
Exploring the synthetic potential of a marine transaminase including discrimination at a remote stereocentre†
Maria
Schwarz‡
ab,
Edel J.
Murphy‡
 a,
Aoife M.
Foley
ab,
David F.
Woods
c,
Ignacio Abreu
Castilla
c,
F. Jerry
Reen
bcd,
Stuart G.
Collins
ab,
Fergal
O'Gara
*bcef and
Anita R.
Maguire
*abg
a,
Aoife M.
Foley
ab,
David F.
Woods
c,
Ignacio Abreu
Castilla
c,
F. Jerry
Reen
bcd,
Stuart G.
Collins
ab,
Fergal
O'Gara
*bcef and
Anita R.
Maguire
*abg
aSchool of Chemistry, Analytical and Biological Chemistry Research Facility, University College Cork, T12 K8AF, Cork, Ireland. E-mail: a.maguire@ucc.ie
bSynthesis and Solid-State Pharmaceutical Centre, University College Cork, T12 K8AF, Cork, Ireland
cBIOMERIT Research Centre; School of Microbiology, University College Cork, T12 K8AF Cork, Ireland. E-mail: f.ogara@ucc.ie
dSchool of Microbiology, University College Cork, T12 K8AF, Cork, Ireland
eHuman Microbiome Programme, School of Pharmacy and Biomedical Sciences, Curtin Health Innovation Research Institute, Curtin University, Perth, WA 6102, Australia
fTelethon Kids Institute, Perth, WA 6008, Australia
gSchool of Pharmacy, University College Cork, T12 K8AF, Cork, Ireland
First published on 23rd October 2020
Abstract
The marine transaminase, P-ω-TA, can be employed for the transamination from 1-aminotetralins and 1-aminoindanes with differentiation of stereochemistry at both the site of reaction and at a remote stereocentre resulting in formation of ketone products with up to 93% ee. While 4-substituents are tolerated on the tetralin core, the presence of 3- or 8-substituents is not tolerated by the transaminase. In general P-ω-TA shows capacity for remote diastereoselectivity, although both the stereoselectivity and efficiency are dependent on the specific substrate structure. Optimum efficiency and selectivity are seen with 4-haloaryl-1-aminotetralins and 3-haloaryl-1-aminoindanes, which may be associated with the marine origin of this enzyme.
Introduction
Chiral amines and related derivatives are common features found in many natural products (e.g. alkaloids), and the prevalence of similar motifs in pharmaceuticals and fine chemicals makes the availability of enantiopure amines as building blocks critically important to the pharmaceutical industry.1,2 For example, the aminotetralin motif (Fig. 1) is present in a variety of pharmaceuticals, such as the anti-depressants sertraline and norsertraline with an α-aminotetralin core, and rotigotine, a treatment for Parkinson's disease containing a β-aminotetralin core unit (Fig. 1).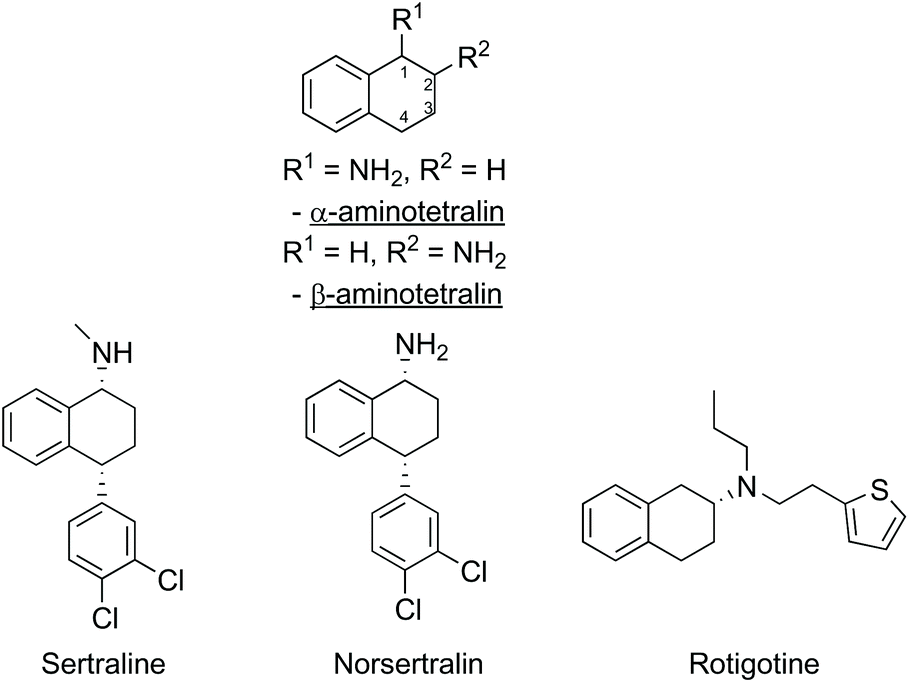 | ||
| Fig. 1 Examples of pharmaceuticals with an α- or β-aminotetralin core. | ||
To develop strategies for the enantioselective synthesis of complex amine building blocks, in addition to controlling the stereochemistry at the amine site, control of the stereochemistry at additional stereocentres in the molecule must also be addressed, and frequently this is more challenging. For example, in the synthesis of sertraline, further to controlling the stereochemistry at C-1 bearing the amino-substituent, the absolute stereochemistry at C-4 must also be controlled in the overall synthetic pathway.
The exquisite chemo-, regio- and stereoselectivity of biocatalysts can open the door to effective synthetic routes to enantiopure compounds, and in addition the use of biocatalysis often has positive benefits in relation to the environmental impact of a process, satisfying 10 of the 12 principles of green chemistry.3 From a synthetic perspective transaminases can provide a route to enantiopure amines, via asymmetric transamination of ketones, or alternatively via kinetic resolution in deamination of racemic amines.4 In the last decade transaminases have attracted considerable attention in synthetic biocatalysis, providing access to enantiopure amines in a synthetically versatile manner, and numerous reports have described their use for API synthesis in the pharmaceutical industry.5–13 An excellent example of the implementation of a transaminase enzyme in the pharma industry is the use of an evolved and then immobilised transaminase for the synthesis of the API Sitagliptin (Januvia®), a Merck drug used in the treatment of diabetes mellitus type 2. This biocatalytic transformation surpasses its chemocatalytic counterpart in terms of selectivity, toxicity and efficiency.14,15
While biocatalysis offers many advantages in enantioselective synthesis, in general, the focus is usually on the control of the absolute stereochemistry at, or near, the site of reaction. Resolution of a remote stereocentre is relatively unusual; however, as an example, the use of hydrolases to mediate the kinetic resolution of a remote stereocentre has been previously reported.16–22 However in almost all the reports of the use of transaminases the focus is on controlling the absolute stereochemistry at the amine site,8,23,24 with just a small number of reports of the resolution of an α-stereocentre using transaminases (e.g.Fig. 2).25,26
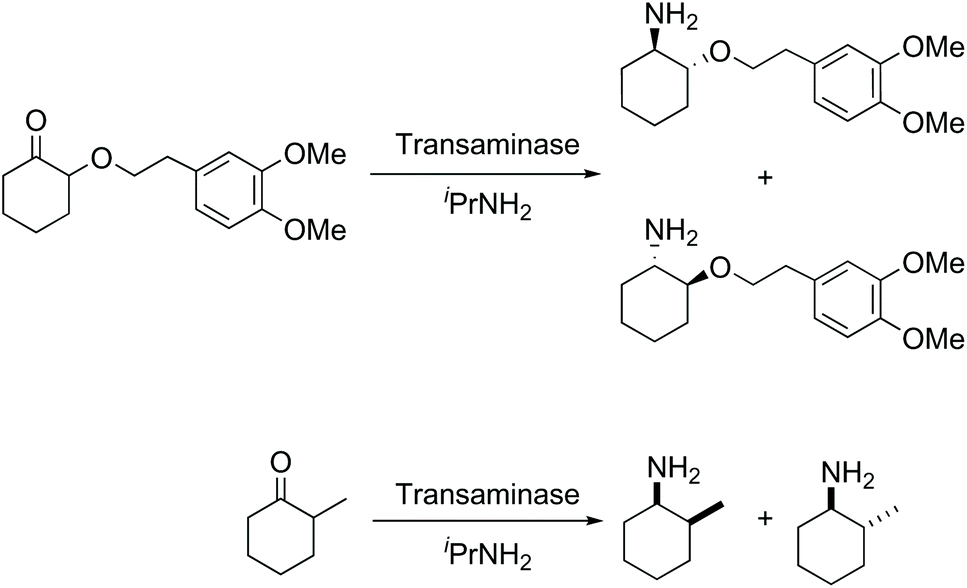 | ||
| Fig. 2 Examples of transaminase mediated asymmetric transamination of a compound with an α-stereocentre.25,26 | ||
The marine environment has provided a wealth of diversity in natural products, bioactive compounds and biocatalysts; developments in (meta)genomic-based technologies, that capture and decode the genetic blueprint of oceanic organisms, without the need to grow or sustain the source organism, have proven a vital tool in marine biodiscovery.27–31 In the context of synthetic chemistry enzymes extracted from the marine environment often display a much higher tolerance when exposed to diverse substrates and reaction media, pressure and heat in comparison to their terrestrial counterparts.32–34
In the first example of transaminase-mediated resolution of a remote stereocentre, we recently reported a marine ω-transaminase P-ω-TA, from a Pseudovibrio species isolated from a marine sponge, identified using genome mining techniques.35P-ω-TA was used effectively for the resolution of amine 1a, a potential intermediate in the synthesis of sertraline. Comparing the marine transaminase to the control transaminase Chromobacterium violaceum (Cv-ω-TA)36 we showed that P-ω-TA discriminates at the remote stereocentre, selectively processing the cis-1a enantiomer over the trans-1a enantiomer (Table 1), unlike Cv-ω-TA which similarly processed both cis- and trans-1a, without noticeable discrimination. To the best of our knowledge, this was the first report of remote stereoselection using an ω-transaminase.35
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35
35
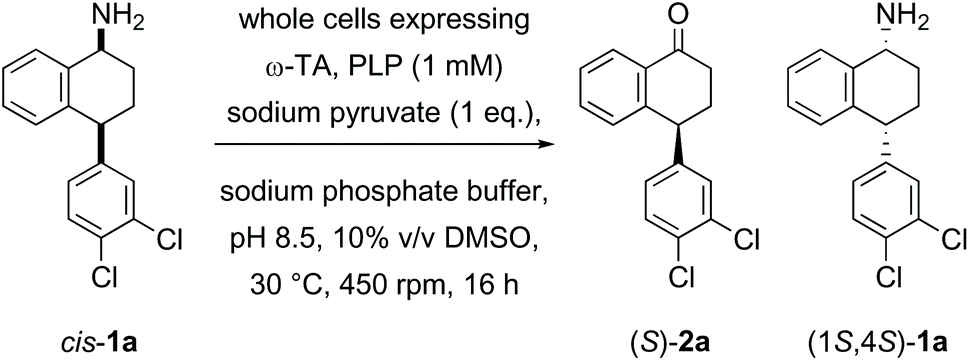
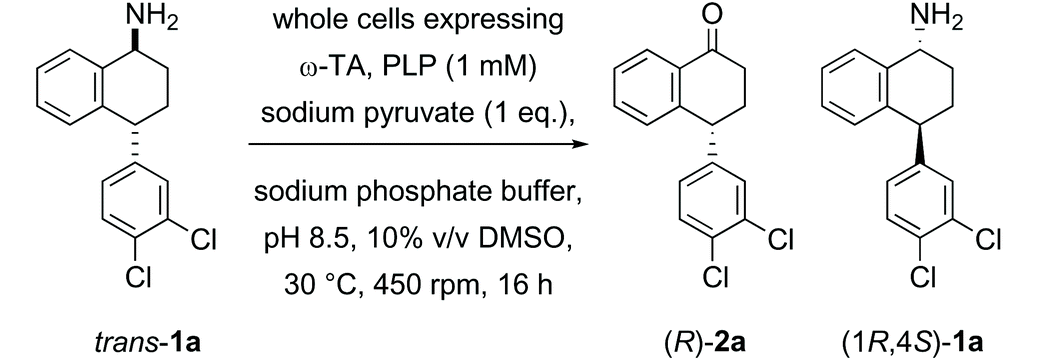
Herein, given the synthetic advantage associated with the remote stereoselection of P-ω-TA, we explore the expansion of the substrate scope beyond 1a to determine the tolerance of P-ω-TA to structural changes within the general framework of the sertraline intermediate 1a, and to specifically explore the influence of variation of the substrate structure on efficiency, enantioselectivity and remote stereoselection. The overall objective was to establish the synthetic potential of P-ω-TA for enantioselective transformation with stereocontrol at the site of reaction in addition to distinguishing the stereochemistry at a remote stereocentre.
Results & discussion
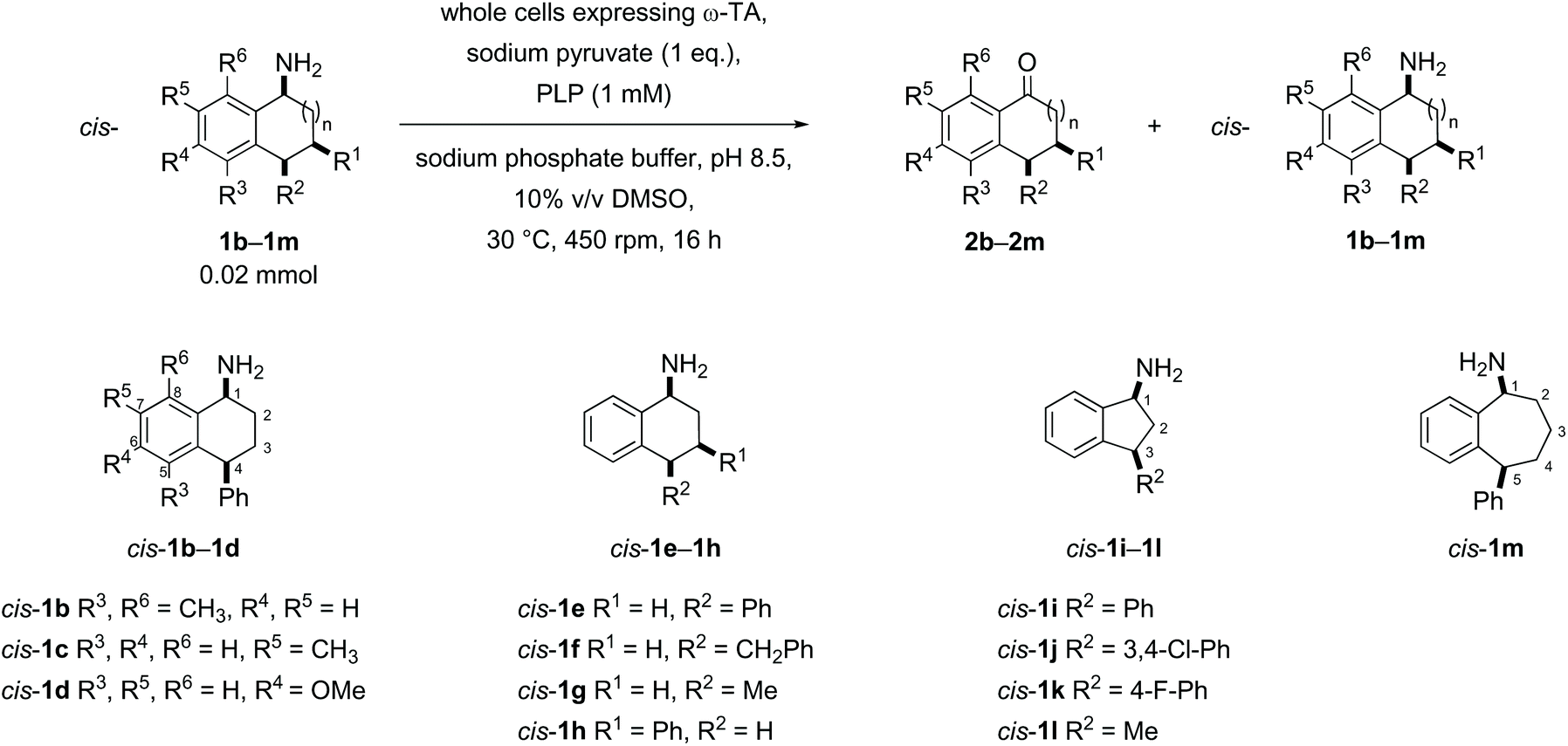
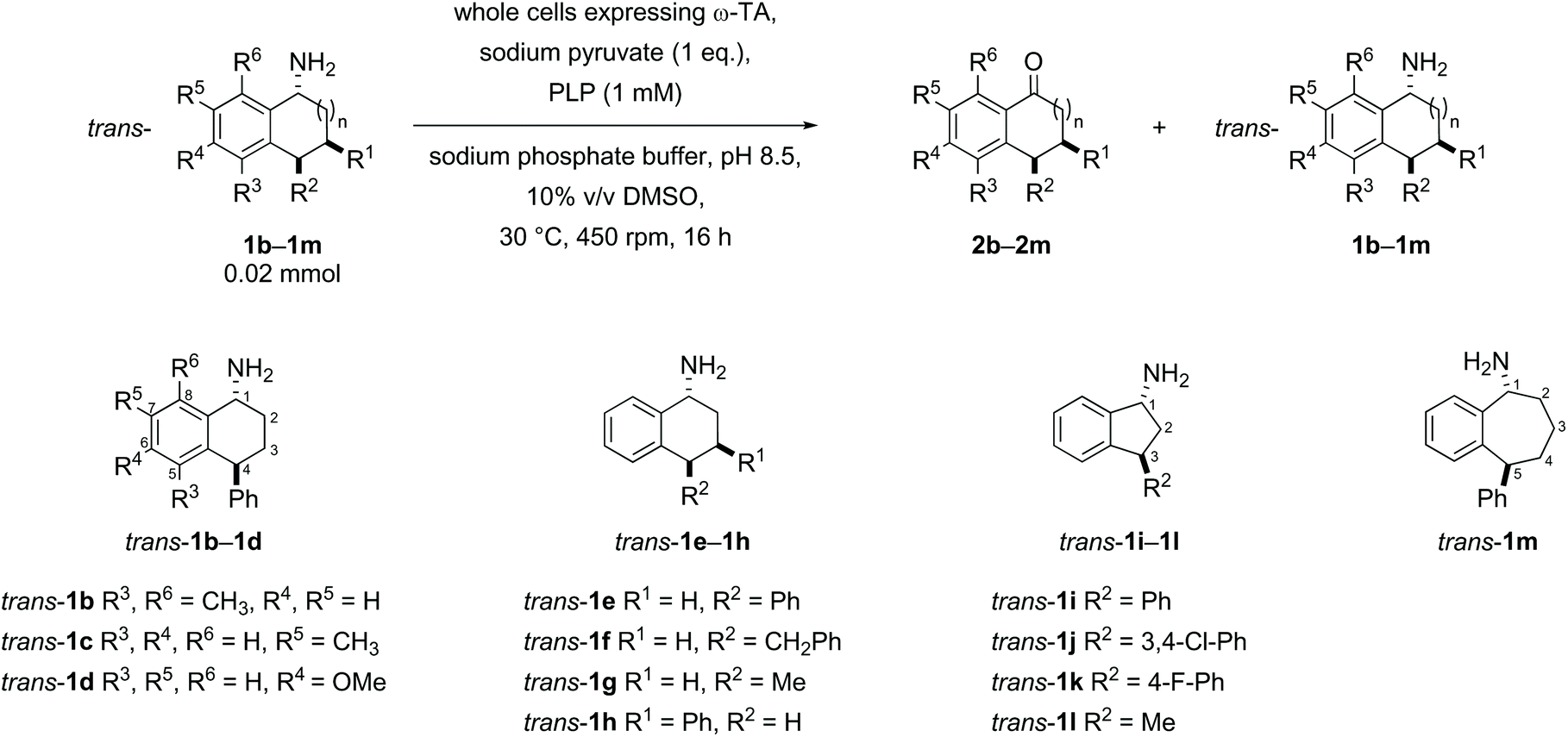
Substrates were designed to explore both steric and electronic effects of substituents on the tetralin core, the impact of ring size and the influence of different aryl and alkyl-substituents in the 3- and 4-position on the transaminase-mediated biotransformations, as summarised in Scheme 1.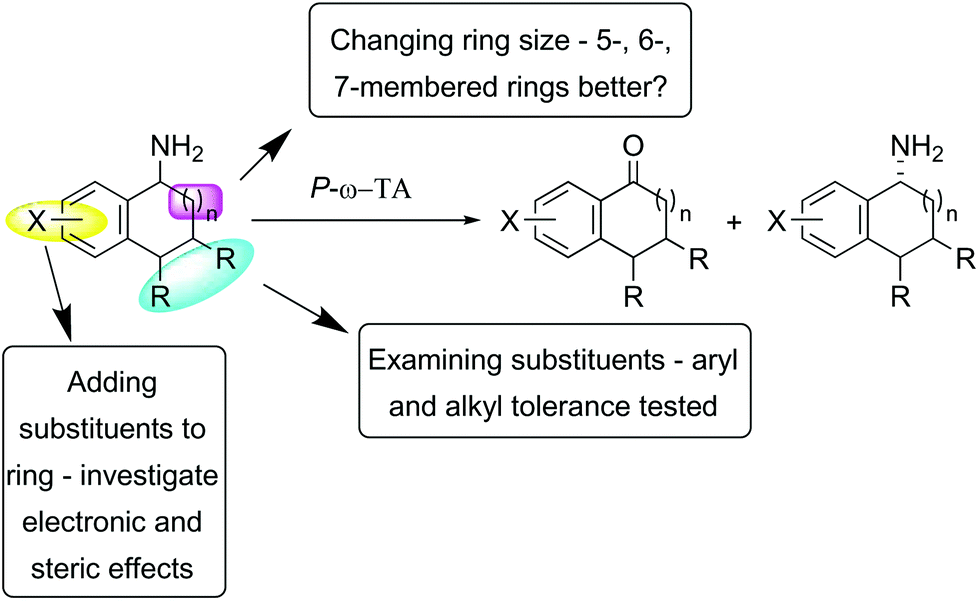 | ||
| Scheme 1 Amine substrates investigated. | ||
Substrate synthesis
Racemic samples of each of the cis- and trans-amines were synthesised in addition to the racemic ketones as standards for chiral HPLC method development. The known ketones 2b–2m were synthesised following literature procedures (details in the ESI†).38–46 Subsequent transformation of each ketone 2b–2m to both diastereomeric amines was undertaken as summarised below in Schemes 2–5, following literature precedent. While amines cis-1e,471h,481j49 and 1l49 are known compounds with spectroscopic characteristics in agreement with literature data, amines 1b, 1c, 1d, trans-1e, 1f, 1g, 1k and 1m are novel and were fully characterised in this work. The hydrochloride salt of cis- and trans-1i has been reported before with only mass spectrometry reported.50 Similarly, the intermediate alcohols 6b and 6c, azides 7b, 7c, 7e, cis-7f, trans-7h, trans-7i and trans-7k, and Boc protected amines 8d and 8m are novel and were fully characterised in this work; other intermediates were known, with spectroscopic characteristics in agreement with reported data.
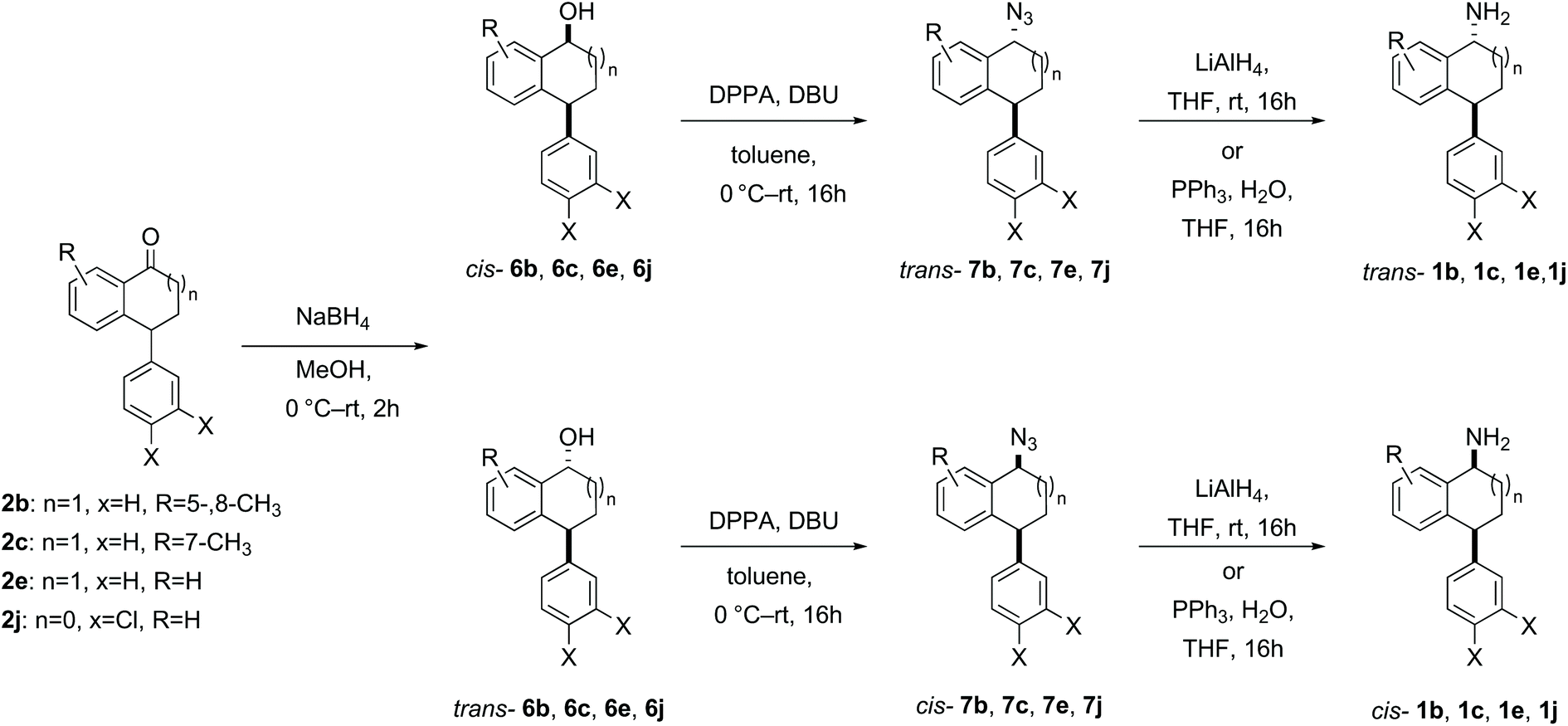 | ||
| Scheme 2 Synthesis of cis- and trans-1b, 1c, 1e, 1j from the corresponding ketones via reduction, azidation with inversion of stereochemistry and subsequent azide reduction. | ||
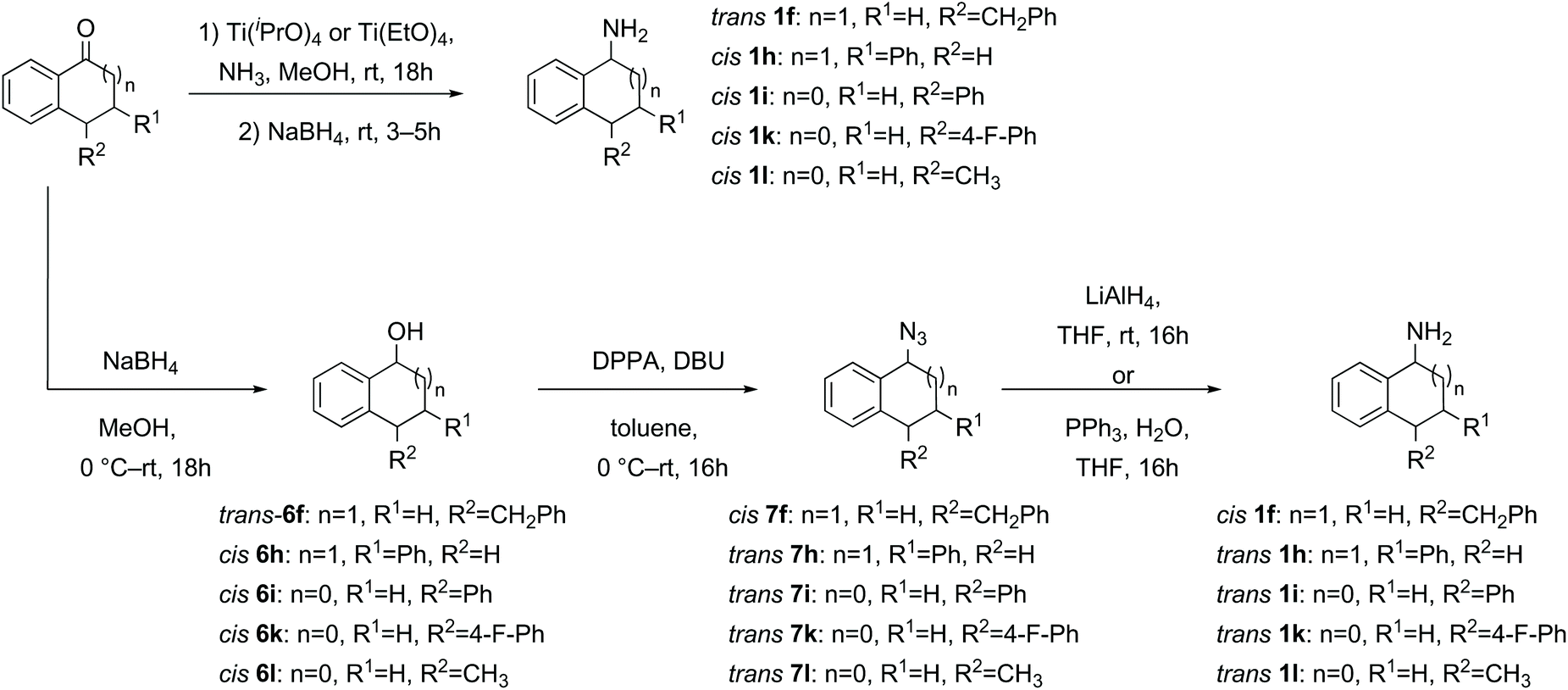 | ||
| Scheme 3 Synthesis of amines via direct reductive amination or via reduction, azidation and azide reduction. | ||
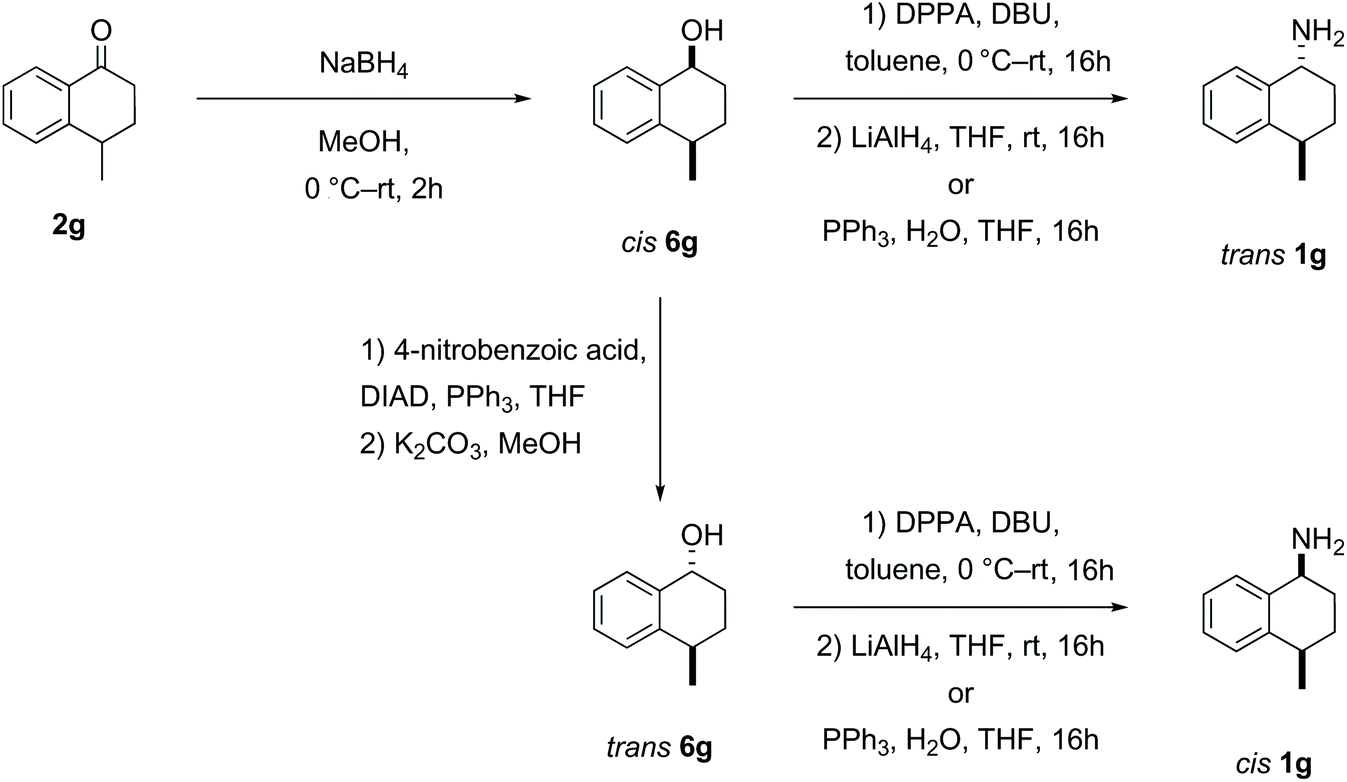 | ||
| Scheme 4 Synthesis of cis- and trans-aminotetralin 1g. | ||
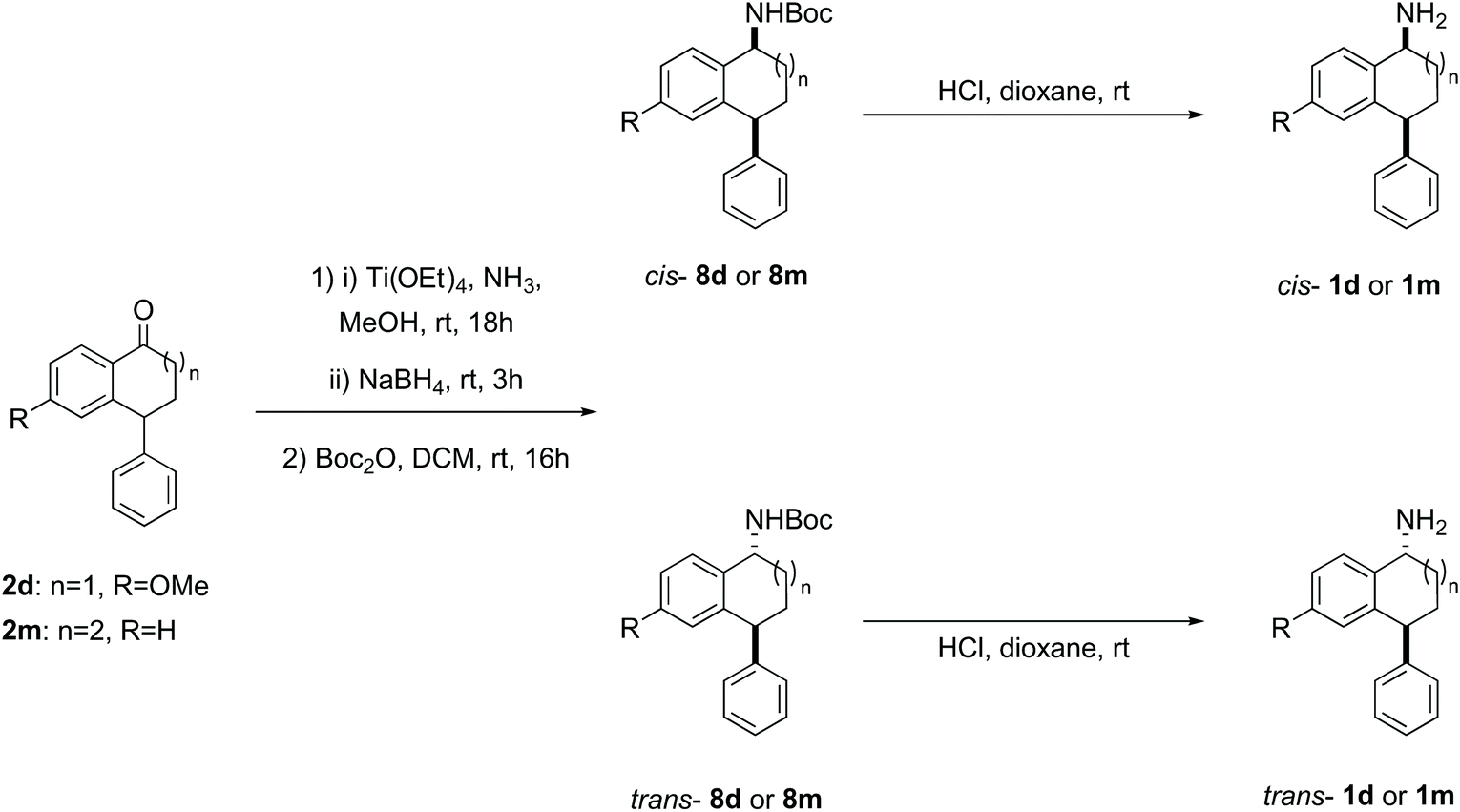 | ||
| Scheme 5 Reductive amination of ketone 2d and 2m with diastereomer separation of the N-Boc derivatives to yield diastereomerically pure amines 1d and 1m. | ||
For the synthesis of each diastereomeric amine 1b, 1c, 1e and 1j, the corresponding ketone was reduced using sodium borohydride following a general procedure,35,51 to give a mixture of diastereomeric alcohols, which were separable by careful chromatography. The reductions generally proceeded cleanly with full conversion; however, in practice, the isolated yields of pure diastereomers was determined by the challenging chromatographic separation of the diastereomers. Each of the diastereomerically pure alcohols 6b, 6c, 6e, 6j were converted to their corresponding azides 7b, 7c, 7e, 7j with clean inversion of stereochemistry using diphenylphosphoryl azide and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) followed by a Staudinger reaction or reduction using LiAlH4 (Scheme 2).35,52
Where the initial sodium borohydride reduction of the ketone afforded primarily one diastereomer and the separation/purification of the minor diastereomer proved difficult, the major diastereomer (trans for 1f and cis for 1h, 1i, 1k and 1l) was subjected to azidation and reduction conditions to synthesise the inverted amine (cis-1f and trans-1h, 1i, 1k and 1l, Scheme 3) by the same method as in Scheme 2.52 The opposite diastereomers could be obtained by direct reductive amination of the ketone through treatment with ammonia in methanol and titanium(IV) isopropoxide or titanium(IV) ethoxide, followed by in situ sodium borohydride reduction furnishing primarily the trans-amine 1f and cis-amine 1h, 1i, 1k and 1l (Scheme 3).35,43 In general the crude amine was converted to the HCl salt, washed, precipitated, recrystallized if necessary and then a salt break furnished the pure amine without column chromatography.
Reduction of the 4-methyl-1-tetralone 2g substrate furnished a mixture of cis- and trans-1g alcohols in a diastereomeric ratio of 55:45 respectively, which could not be separated through flash column chromatography.51 However, the cis-alcohol 6g could be obtained through recrystallization from hexane. The trans-alcohol 6g was accessed by inversion of the stereochemistry via an ester intermediate following a literature procedure.53 The two novel amines cis- and trans-1g were each obtained by azidation and reduction as described above with clean inversion in each case (Scheme 4).52
When the synthesis of amines 1d (p-OMe) and novel 1m (7-membered ring) was attempted following the synthetic sequence outlined in Scheme 2, the corresponding amines were not isolated as single diastereomers. To overcome this problem, the ketones 2d and 2m were subjected instead to reductive amination and the diastereomeric amines in the crude mixtures were protected using Boc-anhydride (Scheme 5).54 The Boc amines 8d and 8m were purified and the diastereomers separated by flash column chromatography on silica gel. The rather low yields can again be attributed to the challenging separation. In addition to making the compounds easier to separate, this approach had the advantage that the Boc protected amines could be used for HPLC method development. The free amines 1d and 1m were readily accessed by deprotection of the Boc amines under acidic conditions. Characterisation of the benzosuberone derived amines 1m and Boc amines 8m is complicated by conformational effects in the 1H and 13C NMR spectra.55
Substrate scope
As our primary objective was to establish a substrate scope for the remote stereoselection, we tested a range of substrates against the biocatalysts P-ω-TA and the control Cv-ω-TA for activity in the thermodynamically favoured deamination reaction using the cis- and trans-amine substrates separately (Tables 2 and 3 respectively). All the biotransformations reported in Tables 2 and 3 below were conducted under the same conditions to enable comparison and insight into the impact of substituents. For each substrate, optimisation of the efficiency and stereoselectivity by adjusting the reaction conditions (e.g. the reaction time, scale up, etc.) could be envisaged. The efficiencies of conversion are recorded in Tables 2 and 3 below in two ways, firstly based on integration of 1H NMR spectra of the crude product mixtures, and secondly by use of Ecalc based on the chiral HPLC data.37 While in most instances there is reasonable alignment, differences may be explained by the challenges in accurately integrating NMR spectra where one of the components is present in a very low level, e.g.Table 2, entry 16 in which case Ecalc perhaps is a better indicator of biocatalyst efficiency.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | TA | Conversion (%) | ee (%) | E 37 | ||
| 1H NMR | E calc 37 | eeS | eeP | ||||
| a It was not possible to accurately estimate the % conversion using 1H NMR spectroscopy. | |||||||
| 1 | cis-1b | P-ω-TA | 0 | — | — | — | — |
| 2 | cis-1b | Cv-ω-TA | 6 | 3 | 1 | 33 | 2 |
| 3 | cis-1c | P-ω-TA | 7 | 6 | 5 | 82 | 10 |
| 4 | cis-1c | Cv-ω-TA | 50 | 51 | >99 | 94 | 170 |
| 5 | cis-1d | P-ω-TA | 28 | 31 | 42 | 93 | 42 |
| 6 | cis-1d | Cv-ω-TA | 51 | 55 | >99 | 82 | 52 |
| 7 | cis-1e | P-ω-TA | —a | 8 | 8 | 93 | 30 |
| 8 | cis-1e | Cv-ω-TA | —a | 16 | 11 | 56 | 4 |
| 9 | cis-1f | P-ω-TA | 4 | 12 | 7 | 50 | 3 |
| 10 | cis-1f | Cv-ω-TA | 61 | 60 | >99 | 65 | 23 |
| 11 | cis-1g | P-ω-TA | 14 | 16 | 18 | 93 | 33 |
| 12 | cis-1g | Cv-ω-TA | 53 | 55 | >99 | 82 | 52 |
| 13 | cis-1h | P-ω-TA | 0 | — | — | — | — |
| 14 | cis-1h | Cv-ω-TA | 0 | — | — | — | — |
| 15 | cis-1i | P-ω-TA | 4 | 14 | 7 | 43 | 3 |
| 16 | cis-1i | Cv-ω-TA | 100 | 71 | 10 | 4 | 1 |
| 17 | cis-1j | P-ω-TA | 46 | 45 | 72 | 87 | 31 |
| 18 | cis-1j | Cv-ω-TA | 51 | 52 | 82 | 77 | 19 |
| 19 | cis-1k | P-ω-TA | 51 | 51 | 84 | 82 | 27 |
| 20 | cis-1k | Cv-ω-TA | 76 | 78 | >99 | 28 | 7 |
| 21 | cis-1l | P-ω-TA | 63 | 42 | 57 | 79 | 3 |
| 22 | cis-1l | Cv-ω-TA | 96 | 89 | 88 | 11 | 15 |
| 23 | cis-1m | P-ω-TA | 27 | 17 | 20 | 95 | 47 |
| 24 | cis-1m | Cv-ω-TA | 53 | 49 | 93 | 96 | 168 |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | TA | Conversion (%) | ee (%) | E 37 | ||
| 1H NMR | E calc 37 | eeS | eeP | ||||
| a It was not possible to estimate enantiopurity accurately from the HPLC data due to the limited extent of conversion. b It was not possible to accurately estimate the % conversion using 1H NMR spectroscopy. | |||||||
| 1 | trans-1b | P-ω-TA | 0 | — | — | — | — |
| 2 | trans-1b | Cv-ω-TA | 0 | — | — | — | — |
| 3 | trans-1c | P-ω-TA | 0 | — | — | — | — |
| 4a | trans-1c | Cv-ω-TA | 10 | — | — | — | — |
| 5a | trans-1d | P-ω-TA | <7 | — | — | — | — |
| 6 | trans-1d | Cv-ω-TA | 52 | 54 | 94 | 82 | 36 |
| 7 | trans-1e | P-ω-TA | 0 | — | — | — | |
| 8 | trans-1e | Cv-ω-TA | —b | 22 | 24 | 87 | 18 |
| 9 | trans-1f | P-ω-TA | 0 | — | — | — | — |
| 10 | trans-1f | Cv-ω-TA | 53 | 54 | >99 | 84 | 60 |
| 11 | trans-1g | P-ω-TA | 18 | 20 | 23 | 94 | 40 |
| 12 | trans-1g | Cv-ω-TA | 51 | 53 | >99 | 89 | 90 |
| 13 | trans-1h | P-ω-TA | 0 | — | — | — | — |
| 14 | trans-1h | Cv-ω-TA | 0 | — | — | — | — |
| 15 | trans-1i | P-ω-TA | <2 | 3 | 1 | 28 | 2 |
| 16 | trans-1i | Cv-ω-TA | <2 | 2 | 1 | 63 | 4 |
| 17 | trans-1j | P-ω-TA | 4 | 6 | 1 | 16 | 1 |
| 18 | trans-1j | Cv-ω-TA | 8 | 11 | 7 | 58 | 4 |
| 19 | trans-1k | P-ω-TA | 0 | — | — | — | — |
| 20 | trans-1k | Cv-ω-TA | 4 | 6 | 4 | 62 | 4 |
| 21 | trans-1l | P-ω-TA | <2 | 5 | 3 | 61 | 4 |
| 22 | trans-1l | Cv-ω-TA | 29 | 30 | 32 | 76 | 10 |
| 23 | trans-1m | P-ω-TA | 0 | — | — | — | — |
| 24 | trans-1m | Cv-ω-TA | 49 | — | — | 92 | — |
Substrates 1b–d illustrate the effects of substituents on the benzene ring of the tetralin core.
• cis- and trans-1b (Tables 2 and 3, entries 1 & 2 respectively)
Examining the outcome of the reactions of cis- and trans-1b, with methyl groups in the 5- and 8-positions, it is clear that the increased steric demand close to the site of reaction completely shuts down any enzyme activity towards cis- and trans-1b (Tables 2 and 3, entries 1 & 2 respectively).
• cis- and trans-1c (Tables 2 and 3, entries 3 & 4 respectively)
With a methyl substituent at the 7-position on the aromatic ring of the tetralin core, the kinetic resolution of cis-1c using P-ω-TA (Table 2, entry 3) displayed diminished activity when compared to the conversion observed for cis-1a, the control Cv-ω-TA showed high activity (50% conversion) and enantioselectivity [>99% enantiomeric excess of the product (eep)] against cis-1c (Table 2, entry 4). Interestingly, in contrast to the conversion of trans-1a, in this case the control enzyme Cv-ω-TA did not react to any significant extent with trans-1c (Table 3, entry 4).
• cis- and trans-1d (Tables 2 and 3, entries 5 & 6 respectively)
To investigate the effect of electron-donating groups on the activity of the novel transaminase, a substrate with a methoxy group in the 6-position 1d was synthesised. The P-ω-TA mediated kinetic resolution of cis-1d (Table 2, entry 5) led to a slightly reduced activity and decreased selectivity, while the activity of Cv-ω-TA (Table 2, entry 6) was unaffected by the presence of the 6-methoxy substituent when compared to cis-1a (Table 1). The activity and selectivity of the known control Cv-ω-TA towards trans-1d (Table 3 entry 6) was comparable to that observed for cis-1d, but for P-ω-TA the activity for trans-substituted substrate 1d (Table 3, entry 5) was much lower than cis-1d.
Substrates 1e–h illustrates the effects of substituents at the remote stereocentre of the tetralin core.
• cis- and trans-1e (Tables 2 and 3, entries 7 & 8 respectively)
In contrast to 1a, use of the unsubstituted C4 phenyl substituent in 1e has an impact on the activity and stereochemical outcome using both transaminases. Focusing initially on Cv-ω-TA it is clear that once again both the cis (Table 2, entry 8) and trans (Table 3, entry 8) isomers are processed with similar efficiencies but with decreased enantioselectivity for cis-1e (56% eep from cis-1e and 87% eep from trans-1e). The activity and selectivity of Cv-ω-TA towards cis-1e is decreased relative to the results observed with the chlorinated derivative (Table 1) cis-1a [with 56% eep2e (Table 2, entry 8) relative to 91% eep2a (Table 1)]. Interestingly Cv-ω-TA processes trans-1e (Table 3) with lower efficiency but slightly higher enantioselectivity when compared to trans-1a (Table 1). With transaminase P-ω-TA, the selectivity for cis-1e (Table 2, entry 7) is similar to that seen with the chlorinated derivative cis-1a [with 93% eep2e (Table 2, entry 7) relative to 96% eep2a (Table 1)] but intriguingly the activity is notably decreased for the formation of 2e (8% Ecalc) from cis-1e compared to 2a (52% Ecalc) from cis-1a. There is no activity observed for P-ω-TA towards trans-1e.
Clearly removing the chloro substituents has an effect on the activity and selectivity of the transaminases. As observed for the previous five substrates a trend is evident whereby the P-ω-TA exhibits selectivity toward the cis-substituted substrates relative to the trans-substituted substrates to a much greater extent than seen with the control transaminase Cv-ω-TA. The impact of substituents on the stereoselectivity patterns is very interesting with both steric and electronic effects evident; the effect of removal of the chloro-substituents from 1a to 1e is particularly notable.
• cis- and trans-1f (Tables 2 and 3, entries 9 & 10 respectively)
To investigate the effect of altered steric demand at the remote stereocentre, the phenyl ring was replaced with a benzyl ring (1f). For the control Cv-ω-TA the activity and selectivity for both cis-1f (Table 2, entry 10) and trans-1f (Table 3, entry 10) were comparable to each other and to cis- and trans-1a (Table 1). Transaminase P-ω-TA displayed no activity towards trans-1f with 0% conversion seen by 1H NMR spectroscopy, and very limited activity toward cis-1f (Tables 3 and 2, entry 9 respectively). Interestingly, the enantiodiscrimination in the transformation of the cis-substituted benzyl derivative is notably lower across both enzymes than that seen with cis-1a (Table 1) but is somewhat comparable to that seen with the phenyl derivative cis-1e (Table 2 entries 7 & 8).
• cis- and trans-1g (Tables 2 and 3, entries 11 & 12 respectively)
Replacing the C-4-aryl group with a less sterically demanding methyl substituent at the C-4 position results in lower activity displayed by P-ω-TA towards cis-1g (Table 2, entry 11) when compared to cis-1a (Table 1). The transaminase P-ω-TA shows low activity towards cis-1g but displays high enantioselectivity 93% ee for 2g (Table 2, entry 11). Notably, the trans-1g isomer is processed by P-ω-TA with increased conversion and selectivity (Table 3, entry 11) when compared to trans-1a (Table 1). Evidently due to the smaller C-4-methyl substituent, both the cis and trans isomers can be accommodated in the active site of the enzyme although cis-1g is processed less efficiently than the C-4-aryl derivatives, cis-1a. Again Cv-ω-TA is very active and enantioselective for both cis-1g and trans-1g (Tables 2 and 3, entry 12 respectively).
• cis- and trans-1h (Tables 2 and 3, entries 13 & 14 respectively)
Moving the phenyl ring to the C-3-postion in 1h is a significant structural change to the substrate. This change allowed us to investigate if the wild-type transaminase active site could accommodate a phenyl substituent at this position in the way it was accommodated at the C-4 position; to the best of our knowledge there are currently no wild type transaminases that can accommodate such substrates. This structural change was not tolerated by either of the ω-transaminases, with 0% conversion across the screen, indicating that the active site of both enzymes (P-ω-TA and Cv-ω-TA) could not accommodate a C-3-aryl substituent irrespective of whether it was cis or trans to the amino substituent (Tables 2 and 3, entries 13 & 14 respectively).
Substrates 1i–l illustrates the effects of substituents on the C3-position of the indane core.
• cis- and trans-1i (Tables 2 and 3, entries 15 & 16 respectively)
Moving from the tetralin core to the indane core structure, the first substrate of this series tested was 1i, which had a phenyl ring at the C3-position on the indane moiety. P-ω-TA exhibited low activity for cis-1i with 4% conversion (Table 2, entry 15); when compared to cis-1e for which a percentage conversion of 51% (Table 2, entry 7) was achieved. The enantiopurity of the ketone formed 2i from cis-1i with P-ω-TA was 43% eep, lower than the enantioselectivity observed for cis-1e, the corresponding tetralin derivative. Notably Cv-ω-TA processed 100% of cis-1i to the ketone 2i, with no enantiodiscrimination, again an unusual outcome (Table 2, entry 16).
The activity of P-ω-TA towards trans-1i (Table 3, entry 15) was very low, as seen with trans-1e, and for the limited amounts processed, the enantioselectivity was very low. The most substantive difference seen between the tetralin 1e and the indane 1i was with the Cv-ω-TA, which processed trans-1e effectively but showed very little catalytic activity towards trans-1i. This is the only example in the study where Cv-ω-TA distinguished between the cis- and trans-substituted substrates, differentiating on the basis of the stereochemistry at the remote site; the observation of both enantiomers of cis-1i being processed without discrimination is notable.
• cis- and trans-1j (Tables 2 and 3, entries 17 & 18 respectively)
Adding two chloro-substituents in the 3- and 4-position of the phenyl ring (1j), analogous to 1a, re-established the remote stereoselection of cis-1j for P-ω-TA and Cv-ω-TA (Table 2, entries 17 & 18), when compared to cis-1i. This result is significant as it is clear that having the dichloro-substituted phenyl ring at the C-4 position of the tetralin core and at the C-3 position of the indane ring is critical for how the substrate binds to the active site of the transaminase but becomes even more evidently important for the indane core structure binding capacity.
In contrast, P-ω-TA showed no activity towards trans-1j (Table 3, entry 17) and Cv-ω-TA displayed very low activity (8% conversion) and moderate enantioselectivity towards trans-1j (58% eep) (Table 3, entry 18). This result for Cv-ω-TA is notable because for trans-1a this control transaminase displays a high level of activity (44% conversion) and enantioselectivity (88% eep). Accordingly, while both enzymes display selectivity for transformation of cis-1j over trans-1j the distinction between the transaminase P-ω-TA and the benchmark Cv-ω-TA is less clear cut with the indane derivative than seen with the original sertraline amine 1a. These results indicate that both enzymes display enantiodiscrimination towards cis-1j over trans-1j.
• cis- and trans-1k (Tables 2 and 3, entries 19 & 20 respectively)
To further probe the effect of substituents in the indane series, a fluoro-substituent was added in the 4-position on the C3-phenyl ring (1k). The P-ω-TA displays good activity and enantioselectivity towards cis-substrate-1k (amine, 84% ees) and product-2k (ketone, 82% eep), giving access to both enantioenriched products (Table 2, entry 19). Adding an electronegative fluorine atom to the phenyl ring at the C-3 position positively impacts both the activity and enantioselectivity of the transaminase towards cis-1k when compared to cis-1i, with the unsubstituted phenyl substituent. Having the fluorine atom on the phenyl ring at the C-3 position allows cis-1k to interact with the active site of P-ω-TA and display similar conversion and enantioselectivity to that of cis-1a and cis-1j. Control transaminase, Cv-ω-TA displays good activity towards cis-1k (78%) with a high degree of enantiopurity 99% ees, but poor enantioselectivity for 2k (ketone, 28% eep). For trans-1kCv-ω-TA displays very low activity with 6% conversion (Table 3, entry 20) and P-ω-TA showed no activity for the trans-substituted substrate 1k (Table 3, entry 19).
Clearly the introduction of the halo-substituents on the C-3 phenyl ring enhances the efficiency of discrimination of cis and trans isomers in addition to the enantiodiscrimination, mirroring the trend seen when comparing 1a to 1e in the tetralin series.
• cis- and trans-1l (Tables 2 and 3, entries 21 & 22 respectively)
The replacement of the phenyl substituent at the C-3 position of the indane ring with a less sterically demanding methyl group has a significant impact on both the activity and selectivity of P-ω-TA toward cis-1l with an increase in activity (Table 2, entry 21) when compared to cis-1i. Clearly the smaller cis-methyl substituent can be accommodated more easily in the active site of the enzyme. Comparing the outcome to that seen for the analogous tetralin derivative cis-1g, it is clear that the enantiodiscrimination is more effective in 1g but less so in 1l highlighting the difference between the way in which the indane and tetralin core structures are accommodated in the active site of the transaminase.
Control transaminase Cv-ω-TA displays very high level of activity for cis-1l coupled with a high degree of enantiopurity for the substrate cis-1l (88% ees) but poor enantiopurity for the product 2l (11% eep). This difference in enantiodiscrimination between the marine P-ω-TA (Table 2, entry 21) and the control Cv-ω-TA (Table 2, entry 22) again highlights the difference in the active sites of each enzyme.
P-ω-TA processes trans-1l with very limited activity and moderate selectivity (Table 3, entry 21). This result, when compared to the results for cis-1l, highlights that the remote stereodiscrimination of the transaminase is retained even though the enantiodiscrimination is decreased with the methyl substituent.
Interestingly, variation of the C-3 substituent had a much greater impact on the enzyme activity and selectivity towards the indane substrates 1i–k relative to the tetralin substrates 1b–h. For the indane series of substrates, the marine transaminase follows a similar trend to that seen with the tetralin series, where it displays activity and enantioselectivity preferences towards the cis-substituted substrates over the trans-substituted substrates while the detailed selectivity patterns vary somewhat. The halogenated aryl substituents offer clear advantages both in terms of efficiency and stereoselectivity, perhaps related to marine origin of the transaminase.30,56
• cis- and trans-1m (Table 2, entries 23, 24 and Table 3, 23, 24 respectively)
The final substrate screened was 1m, with the 6-membered ring increased to a 7-membered ring while retaining a phenyl substituent at the C5-position. Gratifyingly, P-ω-TA exhibited remote stereoselection for benzosuberan 1m with moderate activity and excellent enantioselectivity towards the cis-substrate (Table 2, entry 23) but no activity toward trans-1m (Table 3, entry 23), which is an interesting observation as benzosuberone-based natural products are an important class of medicinal and pharmaceutical compounds.57 For the control transaminase, Cv-ω-TA, it was seen that it could process both cis-1m and trans-1m with high levels of conversion 53% and 49% respectively and also displayed a high degree of enantioselectivity (Tables 2 and 3, entry 24 respectively). Once again, the marine transaminase demonstrates the synthetically useful discrimination of the remote stereocentre which is not possible with Cv-ω-TA.
Conclusion
Overall, the use of P-ω-TA for the deamination of a series of 1-aminotetralins and 1-aminoindanes indicates that this enzyme can be successfully employed across a broader substrate range compared to our initial study using aminotetralin 1a, leading to products with enantiopurity up to 94% ee. It is clear that while 4-substituents are tolerated on the tetralin core, the presence of 3- or 8-substituents inhibits enzyme activity. The substrate scope can be expanded to include 1-indanones and 1-benzosuberones. In general P-ω-TA shows capacity for remote diastereoselection to a much greater extent than the control Cv-ω-TA, although both the stereoselectivity and efficiency is dependent on the precise substrate structure. Given the importance of the 1-aminotetralin and 1-aminoindane core structures the potential of this biocatalyst in enantioselective synthesis is clear. Interestingly, optimum results were obtained with 4-haloaryl 1-aminotetralin, cis-1a and 3-haloaryl 1-aminoindanes, cis-1j and cis-1k which may be linked to the marine origin of this enzyme. In some instances, both the ketone and the amine can be obtained in high enantiopurity highlighting the synthetic utility of this marine biocatalyst (Scheme 6).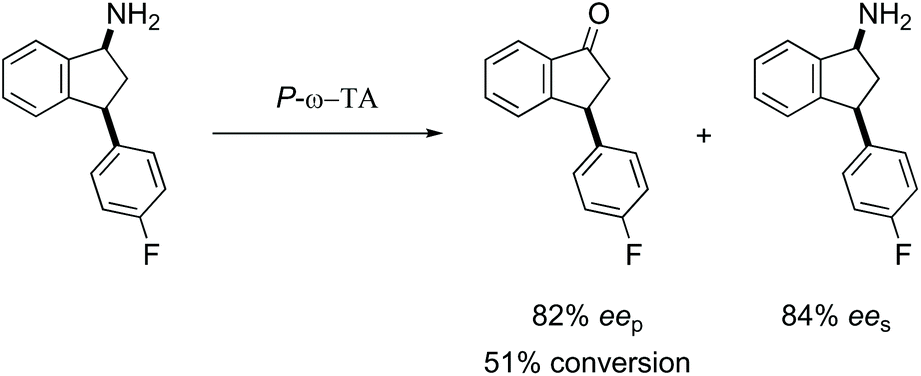 | ||
| Scheme 6 Synthetic utility of P-ω-TA which yielded good enantiopurity in both recovered substrate and product. | ||
Materials and methods
Heterologous expression of P-ω-TA and Cv-ω-TA
Protein expression was preformed following previous protocols.35 In brief, gene specific primers were used to generate a gene amplicon of the transaminases, P-ω-TA and Cv-ω-TA. Directional cloning produced in-frame His-Tag gene fusions in the pET28a expression plasmid. Consensus gene sequence fidelity was analysed by sequencing the insert (Eurofins Genomics) and using the bioinformatic tool, Clustal Omega for local sequence alignment agreement.58 The His-Tag gene fusions in pET28a were introduced into the heterologous host, E. coli BL21 DE RIPL. Cells were cultured with shaking at 180 rpm for 4 h in 37 °C in LB supplemented with 50 μg mL−1 chloramphenicol and 50 μg mL−1 kanamycin. Following this growth phase, the plasmids were induced with 0.5 mM IPTG and incubated for a further 4 h with shaking (180 rpm) in 23 °C. Cells were pelleted using a Sorvall RC series (Thermo Scientific) at 12000 rpm for 10 min, maintained at a temperature of 4 °C.
General procedure for the deamination of amines
E. coli BL21 DE RIPL cells containing the expressed transaminase (30 mg) were suspended in sodium phosphate buffer (50 mM, pH 8.5) in a 15 mL centrifuge tube. The suspension was sonicated (30% intensity) using a probe for 10 s, followed by 30 s on ice. This process was repeated 5 times to lyse the cells. PLP solution (in 50 μL of buffer, final conc. 1 mM) and sodium pyruvate solution (in 50 μL of buffer, overall 1 eq.) were added, followed by the amine substrate (0.02 mmol) dissolved in 100 μL DMSO made up to a total volume of 1 ml, so that the final amine concentration is 20 mM. The solution was shaken at 30 °C, 400 rpm for 16 h. The reaction was stopped through the addition of NaOH (400 μL, 5 M aq. solution). Ethyl acetate (≈4 mL) was added and the tubes were centrifuged to pellet the cells. The organic phase was passed through Na2SO4 in a Pasteur pipette and the solvent was removed in vacuo. The crude products were analysed by 1H NMR and chiral HPLC (detailed methodology is provided in ESI Data†). All experiments were replicated to ensure reproducibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication has emanated from research conducted with the financial support of the Irish Research Council, Thermo Fisher Scientific and GSK under project number EPSPG/2016/41, Synthesis and Solid State Pharmaceutical Centre, funded by Science Foundation Ireland under grant number 12/RC/2275 and with use of equipment provided by Science Foundation Ireland through a research infrastructure from process flow spectroscopy (ProSpect) (grant: SFI 15/RI/3221) (ARM, SGC, MS, EJM, AMF). This research was also supported in part by grants awarded to F. O. G. and F. J. R. by; Enterprise Ireland (CF-2017-0757-P), the Health Research Board/Irish Thoracic Society (MRCG-2018-16), and The Health Research Board (ILP-POR-2019-004). F. O. G. also acknowledges Enterprise Ireland (IP-2015-0390), the European Commission (FP7-PEOPLE-2013-ITN, 607786; FP7-KBBE-2012-6, CP-TP-312184; FP7-KBBE-2012-6, 311975; OCEAN 2011-2, 287589; EU2020-634486-2015), Science Foundation Ireland (SSPC-3, 12/RC/2275; SSPC-2, 12/RC/2275; 13/TIDA/B2625; 12/TIDA/B2411; 12/TIDA/B2405; 14/TIDA/2438; 15/TIDA/2977; SFI09/RFP/BMT2350), the Department of Agriculture and Food (FIRM 11/F009/MabS; FIRM 13/F/516), the Irish Research Council for Science, Engineering and Technology (GOIPG/2014/647), the Health Research Board/Irish Thoracic Society (MRCG-2014-6), the Department of the Marine (BEAU/BIOD/01), Cystic Fibrosis Foundation, USA (OG1710). The authors wish to thank Pat O'Neill for helpful discussions.References
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS.
- T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed.
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585 RSC.
- U. T. Bornscheuer and M. Hohne, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 2012, vol. 19, pp. 779–820 Search PubMed.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 CrossRef CAS.
- C. S. Fuchs, J. E. Farnberger, G. Steinkellner, J. H. Sattler, M. Pickl, R. C. Simon, F. Zepeck, K. Gruber and W. Kroutil, Adv. Synth. Catal., 2018, 360, 768–778 CrossRef CAS.
- E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernández and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1937–1942 CrossRef CAS.
- C. E. Paul, M. Rodríguez-Mata, E. Busto, I. Lavandera, V. Gotor-Fernández, V. Gotor, S. García-Cerrada, J. Mendiola, Ó. De Frutos and I. Collado, Org. Process Res. Dev., 2014, 18, 788–792 CrossRef CAS.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- S. A. Kelly, S. Mix, T. S. Moody and B. F. Gilmore, Appl. Microbiol. Biotechnol., 2020, 104, 4781–4794 CrossRef CAS.
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS.
- N. J. Turner and M. Truppo, in Sustainable Catalysis, ed. P. J. Dunn, K. K. M. Hii, M. J. Krische and M. T. Williams, John Wiley & Sons, Hoboken, 2013, vol. 3, pp. 63–74 Search PubMed.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- K. Rosenthal and S. Lütz, Curr. Opin. Green Sustainable Chem., 2018, 11, 58–64 CrossRef.
- R. E. Deasy, T. S. Moody and A. R. Maguire, Tetrahedron: Asymmetry, 2013, 24, 1480–1487 CrossRef CAS.
- D. P. Gavin, E. J. Murphy, A. M. Foley, I. A. Castilla, F. J. Reen, D. F. Woods, S. G. Collins, F. O'Gara and A. R. Maguire, Adv. Synth. Catal., 2019, 361, 2466–2474 CrossRef CAS.
- R. L. O. R. Cunha, E. A. Ferreira, C. S. Oliveira and Á. T. Omori, Biotechnol. Adv., 2015, 33, 614–623 CrossRef CAS.
- D. P. Gavin, A. Foley, T. S. Moody, U. B. R. Khandavilli, S. E. Lawrence, P. O’Neill and A. R. Maguire, Tetrahedron: Asymmetry, 2017, 28, 577–585 CrossRef CAS.
- E. Hedenström, B.-V. Nguyen and L. A. Silks, Tetrahedron: Asymmetry, 2002, 13, 835–844 CrossRef.
- M. Colombo, M. De Amici, C. De Micheli, D. Pitré, G. Carrea and S. Riva, Tetrahedron: Asymmetry, 1991, 2, 1021–1030 CrossRef CAS.
- J. C. Sih and R. L. Gu, Tetrahedron: Asymmetry, 1995, 6, 357–360 CrossRef CAS.
- Z. Molnár, E. Farkas, Á. Lakó, B. Erdélyi, W. Kroutil, B. G. Vértessy, C. Paizs and L. Poppe, Catalysts, 2019, 9, 438 CrossRef.
- M. L. Contente, M. Planchestainer, F. Molinari and F. Paradisi, Org. Biomol. Chem., 2016, 14, 9306–9311 RSC.
- J. Limanto, E. R. Ashley, J. Yin, G. L. Beutner, B. T. Grau, A. M. Kassim, M. M. Kim, A. Klapars, Z. Liu, H. R. Strotman and M. D. Truppo, Org. Lett., 2014, 16, 2716–2719 CrossRef CAS.
- N. Richter, R. C. Simon, H. Lechner, W. Kroutil, J. M. Ward and H. C. Hailes, Org. Biomol. Chem., 2015, 13, 8843–8851 RSC.
- F. J. Reen, S. Romano, A. Dobson and F. O'Gara, Mar. Drugs, 2015, 13, 4754–4783 CrossRef CAS.
- K.-H. Altmann, Chimia, 2017, 71, 646–652 CrossRef CAS.
- L. Chu, J. Huang, M. Muhammad, Z. Deng and J. Gao, Crit. Rev. Biotechnol., 2020, 40, 571–589 CrossRef CAS.
- M. Parages, J. Gutiérrez-Barranquero, F. Reen, A. Dobson and F. O'Gara, Mar. Drugs, 2016, 14, 62 CrossRef.
- I. A. Castilla, D. F. Woods, F. J. Reen and F. O'Gara, Mar. Drugs, 2018, 16, 21 CrossRef.
- S. Parte, V. L. Sirisha and J. S. D'Souza, in Advances in Food and Nutrition Research, ed. S.-K. Kim and F. Toldrá, Academic Press, 2017, vol. 80, pp. 75–106 Search PubMed.
- J. Rocha-Martin, C. Harrington, A. D. Dobson and F. O'Gara, Mar. Drugs, 2014, 12, 3516–3559 CrossRef.
- F. J. Reen, J. A. Gutierrez-Barranquero, A. D. Dobson, C. Adams and F. O'Gara, Mar. Drugs, 2015, 13, 2924–2954 CrossRef CAS.
- D. P. Gavin, F. J. Reen, J. Rocha-Martin, I. Abreu-Castilla, D. F. Woods, A. M. Foley, P. A. Sánchez-Murcia, M. Schwarz, P. O'Neill, A. R. Maguire and F. O'Gara, Sci. Rep., 2019, 9, 20285 CrossRef CAS.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
- R. Rendy, Y. Zhang, A. McElrea, A. Gomez and D. A. Klumpp, J. Org. Chem., 2004, 69, 2340–2347 CrossRef CAS.
- S. H. Lee, S. J. Park, I. S. Kim and Y. H. Jung, Tetrahedron, 2013, 69, 1877–1880 CrossRef CAS.
- N. G. Nørager, L. L. R. Lorentz-Petersen, L. O. Lyngsø, J. Kehler and K. Juhl, Synlett, 2011, 1753–1755 Search PubMed.
- S. Roesner, J. M. Casatejada, T. G. Elford, R. P. Sonawane and V. K. Aggarwal, Org. Lett., 2011, 13, 5740–5743 CrossRef CAS.
- Z. Ding, S. Xue and W. D. Wulff, Chem. – Asian J., 2011, 6, 2130–2146 CrossRef CAS.
- C. Tyllick, M. F. El-Zohry, M. Li and R. M. Roberts, J. Org. Chem., 1991, 56, 2938–2940 CrossRef CAS.
- G. Leclerc, N. Decker and J. Schwartz, J. Med. Chem., 1982, 25, 709–714 CrossRef CAS.
- L. H. Klemm and G. M. Bower, J. Org. Chem., 1958, 23, 344–348 CrossRef CAS.
- W. A. Denny, R. H. Hutchings, D. S. Johnson, J. S. Kaltenbronn, H. H. Lee, D. M. Leonard, J. B. J. Milbank, J. T. Repine, G. W. Rewcastle and A. D. White, US Pat, 6943183B2, 2001, Pfizer Search PubMed.
- C. Tang, M. Okumura, Y. Zhu, A. R. Hooper, Y. Zhou, Y.-H. Lee and D. Sarlah, Angew. Chem., Int. Ed., 2019, 58, 10245–10249 CrossRef CAS.
- N. R. Wurtz, A. Quoc Viet, S. A. Shaw, B. P. Vokits, E. K. Kick, M. N. Valente, A. K. Dilger, K. B. Pabbisetty and S. Jusuf, US Pat, 20170247396A1, 2017, Bristol Meyers Squibb Search PubMed.
- J. G. A. Walton, D. C. Jones, P. Kiuru, A. J. Durie, N. J. Westwood and A. H. Fairlamb, ChemMedChem, 2011, 6, 321–328 CrossRef CAS.
- J. X. Qiao, C. H. Hu, T. C. Wang and J. Jiang, US Pat, US201261619453P, 2013, Bristol Meyers Squibb Search PubMed.
- L. Shao, F. Wang, S. C. Malcolm, J. Ma, M. C. Hewitt, U. C. Campbell, L. R. Bush, N. A. Spicer, S. R. Engel, L. D. Saraswat, L. W. Hardy, P. Koch, R. Schreiber, K. L. Spear and M. A. Varney, Bioorg. Med. Chem., 2011, 19, 663–676 CrossRef CAS.
- R. Fernandez, A. Ros Laó, A. Magriz, H. Dietrich and J. M. Lassaletta, Tetrahedron, 2007, 2007, 6755–6763 CrossRef.
- M. Kasai, H. Ziffer and J. V. Silverton, Can. J. Chem., 1985, 63, 1287–1291 CrossRef CAS.
- B. Miriyala, S. Bhattacharyya and J. S. Williamson, Tetrahedron, 2004, 60, 1463–1471 CrossRef CAS.
- E. Grunwald and E. Price, J. Am. Chem. Soc., 1965, 87, 3139–3147 CrossRef CAS.
- A. Trincone, Mar. Drugs, 2011, 9, 478–499 CrossRef CAS.
- F. S. Duarte, G. Lach, P. R. C. Martins, G. A. Romeiro and T. C. M. De Lima, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2008, 32, 368–374 CrossRef CAS.
- F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Söding, J. D. Thompson and D. G. Higgins, Mol. Syst. Biol., 2011, 7, 539 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01848a |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2021 |