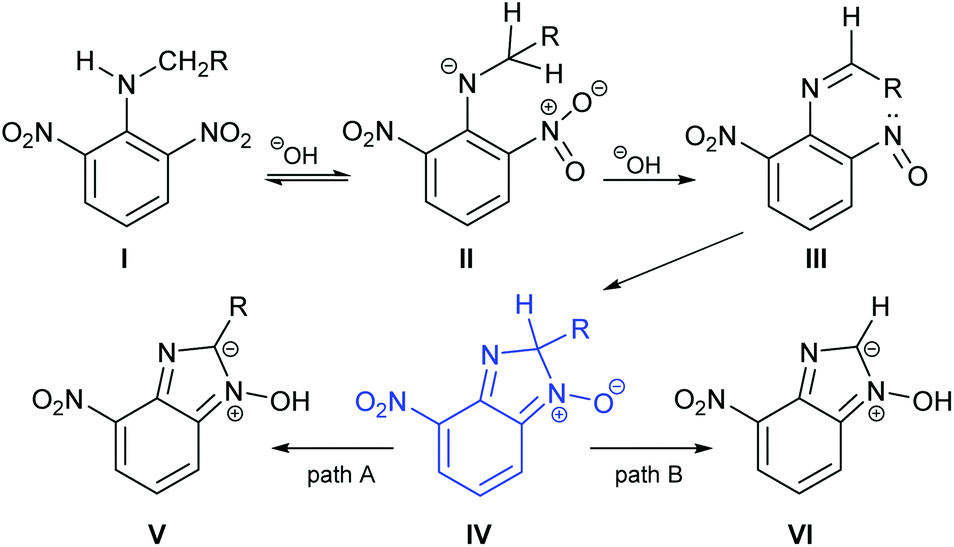 | ||
| Scheme 3 Mechanism for formation of benzimidazole N-oxides highlighting paths A & B. | ||
Our objective was to analyse the energy profiles of paths A and B for each of the amines used in our allied experimental studies. With the knowledge that the rate determining step of the cyclization process occurs prior to fragmentation of intermediate IV,7,8,10 we assumed that paths A and B were directed by thermodynamic factors.
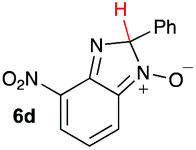
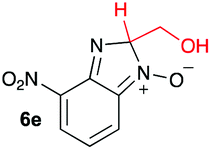
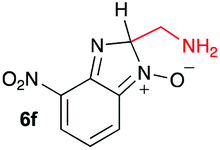
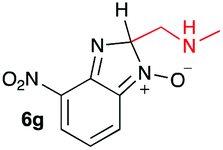
Using intermediate IV as our starting point, we calculated fragmentation energies for path A and path B. In our first pass, we decided to use a simplified approach for our calculations that does not take into account the effect of base on the reaction. For path A, we modelled unimolecular proton loss from IV as the fragmentation reaction, and for path B we modelled unimolecular C–C bond-cleavage. Our results are summarized in Table 2 and show the energy values calculated for C–H and C–C bond-cleavages, taking intermediate IV as the reference and zero energy. Values shown correspond to the energy difference between intermediate IV and the benzimidazole-N-oxide product 4, while the sign indicates if the processes are endothermic (positive sign) or exothermic (negative sign). This formalism was used considering that intermediate IV is expected to be formed independently of the final product. The selectivity towards C–C or C–H bond cleavage is posited to be dependent on the kinetic and thermochemistry of each reaction once IV is generated. For C–H bond-cleavage (path A), there was no notable variation in energy across the different substrates studied, with the exception of the case of 6e, the intermediate arising from the reaction of 1 with ethanolamine (2e) where hydrogen-bonded stability of the cleavage product is possible (Table 2, entry 5). For C–C bond-cleavage (path B) the substituent originating from the parent amine did play a significant role in the process. This is not unexpected, given the anticipated unstable nature of the carbocation intermediate formed in most cases. To probe this further, we inspected key points of the reaction coordinate for path B involving intermediates 6a–6k. The localization of the positive charge on a primary carbon did indeed lead generally to energy profiles disfavouring this path; the energy to break the C–C bond being too high (entries 1–3 and 9–11). As an aside, a rearrangement from a primary to a secondary carbocation is possible along the reaction coordinate in the case of the fragmentation products derived from 6b, 6c, 6j, and 6k (see C–C fragmentation products in ESI†), but this is not sufficient to change the outcome of the reaction from C–H to C–C bond-scission. For 6d, the formation of a phenyl cation disfavours the C–C bond-fragmentation pathway even more (entry 4).
| Entry | Intermediate IV | Energya (kcal mol−1) | |
|---|---|---|---|
| C–H cleavage | C–C cleavage | ||
| a The thermochemistry associated with alternative fragmentation pathways (path A and path B) was calculated assuming that the proton generated in the process subsequently protonates one solvent molecule (ethanol). | |||
| 1 |
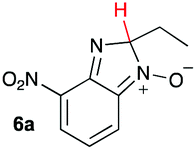
|
16.6 | 62.7 |
| 2 |
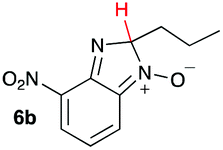
|
16.6 | 51.2 |
| 3 |
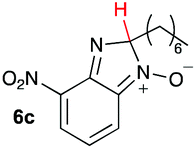
|
16.4 | 37.7 |
| 4 |
|
11.8 | 85.5 |
| 5 |
|
−0.4 | −9.9 |
| 6 |
|
15.8 | −5.2 |
| 7 |
|
15.9 | −7.2 |
| 8 |
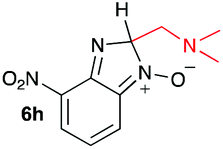
|
15.5 | −4.4 |
| 9 |
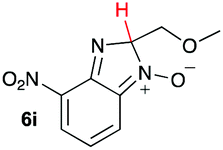
|
15.6 | 29.9 |
| 10 |
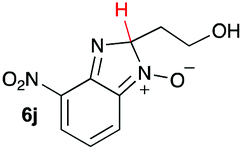
|
10.9 | 21.1 |
| 11 |
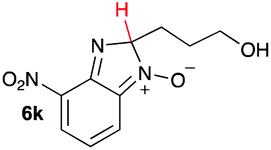
|
15.1 | 20.6 |
When considering the C–C bond-cleavage of intermediates 6e, 6f, and 6g (entries 5–7) both the transition state and product were found to be significantly stabilized. The transition states showed a key hydrogen-bonding interaction between the proton of the heteroatom and the oxygen atom of the benzimidazole-N-oxide moiety; this reducing the energy barrier for the C–C bond-cleavage step (Fig. 2). Further analysis showed that scission of the C–C bond occurs in a concerted fashion with proton transfer between the heteroatom of the substituent and the oxygen of the heterocycle core. As a result, the products of the fragmentation are neutral species: formaldehyde, methanimine and N-methyl-methanimine, for 6e, 6f, and 6g respectively (Fig. 2a, b and c). For 6h (entry 8), a stabilization of the transition state was observed due to the assistance of the N,N-dimethyl group in the incipient formation of the carbocation intermediate; positive charge is shared between the carbon atom and the neighbouring nitrogen via resonance (Fig. 2d).
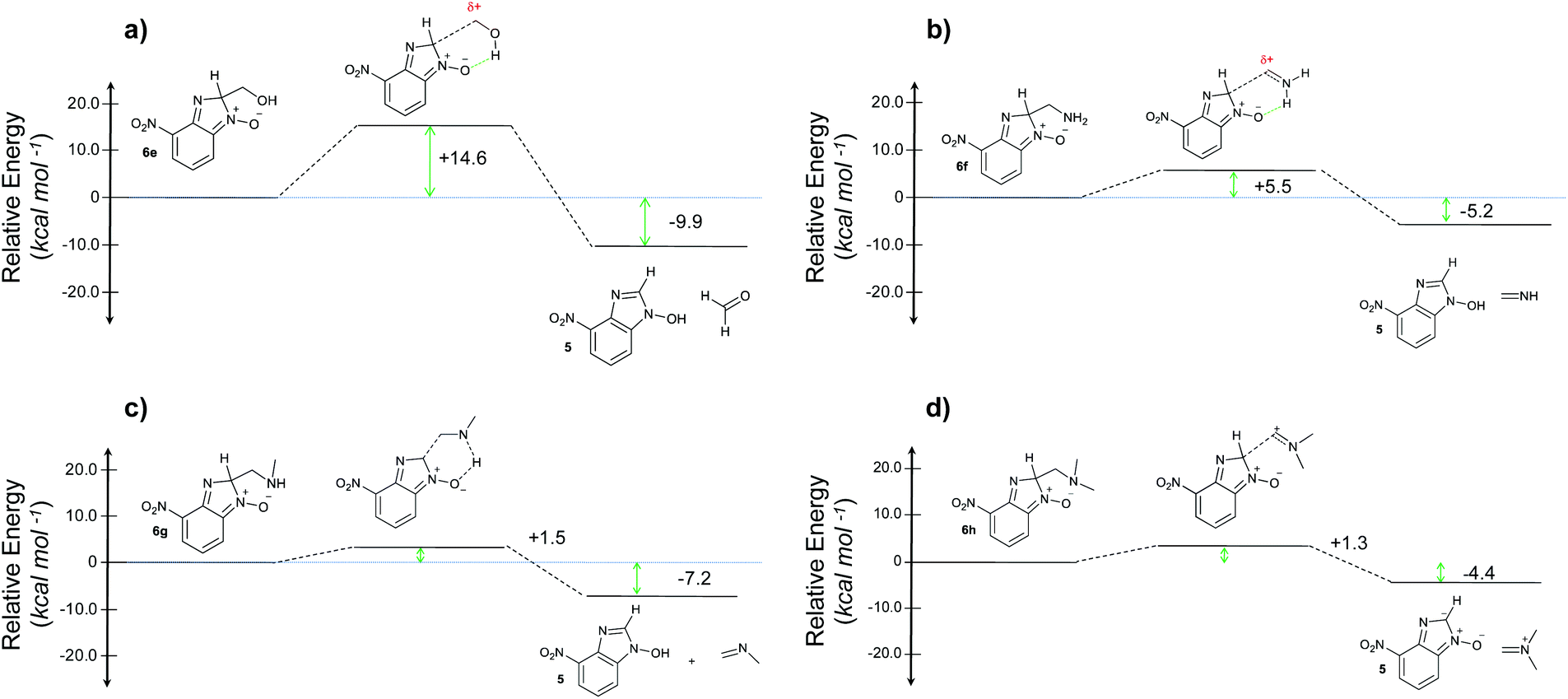 | ||
| Fig. 2 Energy profiles for the reaction with (a) ethanolamine, (b) ethylenediamine, (c) N-methylethylenediamine, and (d) N,N-dimethylethylenediamine. | ||
Our computational study, as well as the experimental results, show that a heteroatom (N or O) in a β-position to the nitrogen of the parent amine plays a fundamental role in the cleavage of the C–C bond in path B. Bringing together what we had gleaned thus far, we posited that the C–C bond scission process could follow at least three different pathways, as shown in Fig. 3. For substituents with hydrogen-containing heteroatoms (N or O) in a β-position to the nitrogen of the parent amine [–OH (6e), –NH2 (6f) –NH(CH3) (6g)], there is the potential for interaction of these hydrogen atoms with the negative charge of the N-oxide oxygen atom, leading to weakening of the C–C bond. This assistance in C–C bond-cleavage occurs through a six-membered transition state where bond breaking and proton transfer is a concerted process (Fig. 3, blue pathway). Since this fragmentation process leads to neutral molecules, this route is energetically favourable. When the heteroatom is not bound to a hydrogen atom but instead is an N,N-dimethyl entity (6h), the methyl groups can contribute to the stability of positive charge on the nitrogen and contribute to the change in hybridization from sp3 to sp2 requisite for the formation of the N,N-dimethylmethyleneammonium cation (Fig. 3, purple pathway). This stabilization appears to be very significant as evidenced by the observation that the transition-state barrier height is the lowest of all the cases studied (Fig. 2d). However, since the fragment generated is cationic, the process is less favoured than that for 6e, 6f, and 6g. Of note is that this behaviour is only observed for substrates like 6h, bearing a tertiary amine moiety, since a similar stabilization is not seen for 6i which contains a methoxy group. This is likely due to the higher capacity of nitrogen to share its lone pair electrons with the vicinal carbon cation as compared to oxygen. As a result, along with 6a–c and 6j–k, C–C bond-cleavage in 6i follows a third possible pathway (Fig. 3, orange pathway) which is significantly higher in energy.
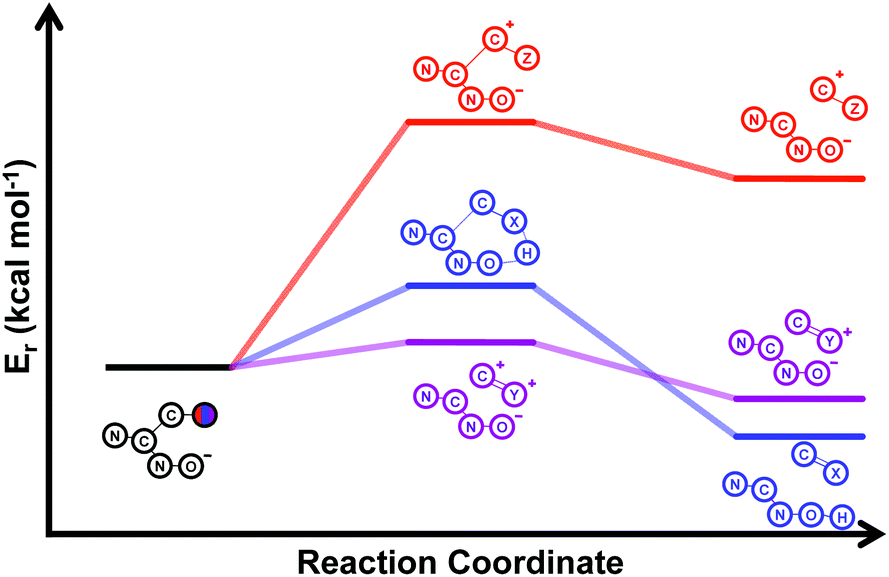 | ||
| Fig. 3 Schematic representation of the three alternative reaction pathways for the C–C bond-cleavage step from intermediate IV, depending on the nature of the substituent: 6a–c and 6i–k – orange pathway, 6e–g – blue pathway, 6h – purple pathway. | ||
Our experimental studies showed that the reaction of 1 with ethanolamine (2e) afforded both the C–H and C–C cleaved products (4e and 5). This competition between the two fragmentation paths is in agreement with the computational results since path B for 6e has a higher activation barrier than that of the other examples studied that showed C–C bond cleavage. The result is also consistent with the reduced stabilization of the –CH2OH group of the transition state geometry compared to –CH2NH2, –CH2NHCH3, and –CH2N(CH3)2 functionalities.
To probe further the bifurcation observed in the case of 6e, we performed a series of calculations taking into account the potential role of the base in the fragmentation process. In the synthesis of the benzimidazole-N-oxides, potassium carbonate is employed. A key step in the reaction is the formation of intermediate II by means of a deprotonation of the dinitroaniline precursor; the carbonate base being converted to bicarbonate. As such, we decided to perform our calculations using bicarbonate as the base component.
Our calculations started with a conformational search since there are a wide range of possible interactions between the bicarbonate anion and 6e. The conformational search showed that the bicarbonate ion had a high affinity for the polar region of 6e, and the main interactions were identified to be hydrogen-bonding in nature. The two most stable conformations differ by only 1 kcal mol−1 and are in equilibrium with each other even at room temperature. One of them, reagent complex A (RC-A), showed a double hydrogen-bond between the bicarbonate anion and 6e. The hydrogen-bonds are from the hydrogen of the sp3 hybridized carbon of the benzimidazole ring and the hydrogen on the CH2OH group of the parent ethanolamine. The other, reagent complex B (RC-B), again showed a double hydrogen-bond between the bicarbonate anion and 6e, this time the oxygen of the N-oxide moiety and the hydrogen on the CH2OH group of the parent ethanolamine are involved. The geometries of RC-A and RC-B as well as the H-bond distances are shown in Fig. 4.
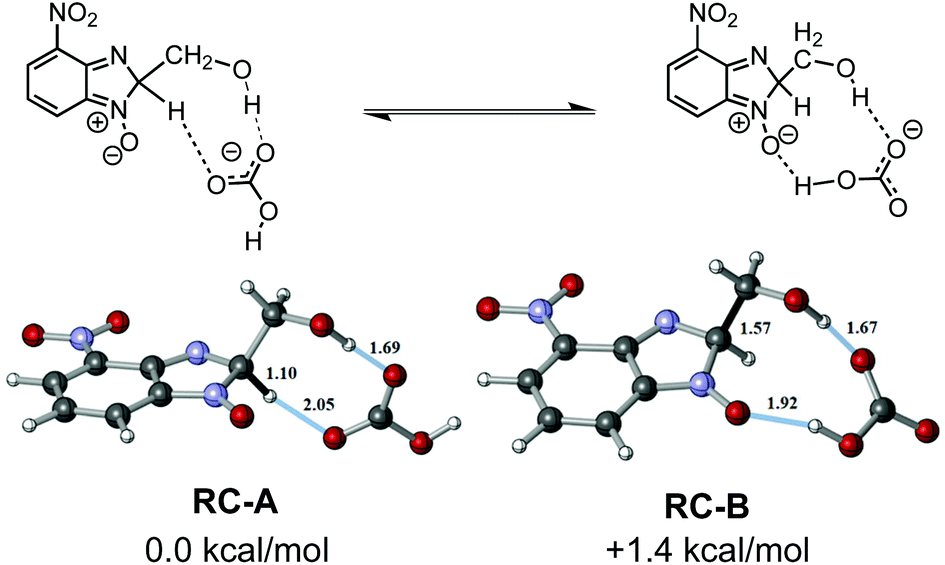 | ||
| Fig. 4 The geometries of reaction complexes A and B, and more relevant bond (denoted in black) and hydrogen-bond (denoted in blue) distances. Energy difference was computed as free energy. | ||
On inspection, the most stable reagent complex (RC-A) has a geometry closely related to C–H bond-cleavage (path A), while RC-B feasibly resembles an intermediate on the path to C–C fragmentation (path B) (see Fig. S12 and S13 in ESI†). We modelled the energy profiles for paths A and B, these being shown in Fig. 5a and b, respectively. The activation barriers for both C–H and C–C bond-cleavage are essentially the same (approximately 6 kcal mol−1 and −22 kcal mol−1, for activation barrier and thermochemistry respectively). The values obtained have been compared across different DFT functionals and larger basis sets (see validation of method and basis set section in the ESI†). All the methods employed agree that both activation barriers are small; C–H fragmentation being slightly more favoured. In the case of thermochemistry C–H fragmentation is also slightly more exothermic than its C–C bond-scission counterpart. In competitive reactions such as this where the two products do not have distinct barriers to formation, analysis is not trivial. Indeed, development of new algorithms to describe the observed selectivity is the subject of current research in theoretical chemistry.17 It is particularly complex in cases were carbocations and their subsequent rearrangements are involved in the mechanism.24 However, in our case a qualitative analysis can be undertaken to probe particular reaction parameters. One such case is the effect of temperature on the outcome of the reaction. From an experimental perspective, performing the reaction of 1 with 2e at 120 °C led to a 40/60 mix of 4e and 5, but reducing the reaction temperature to 80 °C resulted in an inversion of the outcome (60% of 4e and 40% of 5). Given that the activation energies of path A and path B are negligible, we propose that at lower temperature C–H fragmentation is dominant since it is slightly more exothermic. However, under our reaction conditions (elevated temperature), formaldehyde generated in the C–C bond-cleavage process can be released from the reaction mixture due to its volatility, or it can undergo a subsequent reaction, these processes driving the reaction towards C–C bond fragmentation.
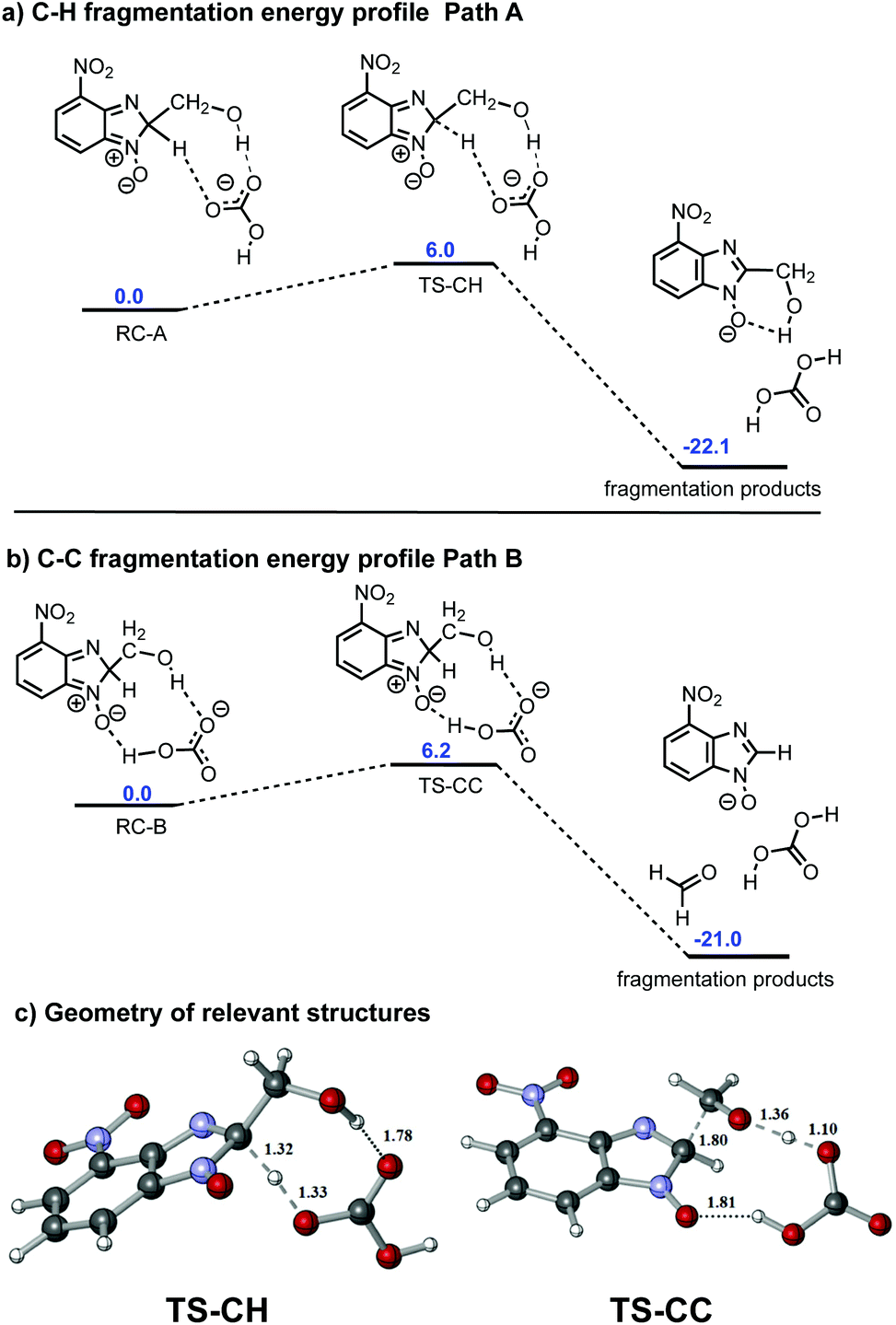 | ||
| Fig. 5 Calculated reaction coordinates for C–H and C–C bond-cleavage observed in the reaction of 1 with ethanolamine (2e). Energy values denoted in blue correspond to free energy values expressed in kcal mol−1 (a) Reaction coordinate for path A; (b) reaction coordinate for path B; (c) optimized geometries for transitions states for C–H (TS-CH) and C–C (TS-CC) bond-cleavage. | ||
To probe our hypothesis further, we performed a series of experiments designed to detect the formaldehyde generated in the reaction of 1 with 2e, predicted from our computational studies. Given that formaldehyde is both found in and generated from a range of consumer products, there are a number of standard analytical methods available for its detection.25,26 We performed the reaction of 1 with 2e in a reaction vessel connected to a bubbler going into a water reservoir, in an approach akin to that use for the analysis of e-cigarette aerosols (see Fig. S5 in ESI†).27 Performing the Tollens’ test on the contents of the water reservoir at the end of the reaction showed a silver mirror, this being indicative of the presence of an aldehyde in solution (see Fig. S6 in ESI†).28 In a second approach, we employed solid-phase microextraction (SPME) (see Fig. S7 in ESI†). Brady's reagent, 2,4-dinitrophenylhydrazine (DNPH),29 can be used as a SPME derivatising agent in order to determine the presence of carbonyl compounds using chromatographic techniques that detect the hydrazone formed.30–32 Employing this approach, and using HPLC-UV as a spectroscopic tool, we detected the 2,4-dinitrophenylhydrazone derivative in the SPME eluted sample from the reaction of 1 with 2e, this confirming the generation of formaldehyde. In order to obtain formaldehyde at a level that was detectable by the techniques we employed, it was necessary to perform the reaction on a scale ten-times that of which we generally operated. This led us to believe that the majority of the formaldehyde formed may be undergoing a Cannizzaro reaction (Scheme 4).28,33,34 The lack of α-H atoms in formaldehyde makes it susceptible to a base-catalysed disproportionation reaction to generate the formate ion and methanol. Comparison of 1H-NMR spectra of the crude product mixture from the reaction of 1 with 2e and a reference reaction between formaldehyde and potassium carbonate under the same heating protocol allowed us to confirm this hypothesis; the formate ion being observed in both cases (see Fig. S8 and S9 in ESI†). This adds further credence to our proposed pathway for the reaction.
 | ||
| Scheme 4 Disproportionation of formaldehyde in basic media (Cannizzaro reaction) producing the formate anion and methanol. | ||
Conclusions
In summary, we have performed experimental and computational studies in order to investigate an unusual carbon–carbon bond cleavage that occurs in the preparation of certain benzimidazole N-oxides from anilines. Our computational model shows good correlation to the results obtained from our experimental observations; C–H, C–C, or a combination of both bond-cleavages are predicted and observed. A key factor determining the outcome of the reaction was found to be the substituents on the amine functionality of the aniline. More specifically, the presence of a heteroatom in the β-position of the amine, and the ability to eject a neutral molecule significantly influences whether C–C bond-scission occurs. The case of ethanolamine was particularly interesting, both C–H and C–C bond-cleavage pathways being observed and so this reaction was studied in more detail both from a computational and experimental perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded in part by the University of Connecticut Program in Accelerated Therapeutics for Healthcare. A fellowship to F. P. by the Fulbright Program and Ministerio de Educación de la Nación Argentina is gratefully acknowledged. We thank Dr James Stuart (University of Connecticut) for providing SPME cartridges and help us with HPLC-UV analysis. In addition, we thank Dr Anthony Provatas and Adam Graichen (University of Connecticut) for guidance regarding in chromatography analytical techniques.Notes and references
- G. G. Allan, C. S. Chopra and T. Mattila, Pestic. Sci., 1972, 3, 153 CrossRef CAS.
- D. Li, P. Wu, N. Sun, Y.-J. Lu, W.-L. Wong, Z. Fang and K. Zhang, Curr. Org. Chem., 2019, 23, 616 CrossRef CAS.
- G. F. dos Santos Fernandes, A. R. Pavan and J. L. dos Santos, Curr. Pharm. Des., 2018, 24, 1325 CrossRef CAS.
- F. Politano, E. I. Buján and N. E. Leadbeater, Chem. Heterocycl. Compd., 2016, 52, 952 CrossRef CAS.
- F. Politano, A. K. Gran-Magano and N. E. Leadbeater, Molecules, 2019, 24, 3639 CrossRef.
- E. I. Buján de Vargas and A. I. Cañas, Tetrahedron Lett., 1996, 37, 767 CrossRef.
- E. I. Buján and M. L. Salum, J. Phys. Org. Chem., 2006, 19, 187 CrossRef.
- M. L. Salum, R. H. de Rossi and E. I. Buján, Eur. J. Org. Chem., 2007, 2007, 2164 CrossRef.
- P. A. Collins Cafiero, C. S. French, M. D. McFarlane, R. K. Mackie and D. M. Smith, J. Chem. Soc., Perkin Trans. 1, 1997, 1375 RSC.
- E. I. Buján, A. I. Cañas and R. H. de Rossi, J. Chem. Soc., Perkin Trans. 2, 2001, 1973 Search PubMed.
- P. A. Nikitina and V. P. Perevalov, Chem. Heterocycl. Compd., 2017, 53, 123 CrossRef CAS.
- C. E. Dykstra, G. Frenkling, K. S. Kim and G. E. Scuseria, Theory and applications of computational chemistry: the first forty years, Elsevier, 1st edn, 2005 Search PubMed.
- E. G. Lewars, Computational Chemistry, Springer International Publishing, Cham, 3rd edn, 2016 Search PubMed.
- J. Rehbein and B. K. Carpenter, Phys. Chem. Chem. Phys., 2011, 13, 20906 RSC.
- D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Çelebi-Ölçüm and K. N. Houk, Angew. Chem., Int. Ed., 2008, 47, 7592 CrossRef CAS.
- S. R. Hare and D. J. Tantillo, Pure Appl. Chem., 2017, 89, 679 CAS.
- S. Lee and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 9210 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef.
- A. Bassani, M. Prato, P. Rampazzo, Q. Ugo and G. Scorrano, J. Org. Chem., 1980, 45, 2263 CrossRef CAS.
- C. Paradisi and G. Scorrano, Acc. Chem. Res., 1999, 32, 958 CrossRef CAS.
- M. Z. Nazer, M. J. Haddadin, J. P. Petridou and C. H. Issidorides, Heterocycles, 1977, 6, 541 CrossRef CAS.
- K. V. N. Esguerra, W. Xu and J.-P. Lumb, Chem, 2017, 2, 533 CAS.
- Y. J. Hong and D. J. Tantillo, Nat. Chem., 2014, 6, 104 CrossRef CAS.
- Facts about formaldehyde|Formaldehyde|US EPA, https://www.epa.gov/formaldehyde/facts-about-formaldehyde#whatcontains, (accessed 15 June 2020).
- T. Su and R. He, in Formaldehyde and Cognition, Springer Netherlands, Dordrecht, 2017, pp. 271–295 Search PubMed.
- J. W. Flora, C. T. Wilkinson, J. W. Wilkinson, P. J. Lipowicz, J. A. Skapars, A. Anderson and J. H. Miller, J. Chromatogr. Sci., 2017, 55, 142 CAS.
- A. I. Vogel, B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's textbook of practical organic chemistry, Longman Scientific & Technical, Harlow, 5th edn, 1989 Search PubMed.
- O. L. Brady and G. V. Elsmie, Analyst, 1926, 51, 77 RSC.
- R. J. Kieber and K. Mopper, Environ. Sci. Technol., 1990, 24, 1477 CrossRef CAS.
- P. a. Martos and J. Pawliszyn, Anal. Chem., 1998, 11, 2311 CrossRef.
- D. Bourdin and V. Desauziers, Anal. Bioanal. Chem., 2014, 406, 317 CrossRef CAS.
- S. Cannizzaro, Justus Liebigs Ann. Chem., 1853, 88, 129 CrossRef CAS.
- C. G. Swain, A. L. Powell, W. A. Sheppard and C. R. Morgan, J. Am. Chem. Soc., 1979, 101, 3576 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, and spectral characterization. See DOI: 10.1039/d0ob01797c |
| This journal is © The Royal Society of Chemistry 2021 |