
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging heterocycle-fused 1,4-benzoquinone to design chemical modulators for both metal-free and metal-bound amyloid-β†
Yelim
Yi
 a,
Kyungmin
Kim
b,
Hakwon
Kim
*b and
Mi Hee
Lim
*a
a,
Kyungmin
Kim
b,
Hakwon
Kim
*b and
Mi Hee
Lim
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: miheelim@kaist.ac.kr
bDepartment of Applied Chemistry, Global Center for Pharmaceutical Ingredient Materials, Kyung Hee University, Gyeonggi-do 1732, Republic of Korea. E-mail: hwkim@khu.ac.kr
First published on 12th March 2025
Abstract
The complex pathology of Alzheimer's disease includes various pathogenic components, such as metal-free amyloid-β (Aβ) and metal-bound Aβ (metal–Aβ). Here we report an effective strategy for developing novel heterocycle-fused 1,4-benzoquinone (BQ) compounds to control the aggregation and toxicity of both metal-free Aβ and metal–Aβ. We designed and synthesized these compounds by fusing BQ with 3-pyrazolone responsible for metal chelation. The compounds' ability to form covalent bonds with Aβ is tuned by the annulation of the BQ moiety and the type, position, and number of substituents on the 3-pyrazolone group. Furthermore, the BQ functionality on the 3-pyrazolone framework can undergo o-hydroxylation, enhancing its metal chelation in a bidentate manner. Our results demonstrate that these heterocycle-fused BQ compounds can redirect the assembly of Aβ into less toxic aggregates by binding to metal ions, modifying Aβ structures in both the absence and presence of metal ions, and promoting oxidative changes to Aβ. This study highlights the importance of structural modifications and optimizations of BQ to leverage its strength of covalently cross-linking to Aβ and overcome its limitations in metal chelation and cytotoxicity, which are critical for designing chemical modulators for metal-free Aβ and metal–Aβ. Our approach offers a novel strategy for developing chemical modulators towards metal-related peptides and proteins as well as therapeutic agents for metal-associated amyloid disorders.
Introduction
Aberrant aggregation of peptides and proteins is linked to numerous pathological conditions.1 The amyloid cascade hypothesis, which suggests that the accumulation of amyloid-β (Aβ) aggregates is a primary cause of Alzheimer's disease (AD), has been extensively studied. This has led to the development of various chemical tools and therapeutic agents aimed at targeting and modulating Aβ aggregation.2,3 Notably, Aβ-directed monoclonal antibodies have been approved by the United States Food and Drug Administration as AD treatment, although concerns about their efficacy and safety persist.4,5 This underscores the importance of the amyloid paradigm in understanding the pathology of AD while highlighting the need for further investigation into other biological factors influencing Aβ aggregation profiles.6,7 High concentrations of metal ions, such as Cu(I/II) and Zn(II), are found in Aβ aggregates in the brains of AD patients.8–16 Metal coordination to Aβ peptides affects their conformation and aggregation pathways, indicating metal-bound Aβ (metal–Aβ) as an additional pathological component of AD.9–21 Therefore, developing chemical reagents that can modulate both metal-free Aβ and metal–Aβ is crucial for addressing the complex pathology of AD.As a means of modifying the aggregation profiles of Aβ, the manipulation of amino acid residues in the peptides has been reported.22–25 As shown in Fig. 1a, 1,4-benzoquinone (BQ), capable of forming a covalent adduct with Aβ, has been demonstrated to alter the assembly routes of Aβ. These reactions, however, are initiated after Aβ accommodates certain small molecules [e.g., N,N-dimethyl-p-phenylenediamine (DMPD), benzene-1,4-diamine, N1-((1H-pyrrol-2-yl)methyl)-N4,N4-dimethylbenzene-1,4-diamine (7), and N1,N1-dimethyl-N4-(thiophen-2-ylmethyl)benzene-1,4-diamine (L1)], which are then chemically transformed into BQ.25,29–31 Moreover, BQ is unable to chelate metal ions, limiting its interaction with metal ions in metal–Aβ complexes. The inherent toxicity of BQ further restricts its biological applications.25,32
 | ||
Fig. 1 Rational design and preparation of heterocycle-fused BQ compounds to modify the aggregation and toxicity of Aβ in the absence and presence of metal ions. (a) Ability of BQ to interact with Aβ and its properties that limit the reactivity of metal–Aβ and biological applications. Structures of monomeric (xx98156![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26) and fibrillar (PDB 2BEG27) Aβ42 are shown according to the Eisenberg hydrophobicity scale (hydrophilic to hydrophobic amino acid residues colored in a gradient from white to red).28 The amino acid residues involved in the self-recognition site and metal coordination are highlighted in red and purple, respectively. (b) Previously reported compounds that can be transformed into BQ. (c) Chemical structures and properties of heterocycle-fused BQ and NQ compounds. 1, 2-(p-Tolyl)-1H-indazole-3,4,7(2H)-trione; 2a, 1-benzyl-2-(p-tolyl)-1H-indazole-3,4,7(2H)-trione; 3, 2-(p-tolyl)-1H-benzo[f]indazole-3,4,9(2H)-trione; 4, 1H-benzo[f]indazole-3,4,9(2H)-trione; 5, 2-methyl-1H-benzo[f]indazole-3,4,9(2H)-trione. (d) Synthetic routes to 1 and 2a. 26) and fibrillar (PDB 2BEG27) Aβ42 are shown according to the Eisenberg hydrophobicity scale (hydrophilic to hydrophobic amino acid residues colored in a gradient from white to red).28 The amino acid residues involved in the self-recognition site and metal coordination are highlighted in red and purple, respectively. (b) Previously reported compounds that can be transformed into BQ. (c) Chemical structures and properties of heterocycle-fused BQ and NQ compounds. 1, 2-(p-Tolyl)-1H-indazole-3,4,7(2H)-trione; 2a, 1-benzyl-2-(p-tolyl)-1H-indazole-3,4,7(2H)-trione; 3, 2-(p-tolyl)-1H-benzo[f]indazole-3,4,9(2H)-trione; 4, 1H-benzo[f]indazole-3,4,9(2H)-trione; 5, 2-methyl-1H-benzo[f]indazole-3,4,9(2H)-trione. (d) Synthetic routes to 1 and 2a. | ||
In this study, we illustrate an effective approach for developing novel heterocycle-fused BQ compounds that can control the aggregation and toxicity of both metal-free Aβ and metal–Aβ. We rationally designed and synthesized heterocycle-fused BQ molecules and probed their effects on the assembly and toxicity of metal-free Aβ and metal–Aβ. Additionally, we determined the molecular-level mechanisms for these reactivities, including their ability to form covalent cross-links with Aβ and chelate metal ions. Our study underscores the significance of structural alterations on the BQ moiety for creating chemical reagents and therapeutic agents with regulatory reactivities towards metal-related peptides and proteins associated with amyloid diseases.
Results and discussion
Design principle and synthesis of compounds
For the covalent adduct formation between Aβ and the BQ moiety, the peptide first needs to recruit BQ-containing compounds. To enhance direct interactions with both metal-free Aβ and metal–Aβ, 3-pyrazolone was fused into the BQ moiety, as illustrated in Fig. 1b. Pyrazolones are a class of pyrazole heterocycles with various chemical and biological properties.33–38 Particularly, the planarity of small molecules containing 3-pyrazolone combined with a benzene ring is suggested to favor the disruption of the β-sheet conformation of Aβ aggregates through hydrophobic interactions.39 The carbonyl group on 3-pyrazolone also participates in hydrogen bonding with Aβ.39 Furthermore, the nitrogen (N) and oxygen (O) donor atoms from the amino and carbonyl groups on 3-pyrazolone can bind to metal ions. Upon fusion with BQ, there are two possible metal-chelation sites: (i) a pair of N and O donor atoms from the amino and carbonyl groups at 1- and 7-positions, respectively; (ii) two O donor atoms from the carbonyl groups at 3- and 4-positions. Unlike the relatively weak metal binding of BQ through its carbonyl O donor atoms, metal-chelation properties of 3-pyrazolone-fused BQ enable a sufficient interaction with metal ions surrounded by Aβ.Through further functionalization of the 3-pyrazolone-fused BQ framework, we rationally designed heterocycle-fused BQ compounds (1 and 2a; Fig. 1b). The incorporation of a p-tolyl group at the 2-position, without interfering with the metal-chelating moieties, generated compound 1 with increased hydrophobicity, which is suitable for hydrophobic interactions with Aβ. Previous studies have shown that a phenyl ring linked to the pyrazole moiety could engage in π–π, π–alkyl, and π–cation interactions with the Phe, Val and Leu, and Lys residues, respectively, located near and in the self-recognition site (Fig. 1a) crucial in the early stages of Aβ aggregation.40,41 Compound 2a, with an additional benzyl group at the 1-position, was devised to further enhance the hydrophobicity of compound 1, thereby improving hydrophobic contacts on Aβ and effectively controlling amyloid fibrillization. Although the benzyl group hinders the metal binding of the N donor atom at the 1-position, compound 2a can still coordinate metal ions via O donor atoms from the carbonyl groups at the 3-, 4-, and 7-positions.
Since α,β-unsaturated carbonyl moieties in BQ are sensitive to nucleophilic attack at the β-carbon (Cβ) atom, a Michael addition reaction can occur between the amino group from the side chain of Lys residues and the Cβ atom at the 5- or 6-position of BQ, resulting in covalent cross-linking.42 To investigate the relevance of 3-pyrazolone-fused BQ in modulating the reactivities with metal-free and metal-associated Aβ, we blocked the Cα and Cβ atoms by fusing a benzene group, creating 3-pyrazolone-fused 1,4-naphthoquinone (NQ) compounds. The annulation of BQ in compound 1 with the benzene ring produced compound 3, which allowed us to examine the role of the 3-pyrazolone-fused BQ in regulating Aβ aggregation mediated by compound 1. Given that the presence of multiple aromatic rings in compounds enhances their ability to interact with Aβ,22,43–49 we further designed compounds 4 and 5 to maintain the same number of aromatic rings as in compound 1 by eliminating the p-tolyl group at the 2-position. In the case of compound 5, the number of donor atoms for hydrogen bonding and metal binding necessary for interacting with metal-free and metal-bound Aβ was preserved by including a methyl group at the 2-position.
As summarized in Fig. 1c, we synthesized heterocycle-fused BQ compounds, 1 and 2a. Compound 1a was diazotized to generate a diazonium salt intermediate, which was then reduced with SnCl2 to synthesize a hydrazine intermediate. Intramolecular cyclization under acidic conditions produced an indazolone compound, 1b.50N-Benzylation of 1b with benzyl chloride under basic conditions yielded an N-benzylated compound, 1c.51 Next, 1d was generated via a Cu(II)-catalyzed Chan–Evans–Lam coupling reaction of 1c with p-tolylboronic acid.52 The removal of the methyl moiety in the methoxy group of 1d using boron tribromide (BBr3) afforded compound 1e.53 Oxidation of 1e with ceric ammonium nitrate led to the synthesis of 2a.54,55 Finally, compound 1 was obtained by the Pd-catalyzed hydrogenolysis of 2a in a quantitative yield.56,57 The synthetic routes and methods for heterocycle-fused NQ compounds (3–5) have been recently described in the patent literature.58 The characterization of the compounds is summarized in Fig. S1–S5.†
Effects of compounds on the aggregation of metal-free Aβ and metal–Aβ
To determine the impact of our compounds on the aggregation of metal-free and metal-bound Aβ, we initially monitored the size distribution of the resultant metal-free Aβ and metal–Aβ species upon incubation with the compounds by gel electrophoresis with western blotting (gel/western blot) using an anti-Aβ antibody (6E10). In the inhibition studies presented in Fig. 2a and S6,† Aβ peptides were incubated with and without the compounds, both in the absence and presence of metal ions, to assess their effects on the formation of Aβ aggregates [Fig. 2a(i)]. As displayed in Fig. 2a(ii), heterocycle-fused BQ compounds, particularly 2a, exhibited significant reactivity towards Aβ aggregation in both the absence and presence of metal ions. | ||
| Fig. 2 Effects of compounds on the aggregation of metal-free and metal-treated Aβ analyzed by two types of experiments [(a) inhibition and (b) disaggregation studies]. (i) Scheme of Aβ aggregation experiments. (ii) Size distribution of the resultant Aβ species probed by gel/western blot using an anti-Aβ antibody (6E10). Lane C: Aβ ± Cu(II) or Zn(II). The original gel images are shown in Fig. S6.† (iii) Morphologies of the resultant Aβ species monitored by TEM. Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-added samples] or pH 6.8 [for Cu(II)-added samples], 150 mM NaCl; 37 °C; 24 h; constant agitation. Scale bar = 200 nm. | ||
Under metal-free conditions, treatment of 1 with Aβ42 decreased the intensity of smearing at 5–11 kDa. Notably, 2a intensified smearing across the gel lane from 5 kDa to 245 kDa. On the other hand, 3 and 4 moderately increased the band intensity at 48–135 kDa, and 5 diminished the signal intensity below ca. 20 kDa. In the presence of Cu(II), the intensity of bands at 35–135 kDa increased upon treatment with 1. The Cu(II)–Aβ42 sample treated with 2a showed an increase in the signal intensity at 5–135 kDa. For Zn(II)–Aβ42, the samples added with heterocycle-fused BQ compounds showed a clearer band at ca. 35 kDa relative to the compound-untreated sample, and 2a additionally intensified the smearing at 5–20 kDa. Regarding heterocycle-fused NQ compounds, the band intensity for metal–Aβ42 species at ca. 35 kDa was slightly enhanced by 3. The signal at the same size was also darkened for Zn(II)–Aβ42 treated with 4. A significant change in the size distribution of metal–Aβ42 was not observed with the addition of 5. The results of the inhibition experiments with Aβ40 also demonstrated the noticeable ability of heterocycle-fused BQ compounds, especially 2a, to alter their aggregation pathways. Compound 2a significantly changed the size distribution of metal-free Aβ40, producing smearing between 5 kDa and 35 kDa, whereas 1 and 3–5 did not induce notable shifts in the size distribution of metal-free Aβ40. Under Cu(II)-present conditions, new discrete bands at 5–11 kDa were detected when 1 and 2a were incubated with Aβ40. Conversely, 3–5 did not modify the size distribution of the aggregates. For the sample of Zn(II)–Aβ40 incubated with 2a, explicit bands between 5 kDa and 11 kDa were revealed, while relatively minor reactivity was observed for the samples upon addition of 1 and 3–5, exhibiting blurred bands right above 5 kDa.
As depicted in Fig. 2a(iii), we further investigated the morphologies of the resultant Aβ aggregates by transmission electron microscopy (TEM), which can image large Aβ aggregates that have difficulty in penetrating the gel matrix.25 Under metal-untreated conditions, all compounds fragmented long fibrils, shown in compound-free Aβ42, into shorter filamentous species. When heterocycle-fused BQ compounds were incubated with Cu(II)–Aβ42, they produced dramatically smaller aggregates with amorphous qualities compared to the compound-untreated sample, which contained larger aggregates. Treatment with 3 also reduced the size of Aβ42 aggregates with Cu(II), but 4 and 5 did not result in considerable morphological and size changes in Cu(II)–Aβ42 aggregates. In the presence of Zn(II), all compounds altered the morphologies of Zn(II)–Aβ42 aggregates from fibrillary to less structured species. In particular, the 2a-treated sample manifested chopped fibrils with small-sized amorphous aggregates. For the samples of Aβ40, a mixture of chopped fibrils with unstructured aggregates was detected by addition of 2a under metal-free conditions, whereas compound-free, 1-, and 3–5-treated samples showed primarily fibrillary species. In the presence of Cu(II), heterocycle-fused BQ compounds generated smaller amorphous aggregates compared to compound-untreated and heterocycle-fused NQ compound-added Cu(II)–Aβ40. In the case of Zn(II)–Aβ40 samples, the formation of thinner filamentous species, without and with smaller less-structured aggregates, was induced by 1 and 2a, respectively, different from the entangled fibrillary species detected in the compound-free sample. Heterocycle-fused NQ compounds facilitated aggregation into less organized structures and generated chopped fibrils in 3-incubated Zn(II)–Aβ40. Collectively, both heterocycle-fused BQ and NQ compounds alter the formation of Aβ aggregates to different degrees.
Moreover, the disaggregation studies were performed to identify the ability of the compounds to disassemble preformed Aβ aggregates or alter their aggregation pathways further, as illustrated in Fig. 2b and S6.† The samples were prepared by pre-incubating Aβ with and without metal ions to form preformed Aβ assembles, followed by treatment with the compounds [Fig. 2b(i)]. As shown in Fig. 2b(ii), both heterocycle-fused BQ and NQ compounds were capable of shifting the size distribution of Aβ42 aggregates under metal-free and metal-present conditions. All compounds increased the intensity of bands at 48–63 kDa without metal ions, with 2a prominently varying the size distribution of metal-free Aβ42 species at 5–63 kDa. In the case of metal–Aβ42 incubated with heterocycle-fused BQ compounds, the signal intensity was enhanced at 20–100 kDa and 5–135 kDa following treatment with 1 and 2a, respectively. Heterocycle-fused NQ compounds slightly elevated the level of Cu(II)–Aβ42 species at 35–100 kDa, and 5 also decreased the signal at 5–11 kDa. In the presence of Zn(II), the size distribution of Aβ42 at 63–135 kDa was shifted by 3–5. For the experiments with Aβ40, incubation of heterocycle-fused BQ compounds remarkably altered the amount of Aβ40 oligomers between 5 kDa and 11 kDa under both metal-free and metal-present conditions, compared to the samples without compounds and those treated with heterocycle-fused NQ compounds, which showed less reactivity.
In the TEM studies depicted in Fig. 2b(iii), 1 and 3 produced thinner metal-free Aβ42 fibrils, compared to relatively thicker and longer fibrillary aggregates observed in the compound-free sample, while 2a, 4, and 5 shortened the fibrils. The size of Cu(II)–Aβ42 aggregates was dramatically reduced by addition of both heterocycle-fused BQ and NQ compounds. Under Zn(II)-treated conditions, all compounds generated thinner filamentous species different from the bundle of fibrillar aggregates of compound-free Zn(II)–Aβ42, and smaller amorphous aggregates were also shown for the samples incubated with 1, 2a, and 3. For Aβ40, both 1 and 2a remarkably truncated fibrillary species displayed in the compound-free sample under metal-free conditions. In the presence of Cu(II), the addition of heterocycle-fused BQ compounds induced the formation of smaller-sized amorphous aggregates. Under Zn(II)-present conditions, relative to Zn(II)–Aβ40 aggregates with fibrillary characteristics, a mixture of amorphous and fibrillary aggregates was monitored upon treatment of 1 with Aβ40 aggregates, and the addition of 2a triggered the formation of chopped fibrils. In contrast, little or no morphological alteration was detected for the samples prepared with heterocycle-fused NQ compounds. Overall, both heterocycle-fused BQ and NQ compounds can disassemble the preformed Aβ aggregates or additionally alter their aggregation pathways in the absence and presence of metal ions to distinct extents. Heterocycle-fused BQ compounds exhibited significant modulatory effects on the aggregation of metal-free and metal-associated Aβ over heterocycle-fused NQ compounds. This indicates that the structural modifications on the BQ moiety, without the fusion of an additional benzene ring at the 5- and 6-positions, are essential for impacting the assembly of Aβ with and without metal ions. Notably, the enhanced ability of 2a relative to 1 in changing the size distribution and morphology of Aβ aggregates under both metal-free and metal-treated conditions suggests that the benzyl ring at the 1-position of 2a contributes to its regulatory reactivity with metal-free Aβ and metal–Aβ.
Mechanistic studies (I): interactions of compounds with metal-free Aβ
To elucidate the distinct reactivities of heterocycle-fused BQ and NQ compounds towards Aβ aggregation, we investigated their interactions with Aβ peptides. Considering that Aβ42 is more pathogenic than Aβ40,59 and the modulative effects of our compounds were more discernible in the samples with Aβ42 rather than Aβ40 (Fig. 2), we focused on analyzing the direct interactions between the compounds and Aβ42 by electrospray ionization–mass spectrometry (ESI–MS). As presented in Fig. 3a, metal-free Aβ42 incubated with 1 and 3–5 showed the peaks assigned to non-covalent adducts of Aβ42 with the compounds at 1589 m/z, 1606 m/z, 1576 m/z, and 1580 m/z, respectively. In the presence of 2a (344 Da), a peak corresponding to the addition of 342 Da to the 3+-charged Aβ42 monomer (4511 Da) was detected at 1619 m/z, likely representing the covalent adduct of 2a with metal-free Aβ42. Further fragmental analysis of the peak through collision-induced dissociation (CID) identified the amino acid residue in Aβ42 involved in the formation of this covalent cross-linking. In tandem MS (ESI–MS2) depicted in Fig. 3b, b fragments smaller than b16 were observed in their native forms. Starting from b16, the b fragments were detected with and without the addition of 342 Da, suggesting Lys16 as the amino acid residue forming a covalent bond with 2a. Native forms of b fragments were no longer monitored beyond b27, indicating that Lys28 could also be a site for covalent cross-linking with 2a. | ||
| Fig. 3 Interaction of compounds with metal-free Aβ42 and 2a's chemical transformation. (a) ESI–MS studies of metal-free Aβ42 with and without compounds. Conditions: [Aβ42] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 7.4; 37 °C; 24 h; constant agitation. The samples were diluted 25-fold with H2O before injection into the mass spectrometer. (b) ESI–MS2 investigations of metal-free Aβ42 with (top; 1620 m/z) and without (bottom; 1505 m/z) covalent bond formation with 2a. (c) Possible interactions of 2a with metal-free Aβ42 (xx98156 (ref. 26)) visualized by docking studies. Nine docked models were obtained with binding energies ranging from −5.9 to −5.5 kcal mol−1, and two representative models are indicated in (i) and (ii). (d) Chemical transformation of 2a monitored by Abs spectroscopy. Conditions: [2a] = 25 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4, 150 mM NaCl; 37 °C; 24 h; no agitation. | ||
Lys residues in several amyloidogenic peptides and proteins have been reported to facilitate both hydrophobic and electrostatic interactions via flexible butyl and ε-ammonium groups in their side chains, respectively, in the intramolecular and intermolecular manner.60,61 More specifically, Lys16, adjacent to the self-recognition site (Fig. 1a), forms a salt bridge with Glu22 in the fibril structure of Aβ62 or is exposed to the solvent, making it available for interacting with other monomers.63 Additionally, Lys28 stabilizes the turn structure of monomeric Aβ through a salt bridge with Asp23, which can mediate the folding and assembly of Aβ.62,64–66 Therefore, the covalent bond formation between the BQ moiety in 2a and Lys residues of Aβ42 could be critical in altering its aggregation pathways.
Interestingly, this phenomenon was observed only in the sample incubated with 2a, not with 1. This implies the involvement of the benzyl ring at the 1-position of 2a in the covalent adduct formation with Aβ42. Based on the potential interactions between 2a and monomeric Aβ42 visualized by docking studies (Fig. 3c), the benzyl group can participate in CH–π interactions with Lys16 and Phe20. This suggests that this benzyl functionality may facilitate the recruitment of 2a by Aβ42, followed by the cross-linking reaction between them. Furthermore, 2a is positioned near the self-recognition and C-terminal regions, encompassing the Lys16 and Lys28 residues as Michael donors for forming covalent bonds with 2a. Multiple hydrogen bonds were also predicted for the carbonyl O atoms located at the 3-, 4-, and 7-positions of 2a with the amide group from the side chain of Gln15 and the backbone amide moieties between Gln15 and Lys16, and Asn27 and Lys28.
Moreover, an additional peak at 1624 m/z, indicating a mass shift of +360 Da from metal-free Aβ42 monomer, appeared upon incubation with 2a. The nature of this peak was first analyzed by examining the chemical transformation of 2a by electronic absorption (Abs) spectroscopy, as shown in Fig. 3d. The optical spectrum of 2a in aqueous solution exhibited three Abs bands at ca. 255 nm, 330 nm, and 440 nm, consistent with those observed for BQ.67–69 After 24 h of incubation, the spectral features at the shortest wavelength diminished, while a hypsochromic shift from ca. 440 nm to ca. 390 nm and a bathochromic shift from ca. 255 nm to ca. 265 nm were monitored, indicative of o-hydroxylation of the BQ moiety in 2a.67–69 Previous studies have proposed that the reaction of BQ with H2O at neutral pH triggers the formation of enolized BQ [4,6-dihydroxycyclohexa-2,4-dien-1-one; BQ(H2O)], which can be rearranged into 1,2,4-trihydroxybenzene (THB) (Fig. S7a†).67–69 Oxidation of BQ(H2O) by BQ and auto-oxidation of THB subsequently produce 2-hydroxy-BQ (2-hydroxycyclohexa-2,5-diene-1,4-dione).67–69 As depicted in Fig. S8,† the mass spectrum of 2a incubated for 24 h in aqueous solution displayed a peak assigned to [2a + 16 + H]+, plausibly denoting 2a substituted with a hydroxyl group (2b; vide infra). Thus, the peak at 1624 m/z in the mass spectrum of 2a-treated metal-free Aβ42 (Fig. 3a) can arise from the non-covalent adduct formation between monomeric Aβ42 and 2a containing a hydroxylated BQ moiety.
In summary, both heterocycle-fused BQ and NQ compounds can directly interact with monomeric Aβ42. We further confirmed that 2a, compared to 1, can participate in the generation of covalent bonds with Aβ42. These suggest that a supplementary benzyl moiety at the 1-position of 2a is required for sufficient contact with Aβ, consequently producing a covalent cross-link to the peptide viaBQ. Moreover, 2a undergoes o-hydroxylation of the BQ moiety and, subsequently, directly interacts with Aβ. Such 2a-driven interactions with metal-free Aβ support its significant reactivity towards Aβ in both the absence and presence of metal ions.
Mechanistic studies (II): interactions of chemically transformed 2a with metal ions, metal-free Aβ, and metal–Aβ
To gain a better understanding of the involvement of chemically transformed 2a in its reactivity towards the aggregation of metal-free Aβ and metal–Aβ, we synthesized compound 2b, which is 2a with a hydroxyl group at the 6-position, and analyzed its interactions with metal ions, metal-free Aβ, and metal–Aβ. As presented in Fig. 4a, compound 1e was dibrominated with Br2, yielding 4,6-dibromo-indazolone and the unexpected 6-bromo-benzoquinone (6-Br-BQ) derivative.70 We assumed that the latter was oxidized by H2O and Br2 derived from 4,6-dibromo-indazolone. Consequently, the reaction conditions were established to synthesize 6-bromo-4,7-dihydroxyindazolone, 2b-i, and 6-Br-BQ derivative, 2b-ii. 2b-i was oxidized to 2b-ii using ceric ammonium nitrate.54,55 The subsequent methoxylation of 2b-ii with sodium methoxide (NaOCH3) resulted in the formation of a 6-methoxy-benzoquinone derivative, 2b-iii.71 Finally, 2b was obtained by the demethylation of 2b-iii using BBr3 in a high yield.72 The characterization of 2b is summarized in Fig. S9.† It is noteworthy that spectral features of 2b in aqueous solution are similar to those of 2a incubated for 24 h, but with greater optical intensity. This observation supports that 2a undergoes chemical transformations into 2b and various other products, including less soluble aggregates (vide infra).67–69,73–75 | ||
| Fig. 4 Preparation of 2b and its ability to interact with metal ions, metal-free Aβ42, and Cu(II)–Aβ42. (a) Synthetic routes to 2b [1-benzyl-6-hydroxy-2-(p-tolyl)-1H-indazole-3,4,7(2H)-trione]. (b) Solution speciation studies of metal–2b (L) complexes. Abs variable-pH titration spectra (top) and the solution speciation diagram (middle) were obtained by titrations of metal–ligand complexes (FM = fraction of species at a given pH). The log β and pM values of metal–ligand complexes are summarized in the table (bottom). The errors in the last digit are shown in parentheses. Charges are omitted for clarity. Conditions: [2b] = 50 μM; [Cu(II) or Zn(II)] = 25 μM; room temperature; I = 0.1 M NaCl. (c) Interactions of 2b with metal-free Aβ42 and Cu(II)–Aβ42 monitored by ESI–MS. Conditions: [Aβ42] = 25 μM; [Cu(II)] = 25 μM; [2b] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 7.4 (for metal-free samples) or pH 6.8 [for Cu(II)-treated samples]; 37 °C; 24 h; constant agitation. The samples were diluted 25-fold with H2O before injection into the mass spectrometer. | ||
o-Hydroxylation of BQ in 2a introduces an additional metal-chelation site, composed of a pair of O donor atoms from hydroxyl and carbonyl groups at the 6- and 7-positions, respectively. To explore the capacity of 2b to bind metal ions in aqueous solution, solution speciation studies with Abs variable-pH titrations were performed. Initially, the solution of 2b (L) was titrated with small aliquots of HCl under metal-free conditions to estimate its acidity constants (Ka). As illustrated in Fig. S10,† two pKa values, 0.627(5) and 4.827(5), were calculated. The solution speciation diagram displayed the presence of anionic (LH-1), neutral (L), and monoprotonated (LH) forms of 2b in the pH range from 2 to 9. The anionic form of the ligand was predicted to predominate at pH 6.8, a condition representing a physiologically acidotic environment where Aβ assembly can be accelerated in the presence of Cu(II),76 and at physiological pH 7.4, accounting for ca. 98% and ca. 100% relative abundance, respectively.
Subsequently, solution speciation experiments of 2b were conducted in the presence of Cu(II) and Zn(II) to identify the metal-to-ligand stoichiometry and ligand's metal-binding affinities. As shown in Fig. 4b, the stability constants (β) were calculated for the formation of metal–2b complexes plausibly present at pH 0.9–7.8. Metal complexes with the 1:2 metal-to-ligand stoichiometry were indicated as a major species at pH 6.8 and pH 7.4. Based on the protonation and metal complexation of 2b at the given pH and concentrations of the ligand and metal ions, the values of pM (−log[Mfree], where [Mfree] denotes the concentration of unchelated metal ions) were computed to predict the relative metal-binding ability of 2b under our experimental conditions.77,78 At pH 6.8 and pH 7.4, 2b presented pCu and pZn values of 7.7 and 5.3, respectively. It should be noted that the metal-binding affinity of 2a could not be experimentally determined due to its chemical transformation in aqueous solution. Instead, we evaluated the metal-binding ability of 6 which is derivatized from 3, which features the substitution of the amino group at the 1-position with a methyl group and the replacement of the p-tolyl group at the 2-position with a phenyl ring.58,79 This compound is relatively stable under our experimental conditions and has potential for metal chelation through two carbonyl O donor atoms in the 3- and 4-positions, similar to 2a; however, no significant spectral change was detected in Abs spectra of the compound upon addition of Cu(II) (Fig. S11†). This corroborates the limited capability of 2a to interact with metal ions under our experimental settings. These results underline that the chemical transformation of 2a imparts the ability to chelate metal ions (Fig. 1b).
To verify the interaction between 2b and Aβ42 with and without metal ions, ESI–MS studies were carried out. As depicted in Fig. 4c and S12,† new peaks at 1651 m/z were detected upon addition of 2b to Aβ42 under both metal-free and metal-present conditions, assigned to [Aβ42 + 2b + DMSO + 3H]3+. In the presence of Cu(II), a peak at 1671 m/z corresponding to [Aβ42 + Cu(II) + 2b + DMSO + H]3+ was observed, indicating the formation of a ternary complex composed of Aβ42, Cu(II), and 2b. This compound also generated a ternary complex with Zn(II)–Aβ42 (Fig. S12†). The pM values of 2b (Fig. 4b), comparable to those of Aβ (ca. 6–12 for pCu at pH 6.5–7.4; ca. 6 for pZn at pH 7.4),11,13,80,81 support such competitive metal coordination of 2b with Aβ. These findings demonstrate that 2b can interact with metal ions as well as Aβ in both the absence and presence of metal ions. Consequently, the chemical transformation of 2a, including o-hydroxylation likely into 2b, enables it to chelate metal ions in a bidentate manner with binding affinities similar to those of Aβ, ultimately impacting the self-assembly of metal–Aβ.
Mechanistic studies (III): impact of 2b on the aggregation of metal-free Aβ and metal–Aβ
The effect of 2b on the assembly of metal-free Aβ and metal–Aβ was further probed by gel/western blot using 6E10 and TEM. In the inhibition studies, the size distribution of Aβ species with and without metal ions was not significantly influenced by 2b, as delineated in Fig. 5a and S6;† however, the impact of 2b on the aggregation of the peptides in the absence and presence of metal ions was discernibly shown in the TEM studies. The resultant Aβ42 and Aβ40 aggregates produced in the presence of 2b were analyzed as shorter fibrils relative to compound-free samples, which showed significant fibrillar aggregates under metal-free and Zn(II)-added conditions, as illustrated in Fig. 5b. Upon incubation of 2b with Cu(II)–Aβ, a mixture of smaller aggregates with amorphous and fibrillary characteristics was generated for Aβ42 and Aβ40, differing from compound-free samples, which mainly displayed larger aggregates. Together, 2b and 2a affect the formation of Aβ aggregates in the absence and presence of metal ions to distinct extents. | ||
| Fig. 5 Impact of 2a and 2b on the formation of metal-free and metal-associated Aβ aggregates. (a) Size distribution of the resultant Aβ species analyzed by gel/western blot using 6E10. Lane C: Aβ ± Cu(II) or Zn(II). The original gel images are shown in Fig. S6.† (b) Morphologies of the resultant Aβ species investigated by TEM. Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-containing samples] or pH 6.8 [for Cu(II)-containing samples], 150 mM NaCl; 37 °C; 24 h; constant agitation. Scale bar = 200 nm. | ||
In the disaggregation studies conducted by gel/western blot using 6E10, as shown in Fig. S6 and S13,† the reactivity of 2b with metal-free and metal-associated Aβ was not clearly monitored. TEM, however, presented that 2b-added Aβ42 and Aβ40 samples included shorter and chopped fibrils, different from compound-unadded samples which exhibited bigger aggregates, under metal-free and metal-added conditions. These findings indicate that both 2b and 2a can disassemble preformed Aβ aggregates or additionally alter their aggregation profiles in the absence and presence of metal ions to varying degrees. Overall, 2b, as the chemically transformed form of 2a, can modulate the aggregation pathways of Aβ in both the absence and presence of metal ions, potentially contributing to the reactivity of 2a with metal-free Aβ and metal–Aβ. Given that Aβ samples incubated with 2a contain various chemically transformed forms of 2a, the reactivity of 2a towards Aβ aggregation with and without metal ions may be influenced not only by 2b but also by other chemically transformed forms, as well as intact 2a (Fig. S7b†). It should be noted that we monitored the formation of 2b′ upon incubation of 2a for 6 h in aqueous media, followed by its disappearance after 24 h, as depicted in Fig. S8.† According to previous studies, compounds with multiple aromatic rings can participate in π–π stacking, leading to the generation of relatively less soluble aggregates that become optically undetectable.71,72 The aggregates present in 2a- and 2b-incubated samples, which differ in composition and relative abundance, may further influence the extent to which 2a and 2b affect the assembly of Aβ. In addition, as shown in Fig. S7c,† a hydroxyl group at the 6-position of 2b enables additional hydrogen bonding with Aβ, potentially altering its reactivity with Aβ in the absence and presence of metal ions compared to 2a.
Mechanistic studies (IV): metal-binding properties of compounds and modifications of Aβ by compounds in the presence of Cu(II)
Apart from 2a, 1 and 3–5 share two possible metal-chelation sites: N and O donor atoms at the 1- and 7-positions, respectively, and two O donor atoms at the 3- and 4-positions. Given that their metal-binding ability can mediate their reactivity with metal–Aβ, metal-binding properties of 1 as a representative molecule were analyzed. Job's method of continuous variation was used to determine the Cu(II)-to-ligand stoichiometry (Fig. S14a†). We mixed 1 and Cu(II) at various ratios while maintaining their total concentration and traced the Abs at 250 nm. The Job plot revealed a maximum intensity at a mole fraction of Cu(II) of ca. 0.3 with an asymmetrical curve shape, suggesting that a mixture of Cu(II)–1 complexes with 1:1 and 1:2 Cu(II)-to-ligand ratios could exist in aqueous solution.82,83 Next, we titrated the aqueous solution of 1 with Cu(II) and monitored the spectral changes at 250 nm (Fig. S14b and c†). These led to estimating the dissociation constants (Kd) of 1 for Cu(II) to be in the micromolar range. Although this Cu(II)-binding affinity is likely insufficient to effectively interact with the primary Cu(II)-binding site in Aβ, which has a nanomolar affinity, the presence of a second Cu(II)-binding site, with an affinity in the micromolar range,10,30,84 implies that the interaction of 1 and 3–5 with Cu(II) at this secondary site in Aβ may still be feasible. It should be noted that the pKa value of 1 and its Zn(II)-binding affinity could not be determined due to the limited spectral change within the pH range monitored for our measurements without dilution effects and upon incubation with Zn(II), respectively, under our experimental conditions (Fig. S15†).
We then examined the samples of metal–Aβ42 incubated with heterocycle-fused BQ or NQ compounds by ESI–MS. As presented in Fig. 6a, 1 and 3–5 formed non-covalent adducts with Aβ42 in the presence of Cu(II), similar to what was observed under metal-free conditions (1589 m/z, 1606 m/z, 1576 m/z, and 1580 m/z for 1, 3, 4, and 5, respectively). 3 additionally indicated the ternary complexation with Aβ42 and Cu(II) (1626 m/z). Under Zn(II)-present conditions, all compounds could also interact with Aβ42 similar to their behavior in the absence of metal ions (Fig. S16†). Interestingly, consecutive peaks appeared in the Cu(II)–Aβ42 samples added with heterocycle-fused BQ compounds at 1510 m/z, 1515 m/z, and 1520 m/z, corresponding to singly, doubly, and triply oxidized Aβ42, respectively. Upon applying the CID energy to the peak at 1510 m/z, as depicted in Fig. 6b, b fragments from b13 were exhibited in their nonoxidized and oxidized forms, indicating that heterocycle-fused BQ compounds oxidized His13, His14, or Met35. The CID of the peaks assigned to doubly and triply oxidized peptides could not be well-resolved under our experimental settings, but three plausible oxidation sites imply that heterocycle-fused BQ compounds simultaneously oxidize two or three of His13, His14, and Met35.
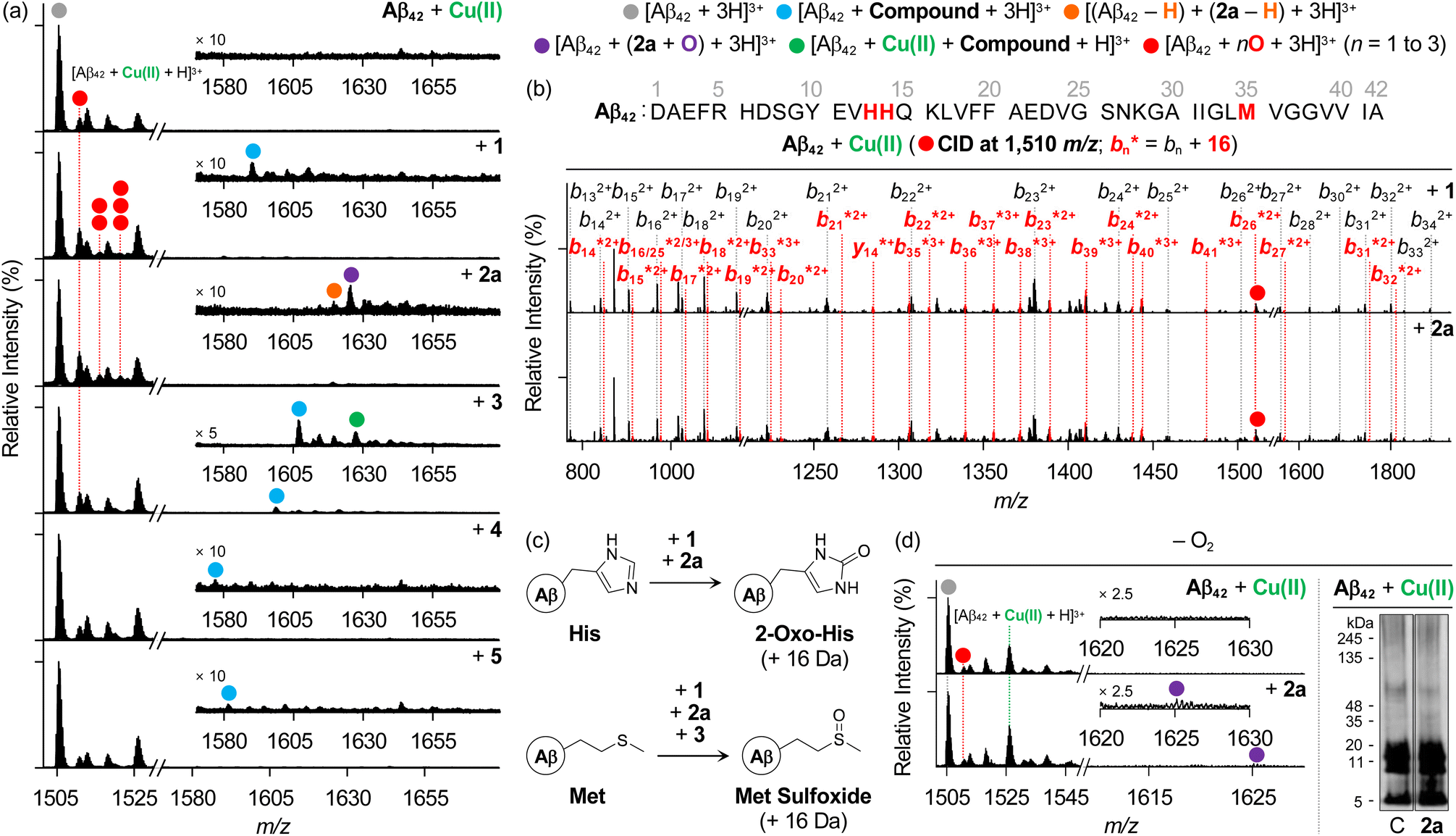 | ||
| Fig. 6 Analysis of Cu(II)–Aβ42 upon addition of compounds in the absence and presence of O2. (a) ESI–MS studies of Cu(II)-added Aβ42 samples with and without compounds under aerobic conditions. The number of red circles represents that of O atoms incorporated into the Aβ42 monomer. Conditions: [Aβ42] = 25 μM; [Cu(II)] = 25 μM; [compound] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 6.8; 37 °C; 24 h; constant agitation. The samples were diluted 25-fold with H2O before injection into the mass spectrometer. (b) ESI–MS2 analysis of the singly oxidized Aβ42 obtained by incubation with heterocycle-fused BQ compounds. (c) Oxidation of His and Met residues observed in this work. (d) Analysis of Cu(II)–Aβ42 samples upon incubation with 2a under anaerobic conditions by ESI–MS (left) and gel/western blot using 6E10 (right). Lanes: (C) Aβ + Cu(II); (2a) C + 2a. The original gel images are shown in Fig. S6.† Conditions: [Aβ42] = 25 μM; [Cu(II)] = 25 μM; [2a] = 25 μM (1% v/v DMSO); 20 mM ammonium acetate, pH 6.8 (for ESI–MS) or 20 mM HEPES, pH 6.8, 150 mM NaCl (for gel/western blot); 37 °C; 24 h; quiescent conditions. The samples were diluted 25-fold with H2O before injection into the mass spectrometer. | ||
As summarized in Fig. 6c, the structural conversion of His into 2-oxo-His, caused by heterocycle-fused BQ compounds, was reported to change the polarity of His, leading to disturbances in electrostatic interactions of Aβ peptides, thereby modifying their assembly profiles.12,85 Additionally, His residues are included in the metal-coordination site in Aβ (Fig. 1a) and, thus, their structural modification affects metal binding to Aβ peptides and their aggregation pathways in the presence of metal ions.11 Furthermore, the oxidative modification of the Met residue results in Met sulfoxide, which can alter the secondary structures across the peptides, reducing their hydrophobic and electrostatic associations.86–89 In the case of the 3-treated sample, a singly oxidized Aβ42 monomer was also shown (Fig. 6a), and Met35 was revealed to be the oxidation site, as detected for the compound-free Cu(II)–Aβ42 sample (Fig. S17†).
To verify the involvement of copper–O2 chemistry in the oxidative transformation of Aβ,90,91 the samples of Cu(II)–Aβ42 incubated with and without 2a under anaerobic conditions were further explored, as shown in Fig. 6d. Distinct from the mass spectra obtained under aerobic conditions (Fig. 6a), there was no notable variation in the signal intensity of the singly oxidized Aβ42 monomer upon treatment with 2a, and the peaks assigned to multiply oxidized peptides were not observed in the absence of O2. Thus, copper–O2 chemistry may promote the oxidative modifications of Aβ in the presence of 2a. Notably, Aβ42 still formed a non-covalent adduct with the chemically transformed 2a (2a containing o-hydroxylated BQ) under anaerobic conditions. In the gel/western blot using 6E10, 2a produced Aβ oligomers of 5–11 kDa. According to the ESI–MS results, Cu(II)-added Aβ42 aggregation could be varied even without O2 by the chemical transformation of 2a and its subsequent interaction with Aβ42. Our biophysical and biochemical observations substantiate the direct interactions of both heterocycle-fused BQ and NQ compounds with Aβ in the presence of metal ions. Heterocycle-fused BQ compounds, particularly 2a, can concurrently oxidize multiple amino acid residues in Aβ in the presence of Cu(II) depending on the availability of O2, changing the aggregation patterns of Cu(II)–Aβ with and without O2 in a distinct manner.
Cytotoxicity of compounds and their influence on Aβ-induced cytotoxicity with and without metal ions
As our compounds affect the aggregation pathways of Aβ in the absence and presence of metal ions (Fig. 2, 5 and S13†), their ability to control the cytotoxicity induced by Aβ with and without metal ions was assessed by the MTT assay [MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] employing human neuroblastoma SH-SY5Y (5Y) cells (Fig. 7a). Prior to conducting cell studies with Aβ, the cytotoxicity of heterocycle-fused BQ and NQ compounds and 2b was evaluated under metal-free and metal-present conditions and compared with that of BQ and NQ. As shown in Fig. 7b, no significant cytotoxicity was observed for heterocycle-fused BQ and NQ compounds and 2b in both the absence and presence of metal ions (cell survival over 90%) at the concentrations used for cell studies with Aβ. In contrast, BQ and NQ severely decreased cell survival by ca. 33–65% regardless of the presence of metal ions. These results demonstrate that the fusion of BQ and NQ on the 3-pyrazole framework, with and without the substitution at the 1- or 2-position or both, as well as the 6-position, can mitigate their cytotoxicity. It should be noted that the cytotoxicity of 4 could not be determined due to its limited solubility under our experimental conditions. | ||
| Fig. 7 Cytotoxicity of compounds under metal-free and metal-treated conditions and their impact on the cytotoxicity induced by metal-free and metal-associated Aβ. (a) Scheme of the experiments. (b) Viability (%) of 5Y cells upon incubation with heterocycle-fused BQ or NQ compounds, 2b, BQ, or NQ with and without metal ions. (c) Influence of heterocycle-fused BQ and NQ compounds and 2b on the cytotoxicity mediated by metal-free and metal-treated Aβ. The MTT assay provided the cell survival (%), calculated relative to that of cells treated with an equivalent amount of the buffered solution (20 mM HEPES, pH 7.4 or pH 6.8, 150 mM NaCl) with 0.2% v/v DMSO. Conditions: [Aβ] = 25 μM; [Cu(II) or Zn(II)] = 25 μM; [compound] = 25 μM (0.2% v/v DMSO); 20 mM HEPES, pH 7.4 [for metal-free and Zn(II)-added samples] or pH 6.8 [for Cu(II)-added samples], 150 mM NaCl. All values are indicated as mean ± SEM. Statistically significant difference from compound-untreated controls was determined by a two-sided unpaired Student's t-test (*P < 0.05, **P < 0.01, or ***P < 0.001). | ||
Moving forward, Aβ species preincubated with and without heterocycle-fused BQ or NQ compounds in the absence and presence of metal ions were introduced into the cells. As illustrated in Fig. 7c, some compounds exhibited substantial mitigating effects on the cytotoxicity triggered by Aβ with and without metal ions. The amelioration of metal-free Aβ42-induced cytotoxicity was achieved by addition of 2a, enhancing cell viability by ca. 10%. When 1 and 2a were incubated with metal–Aβ42, the cell survival increased by ca. 12–17%. Treatment of 2b decreased the cytotoxicity of Cu(II)–Aβ42 by ca. 12%. Cells added with metal-free and metal-associated Aβ42 with heterocycle-fused NQ compounds also showed an increase in cell viability by ca. 6–22%, compared to those treated with compound-free samples. In the case of Aβ40, the cell survival was enhanced upon treatment with 1 in the presence of Zn(II) and 2a with and without metal ions by ca. 23% and ca. 3–7%, respectively. The addition of 2b with Cu(II)–Aβ40 improved cell viability by ca. 6%. Regarding heterocycle-fused NQ compounds, 3 lowered metal–Aβ40-induced cytotoxicity by ca. 3–12%, and 5 also reduced cellular death mediated by Aβ40 under metal-free and Zn(II)-present conditions by ca. 9–10%. Overall, our investigations confirm that heterocycle-fused BQ and NQ compounds and 2b exert cytoprotective effects against metal-free and metal-associated Aβ.
Conclusions
AD is linked to the self-assembly of Aβ in the absence and presence of metal ions, resulting in neurotoxic aggregates observed as deposits in the patients' brains.10–14,19,21,92 In an effort to modulate the aggregation of Aβ, small molecules that can be converted into BQ have been reported.25,29,30,73 Following their chemical transformation, BQ forms covalent adducts with Aβ peptides, with a consequent impact on their assembly routes;25,29,30 however, the inability to chelate metal ions and the significant toxicity of BQ have limited its reactivity with metal–Aβ and its biological applications. In this work, we demonstrate how to effectively design heterocycle-fused BQ compounds that modulate the aggregation of both metal-free Aβ and metal–Aβ by leveraging the strength of BQ (e.g., covalent cross-linking to Aβ) while addressing its weaknesses (e.g., poor metal chelation and toxicity).We rationally designed bidentate ligands by combining BQ with the 3-pyrazolone framework, varying the type, position, and number of substituents on the 3-pyrazolone group without (1 and 2a) and with (3–5) the annulation of BQ with a benzene ring. Our multidisciplinary investigations reveal that heterocycle-fused BQ compounds significantly regulate the aggregation of metal-free Aβ and metal–Aβ, while heterocycle-fused NQ compounds lack this reactivity. These findings underscore the importance of the structural tuning and refinement of the BQ moiety in directing the ability of heterocycle-fused BQ compounds to change the self-assembly pathways of both metal-free Aβ and metal–Aβ. More importantly, 2a can covalently cross-link to Aβ viaBQ with the assistance of the benzyl group at the 1-position for Aβ interaction. The BQ functionality of 2a further undergoes o-hydroxylation, enhancing its ability to chelate metal ions and, consequently, form a ternary complex with metal–Aβ. Moreover, heterocycle-fused BQ compounds simultaneously oxidize multiple amino acid residues in Aβ peptides added with Cu(II) and modify their assembly profiles in both the absence and presence of O2 to varying degrees. Our compounds ultimately reduce the toxicity induced by Aβ with and without metal ions in living cells. Collectively, our findings highlight the structural variations and optimizations of BQ for designing chemical tools to target and control the aggregation and toxicity of both metal-free Aβ and metal–Aβ, as well as developing therapeutic agents to combat metal-entangled pathologies in amyloid diseases. In the near future, detailed structure–activity relationship studies of our compounds will be conducted to identify compounds with the highest efficiency and enhanced properties for biological applications, including metabolic stability and blood–brain barrier permeability, and proceed with their in vivo evaluations.
Data availability
All experimental details and data supporting the findings of this study are available within the paper and its ESI.† The data are also available from the corresponding authors upon reasonable request.Author contributions
Y. Y., H. K., and M. H. L. designed the research. Y. Y. performed gel/western blot, TEM, ESI–MS, docking simulations, and solution speciation studies using Abs spectroscopy and analyzed the data. Y. Y. also conducted cell studies using the MTT assay. K. K. and H. K. synthesized all compounds tested in this work. Y. Y., K. K., H. K., and M. H. L. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government [RS-2022-NR070709 (M. H. L.)] and the GRRC program [GRRC-KyungHee 2023(B01)] of Gyeonggi province, Republic of Korea (H. K.).References
- N. Louros, J. Schymkowitz and F. Rousseau, Nat. Rev. Mol. Cell Biol., 2023, 24, 912 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184 CAS.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306 CrossRef CAS PubMed.
- J. Sevigny, P. Chiao, T. Bussiere, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50 CAS.
- M. Sarazin, J. Lagarde, I. El Haddad, L. C. de Souza, B. Bellier, M. C. Potier, M. Bottlaender and G. Dorothee, Nat. Aging, 2024, 4, 761 Search PubMed.
- Y. Zhang, H. Chen, R. Li, K. Sterling and W. Song, Signal Transduct. Target. Ther., 2023, 8, 248 CrossRef CAS PubMed.
- D. J. Selkoe, Nat. Aging, 2024, 4, 453 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47 CrossRef CAS.
- J. T. Pedersen, J. Ostergaard, N. Rozlosnik, B. Gammelgaard and N. H. Heegaard, J. Biol. Chem., 2011, 286, 26952 CrossRef CAS PubMed.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193 CrossRef CAS PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221 CrossRef CAS PubMed.
- J.-M. Suh, M. Kim, J. Yoo, J. Han, C. Paulina and M. H. Lim, Coord. Chem. Rev., 2023, 478, 214978 CrossRef CAS.
- J. Han, Z. Du and M. H. Lim, Acc. Chem. Res., 2021, 54, 3930 CrossRef CAS PubMed.
- S. Park, C. Na, J. Han and M. H. Lim, Metallomics, 2023, 15, mfac102 CrossRef PubMed.
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Acc. Chem. Res., 2019, 52, 2026 CAS.
- Y. Yi and M. H. Lim, RSC Chem. Biol., 2023, 4, 121 RSC.
- E. Falcone and C. Hureau, Chem. Soc. Rev., 2023, 52, 6595 RSC.
- E. Atrián-Blasco, P. Gonzalez, A. Santoro, B. Alies, P. Faller and C. Hureau, Coord. Chem. Rev., 2018, 371, 38 CrossRef PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205 CrossRef CAS PubMed.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193 CrossRef CAS PubMed.
- A. I. Bush, Trends Neurosci., 2003, 26, 207 CAS.
- M. Hong, M. Kim, J. Yoon, S. H. Lee, M. H. Baik and M. H. Lim, JACS Au, 2022, 2, 2001 CAS.
- C. J. Podracky, C. H. An, A. DeSousa, B. M. Dorr, D. M. Walsh and D. R. Liu, Nat. Chem. Biol., 2021, 17, 317 CAS.
- K. Usui, J. D. Hulleman, J. F. Paulsson, S. J. Siegel, E. T. Powers and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18563 CAS.
- M. Kim, J. Kang, M. Lee, J. Han, G. Nam, E. Tak, M. S. Kim, H. J. Lee, E. Nam, J. Park, S. J. Oh, J. Y. Lee, J. Y. Lee, M. H. Baik and M. H. Lim, J. Am. Chem. Soc., 2020, 142, 8183 CAS.
- W. Yang, B. S. Kim, Y. Lin, D. Ito, J. H. Kim, Y.-H. Lee and W. Yu, bioRxiv, 2021 DOI:10.1101/2021.08.23.457317.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Döbeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342 Search PubMed.
- D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, J. Mol. Biol., 1984, 179, 125 CAS.
- J. S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil, K. J. Korshavn, A. Ramamoorthy, R. Prabhakar, E. J. Merino, J. Shearer, J. Y. Lee, B. T. Ruotolo and M. H. Lim, J. Am. Chem. Soc., 2015, 137, 14785 CAS.
- M. W. Beck, J. S. Derrick, R. A. Kerr, S. B. Oh, W. J. Cho, S. J. C. Lee, Y. Ji, J. Han, Z. A. Tehrani, N. Suh, S. Kim, S. D. Larsen, K. S. Kim, J. Y. Lee, B. T. Ruotolo and M. H. Lim, Nat. Commun., 2016, 7, 13115 CAS.
- J. Han, H. J. Lee, K. Y. Kim, G. Nam, J. Chae and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5160 CrossRef CAS PubMed.
- Y. S. Lin, W. McKelvey, S. Waidyanatha and S. M. Rappaport, Biomarkers, 2006, 11, 14 Search PubMed.
- N. Uramaru, H. Shigematsu, A. Toda, R. Eyanagi, S. Kitamura and S. Ohta, J. Med. Chem., 2010, 53, 8727 CrossRef CAS PubMed.
- X. F. Wei, Y. Huang, Z. Karimi, J. P. Qu and B. M. Wang, J. Org. Chem., 2023, 88, 10190 CrossRef CAS PubMed.
- A. E. Rubtsov, R. R. Makhmudov, N. V. Kovylyaeva, N. I. Prosyanik, A. V. Bobrov and V. V. Zalesov, Pharm. Chem. J., 2002, 36, 608 CrossRef CAS.
- A. M. Asiri and S. A. Khan, Molecules, 2010, 15, 6850 CrossRef CAS PubMed.
- V. Boerlin, B. Maeglin, W. Hagler, M. Kuhn and E. Nuesch, Eur. J. Clin. Pharmacol., 1986, 31, 127 CrossRef CAS PubMed.
- M. Neugebauer, A. Khedr, N. ElRabbat, M. ElKommos and G. Saleh, Biomed. Chromatogr., 1997, 11, 356 CrossRef CAS.
- S. Q. Cong, Y. C. Shi, G. J. Yu, F. Zhong, J. J. Li, J. Liu, C. Y. Ye, Z. H. Tan and Y. Deng, Eur. J. Med. Chem., 2023, 250, 115216 CrossRef CAS PubMed.
- T. Mohamed, A. Shakeri and P. P. N. Rao, Eur. J. Med. Chem., 2016, 113, 258 CrossRef CAS PubMed.
- V. Esposito, R. Das and G. Melacini, J. Am. Chem. Soc., 2005, 127, 9358 CrossRef CAS PubMed.
- N. Shu, L. G. Lorentzen and M. J. Davies, Free Radic. Biol. Med., 2019, 137, 169 CrossRef CAS PubMed.
- Y. L. Han, H. H. Yin, C. Xiao, M. T. Bernards, Y. He and Y. X. Guan, ACS Chem. Neurosci., 2023, 14, 4051 CrossRef CAS PubMed.
- H. T. T. Phan, K. Samarat, Y. Takamura, A. F. Azo-Oussou, Y. Nakazono and M. C. Vestergaard, Nutrients, 2019, 11, 756 CrossRef CAS PubMed.
- H. Kobayashi, M. Murata, S. Kawanishi and S. Oikawa, Int. J. Mol. Sci., 2020, 21, 3561 CrossRef CAS PubMed.
- A. S. DeToma, J. Krishnamoorthy, Y. Nam, H. J. Lee, J. R. Brender, A. Kochi, D. Lee, V. Onnis, C. Congiu, S. Manfredini, S. Vertuani, G. Balboni, A. Ramamoorthy and M. H. Lim, Chem. Sci., 2014, 5, 4851 RSC.
- S. Lee, X. Y. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299 CrossRef CAS PubMed.
- H. J. Lee, K. J. Korshavn, Y. Nam, J. Kang, T. J. Paul, R. A. Kerr, I. S. Youn, M. Ozbil, K. S. Kim, B. T. Ruotolo, R. Prabhakar, A. Ramamoorthy and M. H. Lim, Chem. Eur. J., 2017, 23, 2706 CrossRef CAS.
- Y. Yi, J. Han, M. H. Park, N. Park, E. Nam, H. K. Jin, J. S. Bae and M. H. Lim, Chem. Commun., 2019, 55, 5847 RSC.
- P. I. Eacho, P. S. Foxworthy-Mason, H.-S. Lin, J. E. Lopez, M. K. Mosior and M. E. Richett, World Intellectual Property Organization, WO2004093872A1, 2004 Search PubMed.
- T. M. Sokolenko and L. M. Yagupolskii, Chem. Heterocycl. Compd., 2011, 46, 1335 CrossRef CAS.
- K. Kim, J. H. Kim, H. Choi, B. Lee, J. Lee, K. M. Ok, T. H. Lee and H. Kim, Molecules, 2023, 28, 6706 CrossRef CAS PubMed.
- K. Okamoto and K. Chiba, Org. Lett., 2020, 22, 3613 CrossRef CAS PubMed.
- D. R. Appleton, A. N. Pearce and B. R. Copp, Tetrahedron, 2010, 66, 4977 CrossRef CAS.
- A. Yajima, A. Yamaguchi, F. Saitou, T. Nukada and G. Yabuta, Tetrahedron, 2007, 63, 1080 CrossRef CAS.
- S. M. Bhosale, A. A. Momin, S. Kunjir, P. R. Rajamohanan and R. S. Kusurkar, Tetrahedron Lett., 2014, 55, 155 CrossRef CAS.
- S. M. Bhosale, R. L. Gawade, V. G. Puranik and R. S. Kusurkar, Tetrahedron Lett., 2012, 53, 2894 CAS.
- H. Kim, T. H. Lee, and H. Lee, Korea Patent, KR2023015005A, 2023.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonekheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408 CrossRef CAS PubMed.
- S. Sinha, D. H. J. Lopes, Z. M. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P. Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F. G. Klärner, T. Schrader, C. Y. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958 CAS.
- S. Sinha, D. H. J. Lopes and G. Bitan, ACS Chem. Neurosci., 2012, 3, 473 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. H. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262 CrossRef CAS PubMed.
- S. Zhang, K. Iwata, M. J. Lachenmann, J. W. Peng, S. Li, E. R. Stimson, Y. Lu, A. M. Felix, J. E. Maggio and J. P. Lee, J. Struct. Biol., 2000, 130, 130 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742 CrossRef CAS PubMed.
- A. T. Petkova, W. M. Yau and R. Tycko, Biochemistry, 2006, 45, 498 CrossRef CAS PubMed.
- N. D. Lazo, M. A. Grant, M. C. Condron, A. C. Rigby and D. B. Teplow, Protein Sci., 2005, 14, 1581 CrossRef CAS PubMed.
- O. Fónagy, E. Szabó-Bárdos and O. Horváth, J. Photochem. Photobiol. A, 2021, 407, 113057 CrossRef.
- K. C. Kurien and P. A. Robins, J. Chem. Soc. B, 1970, 855, 10.1039/j29700000855.
- J. Von Sonntag, E. Mvula, K. Hildenbrand and C. Von Sonntag, Chem. Eur. J., 2004, 10, 440 CrossRef CAS PubMed.
- M. Inman, A. Visconti, C. Yan, D. Siegel, D. Ross and C. J. Moody, Org. Biomol. Chem., 2014, 12, 4848 RSC.
- A. Fernández, E. Alvarez, R. Alvarez-Manzaneda, R. Chahboun and E. Alvarez-Manzaneda, Chem. Commun., 2014, 50, 13100 RSC.
- M. E. Jung and R. W. Brown, Tetrahedron Lett., 1981, 22, 3355 CrossRef CAS.
- C. Zhang, Y. Zhang, K. Fan, Q. Zou, Y. Chen, Y. Wu, S. Bao, L. Zheng, J. Ma and C. Wang, CCS Chem., 2022, 4, 2768 CrossRef CAS.
- J. Yang, P. Xiong, Y. Shi, P. Sun, Z. Wang, Z. Chen and Y. Xu, Adv. Funct. Mater., 2020, 30, 1909597 CrossRef CAS.
- F. Toda, M. Senzaki and R. Kuroda, Chem. Commun., 2002, 1778 Search PubMed.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817 CrossRef CAS PubMed.
- W. R. Harris, C. J. Carrano, S. R. Cooper, S. R. Sofen, A. E. Avdeef, J. V. Mcardle and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 6097 CrossRef CAS.
- T. Storr, M. Merkel, G. X. Song-Zhao, L. E. Scott, D. E. Green, M. L. Bowen, K. H. Thompson, B. O. Patrick, H. J. Schugar and C. Orvig, J. Am. Chem. Soc., 2007, 129, 7453 CrossRef CAS PubMed.
- H. Kim, T. H. Lee, and H. Lee, Korea Patent, KR2022036697A, 2022.
- Y. Yi, Y. Lin, J. Han, H. J. Lee, N. Park, G. Nam, Y. S. Park, Y. H. Lee and M. H. Lim, Chem. Sci., 2021, 12, 2456 RSC.
- G. Kim, E. Lelong, J. Kang, J.-M. Suh, N. Le Bris, H. Bernard, D. Kim, R. Tripier and M. H. Lim, Inorg. Chem. Front., 2020, 7, 4222 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792 RSC.
- F. Ulatowski, K. Dabrowa, T. Balakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746 CrossRef CAS PubMed.
- C. Hureau and P. Dorlet, Coord. Chem. Rev., 2012, 256, 2175 CrossRef CAS.
- C. Cheignon, F. Collin, L. Sabater and C. Hureau, Antioxidants, 2023, 12, 472 CrossRef CAS PubMed.
- L. M. Hou, H. Y. Shao, Y. B. Zhang, H. Li, N. K. Menon, E. B. Neuhaus, J. M. Brewer, I. J. L. Byeon, D. G. Ray, M. P. Vitek, T. Iwashita, R. A. Makula, A. B. Przybyla and M. G. Zagorski, J. Am. Chem. Soc., 2004, 126, 1992 CrossRef CAS PubMed.
- A. A. Watson, D. P. Fairlie and D. J. Craik, Biochemistry, 1998, 37, 12700 CrossRef CAS PubMed.
- M. Friedemann, E. Helk, A. Tiiman, K. Zovo, P. Palumaa and V. Tougu, Biochem. Biophys. Rep., 2015, 3, 94 Search PubMed.
- M. Gu and J. H. Viles, Biochim. Biophys. Acta, 2016, 1864, 1260 CAS.
- A. K. Nath, A. Ghatak, A. Dey and S. G. Dey, Chem. Sci., 2021, 12, 1924 CAS.
- G. F. Z. da Silva and L.-J. Ming, Angew. Chem., Int. Ed., 2005, 44, 5501 CrossRef CAS PubMed.
- M. Kim and M. H. Lim, Bull. Korean Chem. Soc., 2021, 42, 1272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S17. See DOI: https://doi.org/10.1039/d4sc06070a |
| This journal is © The Royal Society of Chemistry 2025 |