
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular access to nucleobase GFP-surrogates: pH-responsive smart probes for ratiometric nucleic acid diagnostics†
Keenan T.
Regan‡
a,
Austin
Pounder‡
b,
Ryan E.
Johnson
a,
Makay T.
Murray
b,
Hannah X.
Glowacki
a,
Stacey D.
Wetmore
 *b and
Richard A.
Manderville
*a
*b and
Richard A.
Manderville
*a
aDepartment of Chemistry & Toxicology, University of Guelph, Guelph, Ontario N1G 2W1, Canada. E-mail: rmanderv@uoguelph.ca
bDepartment of Chemistry & Biochemistry, University of Lethbridge, Lethbridge, Alberta T1K 3M4, Canada. E-mail: stacey.wetmore@uleth.ca
First published on 13th March 2025
Abstract
We have utilized a modular on-strand aldol approach to synthesize chalcone-based fluorescent molecular rotors (FMRs) bearing phenolic oxygen donors that mimic the natural tyrosine (Tyr66) chromophore 4-hydroxybenzylidene-imidazolinone (HBI) within green fluorescent proteins (GFPs). Leveraging the FMRs' propensity to undergo non-radiative decay via twisted intramolecular charge transfer upon excitation within certain microenvironments, we have addressed the longstanding issues of poor brightness (εmax × Φfl) and weak turn-on responses for GFP-surrogates within nucleic acids. To demonstrate its potential and lay the groundwork for future applications, these FMRs were incorporated into NarI12 and TBA15 oligonucleotides with canonical (A, C, T, G) or locked nucleic acids (LNAs) (TL, AL) as flanking bases. The resulting duplexes and G-quadruplexes (GQs) were studied using fluorescence spectroscopy, molecular dynamics simulations, and quantum mechanical calculations, yielding a comprehensive understanding of their structural and photophysical properties in DNA, DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RNA, and GQ contexts. Electron-rich chalcones favor neutral phenol excitation (ROH) to afford both phenol (ROH*) and phenolate (RO−*) emission, with the latter generated through an intermolecular excited-state proton transfer process, while electron-deficient chalcones serve as ratiometric excitation indicators, due to their photoacidity. The surrogates display strong turn-on responses (up to 154-fold) in a GQ → duplex topology switch with flanked LNAs, giving Φfl up to 0.58 and molar brightness ∼ 15000 cm−1 M−1 in the duplex. By synergizing the NA sequence and probe, we achieve a switchable ON-to-OFF photoinduced electron transfer, resulting in a 134-fold turn-on emission response to pH. Our findings are the first to optimize the performance of GFP-surrogates as internal nucleobase replacements and suggest multiple ways in which they may be useful tools for NA diagnostics.
:RNA, and GQ contexts. Electron-rich chalcones favor neutral phenol excitation (ROH) to afford both phenol (ROH*) and phenolate (RO−*) emission, with the latter generated through an intermolecular excited-state proton transfer process, while electron-deficient chalcones serve as ratiometric excitation indicators, due to their photoacidity. The surrogates display strong turn-on responses (up to 154-fold) in a GQ → duplex topology switch with flanked LNAs, giving Φfl up to 0.58 and molar brightness ∼ 15000 cm−1 M−1 in the duplex. By synergizing the NA sequence and probe, we achieve a switchable ON-to-OFF photoinduced electron transfer, resulting in a 134-fold turn-on emission response to pH. Our findings are the first to optimize the performance of GFP-surrogates as internal nucleobase replacements and suggest multiple ways in which they may be useful tools for NA diagnostics.
Introduction
Fluorescent proteins have revolutionized bioimaging by enabling the genetic tagging and dynamic monitoring of target proteins in living organisms.1–3 The discovery and development of the green fluorescent protein (GFP) marked a breakthrough in molecular biology, earning the Nobel Prize in Chemistry in 2008.4 GFP and its derivatives have been continually improved to meet the demands of advanced imaging applications. Inspired by the remarkable fluorescent characteristics of nature's GFP, along with its synthetic and genetic variants, numerous research groups have sought to apply GFP-like fluorescent characteristics to nucleic acids (NAs).Fluorescence techniques are indispensable for probing fundamental molecular mechanisms involving NAs – including folding,5 replication, transcription, and translation6,7 – due to their high sensitivity and exceptional spatiotemporal resolution. Beyond monitoring such NA mechanisms, there is an increasing demand for NA-based biosensors, which serve as critical molecular recognition elements for many applications such as those in medicine, food inspection, and environmental monitoring.8 However, since natural nucleotides are essentially nonfluorescent, there is a critical need for suitable emissive analogs that can seamlessly substitute for natural nucleotides, while providing bright and environmentally sensitive emission.
One approach involves labeling NAs through non-covalent interactions, such as intercalation. Following this principle, fluorogenic aptamers have been designed to facilitate the binding and emission intensity of conditionally fluorescent probes to NAs through non-covalent interactions.9–14 The two probes that stand out for these applications are GFP chromophores derived from 4-hydroxybenzylidene imidazolinone (HBI) ligands for Spinach,15 Broccoli,16 Corn17 and Chili18 and thiazole orange (TO) for the mango variants.19 Both HBI10 and TO11 are classic fluorescent molecular rotors (FMRs) with strongly quenched emission in the free state with impressive turn-on fluorescence upon NA binding. However, as free labels, these dyes can suffer from non-specific activation12 and other significant drawbacks, including cytotoxicity.13 Moreover, NA-based fluoromodules can suffer from inadequate fluorophore-binding affinity to the NA, and misfolding tendencies.14,20,21
A second, more robust approach involves covalent modification with internal probes, which is considered the gold standard for NA-based molecular recognition sensing. This method offers site-specific information, while minimizing susceptibility to false readouts.22–28 While hundreds of internal fluorescent probes of various designs have been reported over the decades, the TO family of surrogates called the FIT (forced intercalation) probes probably offer the best light-up responses to hybridization through intercalation of the covalently bound TO surrogate.29–31 In sharp contrast, previous attempts to incorporate HBI analogs into NAs have created internal probes that are tedious to synthesize, with poor Φfl upon hybridization, limiting their practical applications.32–37 For instance, Chowdhury and coworkers developed a chimeric GFP-uracil (GFP-U) probe for incorporation into DNA:PNA hybrid duplexes (Fig. 1A(i)),35 exhibiting a 3-fold turn-on response upon base pairing with adenine. Kanamori and colleagues synthesized dUHBI and dUFBI conjugates, achieving modest emission increases of 5.8-fold and 1.8-fold, respectively, upon triplex formation.36 The most successful prior effort was by Fischer and coworkers, who developed the ONHBI conjugate for RNA hybridization detection.37 Despite achieving a 16-fold turn-on response at 460 nm with a Φfl of 0.52, the probe only offers single-wavelength detection due to the replacement of the phenolic donor with an anisole. Furthermore, the use of a long, flexible non-emissive tether between the chromophore and the dU base limits use as a molecular reporter of location and environment.23
 | ||
| Fig. 1 (A) (i) Previously attempted nucleic acid GFP-surrogates, (ii) TICT schematic with and without nucleic acid hybridization, and (iii) light-up responses for previously attempted nucleic acid GFP-surrogates versus this work. (B) This work employs phenolic O-donor aldehydes to generate phenolic-6HI surrogates structurally related to the GFP chromophore HBI. | ||
Recently, we presented a new modular synthesis approach for surrogate incorporation into NAs that employes a latent enolate handle for aldol condensation with aromatic aldehydes to afford chalcone FMRs.38 We employed 6-hydroxy-indanone (6HI) attached to an acyclic glycerol linker for condensation with aromatic aldehydes containing nitrogen donors (N-donors) to create a family of push–pull FMRs. The resulting probes rival the TO family in terms of probe brightness in duplex DNA (up to 17500 cm−1 M−1), but with much larger Stokes' shifts for superior resolution.38 We have also utilized the N-donors in a fluorescent aptasensor platform for detecting toxic heavy metal ions in G-quadruplex (GQ) motifs.39 Their ease of on-strand synthesis and structural resemblance of the indole chalcone to the tryptophan GFP variant40 suggested the utility of the modular approach for creating oxygen donor (O-donor) mimics of the natural HBI (Fig. 1B). Furthermore, O-donor chalcones were expected to provide diagnostic advantages compared to the N-donor variants, due to their ability to offer dual λex/λem wavelengths, serving as ‘smart’ probes for either ratiometric emission or excitation diagnostics.41,42 This capability provides superior resolution and self-calibration, significantly enhancing the accuracy and reliability of the probe in NA biosensing applications.41,42
Herein, we report the synthesis and properties of the O-donor chalcones, which overcome the previous limitations in covalently labeled GFP-surrogates in NAs. The O-donors were incorporated into NarI12 and TBA15 oligonucleotides with canonical (A, C, T, G) or locked nucleic acids (TL, AL) as flanking bases, enabling the investigation of the physical and optical properties of DNA, DNA:RNA duplexes and GQs. The identity of the nucleobases was found to strongly influence optical properties, with photoinduced electron transfer (PET) quenching playing a pivotal role due to the ability to transition the probe from a PET-ON (quenched emission) to a PET-OFF (strong emission) state by adjusting the NA topology or pH for substantial light-up emission, molar brightness, and Stokes' shifts. The tunability of our design enables the creation of probes that modulate ground-state or excited-state ionization in response to hybridization, significantly outperforming existing GFP-like labels (Fig. 1A(iii)). Our innovative approach not only paves the way for the development of next-generation fluorescent probes with unprecedented precision and versatility, but also affords greater sensitivity and precision in the instruments available in the molecular biologist's toolbox.
Results and discussion
Free dye properties
The O-donor library (Fig. 1B) consists of the parent chalcone (PhOH6HI), which is structurally like HBI, and two fluorinated derivatives (FPhOH6HI and DFPhOH6HI), with DFPhOH6HI serving as a DFHBI mimic. DFHBI has a phenolic pKa of 5.5 and served as the target for the original Spinach RNA aptamer to favor phenolate binding for increased brightness (B ∼ 17500 cm−1 M−1).15 Other members include DMePhOH6HI, which contains electron-donating methyl groups that increase dye lipophilicity and NapOH6HI that extends π-conjugation. The O-donor library was synthesized as free probes through Aldol condensations of the phenolic aldehydes with 6-methoxy-indanone (6MI) and were obtained as E-isomers, as evidenced by NMR38,39 (ESI, Fig. S1–S5†).
The photophysical properties of the O-donor-6MI probes were characterized in MeOH (Table S1 and Fig. S6, ESI†). Excitation of the neutral ROH peak only generated phenolate emission for DFPhOH6MI (564 nm), suggesting excited-state proton transfer (ESPT) in MeOH and an excited-state pKa 10 The other analogs all yielded single ROH* emission (469–538 nm), with no evidence for the phenolate (RO−*) through ESPT, suggesting
10 The other analogs all yielded single ROH* emission (469–538 nm), with no evidence for the phenolate (RO−*) through ESPT, suggesting  values > 0.10 The phenolates emit in the 556–565 nm range in basic MeOH, while the naphtholate of the π-extended NapOH6MI emitted at 698 nm.
values > 0.10 The phenolates emit in the 556–565 nm range in basic MeOH, while the naphtholate of the π-extended NapOH6MI emitted at 698 nm.
PhOH6MI, FPhOH6MI and DFPhOH6HI were sufficiently soluble in aqueous buffer to determine pKa values. The ground-state pKa of PhOH6MI was 8.26, closely matching the pKa value of 8.1 for HBI,43 while FPhOH6MI exhibited a pKa = 7.11, which was reduced to 5.9 for DFPhOH6MI and closely matches the pKa of 5.5 for DFHBI.15 The excited-state  of the parent PhOH6MI was determined using the Förster cycle,44 yielding a value of 1.15 (Table S1†). This value falls within the lower (−1.3) and upper (2.1) estimates for the
of the parent PhOH6MI was determined using the Förster cycle,44 yielding a value of 1.15 (Table S1†). This value falls within the lower (−1.3) and upper (2.1) estimates for the  of HBI.10
of HBI.10
The FMR character of the O-donors was confirmed by comparing their emission intensities in pure MeOH versus a 25:75 MeOH:glycerol mixture (Table S1†). Each phenol exhibited enhanced fluorescence in 25:75 MeOH:glycerol, with relative light-up intensity values (Irel) ranging from 4 to 12.
On-strand DNA synthesis
We first focused on attaching the various phenolic aldehydes to the 6HI handle within NarI12 oligos (Fig. 2). Attempts at promoting condensation using NaOH led to little desired product, as evidenced via HPLC analysis. We hypothesized that this outcome was due to phenolate formation under the strongly basic conditions to diminish aldehyde electrophilicity necessary for aldol condensation. Treatment with piperidine did yield condensation product for PhOH6HI (21%), DMePhOH6HI (28%) and NapOH6HI (42%) surrogates, but failed to yield product for the two fluorinated analogs. Notably, when the weaker amine base morpholine (pKb ∼ 6) was employed, both FPhOH6HI and DFPhOH6HI were obtained in ∼30% yield, while yields of the other surrogates increased to 50–60%. Thus, morpholine was utilized for the synthesis of the O-donor library (see ESI† for HPLC chromatograms (Fig. S8†) and MS of modified oligos (Table S2†)).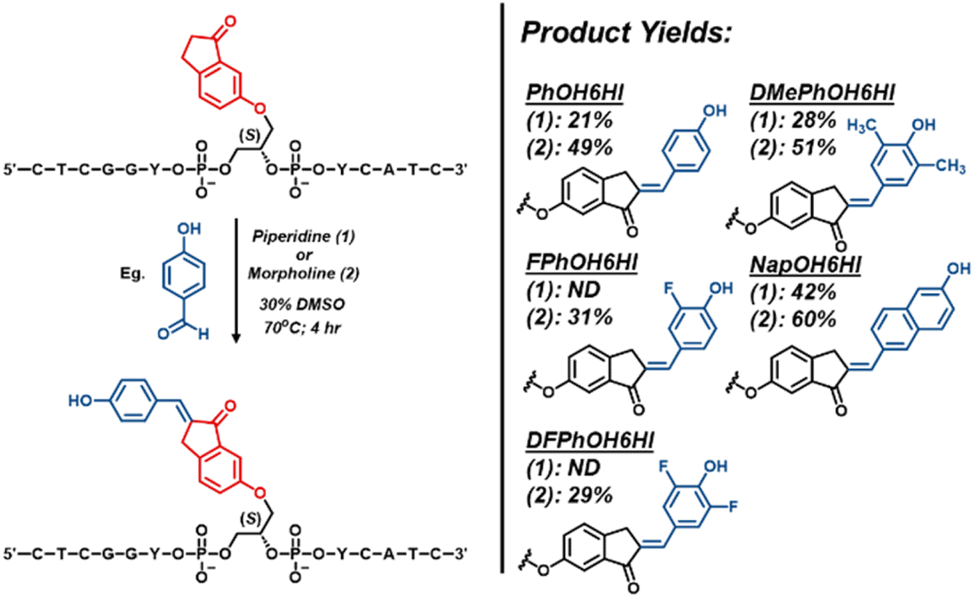 | ||
| Fig. 2 General scheme for the on-strand aldol condensation using either piperidine (1) or morpholine (2), with respective surrogate structures and yields from HPLC analysis. | ||
GFP-surrogates in TXT NarI12
The fluorescence of the O-donor library was initially characterized and studied in the single-strand (SS) context using 12-mer TXT NarI12 sequences 5′-CTCGGT-X-TCATC-3′ (X = probe) in PBS buffer at pH 7.4. The modified oligos were also hybridized to three different complementary strands (full-length (FL) DNA with X paired opposite dC, an N-1 DNA complement with the surrogate present as a central bulge, and a FL 2′-OMe-RNA strand with the probe paired opposite C for direct comparison to the FL DNA duplex) for comparison to the SS. Melting temperatures and photophysical properties of the 15 modified duplexes are summarized in Table S3.† For PhOH6HI, DMePhOH, and NapOH, the buffer conditions favor the neutral ROH with single excitation bands at 390–407 nm in both the SS and duplexes. These neutral probes did not dramatically impact duplex stability, with ΔTm values ranging from +1.3 to −4.6 °C compared to the native helix containing X = G paired with C. All three probes exhibited relatively strong ROH* emission in the 495–565 nm region when paired with the RNA complement to produce the hybrid duplex, as exemplified for PhOH6HI in Fig. 3A (green trace, Φfl = 0.27, B = 7130 cm−1 M−1). The emission of PhOH6HI in the hybrid DNA:RNA duplex represents a 20-fold light-up response compared to the SS and 3.2-fold compared to the FL DNA duplex. A similar light-up response was noted for the DMePhOH probe (Fig. S9†), which afforded a 17-fold light-up compared to its emission in the SS, with Φfl = 0.34, B = 9040 cm−1 M−1 (Table S3†). The π-extended NapOH6HI displayed comparatively dim fluorescence with a 7-fold turn-on response with B = 2520 cm−1 M−1 (Fig. S10 and Table S3†).
 | ||
| Fig. 3 (A) Emission spectra of PhOH6HI (SS vs. FL DNA duplex vs. DNA:RNA hybrid duplex, green trace) highlighting hybridization impact on relative emission intensity, λex = 390 nm. (B) Excitation/emission spectra of PhOH6HI (SS vs. FL DNA vs. N-1 DNA, blue trace) highlighting hybridization impact on ESPT to afford PhO−* emission from PhOH excitation at 380 nm. (C) Excitation/emission spectra of FPhOH6HI (SS vs. FL DNA) highlighting ratiometric excitation response to hybridization at pH 7.4, excitation spectra recorded with λem = 570 nm. (D) Excitation spectra (λem = 570 nm) for FPhOH6HI displaying phenolate (FPhO−) excitation in the SS (black trace) to predominately phenol (FPhOH) excitation in the DNA:RNA hybrid duplex (green traces) at pH 7.4. (E) Excitation/emission spectra of DFPhOH6HI (SS vs. FL DNA at pH 7.4 and 6.0) highlighting ratiometric excitation response to hybridization at pH 6.0. (F) Hybridization impact on excitation spectra for DFPhOH6HI at pH 7.4 displaying phenolate (DFPhO−) excitation in the SS (black trace) to progressive increases in phenol (DFPhOH) excitation upon hybridization to afford the N-1 DNA (blue trace), FL DNA (purple trace) and DNA:RNA hybrid duplex (green trace). | ||
Both PhOH6HI (Fig. 3B) and DMePhOH6HI (Fig. S9†) displayed dual emission with separate peaks for the phenol and phenolate, with the intensity ratio (IROH*/IRO−*) changing with probe microenvironment for a ratiometric emission response. For PhOH6HI, formation of the N-1 duplex afforded RO−* emission at 570 nm with greater intensity than PhOH* emission at 493 nm (Fig. 3B, blue trace, IROH*/IRO−* = 0.8), demonstrating preferential ESPT of the PhOH6HI probe in the N-1 duplex. In the SS (black trace), the emission intensity slightly favored ROH* (IROH*/IRO−* = 1.5), with the ROH* intensity further increasing in the FL DNA complement (purple trace, IROH*/IRO−* = 2.0). For DMePhOH6HI the IROH*/IRO−* ratios differ in the various microenvironments compared to the PhOH6HI response (Fig. S9†). In the SS, phenolate emission at 588 nm is slightly favored over ROH emission at 505 nm (IROH*/IRO−* = 0.75). In the FL DNA duplex, RO−* emission shifts to 584 nm with reduced intensity compared to ROH* at 505 nm (IROH*/IRO−* = 2.3). In the N-1 duplex, phenolate emission is more prominent and red-shifted to 595 nm than in the FL duplex, but with reduced intensity compared to ROH* (IROH*/IRO−* = 1.4). In the DNA:RNA hybrid, the DMePhOH6HI continues to exhibit RO−* emission at 592 nm, but ROH* was strongly favored (IROH*/IRO−* = 3.2).
Tuning the probe properties through addition of ortho-F substituents to PhOH6HI lowers both the ground-state and excited-state pKa values. Consequently, the fluorinated surrogates, FPhOH6MI and DFPhOH6HI, function as ratiometric excitation indicators during hybridization with dual ROH/RO− excitation, but single RO−* emission, due to their photoacidity.
Because of their differential ground-state pKa values (Table S1†), ratiometric excitation operates at differing pH ranges, with hybridization increasing the phenolic ground-state pKa at constant pH, which aligns with the expected ΔpKa values for phenol in water versus less-polar media (pKa ∼ 10 in water versus 14 in MeOH). Exploitation of the pKa switch reduces background SS emission through selective phenol excitation, enabling strong turn-on emission response with a large Stokes' shift (Δν), driven by ESPT.
At pH 7.4, FPhOH6MI functions effectively as a ratiometric excitation probe (pKa of 7.11, Table S1†), primarily existing as phenolate (FPhO−) in the SS, showing excitation at 445 nm and phenolate emission at 570 nm (Fig. 3C). Upon hybridization to afford the FL DNA duplex, there is a shift to neutral FPhOH, evidenced by a strong increase in excitation maxima at 378 nm and a reduction in phenolate excitation intensity (Fig. 3C, IROH/IRO− = 5.3). Despite this, FPhOH excitation at 378 nm still predominately affords phenolate emission at 575 nm (with residual phenol emission at ∼490 nm), due to its photoacidity for a 6-fold light-up response (B = 3900 cm−1 M−1, Table S3†). RNA titration (1–3 equiv.) at pH 7.4 shows a similar response, with an isosbestic point at ∼420 nm and an IROH/IRO− of 7.8 after adding 3 equiv. of RNA (Fig. 3D).
For DFPhOH6HI, ratiometric excitation is optimal at pH 6.0. The dual excitation in the SS form shifts toward phenol (370 nm, Fig. 3E), and hybridization to form the FL DNA duplex reduces phenolate intensity. At pH 7.4, DFPhOH6HI favors phenolate in the SS with λmax ∼ 450 nm (Fig. 3F). The shift in excitation wavelengths from the SS (black trace) to duplex forms clearly provide insight into the hydrophobicity of the probe environment in the different duplex structures, with DNA:RNA ≫ FL DNA > N-1 DNA. Upon hybridization to the N-1 complement phenolate intensity increases (blue trace), with slight formation of the neutral phenol (IROH/IRO− = 0.24). In the FL DNA duplex (purple trace), DFPhOH formation is more prominent (IROH/IRO− = 0.38); however, upon DNA:RNA hybrid duplex formation (green trace), the neutral DFPhOH peak at 370 nm is clearly dominant over the DFPhO− peak (IROH/IRO− = 1.54).
Molecular dynamics (MD) simulations provided detailed insights into the ability of PhOH6HI to minimally disrupt the duplex and its ESPT behavior in various DNA microenvironments (Fig. 4). The PhOH6HI surrogate minimally disrupts duplex stability, with only slightly diminished π-stacking interactions compared to the canonical system. The bent shape of PhOH6HI closely matches the natural curvature of the DNA helix, allowing for seamless integration into the structure. In the FL duplex, PhOH6HI intercalates between the dA:dT flanking base pairs, with the opposing dC predominantly displaced into the major groove (Fig. 4A), maintaining a stable conformation over time as confirmed by RMSD and RMSF analysis (Fig. S28†). The acceptor component of PhOH6HI maintained strong π-stacking with the flanking bases (dT6 (91.1 ± 2.6%), dT8 (99.5 ± 0.1%)), while the donor phenolic group had reduced stacking interactions (dA17 (81.6 ± 9.8%), dA19 (73.3 ± 19.2%)) compared to the canonical system. During the simulation, the phenolic moiety formed a persistent H-bond to O4′ of dC18 (59.6 ± 33.4%).
 | ||
| Fig. 4 Representative MD structures of Narl12 DS containing PhOH6HI (white), images are side views from the major groove with water, ions, and nonpolar hydrogen atoms hidden for clarity. (A) PhOH6HI opposite dC18 (pink) in FL DNA, (B) PhOH6HI opposite N-1 strand, and (C) PhOH6HI opposite C18 (pink) in DNA:RNA hybrid. (D) Average co-planar angle and number of 90° rotations for FL, N-1, and RNA:DNA systems. A 100 ns moving average is superimposed onto the raw data to highlight trends. (E) MD-determined solvent accessible surface areas for (i) PhOH6HI in FL DNA, (ii) PhOH6HI in N-1 DNA, and (iii) PhOH6HI in DNA:RNA hybrid, and the percent shielding of the phenolic oxygen atom. | ||
In the N-1 duplex (Fig. 4B), the phenolic donor displayed more persistent π-stacking interactions with the flanking bases (dA17 (93.1 ± 1.2%), dA18 (98.5 ± 0.8%)) compared to the FL duplex, without impacting the π-stacking interactions of the indanone acceptor (Fig. 4Avs.Fig. 4B). The absence of the opposing dC18 allowed the phenolic OH group to hydrogen bond with O4′ (15.9 ± 3.0%) and O5′ (23.3 ± 4.2%) of dA18. Consequently, in the N-1 duplex, the phenolic donor of PhOH6HI is positioned closer to the complementary phosphate backbone and is more solvent-exposed than in the FL duplex, both of which are key factors in promoting ESPT. The free PhOH6MI probe exhibited a 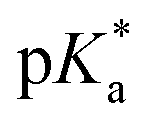 value of 1.15 and proton migration from excited PhOH6HI is expected to involve the phosphate group (pKa ∼ 1.5). The phosphate is less basic than a carboxylate in the β-barrel of GFP, strongly influencing ESPT efficiency (phenolate emission in GFP is strongly preferred vs. a slight preference for phenolate emission in the N-1 duplex).
value of 1.15 and proton migration from excited PhOH6HI is expected to involve the phosphate group (pKa ∼ 1.5). The phosphate is less basic than a carboxylate in the β-barrel of GFP, strongly influencing ESPT efficiency (phenolate emission in GFP is strongly preferred vs. a slight preference for phenolate emission in the N-1 duplex).
The MD structure of the DNA:RNA hybrid (Fig. 4C) displays greater helical twist, with a more A-form structure (Table S6†). A kink in the backbone promotes strong hydrogen bonding between the phenolic OH and the complementary backbone (99.7 ± 0.1% with OP1/OP2 of C18) and very persistent π-stacking with the interstrand bases (A17 (98.8 ± 0.2%), A19 (99.6 ± 0.0%)).
The enhanced quantum yield observed upon transitioning from the FL complement to the DNA:RNA hybrid can be rationalized by assessing the rigidity of the dye within its microenvironment. In the S1 state, 90° twisting of the probe is energetically favourable and leads to a twisted intramolecular charge transfer (TICT) nonemissive state, as evaluated with time-dependent density functional theory (TD-DFT) (Fig. S20†). The DNA:RNA hybrid exhibits a more pronounced helical twist and reduced rise, which likely impedes the formation of a TICT state by the FMR and thus preserves the quantum yield.34 Analysis of the co-planar angle (θ) between the two aryl moieties of PhOH6HI during the MD simulations indicates that PhOH6HI maintains an average angle of 31.3 ± 12.6° in the FL helix, while this angle decreases to 20.5 ± 12.5° in the N-1 helix (Fig. 4D). Upon RNA complementation, the dye experiences further rigidification, adopting an average angle of 13.3 ± 8.3°. As such, the probes environment plays a crucial role in preserving the quantum yield by restricting its propensity to achieve a TICT state.
Analysis of the solvent accessibility of PhOH6HI parallels the spectral data in the three duplex microenvironments (Fig. 4E). Compared to a free solvent-exposed PhOH6HI (133.44 Å2), hybridization with an N-1 complement leads to mild encapsulation (99.71 ± 7.73 Å2), with further decreasing solvent exposure observed for the FL complement (90.18 ± 17.30 Å2) and the DNA:RNA hybrid (90.00 ± 5.60 Å2), which is clearly displayed in the percent of phenolic oxygen solvent shielding. Decreased solvent exposure reduces ESPT with water and phenolate emission. This observation is consistent with more A-form duplex character for the DNA:RNA hybrid, as A-form helices are more compact and less hydrated than B-form DNA.45,46
Flanking bases and pH: controlling PET response
In the FL DNA duplex, PhOH6HI exhibited strong sensitivity to the nature of the flanking bases. Specifically, minimal emission occurred when the surrogate was flanked by dCs or dGs, suggesting PET quenching, but displayed usable fluorescence in AXA and TXT sequences, with the brightest emission observed in the TXT sequence (Φfl = 0.21, 101-fold brighter than CXC, Fig. 5A). The emission spectrum of the FL TXT DNA duplex peaked at 493 nm for the neutral ROH*, with a shoulder at 562 nm corresponding to RO−* emission, due to ESPT in the FL duplex (Fig. 5A). | ||
| Fig. 5 (A) Emission spectra of PhOH6HI in the FL NarI12 DNA duplex, highlighting the effect of flanking sequence on relative emission intensity (excitation maxima 390 nm). (B) Emission spectra of DFPhOH6HI in the FL NarI12 DNA duplex, highlighting the effect of flanking sequence on relative emission intensity (excitation maxima 450 nm). (C) Emission spectra of DFPhOH6HI in GXG NarI12 as a function of pH (5.5–8.0, excitation maxima 450 nm). (D–F) Minimum energy structures for DFPhO−6HI (D), PhOH6HI (E), and DFPhOH6HI (F), flanked by dG (purple), as evaluated at the SMD-ωB97X-D/Def2-TZVP//SMD-ωB97X-D/Def2-SVP (water) level of theory with their HOMO − 1, HOMO, and LUMO (H − 1, H, L) energy levels (isovalue = 0.02). The oscillator strength f (S0 → S1) was determined through a TD-DFT calculation at the SMD-ωB97X-D/Def2-TZVP (water) level of theory. | ||
The DFPhOH6HI probe also displayed the brightest emission in the TXT sequence (Fig. 5B, Φfl = 0.15), but was far more resistant to PET quenching in CXC and GXG sequences compared to PhOH6HI. In the CXC SS, the pKa of DFPhOH6HI is 6.5 for a ΔpKa of +0.6 compared to the free DFPhOH6MI dye (pKa = 5.9, Table S1†). Thus, at pH 7.4 in PBS buffer, the probe is predominately deprotonated with phenolate excitation at ∼450 nm to afford phenolate emission at ∼570 nm. This PET-OFF response to CXC and GXG flanking sequences by DFPhOH6HI at pH 7.4 suggested that the higher HOMO energy for the phenolate played a key role, and lowering the pH to promote phenol excitation at ∼370 nm would transition DFPhOH6HI into a PET-ON state, as noted for the neutral PhOH6HI. To test this hypothesis, the pH response of DFPhOH6HI was established in the GXG sequence (Fig. 5C). The emission spectra were obtained using phenolate excitation at 450 nm. At pH 5.5, the probe absorbs at ∼370 nm and excitation at 370 nm afforded dim phenolate emission at ∼570 nm due to PET quenching, as anticipated. The pH-responsive nature of DFPhOH6HI highlighted in Fig. 5C is dramatic (Irel = 134-fold from pH 5.5–8.0) because it involves both PET quenching and ratiometric excitation (change in probe excitation wavelength as a function of pH).
To better understand the fluorescence-quenching mechanism of these probes and the influence of flanking bases, we performed density functional theory (DFT) calculations on the phenolic states of PhOH6HI and DFPhOH6HI versus the phenolate of DFPhOH6HI (Fig. 5D). Using TD-DFT, we identified the molecular orbitals (MOs) responsible for the observed S0 → S1 transitions. When the probes were flanked by dG (Fig. 5D), the neutral PhOH6HI and DFPhOH6HI exhibited strong transitions from the HOMO − 1 to the LUMO. Upon deprotonation, the HOMO energy level of DFPhOH6HI was raised, resulting in a HOMO to LUMO transition upon photoexcitation. Consequently, in their neutral states, these dyes undergo rapid acceptor-PET quenching, as the HOMO—centered on the dG—lies higher in energy than the hole generated during photoexcitation. In contrast, the deprotonated DFPhOH6HI is resistant to acceptor-PET quenching, as the HOMO energy is raised above that of the dG.
When the probes were flanked by dT (Fig. S19†), both neutral and deprotonated dyes exhibited strong HOMO to LUMO transitions upon photoexcitation. These systems are resistant to acceptor-PET quenching by the flanking nucleobases, allowing for observable fluorescence. This suggests that the fluorescence-quenching behavior is highly dependent on the specific flanking base, with dT effectively minimizing quenching and dG promoting quenching, unless the probe is deprotonated.
Thus, it becomes clear how RNA aptamer–fluorogen systems, like Spinach,15 induces the fluorescence of the GFP-like DFHBI ligand without severe PET quenching. At neutral pH, free DFHBI (pKa = 5.5) predominantly populates the deprotonated, phenolate form, raising the HOMO energy to avoid acceptor-PET quenching by the GQ on which it stacks with. The GQ simply serves as a floor to support the probe with neighboring nucleotides, which stabilizes the planar conformation of DFHBI.
GQ → duplex exchange in TBA
The optical performance of the GFP-surrogates was also tested in a GQ-folding sequence given that free DFHBI analogs have displayed strong emission when bound to GQ-folding RNA aptamers.15,16 For these experiments, the O-donors were inserted into the G8-site of the 15-mer thrombin-binding aptamer (5′-GGTTGG-TG8T-GGTTGG-3′, TBA15). TBA15 folds into an antiparallel GQ in the presence of K+ with the G8-site undergoing π-stacking interactions with the G1–G6–G10–G15 tetrad.47 Therefore, we expected that the neutral probes, PhOH6HI and DMePhOH6HI, would be present in a PET-ON state with quenched fluorescence at the G8-site of TBA, while DFPhOH6HI would be in a PET-OFF state at pH 8.0, due to formation of the phenolate. Furthermore, the G8-site is flanked by Ts to represent a TXT sequence context when the strand is paired with the complementary DNA or RNA, suggesting that the neutral PhOH6HI and DMePhOH6HI probes would transition from a PET-ON state in the GQ to a PET-OFF state in the duplex for a strong light-up emission response.Our TBA studies were conducted in Tris–HCl (25 mM Tris, 50 mM KCl, 50 mM MgCl2, pH 8.0), which resemble the buffer conditions optimized by Nie and coworkers for studying DNA GQ binding by free DFHBI analogs,48 and included the best O-donors from our NarI12 studies. Notably, replacing the dG8 nucleoside with the GFP-surrogates increased the thermal stability of the native K+-templated GQ, which had a Tm of 40.1 °C (Table S5, ESI†). As anticipated, the DFPhOH6HI surrogate afforded the brightest fluorescence with phenolate excitation at 437 nm and emission at 565 nm (Fig. 6A). The excitation spectrum also displayed a strong energy transfer (ET) peak at ∼260 nm (see Fig. S35, ESI†), indicating efficient stacking interactions of DFPhOH6HI with the G-tetrad.49 CD spectra confirmed the formation of the antiparallel GQ structure in the G8-DFPhOH6HI-TBA15 sample compared to the native TBA15 (Fig. S16, ESI†). Replacement of DFPhOH6HI with FPhOH6HI reduced fluorescence intensity with an Irel of 0.35 compared to DFPhOH6HI (Fig. 6A, teal vs. pink trace). The two neutral phenolic probes (PhOH6HI and DMePhOH6HI) both provided strongly PET quenched emission by the neighbouring Gs (black trace for DMePhOH (Fig. 6A, Irel = 0.07), blue trace for PhOH6HI (Irel = 0.05)).
 | ||
| Fig. 6 (A) Excitation/emission spectra for DFPhOH6HI (pink trace), FPhOH6HI (teal trace), DMePhOH6HI (black trace), and PhOH6HI (blue trace) at G8 of TBA15 recorded in Tris–HCl pH 8.0, 50 mM KCl, 50 mM MgCl2. Irel given in the inset. (B) Fluorescence emission spectra highlighting turn-on responses to hybridization by PhOH6HI at the G8-site of TBA15 in the GQ vs. DNA FL duplex (red trace), DNA:RNA hybrid (blue trace), and DNA:RNA–AL hybrid (purple trace) in Tris–HCl pH 8.0, 50 mM KCl and 50 mM MgCl2. Representative MD structures of TBA15 containing PhOH6HI (white) in (C) DNA:RNA hybrid, (D) PhOHDNA:RNA–AL hybrid, (E) DNA:RNA–TL hybrid, and (F) BA15 GQ K+. Water, ions, and nonpolar hydrogen atoms are hidden for clarity. (G) Average co-planar angle and number of 90° rotations for TBA15 GQ K+, TBA DNA:RNA–AT, TBA DNA:RNA–AL, and TBA RNA:DNA. A 100 ns moving average is superimposed on the raw data to highlight trends. (H) MD-determined solvent accessible surface area for PhOH6HI (white) for TBA GQ K+, and percent solvent shield occupancy for the phenolic oxygen. | ||
For probe emission in TBA15 duplex structures, we focused on the neutral phenols PhOH6HI and DMePhOH6HI, which show quenched emission in the TBA15 GQ. These surrogates also provided the brightest emission in NarI12 DNA:RNA hybrid duplexes (Table S3†), suggesting their ability to serve as effective light-up probes for GQ → duplex topology switching. We also examined the impact of neighboring “Locked Nucleic Acids” (LNAs) as flanking bases, either within the modified TBA strands as locked-Ts (TL) or in the complementary RNA strand as locked-As (AL). Seitz and coworkers established that the fluorescence of FIT probes strongly increase when placed adjacent to an LNA rather than a native DNA base.30 However, since the best responding FIT probe quinoline blue (QB-FIT) is a symmetrical monomethine cyanine dye they placed a single TL base adjacent to the surrogate in the modified strand. Since the O-donors are non-symmetric, we tested both TL and AL, expecting that AL in the complementary strand would be more effective at increasing probe emission through stacking interactions directly with the more flexible phenolic donor to increase its rigidity.
Both PhOH6HI and DMePhO6HI provided strong ROH* light-up responses in the duplex compared to the probe in the GQ (Table 1). The largest Irel values were observed upon formation of the DNA:RNA–AL hybrid duplex with PhOH6HI showing a 144-fold light-up response (Fig. 6B) with Φfl = 0.46, B = 13900 cm−1 M−1 (Table 1). In the DNA:RNA hybrid lacking AL (Fig. 6C), PhOH6HI still provided a 105-fold light-up response (Fig. 6B, blue trace) with B = 10800 cm−1 M−1. The photophysical parameters show that the AL nucleobase increased probe brightness by enhancing εmax (Table 1). The brightest emission for both probes was produced in the DNA:RNA–(TL/AL) hybrid duplexes containing LNAs in both strands. For the PhOH6HI surrogate, B was 14700 cm−1 M−1, although the Irel was only 16-fold due to enhanced emission of PhOH6HI in the GQ containing the TL nucleobase. The Φfl = 0.58 for DMePhOH6HI in the TBA15 DNA:RNA–(TL/AL) hybrid duplex (Table 1) was the highest value recorded throughout our studies. The DMePhOH6HI probe also displayed the strongest light-up response of 154-fold upon hybridization with the RNA–AL complement (Table 1).
:RNA hybrid duplexes
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Xa | DSb | T m (ΔTm)c (°C) | λ ex ROHd | λ em ROH*e | I rel | Φ fl | ε max | B |
|
a See Fig. 1 for surrogate abbreviations.
b Double strand, RNA complements contain 2′-OMe groups.
c
T
m values in °C of TBA15 (5′-GGTTGGT-X-TGGTTGG) DS (2 μM) measured at 260 nm in Tris–HCl, pH 8.0, 50 mM KCl, 50 mM MgCl2 heating rate of 0.5 °C min−1, errors are ±1 °C; ΔTm = Tm (modified DS with probe X) – 62.5 °C (Tm of native TBA15 DS containing X = G paired with C) or – 63.8 °C (Tm of native DNA:RNA hybrid DS).
d Excitation maximum in nm for the phenol (ROH).
e Emission maximum in nm for the phenol (ROH*).
f Relative emission intensity of the O-donors in the DS versus GQ.
g Fluorescence quantum yield of probes in the DS measured by the comparative method using quinine sulfate (Φfl = 0.56) in 0.1 M H2SO4, errors are ±5% obtained from three measurements.
h Probe absorption coefficients in DS (10−3 cm−1 M−1).
i Brightness (Φfl × εmax), cm−1 M−1.
|
||||||||
| PhOH | DNA | 59.8 (−2.7) | 390 | 494 | 75 | 0.38 | 21.0 | 7980 |
| PhOH | RNA | 54.4 (−9.4) | 387 | 490 | 105 | 0.45 | 24.0 | 10800 |
| PhOH | AL | 62.6 (−1.2) | 390 | 490 | 144 | 0.46 | 30.0 | 13900 |
| PhOH | TL | 57.7 (−6.1) | 385 | 495 | 10 | 0.37 | 25.0 | 9280 |
| PhOH | TL/AL | 63.4 (−0.4) | 385 | 490 | 16 | 0.49 | 29.0 | 14700 |
| DMePhOH | DNA | 62.3 (−0.2) | 400 | 493 | 85 | 0.17 | 15.0 | 2500 |
| DMePhOH | RNA | 56.8 (−7.0) | 400 | 500 | 120 | 0.29 | 19.0 | 5600 |
| DMePhOH | AL | 57.8 (−6.0) | 400 | 500 | 154 | 0.51 | 19.8 | 10100 |
| DMePhOH | TL | 64.4 (0.6) | 395 | 505 | 35 | 0.44 | 19.0 | 8410 |
| DMePhOH | TL/AL | 62.3 (−1.5) | 405 | 505 | 55 | 0.58 | 19.0 | 11070 |

MD simulations suggest increased π-stacking for the PhOH6HI surrogate in the hybrid containing AL (Fig. 6Cvs.Fig. 6E). In the FL DNA:RNA hybrid, the phenolic donor protrudes toward the minor groove, forming relatively poor π-stacking interactions with the flanking As (A22 69.1 ± 4.7% and A24 62.6 ± 7.1%, Fig. 6C). Upon locking the sugar, the π-stacking interaction between the phenolic donor and the A residues increases by 24.7% and 30.1%, respectively (Fig. 6E).
The MD simulations of PhOH6HI at the G8-site in the loop region of the antiparallel GQ (Fig. 6F) revealed direct stacking interactions with the G-tetrad and neighboring T residues (T7 and T9). Compared to its behavior in duplex microenvironments (80.34 ± 16.78 Å2, TBA) (Fig. 6H), PhOH6HI in the GQ has greater solvent exposure (100.28 ± 11.37 Å2). Most notably, the photoacidic phenolic oxygen experienced much less solvent shielding compared to a duplex environment, which supports its deprotonation (Fig. 6H).
The influence of the transition from GQ to duplex topology, alongside the incorporation of LNA flanking bases, on quantum yield was evaluated by analyzing the rigidity of the dye. For the GQ structure, MD simulations revealed an average co-planar angle of 23.9 ± 14.0°. Despite this seemingly planar configuration, the dye exhibited significant conformational dynamism, with a frequency of 90° flips reaching 23 flips per μs. This behavior suggests that while the dye maintains adequate π-stacking interactions with the G-tetrad, it attains 90° TICT states. More importantly, the reductive PET quenching by the G-tetrad, as predicted by DFT calculations (Fig. 5), is the primary factor contributing to its poor fluorescence. Upon transitioning to a duplex topology, the dye's rigidity is slightly decreased, with the average co-planar angle of PhOH6HI measured at 37.6 ± 15.0° for the DNA:RNA complex, which increases to 44.7 ± 14.5° in DNA:RNA–TL. Despite this, the quantum yield is increased because of the inhibition of PET quenching. When the interstrand alignment is constrained in DNA:RNA–AL, a substantial reduction in the co-planar angle to 14.4 ± 10.3° is observed, effectively eliminating the dye's capability to achieve 90° TICT states, leading to the increased quantum yield observed among the duplex systems. Despite the initial turn-on response going to SS to DS being driven by increased dye rigidity and reduced access to TICT states, the marked enhancement in fluorescence observed during the GQ-to-duplex transition is primarily due to the elimination of PET quenching by the G-tetrad (Fig. S56†).
O-donor chalcone performance as GFP-surrogates and comparison to other FMR and smart surrogates for NA diagnostics
Our combined experimental and computational study overcomes and addresses longstanding challenges in developing GFP-surrogates for use as internal NA replacements.32–37 Standout probe features include the performance of PhO6HI and DMePhOH6HI (Fig. 7) for monitoring GQ → duplex exchange with Irel values > 100-fold upon duplex formation with B > 10000 cm−1 M−1 (Table 1). This topology switch is a NA nanodevice capable of an extension-contraction movement and plays a critical role in many NA diagnostic applications.50,51 Furthermore, there is much interest in understanding how GQ structures regulate transcription,52 and GFP-surrogates could serve as mechanistic tools to monitor GQ → duplex exchange mediated by polymerase bypass. The second standout feature was the pH-responsive nature of DFPhOH6HI in the GXG duplex with Irel > 100-fold upon increasing the pH from 5.5 to 8.0 (Fig. 5C). Most NA pH-sensors rely on i-motif formation with the probe responding to the change in NA topology induced by dC protonation.53 The outstanding pH-response by DFPhOH6HI is of potential utility for monitoring the endosomal escape of NA therapeutics.54
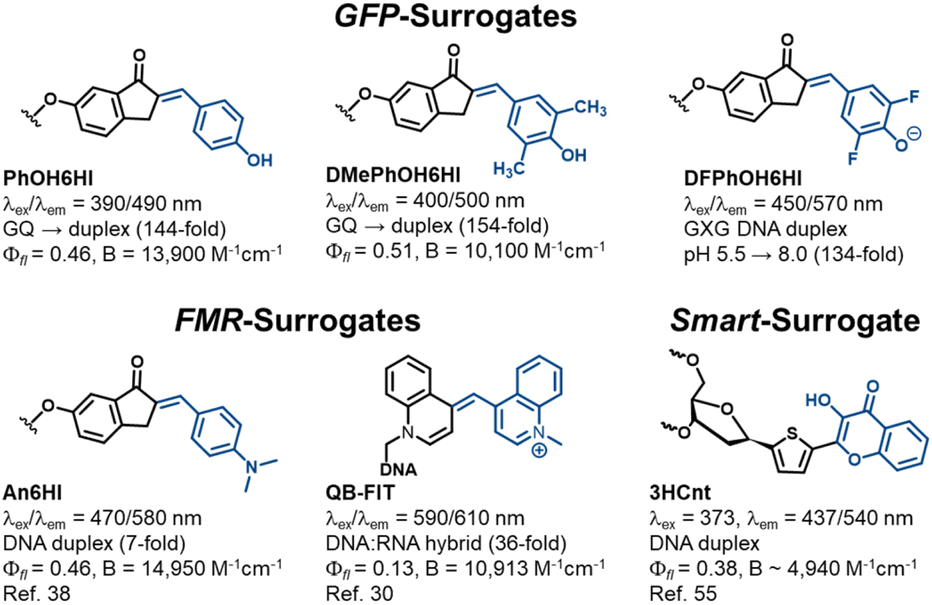 | ||
| Fig. 7 Performance of GFP-surrogates versus other FMR and smart surrogates used for NA diagnostics. | ||
Competitive light-up FMR-surrogates include An6HI that can be regarded as the parent N-donor chalcone,38 and QB-FIT developed by the Sietz laboratory (Fig. 7).30 Similar to the GFP-surrogates these probes have a bent structure with a single methine bridge separating the donor/acceptor groups for optimal fit within the duplex, with minimal impact on stability. Both N-donor chalcones and the FIT probes provide single-wavelength turn-on λex/λem with little sensitivity to the nature of the flanking bases, making them universal turn-on hybridization probes. The big advantage of QB-FIT is its strong turn-on fluorescence upon hybridization with the demonstrated ability to detect RNA in live cells.30 It is also responsive to i-motif formation within a dC-rich “proton aptamer” providing a 5-fold emission enhancement upon lowering the pH from 7.0 to 5.6.31 Clearly this response is not competitive to the pH-responsive nature of DFPhOH6HI in duplex DNA (134-fold, Fig. 5C). In addition, the An6HI probe cannot be employed to distinguish a GQ from a duplex due to bright fluorescence in both topologies.39
In terms of ‘smart’ probes for ratiometric diagnostics the thiophene-appended 3-hydroxychromone surrogate (3HCnt, Fig. 7) serves as a ratiometric emissive sensor with dual wavelengths at 437/540 nm (N*/T*) following excitation at 373 nm.55 The surrogate has a destabilizing impact on duplex DNA stability but can monitor probe microenvironment through changes in the intensity ratio of the two emissive peaks (IN*/IT*). The free probe has εmax of 13000 M−1 cm−1 and this value was used to estimate a probe brightness of 4940 cm−1 M−1 in a TXT duplex. This value probably overestimates probe brightness in the helix by 25–40%,56 given that εmax values for UV-absorbent bases decrease significantly in oligonucleotides. The superior brightness of the GFP-surrogates and ease of modular synthesis is a competitive advantage.
Experimental
All experimental procedures, HPLC chromatographs, MS, NMR spectra, detailed computational methods are found in the ESI.†Conclusions
In summary, our aldol modular approach effectively accommodates the incorporation of phenolic O-donors into nucleic acids, which serve as GFP-surrogates. The electron-rich PhOH6HI and DMePhOH6HI are ratiometric emission indicators, exhibiting both phenol and phenolate emission upon single-wavelength phenol excitation due to an ESPT process. These probes display strongly quenched emission when flanked by Gs or Cs due to a PET process. Thus, within a GQ to duplex topology switch, these surrogates transition from a PET-ON to a PET-OFF state to afford a strong turn-on fluorescence response (up to 158-fold), with B ∼ 10000–15000 cm−1 M−1 using LNA flanking bases.
Tuning the phenolic donor by attaching ortho-F substituents lowers both the ground-state and excited-state phenolic pKa to create ratiometric excitation probes, which are highly useful for eliminating background emission upon hybridization. At constant pH, both FPhOH6HI and DFPhOH6HI transition from the phenolate in the solvent-exposed SS into the phenol upon hybridization due to the decrease in polarity to favor the neutral phenol within the duplex. Excitation of the phenol still affords phenolate emission due to the photoacidity of the fluorinated surrogates. Their differences in ground-state pKa allows tuning excitation ratiometry, with FPhOH6HI working at pH 7.4 and DFPhOH6HI at pH 6.0. The DFPhOH6HI surrogate also displays strong sensitivity to pH within duplex DNA when flanked by G residues. At pH 5.5, the phenol is in a PET-ON state with strongly quenched emission. Increasing the pH to 8.0 affords the phenolate, which is in a PET-OFF state due to the increase in the HOMO energy level of the phenolate compared to the neutral phenol to afford a 134-fold increase in emission intensity upon transitioning from pH 5.5 to 8.0. These findings not only validate the efficacy of our GFP-surrogate design, but also open new avenues for applying chalcone FMRs in NA research, as their innate tunability will foster numerous applications in biosensing, therapeutics, and NA nanotechnology.
Data availability
All relevant data are presented in the article and the ESI.†Author contributions
Keenan T. Regan: investigations, data curation, writing – original draft. Austin Pounder: investigations, data curation, writing – original draft. Ryan E. Johnson: data curation. Makay T. Murray: data curation. Hannah X. Glowacki: data curation. Stacey D. Wetmore: supervision, funding acquisition, writing – review & editing. Richard A. Manderville: supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (grant 2016-04568) to S. D. W., and grant 2018-400136 to R. A. M. A. P. thanks Alberta Innovates for a PDF fellowship and M. T. M. thanks the RNA Innovation (NSERC CREATE) program and University of Lethbridge for graduate scholarships. S. D. W., A. P., and M. T. M. also thank computational resources provided by the Digital Research Alliance of Canada (the Alliance), and the Canada Research Chair Program for supporting this research.References
- R. N. Day and M. W. Davidson, The Fluorescent Protein Palette: Tools for Cellular Imaging, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- R. M. Hoffman, The Multiple Uses of Fluorescent Proteins to Visualize Cancer in Vivo, Nat. Rev. Cancer, 2005, 5, 796–806 CrossRef CAS PubMed.
- R. H. Newman, M. D. Fosbrink and J. Zhang, Genetically Encodable Fluorescent Biosensors for Tracking Signaling Dynamics in Living Cells, Chem. Rev., 2011, 111, 3614–3666 CrossRef CAS.
- R. Y. Tsien, Constructing and Exploiting the Fluorescent Protein Paintbox (Nobel Lecture), Angew. Chem., Int. Ed., 2009, 48, 5612–5626 CrossRef CAS PubMed.
- F. D. Steffen, M. Khier, D. Kowerko, R. A. Cunha, R. Börner and R. K. O. Sigel, Metal Ions and Sugar Puckering Balance Single-Molecule Kinetic Heterogeneity in RNA and DNA Tertiary Contacts, Nat. Commun., 2020, 11, 104 CrossRef CAS PubMed.
- S. Ray, J. R. Widom and N. G. Walter, Life under the Microscope: Single-Molecule Fluorescence Highlights the RNA World, Chem. Rev., 2018, 118, 4120–4155 CrossRef CAS PubMed.
- P. Ghosh, H. M. Kropp, K. Betz, S. Ludmann, K. Diederichs, A. Marz and S. G. Srivatsan, Microenvironment-Sensitive Fluorescent Nucleotide Probes from Benzofuran, Benzothiophene, and Selenophene as Substrates for DNA Polymerases, J. Am. Chem. Soc., 2022, 144, 10556–10569 CrossRef CAS PubMed.
- Y. Du and S. Dong, Nucleic Acid Biosensors: Recent Advances and Perspectives, Anal. Chem., 2017, 89, 189–215 CrossRef CAS PubMed.
- R. E. Johnson, A. Pounder, J. van der Zalm, A. Cheng, I. J. Bell, T. J. Van Raay, S. D. Wetmore and R. A. Manderville, Thieno[3,2-b]thiophene for the Construction of Far-Red Molecular Rotor Hemicyanines as High-Affinity DNA Aptamer Fluorogenic Reporters, Anal. Chem., 2024, 96, 16252–16259 CrossRef CAS PubMed.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, Conformationally Locked Chromophores as Models of Excited-State Proton Transfer in Fluorescent Proteins, J. Am. Chem. Soc., 2012, 134, 6025–6032 CrossRef CAS PubMed.
- G. L. Silva, V. Ediz, D. Yaron and B. A. Armitage, Experimental and Computational Investigation of Unsymmetrical Cyanine Dyes: Understanding Torsionally Responsive Fluorogenic Dyes, J. Am. Chem. Soc., 2007, 129, 5710–5718 CrossRef CAS PubMed.
- S. R. Meech, Excited State Reactions in Fluorescent Proteins, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC.
- J. C. Liao, J. Roider and D. G. Jay, Chromophore-Assisted Laser Inactivation of Proteins Is Mediated by the Photogeneration of Free Radicals, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2659–2663 CrossRef CAS PubMed.
- K. Y. Han, B. J. Leslie, J. Fei, J. Zhang and T. Ha, Understanding the Photophysics of the Spinach–DFHBI RNA Aptamer–Fluorogen Complex To Improve Live-Cell RNA Imaging, J. Am. Chem. Soc., 2013, 135, 19033–19038 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, RNA Mimics of Green Fluorescent Protein, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, Broccoli: Rapid Selection of an RNA Mimic of Green Fluorescent Protein by Fluorescence-Based Selection and Directed Evolution, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- K. D. Warner, L. Sjekloca, W. Song, G. S. Filonov, S. R. Jaffrey and A. R. Ferre-D'Amare, Nat. Chem. Biol., 2017, 13, 1195–1201 CrossRef CAS PubMed.
- C. Steinmetzger, N. Palanisamy, K. R. Gore and C. Höbartner, A Multicolor Large Stokes Shift Fluorogen-Activating RNA Aptamer with Cationic Chromophores, Chem.–Eur. J., 2019, 25, 1931–1935 CrossRef CAS PubMed.
- E. V. Dolgosheina, S. C. Jeng, S. S. Panchapakesan, R. Cojocaru, P. S. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, RNA Mango Aptamer-Fluorophore: A Bright, High-Affinity Complex for RNA Labeling and Tracking, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed.
- R. L. Strack, M. D. Disney and S. R. A. Jaffrey, Superfolding Spinach2 Reveals the Dynamic Nature of Trinucleotide Repeat–Containing RNA, Nat. Methods, 2013, 10, 1219–1224 CrossRef CAS PubMed.
- L. M. Tolbert, A. Baldridge, J. Kowalik and K. M. Solntsev, Collapse and Recovery of Green Fluorescent Protein Chromophore Emission through Topological Effects, Acc. Chem. Res., 2012, 45, 171–181 CrossRef CAS PubMed.
- R. A. Manderville and S. D. Wetmore, C-Linked 8-Aryl Guanine Nucleobase Adducts: Biological Outcomes and Utility as Fluorescent Probes, Chem. Sci., 2016, 7, 3482–3493 RSC.
- W. Xu, K. M. Chan and E. T. Kool, Fluorescent Nucleobases as Tools for Studying DNA and RNA, Nat. Chem., 2017, 9, 1043–1055 CrossRef CAS PubMed.
- M. Bood, A. F. Füchtbauer, M. S. Wranne, J. J. Ro, S. Sarangamath, A. H. El-Sagheer, D. L. M. Rupert, R. S. Fisher, S. W. Magennis, A. C. Jones, F. Höök, T. Brown, B. H. Kim, A. Dahlén, L. M. Wilhelmsson and M. Grøtli, Pentacyclic Adenine: A Versatile and Exceptionally Bright Fluorescent DNA Base Analogue, Chem. Sci., 2018, 9, 3494–3502 RSC.
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Probing of Nucleic Acid Structures, Dynamics, and Interactions with Environment-Sensitive Fluorescent Labels, Front. Chem., 2020, 8, 112 CrossRef CAS.
- D. Dziuba, Environmentally Sensitive Fluorescent Nucleoside Analogues as Probes for Nucleic Acid – Protein Interactions: Molecular Design and Biosensing Applications, Methods Appl. Fluoresc., 2022, 10, 044001 CrossRef CAS.
- A. Karimi, R. Börner, G. Mata and N. W. Luedtke, A Highly Fluorescent Nucleobase Molecular Rotor, J. Am. Chem. Soc., 2020, 142, 14422–14426 CrossRef CAS.
- Y. Tor, Isomorphic Fluorescent Nucleosides, Acc. Chem. Res., 2024, 57, 1325–1335 CrossRef CAS.
- F. Hövelmann and O. Seitz, DNA Stains as Surrogate Nucleobases in Fluorogenic Hybridization Probes, Acc. Chem. Res., 2016, 49, 714–723 CrossRef.
- F. Hövelmann, I. Gaspar, J. Chamiolo, M. Kasper, J. Steffen, A. Ephrussi and O. Seitz, LNA-Enhanced DNA FIT-Probes for Multicolour RNA Imaging, Chem. Sci., 2016, 7, 128–135 RSC.
- S. B. Ebrahimi, D. Samanta, H. F. Cheng, L. I. Nathan and C. A. Mirkin, Forced Intercalation (FIT)-Aptamers, J. Am. Chem. Soc., 2019, 141, 13744–13748 CrossRef CAS PubMed.
- T. Stafforst and U. Diederichsen, Synthesis of Alaninyl and N-(2-Aminoethyl)Glycinyl Amino Acid Derivatives Containing the Green Fluorescent Protein Chromophore in Their Side Chains for Incorporation into Peptides and Peptide Nucleic Acids, Eur. J. Org Chem., 2007, 2007, 899–911 CrossRef.
- U. Wenge and H.-A. Wagenknecht, Synthetic GFP Chromophore and Control of Excited-State Proton Transfer in DNA: An Alternative for Fluorescent DNA Labels with Large Apparent' Shifts, Synthesis, 2011, 2011, 502–508 CrossRef.
- J. Riedl, P. Ménová, R. Pohl, P. Orság, M. Fojta and M. Hocek, GFP-like Fluorophores as DNA Labels for Studying DNA–Protein Interactions, J. Org. Chem., 2012, 77, 8287–8293 CrossRef CAS PubMed.
- M. Chowdhury, J. A. Turner, D. Cappello, M. Hajjami and R. H. E. Hudson, Chimeric GFP–Uracil Based Molecular Rotor Fluorophores, Org. Biomol. Chem., 2023, 21, 9463–9470 RSC.
- T. Kanamori, A. Takamura, N. Tago, Y. Masaki, A. Ohkubo, M. Sekine and K. Seio, Fluorescence Enhancement of Oligodeoxynucleotides Modified with Green Fluorescent Protein Chromophore Mimics upon Triplex Formation, Org. Biomol. Chem., 2017, 15, 1190–1197 RSC.
- A. Saady, V. Böttner, M. Meng, E. Varon, Y. Shav-Tal, C. Ducho and B. Fischer, An Oligonucleotide Probe Incorporating the Chromophore of Green Fluorescent Protein Is Useful for the Detection of HER-2 MRNA Breast Cancer Marker, Eur. J. Med. Chem., 2019, 173, 99–106 CrossRef CAS PubMed.
- R. E. Johnson, M. T. Murray, L. J. Bycraft, S. D. Wetmore and R. A. Manderville, A Modular Aldol Approach for Internal Fluorescent Molecular Rotor Chalcone Surrogates for DNA Biosensing Applications, Chem. Sci., 2023, 14, 4832–4844 RSC.
- R. E. Johnson, M. T. Murray, D. J. Roby, L. J. Bycraft, Z. R. Churcher, S. Yadav, P. E. Johnson, S. D. Wetmore and R. A. Manderville, Unlocking Pb2+ Sensing Potential in a DNA G-Quadruplex via Loop Modification with Fluorescent Chalcone Surrogates, ACS Sens., 2023, 8, 4756–4764 CrossRef CAS PubMed.
- M. A. Rizzo, G. H. Springer, B. Granada and D. W. Piston, An Improved Cyan Fluorescent Protein Variant Useful for FRET, Nat. Biotechnol., 2004, 22, 445–449 CrossRef CAS PubMed.
- A. P. Demchenko, The Concept of λ-Ratiometry in Fluorescence Sensing and Imaging, J. Fluoresc., 2010, 20, 1099–1128 CrossRef PubMed.
- A. P. Demchenko, Dual Emission and its λ-Ratiometric Detection in Analytical Fluorimetry. Pt. I. Basic Mechanisms of Generating the Reporter Signal, Methods Appl. Fluoresc., 2023, 11, 033002 CrossRef CAS PubMed.
- M. Kneen, J. Farinas, Y. Li and A. S. Verkman, Green Fluorescent Protein as a Noninvasive Intracellular pH Indicator, Biophys. J., 1998, 74, 1591–1599 CrossRef CAS PubMed.
- T. Förster, Die PH-Abhängigkeit Der Fluoreszenz von Naphthalinderivaten, Z. Elektrochem. Angew. Phys. Chem., 1950, 54, 531–535 CrossRef.
- J. I. Gyi, G. L. Conn, A. N. Lane and T. Brown, Comparison of the Thermodynamic Stabilities and Solution Conformations of DNA·RNA Hybrids Containing Purine-Rich and Pyrimidine-Rich Strands with DNA and RNA Duplexes, Biochemistry, 1996, 35, 12538–12548 CrossRef CAS PubMed.
- D. Venkateswarlu, K. E. Lind, V. Mohan, M. Manoharan and D. M. Ferguson, Structural Properties of DNA:RNA Duplexes Containing 2′-O-Methyl and 2′-S-Methyl Substitutions: A Molecular Dynamics Investigation, Nucleic Acids Res., 1999, 27, 2189–2195 CrossRef CAS PubMed.
- P. Schultze, R. F. Macaya and J. Feigon, Three-Dimensional Solution Structure of the Thrombin-Binding DNA Aptamer d(GGTTGGTGTGGTTGG), J. Mol. Biol., 1994, 235, 1532–1547 CrossRef CAS.
- G. Feng, C. Luo, H. Yi, L. Yuan, B. Lin, X. Luo, X. Hu, H. Wang, C. Lei, Z. Nie and S. Yao, DNA Mimics of Red Fluorescent Proteins (RFP) Based on G-Quadruplex-Confined Synthetic RFP Chromophores, Nucleic Acids Res., 2017, 45, 10380–10392 CrossRef CAS PubMed.
- D. E. Armstrong-Price, P. S. Deore and R. A. Manderville, Intrinsic “Turn-On” Aptasensor Detection of Ochratoxin A Using Energy-Transfer Fluorescence, J. Agric. Food Chem., 2020, 68, 2249–2255 CrossRef CAS PubMed.
- P. Alberti and J.-L. Mergny, DNA Duplex–Quadruplex Exchange as the Basis for a Nanomolecular Machine, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1569–1573 CrossRef CAS PubMed.
- P. S. Deore, M. D. Gray, A. J. Chung and R. A. Manderville, Ligand-Induced G-Quadruplex Polymorphism: A DNA Nanodevice for Label-Free Aptasensor Platforms, J. Am. Chem. Soc., 2019, 141, 14288–14297 CrossRef CAS PubMed.
- J. Robinson, F. Raguseo, S. P. Nuccio, D. Liano and M. Di Antonio, DNA G-Quadruplex Structures: More Than Simple Roadblocks to Transcription?, Nucleic Acids Res., 2021, 49, 8419–8431 CrossRef CAS PubMed.
- M. N. Mattath, D. Ghosh, S. Pratihar, S. Shi and T. Govindaraju, Nucleic Acid Architectonics for pH-Responsive DNA Systems and Devices, ACS Omega, 2022, 7, 3167–3176 CrossRef CAS.
- M. R. Herling and I. J. Dmochowski, Ratiometric, pH-Sensitive Probe for Monitoring SiRNA Delivery, J. Am. Chem. Soc., 2023, 145, 9417–9422 CrossRef CAS PubMed.
- D. Dziuba, V. Y. Postupalenko, M. Spadafora, A. S. Klymchenko, V. Guérineau, Y. Mély, R. Benhida and A. Burger, A Universal Nucleoside with Strong Two-Band Switchable Fluorescence and Sensitivity to the Environment for Investigating DNA Interactions, J. Am. Chem. Soc., 2012, 134, 10209–10213 CrossRef CAS PubMed.
- M. B. Turner, J. M. Cizmic, D. B. Rosansky, J. Ceja, M. Patterson, S. Kilcoyne, K. Thurber, G. Kim, T. J. Dwyer and B. W. Purse, Sequence-Specific Fluorescence Turn-On Sensing of RNA by DNA Probes Incorporating the Tricyclic Cytidine Analogue DEAtC, Bioconjugate Chem., 2023, 34, 1061–1071 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, HPLC, MS, NMR spectra, detailed computational methods. See DOI: https://doi.org/10.1039/d4sc07994a |
| ‡ K. T. R. and A. P. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |