DOI:
10.1039/D5SC00064E
(Edge Article)
Chem. Sci., 2025,
16, 6458-6467
Nitrene-mediated aminative N–N–N coupling: facile access to triazene 1-oxides†
Received
7th January 2025
, Accepted 9th March 2025
First published on 10th March 2025
Abstract
Significant progress has been made in metal-catalyzed cross-coupling reactions over the past few decades. However, the development of innovative aminative coupling strategies remains highly desirable. Herein, we report a nitrene-mediated aminative N–N–N coupling reaction that leverages an anomeric amide as a key reagent to bridge amines with nitrosoarenes. This strategy enables the in situ generation of an aminonitrene intermediate, which is efficiently intercepted by nitrosoarenes, providing a direct, mild, and highly efficient route to triazene 1-oxides. Mechanistic investigations reveal that the N-substituents of the amine play a crucial role in modulating the reactivity of the aminonitrene intermediate. Complementary computational studies further indicate that aminonitrene acts as a nucleophile, while nitrosobenzene serves as an electrophile. Notably, aminonitrene–nitrosoarene coupling is significantly favored due to a substantial reduction in distortion energy, effectively outcompeting the nitrene dimerization pathway.
Introduction
Transition metal-catalyzed (TMC) coupling reactions, including the Suzuki–Miyaura, Sonogashira, and Buchwald–Hartwig reactions, have become indispensable tools for constructing C–C and C–N bonds by coupling nucleophiles and electrophiles (Nu–E, Fig. 1Ai). These methodologies are widely employed in synthesizing natural products, pharmaceuticals and agrochemicals.1–4 Complementarily, TMC oxidative coupling reactions enable the direct coupling of two nucleophiles (Nu1–Nu2, Fig. 1Aii), offering an efficient strategy to expand chemical space and diversify molecular architecture.5–9 Despite these advances, aminative coupling reactions that simultaneously link nitrogen atoms with two reactants remain highly challenging and are virtually unexplored. More recently, Liu and co-workers reported a Pd-catalyzed aminative Suzuki–Miyaura coupling, realizing a formal nitrene insertion process between nucleophilic and electrophilic partners (Nu–N*–E, Fig. 1Aiii).10 However, aminative coupling between two partners (Nu1–N*–Nu2 or Nu1–N*–E, Fig. 1Aiv) still remains highly demanding.11 The primary obstacle lies in the inherent high electronegativity of nitrogen species. Upon installing nitrogen onto the first nucleophile (Nu1), the resulting nitrogen intermediate typically retains its nucleophilic character. Without effective in situ activation strategies to convert nucleophilic nitrogen into a more reactive nitrogen species, such as a nitrene or aminyl intermediate, subsequent coupling with a second reagent is hindered (Fig. 1B, top).12–18 Therefore, developing an aminative coupling strategy that overcomes these limitations represents a significant unmet need in synthetic chemistry.
 |
| | Fig. 1 A summary of transition-metal catalyzed coupling reactions and nitrene-mediated aminative N–N–N coupling. (A) State-of-the-art in coupling reaction. (B) Our aminative coupling hypothesis. (C) Established reactivity of amines with anomeric amides. (D) Nitrene-mediated aminative N–N–N coupling (this work). | |
Nitrenes, characterized by their electron-deficient nature and possessing only six valence electrons, are versatile reactive intermediates widely employed in synthetic chemistry.19 Traditional methods of nitrene generation generally involve forming nitrene intermediates from precursors under conditions of light, heat, or transition metal catalysis, followed by reactions with C–H bonds, unsaturated π-systems, and heteroatomic compounds.20–33 However, these methods are typically limited to reactions with a single nucleophile. Aminonitrenes (1,2-diazenes) are a special class of reactive intermediates traditionally generated through the oxidation of 1,1-disubstituted hydrazines or decomposition of 1,1-disubstituted 2-sulfonyl hydrazine salts.19 These methods often require multistep procedures involving stoichiometric amounts of hazardous reagents such as mercuric oxide or lead tetraacetate, significantly constraining their utility.34 Moreover, aminonitrenes predominantly exhibit tendencies towards dimerization to tetrazenes, addition to unsaturated systems, or fragmentation reactions.35–50 In 1991, Glover et al. hypothesized the formation of an aminonitrene intermediate in the reaction of N-methyl aniline with anomeric amides, leading to tetrazene (Fig. 1C).51–55 More recently, the Levin,56–58 Lu,59–61 and other groups62–64 have leveraged aminonitrene intermediates for molecular editing through nitrogen extrusion, facilitating radical-mediated coupling to form new chemical bonds (Fig. 1C). Building on these seminal studies and our prior work on nitrene-mediated N–X (X = N, P, S) coupling reactions,65–69 we envisioned that the in situ generated aminonitrene could serve as a key reaction intermediate for coupling with another reagent to achieve a formal aminative coupling. Herein, we present a mild and efficient nitrene-mediated aminative N–N–N coupling reaction for the synthesis of triazene-1-oxide from an amine (Nu), anomeric amide (N*), and nitrosobenzene (E) (Fig. 1D). The reaction initiates with the in situ formation of an aminonitrene intermediate from the amine and anomeric amide, which is subsequently intercepted by nitrosobenzene. This strategy features several advantages, including one-step assembly, mild conditions, and broad substrate scope. Mechanistic studies reveal that N-substituents dictate the fate of the aminonitrene intermediate. The computational investigation further elucidates that the aminonitrene coupling with aryl nitroso benefits from significantly lower distortion energy than aminonitrene dimerization, thereby selectively favoring the desired aminative N–N–N coupling pathway.
Results and discussion
Triazene-1-oxides are crucial compounds with applications ranging from functional molecules to biological agents.70,71 They are used in metal determination in analytical chemistry and as intermediates in organic synthesis. They exhibit significant biological activities, including serving as fluorescence sensors and possessing antibacterial, antileukemic, anticancer, and antitrypanosomal properties.71–77 Traditional methods for synthesizing triazene-1-oxides involve two main approaches: (i) diazotization of primary aromatic amines followed by nucleophilic substitution with hydroxylamine78–80 and (ii) N-nitrosation of amines, reduction to hydrazine, and subsequent reaction with nitrosobenzene under oxidative conditions.81–84 These methods are limited by the requirement of excess amines, narrow substrate compatibility, multiple reaction steps, and the use of highly toxic metals.78–84 Currently, approaches are primarily restricted to synthesizing triazene-1-oxides from primary aromatic amines.71–82 As a result, triazene-1-oxides derived from more commonly encountered alkyl amines are exceedingly rare. This scarcity significantly hinders the exploration of their biological activities and functional properties. Therefore, developing a direct and efficient method for synthesizing triazene-1-oxides from alkyl amines is highly desirable. In our previous studies, we developed nitrene-mediated N–N coupling reactions,65–67 where aromatic amine compounds coupled with electron-deficient nitrenes under metal catalysis to form N–N65,67 and N–N–N67 linkages. Our ongoing research seeks to expand the scope of nitrene-mediated N-heteroatom bond formation by developing a strategy for N–N–N coupling, leveraging aminonitrenes in conjunction with another nitrogen-based reagent. Inspired by Glover's51–55 and other related studies,85–88 we hypothesized that amines could act as nucleophiles, initially undergoing an SN2 reaction with an anomeric amide to form an unstable N-alkoxy-hydrazinamide intermediate. This intermediate would then undergo a concerted rearrangement, yielding alkyl esters and aminonitrene species.53 Although previous reports suggest that aminonitrenes are unstable at room temperature and tend to dimerize to tetrazene, we propose that by introducing a second reagent, it might be possible to capture the highly reactive aminonitrene intermediate, thereby facilitating formal aminative coupling (Fig. 2).
 |
| | Fig. 2
a All reactions were conducted at a 0.3 mmol scale with amine 1 (0.3 mmol), anomeric amide 2 (0.36 mmol), nitrosobenzene 3 (0.45 mmol), and base (0.6 mmol) in 6.0 mL solvent. Yields were determined by NMR analysis of the reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard. b Isolated yield. c Accompanied by the formation of azoxybenzene. N.D. = not detected. DABCO = 1,4-diazabicyclo[2.2.2]octane. d The neat condition was highly exothermic, leading to vigorous gas evolution and resulting in a messy reaction mixture. | |
Reaction development
Piperazine, a privileged structure motif in numerous pharmaceutical compounds, was selected as a model substrate for this study. Initial investigations demonstrate that the reaction of 1-phenylpiperazine (1), anomeric amide (2a), and nitrosobenzene (3) in the presence of DABCO as a base in tetrahydrofuran (THF) yielded triazene-1-oxide (4) in 88% NMR yield (condition A, 85% isolated yield). A trace amount of tetrazene (5) was observed as a byproduct from aminonitrene dimerization.51–55 Notably, the intramolecular C–C coupling byproduct 6, resulting from dinitrogen extrusion, was not detected, likely due to the difficulty of fragmentation to a relatively stable radical species. Solvent screening revealed a significant influence on reaction efficiency. Low-polarity halogenated alkanes, including chloroform, dichloromethane, and tetrachloroethane, provided poor yields of the desired product 4 (∼20% yield) alongside a minor formation of byproduct 5 (∼5%, entry 2 and see the ESI† for more details). Consistent with Glover's previous reports, weakly protic solvents such as methanol and ethanol afforded 4 in only 20% yield, accompanied by 21% yield of byproduct 5 (entry 3).53 In contrast, more acidic solvents like trifluoroethanol (TFE) and hexafluoroisopropanol (HFIP) resulted in the alcoholysis of 2a to N-(benzyloxy)acetamide, completely suppressing the formation of 4 and 5 (entry 4). Ether solvents, including diethyl ether, 1,4-dioxane, methyl tert-butyl ether (MTBE), and THF, gave moderate to good yields of 4, with THF emerging as the optimal solvent, delivering an 88% yield (entries 1 and 5, see the ESI† for more details). Additionally, the reaction system exhibited strong water compatibility. Adding 3–10 equivalents of water or employing a THF–water co-solvent system (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, entry 6 and ESI†) still produced product 4 in good yield. However, the reaction concentration significantly impacted the yield (entries 7–8); under neat conditions, the reaction exclusively produced tetrazene 5, with no detectable 4 (entry 7). Temperature variation revealed that higher temperatures induced more side reactions, while room temperature conditions were more conducive to forming the desired aminative coupling product (entries 9–10). Notably, in the absence of nitrosobenzene 3, tetrazene byproduct 5 was obtained in 46% yield (entry 11). Remarkably, the desired product was still obtained in 71% yield even without DABCO (entry 12, condition B, see the ESI† for more details). Variation of the anomeric amides 2a–2f identified 2a as the most effective reagent, underscoring its critical role in enabling this aminative coupling reaction.
:1 v/v, entry 6 and ESI†) still produced product 4 in good yield. However, the reaction concentration significantly impacted the yield (entries 7–8); under neat conditions, the reaction exclusively produced tetrazene 5, with no detectable 4 (entry 7). Temperature variation revealed that higher temperatures induced more side reactions, while room temperature conditions were more conducive to forming the desired aminative coupling product (entries 9–10). Notably, in the absence of nitrosobenzene 3, tetrazene byproduct 5 was obtained in 46% yield (entry 11). Remarkably, the desired product was still obtained in 71% yield even without DABCO (entry 12, condition B, see the ESI† for more details). Variation of the anomeric amides 2a–2f identified 2a as the most effective reagent, underscoring its critical role in enabling this aminative coupling reaction.
Substrate scope
Upon establishing the optimal conditions, we extended the substrate scope of this reaction (Fig. 3). First, N-substituted piperazines, such as N-Boc, afforded the target product 7 in 83% yield. The lactam-containing piperazine derivative was also well tolerated, furnishing product 8 in 85% yield. Morpholine and thiomorpholine derivatives yielded the desired products, 9 and 10, in moderate to good yields. Similarly, substituted piperidines 11, 12, and 15 produced the desired products in moderate to good yields, notably tolerating free –OH and ether groups. Moreover, piperazines with N-substituted heterocycles at the 1-position (13–14) also provided the desired products in good to excellent yields. Furthermore, as demonstrated in examples 16–21, five- to eight-membered cyclic amines successfully afforded the desired products in moderate yields. Unfortunately, azetidine failed to yield the target product and predominantly underwent nitrogen extrusion.89,90 These results were likely due to the high strain in small rings, which shortens the lifetime of the aminonitrene intermediate and favors fragmentation over the desired aminative N–N–N coupling. Interestingly, simple, acyclic, less sterically hindered secondary amines, such as N-methylcyclohexylamine and dipropylamine, produced the desired products 22–23 in moderate yields. In contrast, bulkier secondary amines, like diisopropylamine, led only to the recovery of the starting material without any desired product formation (see the ESI† for more information). This outcome may stem from the steric hindrance of these amines, which likely prevents the initial SN2-like reaction with the anomeric amide.56 Unfortunately, primary amines, including primary aromatic amines and aliphatic amines, were incompatible with this reaction, yielding only deamination byproducts rather than the desired aminative N–N–N coupling product (see the ESI† for more details).57 Notably, natural products and pharmaceutical molecules, including cytisine, vortioxetine, amoxapine, and paroxetine, gave the desired products (24–27) in moderate to good yields. Next, we investigated the scope of various amines and aromatic nitroso compounds. For nitrosoarenes, substituents with varied electronic properties on the benzene ring motif, such as 4-Cl, 2-Et, 2-Bn, 3-COOMe, and 4-Ph, efficiently underwent aminative coupling with cyclic amines containing N-Ph, –COOMe, N-piperonyl and N-Bn to yield desired products 28–32 and 36 in moderate yields. X-ray crystallography of product 28 confirmed the accuracy of the N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(O) linkage.91 The NN moiety adopts the Z configuration in all products. Additionally, non-covalent interaction analysis92 revealed dual C–H⋯ O interactions involving nearby C–H bonds (Fig. 3, see the ESI† for more details). Interestingly, bridged-bicyclo-amine reacted with NHBoc-containing nitrosobenzene to give aminative coupling products 33 in 58% yield. Alkene-containing piperidines were well tolerated, yielding product 34 in 61% yield. Additionally, CF3-containing multi-substituted nitrosobenzenes coupled with piperazine derivatives to form 35 in good yield. A notable example includes galactose-containing nitrosobenzene efficiently coupling with the pharmaceutical molecule amoxapine, yielding product 37 in 75% yield. Surprisingly, free-COOH containing ciprofloxacin also furnished product 38 in 49% yield. Furthermore, ibuprofen and adapalene-derived nitrosobenzenes coupled with ceritinib and palbociclib, affording 39 and 40 in 56% and 40% yields, respectively. These examples further illustrate the potential of this method for the coupling of pharmaceutical molecules.
N(O) linkage.91 The NN moiety adopts the Z configuration in all products. Additionally, non-covalent interaction analysis92 revealed dual C–H⋯ O interactions involving nearby C–H bonds (Fig. 3, see the ESI† for more details). Interestingly, bridged-bicyclo-amine reacted with NHBoc-containing nitrosobenzene to give aminative coupling products 33 in 58% yield. Alkene-containing piperidines were well tolerated, yielding product 34 in 61% yield. Additionally, CF3-containing multi-substituted nitrosobenzenes coupled with piperazine derivatives to form 35 in good yield. A notable example includes galactose-containing nitrosobenzene efficiently coupling with the pharmaceutical molecule amoxapine, yielding product 37 in 75% yield. Surprisingly, free-COOH containing ciprofloxacin also furnished product 38 in 49% yield. Furthermore, ibuprofen and adapalene-derived nitrosobenzenes coupled with ceritinib and palbociclib, affording 39 and 40 in 56% and 40% yields, respectively. These examples further illustrate the potential of this method for the coupling of pharmaceutical molecules.
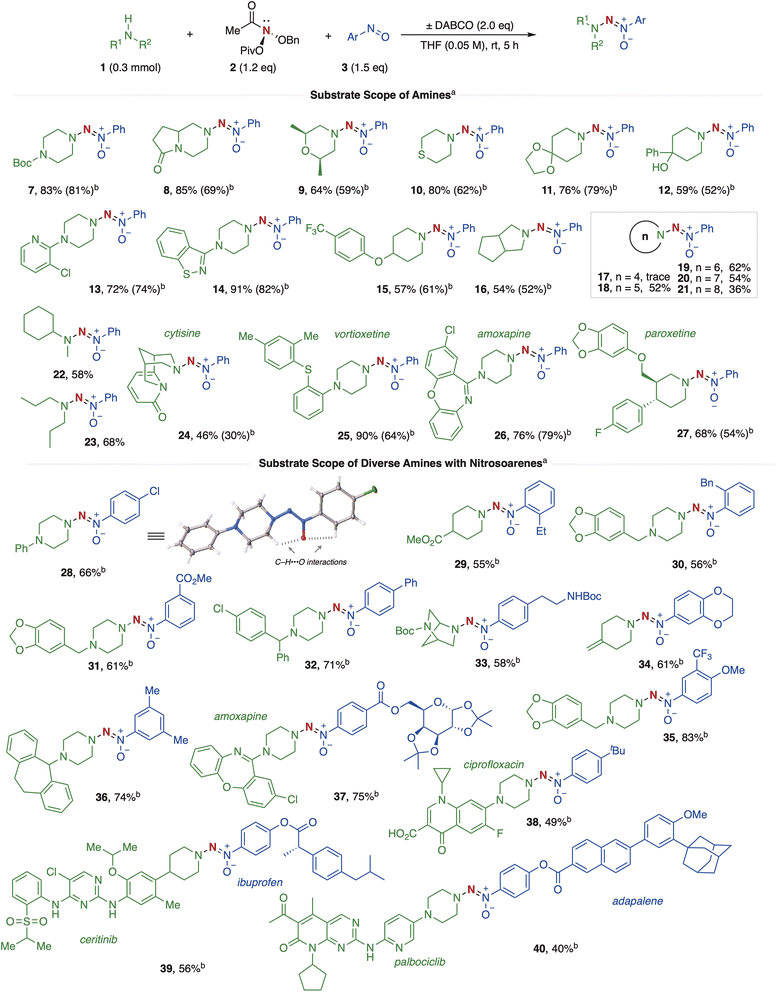 |
| | Fig. 3 Substrate scope of aminative N–N–N coupling. a Reactions were conducted at a 0.3 mmol scale, isolated yield. b Condition B: without DABCO. | |
Synthetic application
To demonstrate the synthetic utility of this method, we carried out a gram-scale synthesis of the model reaction (Fig. 4a). Remarkably, compound 4 was obtained in an impressive 77% yield (3.25 g) in the gram-scale synthesis, highlighting the efficiency of our approach. In contrast, traditional synthetic methods require the nitrosation of the amine, followed by reduction with lithium aluminum hydride (LAH) to the hydrazine and subsequent oxidation with stoichiometric lead tetraacetate in the presence of nitrosobenzene 3. This conventional route yields only 12% of compound 4 and 56% of tetrazenes as byproducts (Fig. 4b, see the ESI† for more details).82 Moreover, triazene 1-oxide 4 can be efficiently reduced to the corresponding triazene derivative 41 under either Pd/C/H2 or LAH conditions, achieving high yields of 76% and 66%, respectively (Fig. 4c). Notably, piperazine 42 could not undergo double N–N–N coupling to afford product 43 under standard conditions. However, by introducing a Boc-protecting group on one nitrogen atom prior to the first aminative N–N–N coupling, followed by Boc deprotection with hydrochloric acid and a subsequent second aminative N–N–N coupling, product 43 was successfully obtained in a four-step sequence with an overall yield of 57% (Fig. 4c). These results underscore the robustness and versatility of our synthetic strategy, making it a superior alternative to conventional methods to prepare triazene 1-oxides and unsymmetrical triazenes.81,83,84,93
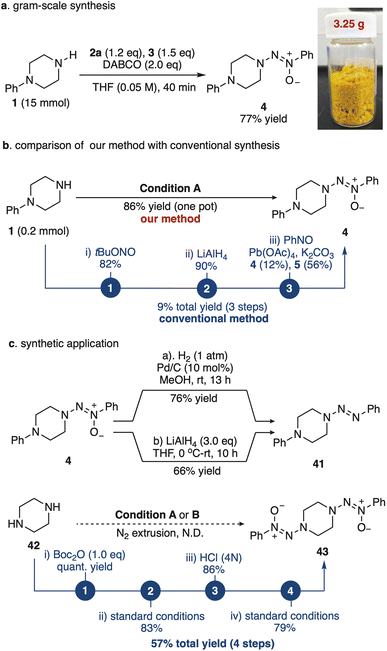 |
| | Fig. 4 The gram scale synthesis and synthetic applications. (a) Gram-scale synthesis of 4. (b) Comparison of our method with conventional synthesis. (c) Synthetic application. | |
Mechanistic studies
The mechanism of this aminative coupling was thoroughly investigated through control experiments combined with density functional theory (DFT) calculations. As shown in Fig. 5a, the addition of tetramethylpiperidine 1-oxyl (TEMPO) and butylated hydroxytoluene (BHT) had minimal impact on the yield of 4, which remained at 80% and 79% yields, respectively. This observation suggests that the reaction likely proceeds via a non-radical pathway, given that the yields of 4 were largely unaffected by these radical scavengers (see the ESI† for more details). Moreover, in the absence of nitrosobenzene 3, byproduct 5 was also isolated in yields of 39% and 46%, respectively, providing additional evidence for the non-radical nature of this transformation. Additionally, we explored N-substituents’ effect on the aminonitrene species' reactivity. Drawing from the established radical stability order94 (allylic > benzylic > 3° > 2° > 1° > methyl > vinyl) and previous related studies,56–58 we selected allylic and benzylic substrates to investigate how these substituents affect and control the aminonitrene's fate. As shown in Fig. 5b, these results suggest the following conclusions: (1) the formation of a benzylic radical is crucial for the fragmentation process that leads to the C–C coupling products. (2) Conversely, allylic radicals alone are ineffective in facilitating C–C bond formation via radical recombination. Notably, when using 1d as substrate, the intramolecular C–C coupling product (37% yield) and the desired triazene-1-oxide (45%) were obtained. Furthermore, with 1e as substrate, the desired triazene-1-oxide was exclusively produced in 59% yield. These findings suggest that the N-alkyl group with lower radical stability can prolong the lifetime of the aminonitrene intermediate, allowing it to persist long enough to undergo an intermolecular reaction, thereby forming either triazene-1-oxide or tetrazenes.51–58
 |
| | Fig. 5 Radical trapping and N-substituent effects studies. (a) Radical trapping experiments. (b) N-Substituent effects. | |
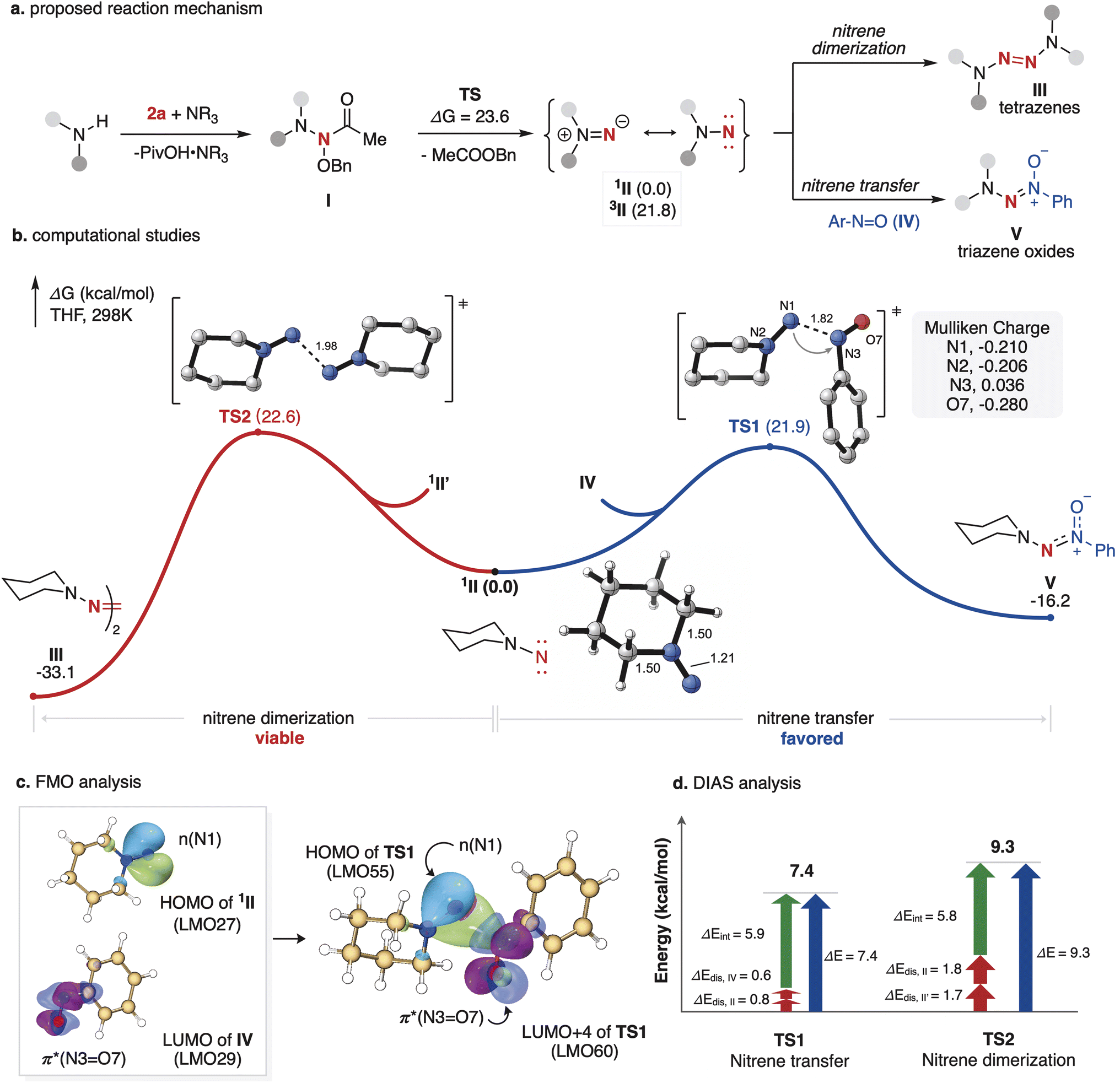
Based on our preliminary mechanistic studies and previous related studies, we proposed the mechanism for this aminative coupling reaction. As depicted in Fig. 6a, the amine (Nu1) undergoes an SN2 reaction with anomeric amide 2 under basic conditions, forming the unstable intermediate I, which is likely a transient nitrene precursor. Subsequently, intermediate I undergoes a concerted rearrangement, resulting in the extrusion of benzyl acetate to generate aminonitrene II, which further reacts with nitrosobenzene to produce triazene-1-oxide IV or dimerizes to tetrazenes III. To gain a deeper insight into the aminative coupling mechanism, density functional theory (DFT) calculations were performed at the SMD(THF)-M06-2X/Def2-TZVPP//M06-2X/Def2-SVP level of theory. Aminonitrene II is generated from I by traversing TS with a 23.6 kcal mol−1 energy barrier. Setting singlet aminonitrene 1II as the zero energy point, it was found that singlet aminonitrene 1II is energetically more stable than its triplet counterpart by 21.8 kcal mol−1 (see the ESI† for more details). The optimized structural analysis of aminonitrene 1II shows that the Mayer bond order (MBO) of N1N2 is 1.76, indicating that the N1N2 bond is best viewed as a double bond. The near planar geometry of the N1–C3–C4–N2 moiety, with a dihedral angle of 0.73°, further supports this (see the ESI† for more details). Frontier molecular orbital (FMO) analysis92 revealed that the highest occupied molecular orbital (HOMO) of aminonitrene (1II) is primarily composed of the lone pair on N1, indicating its nucleophilic character.35 Meanwhile, the lowest unoccupied molecular orbital (LUMO) of IV is predominantly localized on the O7 and N3 atoms, best described as a π* (O7N3) orbital, suggesting its electrophilic nature (Fig. 6c). As shown in Fig. 6b, the nucleophilic attack of aminonitrene 1II on nitrosobenzene IV proceeds via the transition state TS1, with an energy barrier of 21.9 kcal mol−1. Mulliken charge population analysis reveals that the N1 atom in 1II has a charge of −0.210, while the N3 atom in IV has a charge of 0.036. These values indicate that 1II acts as a nucleophile,35 whereas IV functions as an electrophile, consistent with the frontier molecular orbital (FMO) analysis (Fig. 6c). In comparison, the nitrene dimerization through TS2 shows a slightly higher energy barrier of 22.6 kcal mol−1 (ΔΔG(TS2vs.TS1) = 0.7 kcal mol−1), consistent with our experimental observations that the formation of small amounts of tetrazenes always accompanies the reaction. Furthermore, the distortion/interaction activation strain (DIAS) analysis95 reveals that the electronic energy difference between TS1 and TS2 is minimal (1.9 kcal mol−1), aligning well with the DFT calculations. Although TS1 and TS2 exhibit similar interaction energies (ΔEint), their distortion energies differ significantly (ΔEdist = 1.4 kcal mol−1 and 3.5 kcal mol−1, respectively), indicating that the reactivities of aminonitrene are primarily governed by the distortion energy (Fig. 6d).
 |
| | Fig. 6 Mechanistic studies. (a) Proposed reaction mechanism. (b) Computational studies of nitrene dimerization (red line) and nitrene transfer (blue line). (c) Frontier molecular orbital (FMO) analysis (isovalue = 0.05). (d) Distortion/interaction activation strain analysis. | |
Conclusions
In conclusion, we have developed a nitrene-mediated aminative N–N–N coupling that operates under mild, environmentally benign conditions with a broad substrate scope, facilitating the efficient and selective formation of triazene-1-oxide. Mechanistic studies highlight the critical role of N-substituents on the amine in dictating the fate of the aminonitrene intermediate. Furthermore, computational studies reveal that aryl nitroso and aminonitrene coupling require much lower distortion energy than the competing nitrene dimerization pathway, favoring the desired aminative coupling. This novel methodology provides a versatile tool for the synthesis of triazene-1-oxides. It expands the chemical space for aminative coupling strategies, holding significant potential for application in drug discovery and molecular engineering.
Data availability
The X-ray crystallographic data for compound 28 have been deposited in the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe with CCDC no. 2397933, and can be obtained free of charge from the CCDC viahttp://www.ccdc.cam.ac.uk/getstructures. All other data supporting the findings of this study are available within the article and its ESI.†
Author contributions
S. Zhu conducted most of the reaction optimization, structural determination of products, and preparation of the ESI.† H.-R. Zhang, B.-Y. Sun, and Z.-Q. Bai contributed to the expansion of the substrate scope. H. Wang performed the computational studies. G. He and G. Chen supervised part of the experimental studies. H. Wang conceived the initial ideas for this project, provided overall supervision, and prepared most of the manuscript.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFA1508800 and 2022YFA1504303), the National Natural Science Foundation of China (22371144 and 92256302), the Frontiers Science Center for New Organic Matter (63241101), and the Fundamental Research Funds for the Central Universities (63243107 and 63241208).
References
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
-
S. E. Denmark and R. F. Sweis, Metal-catalyzed cross-coupling reactions, Wiley-VCH, Weinheim, 2004, pp. 163–216 Search PubMed.
-
I. D. Kostas, Transition metal catalyzed cross-coupling reactions, MDPI – Multidisciplinary Digital Publishing Institute, Basel, Switzerland, 2021 Search PubMed.
- A. Lei, C. Liu and L. Jin, Synlett, 2010, 2527–2536 CrossRef.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
-
C. He, H. Zhang, W. Liu, C. Liu, W. Shi and A. Lei, Oxidative cross-coupling reactions, Wiley, Weinheim, Germany, 2016 Search PubMed.
-
A. Lei, Transition metal catalyzed oxidative cross-coupling reactions, Springer, Berlin, Heidelberg, 2019 Search PubMed.
- P. Onnuch, K. Ramagonolla and R. Y. Liu, Science, 2024, 383, 1019–1024 CrossRef CAS.
- C. Stein, J. L. Tyler, J. Wiener, F. Boser, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2024, 63, e202418141 Search PubMed.
- V. C. M. Gasser, S. Makai and B. Morandi, Chem. Commun., 2022, 58, 9991–10003 RSC.
- L. G. O'Neil and J. F. Bower, Angew. Chem., Int. Ed., 2021, 60, 25640–25666 CrossRef PubMed.
- E. Jarvo and T. Barker, Synthesis, 2011, 2011, 3954–3964 CrossRef.
- D. Jinan, P. P. Mondal, A. V. Nair and B. Sahoo, Chem. Commun., 2021, 57, 13495–13505 RSC.
- C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- A. I. Olivos Suarez, V. Lyaskovskyy, J. N. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef PubMed.
-
W. Lwowski, Nitrenes, John Wiley & Sons, Inc., Weinheim, 1970 Search PubMed.
- G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed.
- J. Qin, Z. Zhou, T. Cui, M. Hemming and E. Meggers, Chem. Sci., 2019, 10, 3202–3207 RSC.
- H. Shi and D. J. Dixon, Chem. Sci., 2019, 10, 3733–3737 RSC.
- H. M. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- J. L. Roizen, M. Harvey and J. D. Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- J. Egger and E. M. Carreira, Nat. Prod. Rep., 2014, 31, 449–455 RSC.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526–1532 RSC.
- Y.-D. Du, C.-Y. Zhou, W.-P. To, H.-X. Wang and C.-M. Che, Chem. Sci., 2020, 11, 4680–4686 RSC.
- M. Ju and J. M. Schomaker, Nat. Rev. Chem., 2021, 5, 580–594 CrossRef CAS PubMed.
- B. Du, C. M. Chan, C. M. Au and W. Y. Yu, Acc. Chem. Res., 2022, 55, 2123–2137 CrossRef CAS PubMed.
-
C. M. Che, V. K. Y. Lo and C. Y. Zhou, Comprehensive Organic Synthesis II, 2014, pp. 26–85 Search PubMed.
- M. N. Cosio and D. C. Powers, Nat. Rev. Chem., 2023, 7, 424–438 CrossRef CAS PubMed.
- A. Fanourakis and R. J. Phipps, Chem. Sci., 2023, 14, 12447–12476 RSC.
- T. A. Trinh, S. Cherempei, D. S. Rampon and J. Schomaker, Chem. Sci., 2025, 16, 4796–4805 RSC.
- M. A. Kuznetsov and B. V. Ioffe, Russ. Chem. Rev., 1989, 58, 732–746 CrossRef.
- B. V. Ioffe and M. A. Kuznetsov, Russ. Chem. Rev., 1972, 41, 131–145 CrossRef.
- R. S. Atkinson and C. W. Rees, J. Chem. Soc., 1969, 772–778 CAS.
- D. J. Anderson, D. C. Horwell and C. W. Rees, J. Chem. Soc., 1970, 576–582 CAS.
- R. S. Atkinson and J. R. Malpass, J. Chem. Soc., Perkin Trans. 1, 1977, 2242–2249 RSC.
- P. B. Dervan and T. Uyehara, J. Am. Chem. Soc., 1979, 101, 2076–2082 CrossRef CAS.
- W. D. Hinsberg III and P. B. Dervan, J. Am. Chem. Soc., 1979, 101, 6142–6144 CrossRef.
- T. Siu and A. K. Yudin, J. Am. Chem. Soc., 2002, 124, 530–531 CrossRef CAS PubMed.
- J. Li, J.-L. Liang, P. W. H. Chan and C.-M. Che, Tetrahedron Lett., 2004, 45, 2685–2688 CrossRef CAS.
- R. D. Richardson, M. Desaize and T. Wirth, Chem.–Eur. J., 2007, 13, 6745–6754 CrossRef CAS PubMed.
- J. K. Mitchell, W. A. Hussain, A. H. Bansode, R. M. O'Connor and M. Parasram, J. Am. Chem. Soc., 2024, 146, 9499–9505 CrossRef CAS PubMed.
- D. M. Lemal and T. W. Rave, J. Am. Chem. Soc., 1965, 87, 393–394 CrossRef CAS.
- C. G. Overberger, J. G. Lombardino and R. G. Hiskey, J. Am. Chem. Soc., 1957, 79, 6430–6435 CrossRef CAS.
- R. L. Hinman and K. L. Hamm, J. Am. Chem. Soc., 1959, 81, 3294–3297 CrossRef CAS.
- D. M. Lemal, T. W. Rave and S. D. Mcgregor, J. Am. Chem. Soc., 1963, 85, 1944–1948 CrossRef CAS.
- G. K. Goga and J.-P. A, J. Am. Chem. Soc., 1969, 91, 4323–4324 CrossRef.
- P. G. Schultz and P. B. Dervan, J. Am. Chem. Soc., 1982, 104, 6660–6668 CrossRef CAS.
- J. J. Campbell and S. A. Glover, J. Chem. Soc., Perkin Trans. 2, 1991, 2067–2079 RSC.
- J. J. Campbell and S. A. Glover, J. Chem. Soc., Perkin Trans. 2, 1992, 1661–1663 RSC.
- S. A. Glover, Tetrahedron, 1998, 54, 7229–7271 CrossRef CAS.
- J. J. Campbell and S. A. Glover, J. Chem. Res., 1999, 474–475 CAS.
- S. A. Glover, G. Mo, A. Rauk, D. J. Tucke and P. Turner, J. Chem. Soc., Perkin Trans. 2, 1999, 2053–2058 RSC.
- S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS.
- K. J. Berger, J. L. Driscoll, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 17366–17373 CrossRef CAS.
- J. Masson-Makdissi, R. F. Lalisse, M. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2024, 146, 17719–17727 CrossRef CAS PubMed.
- X. Zou, J. Zou, L. Yang, G. Li and H. Lu, J. Org. Chem., 2017, 82, 4677–4688 CrossRef CAS PubMed.
- H. Qin, W. Cai, S. Wang, T. Guo, G. Li and H. Lu, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS PubMed.
- T. Guo, J. Li, Z. Cui, Z. Wang and H. Lu, Nat. Synth., 2024, 3, 913–921 CrossRef CAS.
- C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, J. Am. Chem. Soc., 2021, 143, 18864–18870 CrossRef CAS PubMed.
- B. A. Wright, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. E. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef CAS.
- M. Gauthier, J. B. M. Whittingham, A. Hasija, D. J. Tetlow and D. A. Leigh, J. Am. Chem. Soc., 2024, 146, 29496–29502 CrossRef CAS PubMed.
- H. Wang, H. Jung, F. Song, S. Zhu, Z. Bai, D. Chen, G. He, S. Chang and G. Chen, Nat. Chem., 2021, 13, 378–385 CrossRef CAS PubMed.
- F. Song, S. Zhu, H. Wang and G. Chen, Chin. J. Org. Chem., 2021, 41, 4050–4058 CrossRef CAS.
- S.-Y. Zhu, W.-J. He, G.-C. Shen, Z.-Q. Bai, F.-F. Song, G. He, H. Wang and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202312465 CrossRef CAS PubMed.
- Z. Bai, F. Song, H. Wang, W. Cheng, S. Zhu, Y. Huang, G. He and G. Chen, CCS Chem., 2022, 4, 2258–2266 CrossRef CAS.
- Z. Bai, S. Zhu, Y. Hu, P. Yang, X. Chu, G. He, H. Wang and G. Chen, Nat. Commun., 2022, 13, 6445 CrossRef CAS PubMed.
-
D. E. V. Wilman, Proceedings of the 3rd International Symposium on the Biological Oxidation of Nitrogen in Organic Molecules, Ellis Horwood, Chichester, 2003, pp. 297–302 Search PubMed.
- M. A. Martins, P. R. Salbego, G. A. de Moraes, C. R. Bender, P. J. Zambiazi, T. Orlando, A. B. Pagliari, C. P. Frizzo and M. Hörner, CrystEngComm, 2018, 20, 96–112 RSC.
- G. M. de Oliveira, M. Hörner, A. Machado, D. F. Back, J. H. Monteiro and M. R. Davolos, Inorg. Chim. Acta, 2011, 366, 203–208 CrossRef.
- M. F. G. Stevens, A. Gescher and C. P. Turnbull, Biochem. Pharmacol., 1979, 28, 769–776 CrossRef CAS PubMed.
- Z. Rachid, M. Macphee, C. Williams, M. Todorova and B. J. Jean-Claude, Bioorg. Med. Chem. Lett., 2009, 19, 5505–5509 CrossRef CAS PubMed.
- H. Aneetha, J. Padmaja and P. S. Zacharias, Polyhedron, 1996, 15, 2445–2451 CrossRef CAS.
- B. A. Iglesias, D. F. Back, M. Hörner, E. R. Crespan and F. Broch, J. Organomet. Chem., 2014, 752, 12–16 CrossRef CAS.
- K. Alizadeh, B. Rezaei and E. Khazaeli, Sens. Actuators, B, 2014, 193, 267–272 CrossRef CAS.
- K. Vaughan, L. M. Cameron, S. Christie and M. J. Zaworotko, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1985–1988 CrossRef.
- A. J. R. W. A. dos Santos, P. Bersch, H. P. M. de Oliveira, M. Hörner and G. L. Paraginski, J. Mol. Struct., 2014, 1060, 264–271 CrossRef.
- D. Enders, C. Rijksen, E. Bremus-Köbberling, A. Gillner and J. Köbberling, Tetrahedron Lett., 2004, 45, 2839–2841 CrossRef CAS.
- E. Bamberger and A. Stiegelmann, Ber. Dtsch. Chem. Ges., 1899, 32, 3554–3560 CrossRef CAS.
- L. Hoesch and B. Köppel, Helv. Chim. Acta, 1981, 64, 864–889 CrossRef CAS.
- S. G. Zlotin, O. V. Prokshits, N. F. Karpenko and O. A. Lukyanov, Russ. Chem. Bull., 1990, 39, 1526–1528 CrossRef.
- R. Moriarty, T. Hopkins, I. Prakash, B. Vaid and R. Vaid, Synth. Commun., 1990, 20, 2353–2357 CrossRef CAS.
- In our previous work, we found that the [IrCp*Cl2]2-catalyzed N–N coupling reaction using dioxazolone as a nitrene precursor with nitrosoarene demonstrates efficient reactivity.
- B. G. Cai, C. Empel, W. Z. Yao, R. M. Koenigs and J. Xuan, Angew. Chem., Int. Ed., 2023, 62, e202312031 CrossRef CAS PubMed.
- Z.-L. Chen, Q.-Q. Li, A. Studer and J. Xuan, Sci. China: Chem., 2024, 36, 110425 Search PubMed.
- W. Huang, H. Yan, Q. Wang, J. Chen, Y. Luo and Y. Xia, Org. Chem. Front., 2024, 11, 2561–2565 RSC.
- C. L. Bumgardner, K. J. Martin and J. P. Freeman, J. Am. Chem. Soc., 1963, 85, 97–99 CrossRef CAS.
- J. P. Freeman and W. H. Graham, J. Am. Chem. Soc., 1967, 89, 1761–1762 CrossRef CAS.
- Deposition Number 2397933† (for 28) contains the supplementary crystallographic data for this paper.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- G. Kiefer, T. Riedel, P. J. Dyson, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 302–305 CrossRef CAS.
-
M. S. Sherburn, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley-VCH, 2012, pp. 1–23 Search PubMed.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Gong
Chen
ab,
Gong
Chen