
Challenges in membrane electrode assemblies at elevated temperatures for proton exchange membrane fuel cells: a review
Caizheng
Yue
 ab,
Weibo
Zheng
*ab,
Qiuya
Wang
c,
Zhendong
Wang
ab,
Bing
Li
ab,
Cunman
Zhang
ab and
Pingwen
Ming
*ab
ab,
Weibo
Zheng
*ab,
Qiuya
Wang
c,
Zhendong
Wang
ab,
Bing
Li
ab,
Cunman
Zhang
ab and
Pingwen
Ming
*ab
aSchool of Automotive Studies, Tongji University, Shanghai, 201804, China. E-mail: zhengweibo@tongji.edu.cn; pwming@tongji.edu.cn
bClean Energy Automotive Engineering Center, Tongji University, Shanghai, 201804, China
cSchool of Materials Science and Engineering, East China University of Science and Technology, Shanghai, 200237, China
First published on 20th May 2025
Abstract
Elevated temperatures within membrane electrode assemblies (MEAs) have gained considerable attention as the development of proton exchange membrane fuel cells (PEMFCs) has progressed. For PEMFCs to achieve widespread commercialization, ensuring the stable operation of MEAs at elevated temperatures is essential, but elevated temperatures create several challenges that can negatively impact a PEMFC's performance and lifespan. These challenges include limited mass transfer, charge conduction, and electrochemical reactions, as well as difficulty managing water and gas flows within a system. Elevated temperatures also induce the accelerated degradation of materials and structures, thereby affecting the long-term durability of various MEA components. This review provides an in-depth analysis of these challenges, describing the causes and classification of elevated temperature states within a MEA and the critical changes in transport processes and load response characteristics that they cause. It also discusses the effects of elevated temperatures on the material properties and structural integrity of MEA components (catalyst layer, proton exchange membrane, and gas diffusion layer), and the resulting performance loss mechanism. Recent progress in overcoming challenges caused by the elevated temperatures is also summarised. Finally, based on the identified challenges and current research, this review emphasises the significant role of heat in suboptimal MEA performance and lifespan and offers valuable insights into future directions for optimizing MEA performance and longevity at elevated temperatures. This provides guidance for the further development of PEMFCs for applications at elevated temperatures.
Broader contextThe global environmental challenges associated with the widespread use of fossil fuels underscore the urgent need for sustainable energy solutions. Hydrogen energy, particularly through the proton exchange membrane fuel cell (PEMFC), presents a clean and efficient alternative. The operation of the membrane electrode assemblies (MEAs) at elevated temperatures has garnered increasing attention as PEMFC technology progresses. However, the performance and longevity of PEMFCs under such conditions remain significantly limited, primarily due to the inability of current operating strategies and cell designs to effectively address the new challenges faced by MEA performance at elevated temperatures. A comprehensive understanding of the effects of elevated temperatures on the internal transport processes and degradation mechanisms of MEA components is an essential prerequisite for improving PEMFC operation under such conditions. In this review, we thoroughly analyze the causes, classification, and impacts of the elevated temperature state in the MEA, highlighting the associated challenges in MEA transport processes and component durability and summarizing the recent progress in overcoming elevated temperature limitations. We also propose valuable insights into future directions for improving performance at elevated temperatures, which are expected to be implemented in next-generation PEMFCs. |
1. Introduction
The environmental challenges associated with the widespread combustion of fossil fuels are global in scope and require international collaboration. In response, the development of sustainable green energy sources has become critical, with hydrogen energy emerging as a clean and efficient alternative. Proton exchange membrane fuel cells (PEMFCs) are promising power sources that directly convert chemical energy stored in hydrogen molecules into electrical energy through electrochemical reactions. Their advantages, including zero emissions, higher energy conversion efficiencies, and rapid responses, have driven their applications in fields such as vehicles, submarines, and drones.1–3As the power density of fuel cells increases, the heat generation rate of the fuel cell stacks also rises significantly, creating challenges for heat dissipation within the operating temperature range from 60 °C to 80 °C.4,5 Increasing the operating temperature has been proposed as a solution to this issue, particularly for high-power PEMFC stacks subjected to long-term operation under heavy-duty conditions. Both theoretical analyses and experimental studies have indicated that operating PEMFCs above 100 °C offers several advantages, such as improved reaction kinetics, simplified water management (only water vapour when exceeding 100 °C), and greater CO tolerance.6 Projections by Japan's New Energy and Industrial Technology Development Organization (NEDO) have suggested that fuel cell operating temperatures will rise to 105 °C by 2030 and 120 °C by 2040.7 Therefore, higher operating temperatures have become a favourable option in the development of PEMFCs. Efforts to enable stable and efficient PEMFC operation at elevated temperatures include the development of modified perfluorosulfonic acid (PFSA) membranes with improved thermal stability and water retention capacities, as well as the use of graphitised carbon nanotube supports that are thermally stable at elevated temperatures.8,9 However, the performance and lifetimes of PEMFCs are still significantly limited at elevated operating temperatures, which present challenges to the stable and efficient operation of the membrane electrode assemblies (MEAs), the core components of PEMFCs.
The output performance of PEMFCs, including their steady-state response characteristics (such as polarization and power density curves) and dynamic response characteristics (such as voltage overshoot and recovery time), is significantly limited at elevated temperatures. This is primarily due to the inability of existing materials or operational strategies to accommodate the substantial alterations in transport processes within the MEA caused by increased temperatures. For instance, the lower water content of ionomers within the proton exchange membranes (PEMs) and the catalyst layer (CL) at temperatures near or above the boiling point of water significantly reduces the proton conductivity.10–14 Increasing the inlet humidity can theoretically mitigate excessive dehydration of ionomers by maintaining a sufficient environmental humidity.15 However, excess water vapour entering the PEMFC lowers the partial pressure of reactant gases, thereby reducing the reaction rate at active sites and increasing the polarization.16 Consequently, at elevated temperatures, the coupling between reactant gases and water vapour is intensified, which may render the current water and gas management strategies for maintaining PEMFC performance at low temperatures ineffective. This issue stems from the limited systematic understanding of the transport processes within the MEA at elevated temperatures. Moreover, elevated temperatures can influence the transport processes by modifying critical structural features, such as the three-phase boundary (TPB) and charge transport pathways, primarily due to changes in the physical properties of the ionomer.17–19 In elevated temperature environments, the dynamic load response of the PEMFC becomes more complex and difficult to control due to significant variations in the water content, reactants, and charged species.20,21 Thus, it is imperative to systematically analyse critical changes in the transport processes within the MEA due to elevated temperatures to ensure steady-state and dynamic load response characteristics of PEMFCs at elevated temperatures.
Due to the accelerated degradation of MEA components at elevated temperatures, MEA components must have a high thermal stability to ensure the long-term stable operation of PEMFCs in such environments. Ruiu et al.22 demonstrated that short-term temperature cycling between 90 °C and 120 °C caused more severe degradation of the MEA components than long-term stable operation under automotive-relevant conditions at 80 °C. This led to a fivefold increase in the irreversible performance loss rate of the PEMFC. Factors such as a high electrode potential, humidity fluctuations, and attack by free radicals are commonly considered to be the primary drivers of MEA degradation, while the role of elevated temperatures has often been overlooked.4,23,24 Elevated temperatures induce structural changes in the materials of MEA components and accelerate other degradation processes within the MEA, both of which result in a more severe, complex, and highly coupled degradation mechanism of the MEA at elevated temperatures. This makes it essential to have a clear understanding of the mechanisms by which elevated temperature degrades MEA components, along with identifying the key degradation factors and characteristics. This knowledge will provide a critical theoretical foundation for developing and selecting materials for MEAs with enhanced stability for extending the service life of PEMFCs that operate at elevated temperatures. However, a systematic understanding of the elevated temperature-induced degradation mechanism in the MEA is lacking.
Hence, this review summarises the challenges associated with MEAs at elevated temperatures in PEMFCs. First, the factors contributing to elevated temperatures and the classification of elevated temperature states in the MEA are discussed. Subsequently, critical alterations in the transport processes, covering reaction gas and water transport, charged species conduction, and load response characteristics, including steady-state and dynamic responses at elevated temperatures, are analysed. Then, the durability challenges of materials and structures in the MEA, focusing on CL, PEM, and gas diffusion layer (GDL), as well as their impacts on PEMFC performance loss are summarised. Moreover, recent progress in overcoming the challenges due to elevated temperatures is summarised, followed by recommendations for future research.
2. Description of elevated temperature states in the MEA
2.1 Causes of elevated temperature states
To better understand how elevated temperatures occur in MEAs, it is necessary to first outline the fundamental operating principle of PEMFCs. As PEMFC technology has advanced, the design of typical fuel cells has evolved into a multilayer structure composed of multiple functional components. As shown in Fig. 1(a), a single PEMFC generally consists of a MEA, an anode bipolar plate (ABP), and a cathode bipolar plate (CBP), with both plates containing gas channels and coolant channels. This review discusses an MEA without a sealed frame and mainly includes the anode gas diffusion backing, anode microporous layer, anode catalyst layer (ACL), proton exchange membrane (PEM), cathode catalyst layer (CCL), cathode microporous layer, and cathode gas diffusion backing. Electricity generation is driven by half-electrode reactions, specifically the hydrogen oxidation reaction (HOR) at the ACL and the oxygen reduction reaction (ORR) at the CCL.23 To facilitate these reactions, various reactants must be transported to the reaction sites. Hydrogen (H2) and oxygen (O2) are first supplied to the fuel cell and distributed throughout the MEA through gas channels. H2 passes through the anode gas diffusion backing and anode microporous layer to reach the ACL, where the HOR occurs and releases electrons and protons. The electrons travel through an external circuit to generate electricity, while the protons are conducted by the ionomer on the catalyst, through the PEM, and finally to the CCL, where the ORR occurs and produces water (H2O) through a four-electron transfer mechanism.25 | ||
| Fig. 1 Illustration of the reason for the MEA maintaining the elevated temperature states. (a) The fundamental operating principle of PEMFCs. (b) Enhanced reaction kinetics. (c) Simplified water and heat management. (d) Improved tolerance of the catalyst to CO impurities. (e) Massive heat generation within the MEA. (f) Low heat dissipation capability of the MEA components. | ||
Electrochemical reactions, charge-transfer processes, and the phase change of water products release heat.23 Due to the variety, uneven distribution, and different magnitudes of heat sources within a PEMFC, it is typically necessary to control the operating temperature using fluids such as water or ethylene glycol to maintain a desired temperature and ensure reliable electrochemical reactions. The MEA component is not a perfect thermal conductor, resulting in a temperature gradient between the MEA and the external operating environment.23 This causes the internal temperature of the MEA to be higher than the external operating temperature. The temperature of the MEA is influenced by two factors: the external operating temperature, where a higher operating temperature will raise the overall PEMFC temperature, and the temperature gradient between the MEA and its external surroundings, where the configuration of the MEA causes heat accumulation, further elevating its internal temperature. Sections 2.1.1 and 2.1.2 will discuss how these two factors jointly contribute to the elevated temperatures within the MEA.
Sluggish reaction kinetics create activation overpotentials during the half-electrode reactions, particularly the ORR, which is a major contributor to output voltage losses in PEMFCs. Elevated temperatures can significantly accelerate the reduction of adsorbed oxygen.26–28 Yano et al.29 demonstrated that the apparent rate constant of the ORR on Pt in 0.1 M HClO4 increased exponentially between 30 °C and 110 °C, in accordance with the Arrhenius equation. There was a 3.6-fold increase in the reaction rate when the temperature rose from 80 °C to 120 °C, which improved the electrode reaction kinetics and enhanced the output performance, as shown in Fig. 1(b).
As shown in Fig. 1(c), when operating at temperatures ≤80 °C, PEMFCs often struggle with inadequate heat removal using radiator technologies in transport vehicles, especially in heavy-duty fuel cell vehicles. This issue necessitates specialised cooling systems, increasing overall costs, but the heat transfer rate improves due to a larger temperature gradient between the fuel cell and the external environment as the operating temperature of the PEMFC increases. This improvement allows the use of existing cooling systems in transport vehicles, enhancing mass-specific energy densities, weight efficiency, and overall energy performance.30–32 Moreover, when operating at ≤80 °C under atmospheric pressure, PEMFCs operate with a dual-phase water system (liquid water and water vapour) to maintain proper humidification within the PEM and prevent flooding in the CL. This presents challenges for water management33–35 that are not present at higher temperatures (>100 °C), where only a single gaseous phase exists. This avoids the flooding issue, resulting in a more straightforward water management system.
Trace amounts of CO in hydrogen feed gas can significantly reduce a PEMFC's performance due to its strong adsorption onto Pt.31 At higher temperatures, however, the adsorption of CO on Pt is reduced, which improves the CO tolerance,8,36 as depicted in Fig. 1(d). For instance, the CO tolerance increases from 10–20 ppm at 80 °C to 1000 ppm at 130 °C and can even reach 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm at 200 °C.37 This eliminates multiple stages of fuel processing and gas cleaning, thereby enabling the use of more cost-effective fuel.
000 ppm at 200 °C.37 This eliminates multiple stages of fuel processing and gas cleaning, thereby enabling the use of more cost-effective fuel.
Regarding heat generation, for a PEMFC with an efficiency of 50%, approximately 45% of energy is released as heat.79,80 Heat generation has a more significant effect on fuel cells operating at high current densities, as depicted in Fig. 1(e). This substantial heat generation mainly arises from the combined contributions of multiple heat sources, including reversible reaction heat (Qrev), irreversible reaction heat (Qirrev), and Joule heat (Qj).23Fig. 2(a) depicts the influence of the key factors for massive heat generation, where the colour intensity of the arrow is directly proportional to the magnitude of the applied factor. Lighter colours indicate a lower magnitude, whereas darker colours indicate a greater magnitude. Existing commercial Pt/C catalysts do not achieve outstanding power generation efficiency (i.e., catalyst activity),81 leading to the release of a large amount of reaction heat. Furthermore, increasing the current density releases more reaction heat and Qj due to the higher chemical reaction intensity76,82,83 and charged species conduction,76 respectively. Decreasing the catalyst loading can increase Qirrev due to significantly higher gas transport resistance.84–86 Furthermore, the harsh working conditions of the PEMFC itself and reaction flow such as slow electron and proton conduction capacities, poor intercomponent contact, severe gas shortage,50,78 and insufficient humidification may also generate additional heat.
 | ||
| Fig. 2 (a) The influence rule of the key factors for massive heat generation. (b) Statistical results for the thermal conductivities of PEMFC components including the CL,23,38–52 microporous layer (MPL),23,38,40,42–47,49,51–54 gas diffusion backing (GDB),23,38,40,42–47,49,51,52,55–72 and bipolar plate (BP).23,38,40,42–47,49,51,52,73 (c) Statistical results for the temperature differences between the PEM and the CL, GDL and coolant.42,46,49,50,52,54,56,74–78 | ||
The magnitude of thermal conductivity or thermal conduction resistance usually increases the heat conduction capacity. Statistical analysis of the thermal conductivities for PEMFC components participating in heat dissipation in the through-plane direction was conducted using 37 relevant studies from 2008 to now, and the results are illustrated in Fig. 2(b). The median thermal conductivities of the CL, microporous layer (MPL), gas diffusion backing (GDB), and bipolar plate (BP) were respectively 0.21, 0.10, 0.42, and 16.65 W m−1 K−1. Except for the BP, the thermal conductivities of other components were so low that they were equivalent to those of some thermal insulation materials. This shows that it is difficult to remove heat generated inside the MEA, as demonstrated in Fig. 1(f). Statistical results of 12 relevant studies from 2013 to 2024 show that the average temperature difference between the PEM and CL was 10.41 K, while that between the GDL and coolant was 9.31 K, as illustrated in Fig. 2(c). Therefore, based on the current MEA configuration, the actual temperature in the MEA will be higher, especially under harsh working conditions.
2.2. Classification of elevated temperature states
NEDO has established 120 °C as the maximum operating temperature target for sulfonic acid-based PEMFCs,7 implying that elevated temperature operation can range from 80 °C to 120 °C. Considering an approximate 10 °C temperature difference between the MEA and coolant, as shown in Fig. 2(c), the actual MEA temperature may reach 90–130 °C. Moreover, during fuel cell operation at 80 °C, local hotspots caused by membrane pinholes may raise local temperatures to around 140 °C, creating temperature gradients of up to 60 °C,87 but this result has only been obtained using qualitative analysis of thermochromic pigments. To more comprehensively investigate the effects of elevated temperatures, this review investigated the transport and degradation mechanisms in the MEA over the broader temperature range of 90–180 °C.Within the elevated temperature range, both the water phase and ionomer in the MEA undergo significant phase transitions, as depicted in Fig. 3. When the temperature exceeds the boiling point of water (∼110 °C at 143.27 kPa), liquid water converts to vapour, which drastically changes the internal hydration environment of the MEA. This shift affects material utilization and transport-reaction processes. In the vapour phase, the absence of liquid water films on Pt catalyst surfaces reduces the electrochemical surface area (ECSA).88 Ionomer hydration/dehydration causes deformation, leading to a redistribution of the carbon support and disruption of electron network pathways.17 Schroeder's paradox highlights that an ionomer absorbs less water when it contacts saturated vapour than liquid water,89 leading to diminished proton conductivity. Moreover, changes in the water environment also influence the degradation of the MEA components. As the temperature rises beyond the ionomer's glass transition temperature (∼130 °C), its chain mobility increases, which accelerates ionomer instability.90 Based on these behaviours and current research processes, the MEA's elevated temperature range discussed in this review is classified into three regimes: (1) sub-boiling at moderate temperatures (90–110 °C), defined as the (M) regime; (2) super-boiling at sub-glass transition temperatures (110–130 °C), defined as the (H) regime, and (3) super-glass transition at extreme temperatures (130–180 °C), defined as the (E) regime. Correspondingly, operating temperatures are grouped into two ranges: 80–100 °C and 100–120 °C, corresponding to regimes (M) and (H), respectively. Regime (E) corresponds to fault conditions, such as local hotspots. The respective changes in MEA transport characteristics and degradation behaviours across the three different elevated temperature regimes are discussed in the following section.
 | ||
| Fig. 3 Classification of the elevated temperature range in the MEA. | ||
3. Critical changes in transport processes and load responses at elevated temperatures
Half-electrode reactions are how PEMFCs power electrical appliances and require a sufficiently fast transport process to guarantee that the PEMFC has the desired steady and dynamic load response capacity. The major transport and electrochemical processes in a MEA are listed in Table 1. The transport processes refer to the movements of reactive species within the different media of a PEMFC. There are two transport processes involved: (i) mass transport processes, including the transport of reactant gases (H2 and O2) from the BP towards the CL, and water (gas or liquid) transport from the BP to CL, from the CL to BP, and within the PEM and (ii) charged species conduction, including proton conduction from the ACL to CCL through the PEM layer and electron conduction from the ACL to CCL through the conductive network. Key changes in these transport processes and the load response characteristics at elevated temperatures are summarised in the following sections.| Component | Role | Transport and electrochemical processes |
|---|---|---|
| CL | Assist HOR and ORR | Proton conduction in the ionomer network |
| Electron conduction in the carbon network | ||
| Water transport in the pore and ionomer networks | ||
| Hydrogen/oxygen diffusion in the pore network and across the ionomer thin film | ||
| HOR: H2 → 2H+ + 2e− | ||
| ORR: 1/2O2 + 2e− + 2H+ → H2O | ||
| PEM | Permit flow of protons from the anode to the cathode; stop electrons from traveling | Water transport (electro-osmotic drag, back diffusion, and hydraulic permeation) |
| Proton conduction through the PEM | ||
| GDL | Ascertain gas diffusion from flow fields to active locations | Hydrogen and water vapor transport (usually diffusion dominates) in the anode |
| Drain the water | Oxygen and water vapor transport (usually diffusion dominates) in the cathode | |
| Liquid water transport | ||
| Electron conduction |
3.1. Gas and water mass transport
As illustrated in Fig. 4(a) and (b), compared with low-temperature operation, elevated temperature environments significantly alter the MEA transport of reactant gases and water. Tables 2 and 3 summarise the gas and water transport characteristics at different temperatures, respectively. At elevated temperatures, intrinsic gas diffusivities increase, with the oxygen diffusion coefficient rising from 3.896 × 10−6 m2 s−1 at 80 °C to 5.347 × 10−6 m2 s−1 at 90 °C.20 The hydrogen diffusivity in the ionomer phase increased from 2.59 cm2 s−1 at 80 °C to 3.48 cm2 s−1 at 95 °C.95 Song et al.99 indicated that the apparent diffusion-limited current density for the cathode side increased from 1.32 to 2.76 A cm−2, and the apparent diffusion-limited current density for the anode side increased from 3.01 to 9.63 A cm−2 as the cell temperature rose from 23 °C to 120 °C. This implies enhanced reactant gas transport at elevated temperatures, and this effect is more evident for hydrogen transport. Modelling calculations and experimental impendence analysis have also shown that gas transport is enhanced at elevated temperatures,13,14,20,91,96,100 as shown in Fig. 5(b). Notably, elevated temperature also reduces the solubility of reactant gases in the ionomer, with oxygen solubility undergoing a 1.1-fold decrease from 9.34 × 10−6 mol cm−3 at 30 °C to 4.43 × 10−6 mol cm−3 at 80 °C. Zhang et al.101 discovered that the gas transfer resistance increased when the cell temperature was raised from 160 °C to 200 °C, and they emphasised the importance of reactant gas solubility in the ionomer film in the CL. The permeability of reactant gases through the ionomer film is the product of the diffusion coefficient and solubility. Therefore, a higher gas diffusion resistance is observed when the effect of the reduced gas solubility is larger than the effect of the increased gas diffusivity.101 However, this effect becomes significant only at cell operating temperatures above 160 °C. | ||
| Fig. 4 Evolution patterns of the transport processes and load responses at elevated temperatures. (a) Gas transport. (b) Water transport. (c) Effects of other operation parameters on the gas and water transport. (d) Proton conduction. (e) Electron conduction. (f) Load responses. | ||
| Serial number | Operation conditions (anode/cathode) | Items | Values | Ref. |
|---|---|---|---|---|
| 1 | — | Oxygen diffusion coefficient | 2.694 × 10−6 m2 s−1 @ 60 °C | 20 |
| 3.896 × 10−6 m2 s−1 @ 80 °C | ||||
| 5.347 × 10−6 m2 s−1 @ 90 °C | ||||
| 2 | — | Oxygen diffusion coefficient in hydrated Nafion membranes | 9.95 × 10−7 cm2 s−1 @ 30 °C | 94 |
| 8.70 × 10−6 cm2 s−1 @ 80 °C | ||||
| 3 | — | Oxygen solubility in hydrated Nafion membranes | 9.34 × 10−6 mol cm−3 @ 30 °C | |
| 4.43 × 10−6 mol cm−3 @ 80 °C | ||||
| Hydrogen diffusivity in the Nafion 117 membrane | 2.59 cm2 s−1 @ 80 °C | |||
| 3.48 cm2 s−1 @ 95 °C | 95 | |||
| 4 | Total pressure: 200 kPa, RH: 0.7; | Gas partial pressure | H2O: | 16 |
| 33 kPa @ 80 °C | ||||
| 71 kPa @ 100 °C | ||||
| 139 kPa @ 120 °C | ||||
| O2: | ||||
| 35 kPa @ 80 °C | ||||
| 27 kPa @ 100 °C | ||||
| 13 kPa @ 120 °C | ||||
| N2: | ||||
| 132 kPa @ 80 °C | ||||
| 102 kPa @ 100 °C | ||||
| 48 kPa @ 120 °C | ||||
| 5 | Back pressure: 150 kPa | Gas partial pressure | H2O: | 91 |
| 51.87 kPa @ 80 °C | ||||
| 85.13 kPa @ 120 °C | ||||
| O2: | ||||
| 14.81 kPa @ 80 °C | ||||
| 60.28 kPa @ 120 °C | ||||
| 6 | RH: 0.4/0.4, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5; | Cell mass transfer resistance | At 1.8 A cm−2 | 96 |
| 54.657 mΩ cm2 @ 80 °C | ||||
| 41.889 mΩ cm2 @ 90 °C | ||||
| 29.534 mΩ cm2 @ 95 °C | ||||
| 7 | RH: 1.0/1.0, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5; | Cell mass transfer resistance | At 1.8 A cm−2: | 96 |
| 68.945 mΩ cm2 @ 80 °C | ||||
| 48.232 mΩ cm2 @ 90 °C | ||||
| 34.122 mΩ cm2 @ 95 °C | ||||
| 8 | Back pressure: 100 kPa | Cell mass transfer resistance | 137.3 s m−1 @ 80 °C | 91 |
| 111.68 s m−1 @ 100 °C | ||||
| 85.75 s m−1 @ 120 °C | ||||
| 9 | RH: 1.0/1.0, back pressure (psig): 30/30, flow rate (SLPM): 0.3/1.0; | Cell mass transfer resistance | At 0.91 A cm−2: | 13 |
| 0.077 Ω cm2 @ 80 °C | ||||
| 0.072 Ω cm2 @ 100 °C | ||||
| 0.072 Ω cm2 @ 120 °C |
| Serial number | Items | Values | Ref. |
|---|---|---|---|
| 1 | Net water transport coefficient in the Nafion 212 R membrane | At 1.1 A cm−2: | 93 |
| 0.016 @ 80 °C | |||
| 0.103 @ 100 °C | |||
| 0.394 @ 120 °C | |||
| 2 | Net water transport coefficient in the Aquivion E87-05S membrane | At 0.9 A cm−2: | 93 |
| −0.028 @ 80 °C | |||
| 0.080 @ 100 °C | |||
| 0.398 @ 120 °C | |||
| 3 | Water content | At 1.0 A cm−2: | 97 |
| Anode side (8.166 @ 70 °C, 8.537 @ 80 °C, 5.059 @ 90 °C) | |||
| Cathode side (15.435 @ 70 °C, 14.394 @ 80 °C, 7.836 @ 90 °C) | |||
| Average (11.410 @ 70 °C, 11.182 @ 80 °C, 6.378 @ 90 °C) | |||
| 4 | Water content in the membrane | At 0.75 V: | 98 |
| 7.922 @ 70 °C | |||
| 7.862 @ 80 °C | |||
| 7.803 @ 90 °C | |||
| At 0.65 V: | |||
| 6.782 @ 70 °C | |||
| 6.748 @ 80 °C | |||
| 6.688 @ 90 °C | |||
| At 0.45 V: | |||
| 5.407 @ 70 °C | |||
| 5.252 @ 80 °C | |||
| 5.008 @ 90 °C | |||
| At 0.27 V: | |||
| 4.784 @ 70 °C | |||
| 4.530 @ 80 °C | |||
| 4.155 @ 90 °C | |||
| 5 | Water saturation in the cathode GDL | At 0.75 V: | 98 |
| 0.088 @ 70 °C | |||
| 0.089 @ 80 °C | |||
| 0.082 @ 90 °C | |||
| At 0.65 V: | |||
| 0.133 @ 70 °C | |||
| 0.134 @ 80 °C | |||
| 0.128 @ 90 °C | |||
| At 0.45 V: | |||
| 0.148 @ 70 °C | |||
| 0.158 @ 80 °C | |||
| 0.138 @ 90 °C | |||
| At 0.27 V: | |||
| 0.151 @ 70 °C | |||
| 0.156 @ 80 °C | |||
| 0.133 @ 90 °C | |||
| 6 | Water generation rate in the cathode side | At 60 °C: | 88 |
| 4.47 ml h−1 @ a dew point temperature of 30 °C | |||
| 6.71 ml h−1 @ a dew point temperature of 60 °C | |||
| 12.20 ml h−1 @ a dew point temperature of 90 °C | |||
| At 90 °C: | |||
| 1.39 ml h−1 @ dew point temperature of 30 °C | |||
| 8.50 ml h−1 @ a dew point temperature of 60 °C | |||
| 11.97 ml h−1 @ a dew point temperature of 90 °C | |||
| 7 | Water content in the membrane | 6.571 @ 80 °C | 91 |
| 6.511 @ 100 °C | |||
| 6.029 @ 120 °C |
 | ||
| Fig. 5 (a) Partial pressure change patterns of O2 and water vapor as the cell temperature increases.16 (b) The impacts of operating temperature (OT) on the water transfer: lower OT and higher OT.20 (c) Molar concentration distribution of oxygen at 120 °C and different pressures.91 (d) X-ray imaging of MEAs with 1.5 A cm−2 at 100 and 300 kPa (abs.) and 60, 80, 100 °C and with 3.0 A cm−2 at 300 kPa (abs.) and 60, 80, 100 °C.92 (e) Water generation rate at different temperatures.88 (f) Effects of operating parameters (temperature, stoichiometric ratio of air, and anode RH) on the net water transport coefficient (αNMD) for cells with the Aquivion membrane.93 | ||
The vapour saturation pressure rises exponentially with the temperature102 because water vapourises at elevated temperatures, increasing its partial pressure in the MEA mass transfer structure. This causes a significant drop in the partial pressure of the reactant gases, particularly on the cathode side at high current densities (Fig. 5(a)). Tang et al.13 reported that the mass transfer resistances were equal at 100 °C and 120 °C, implying a balance between the increased gas diffusivity in the diffusion medium and the reduced gas partial pressure and solubility in the ionomer phase for overall gas transport. Butori et al.16 found that the partial pressure of oxygen decreased significantly from 27 kPa to 13 kPa when the temperature rose from 100 °C to 120 °C, the total air pressure remained constant at 200 kPa, and the relative humidity (RH) remained constant at 70%. This is the main cause of cell voltage losses at a fixed current density.103 Consequently, the significant role of the decreased gas partial pressure and solubility in the gas transport process at elevated temperature must be considered, especially in the (H) regime due to the complete conversion from liquid water to water vapour. Therefore, at elevated temperatures, the coupling between reactant gas and water transport becomes stronger, mainly due to the increased vapourization of liquid water.
Furthermore, this significant phase transition causes the equilibrium between the membranous water and the liquid water and water vapour in the pore structure to be disrupted at lower temperatures, especially in the (H) regime, where only the equilibrium between the membranous water and water vapour exists. This results in a lower ionomer water content based on Schroeder's paradox.89 As depicted in Fig. 5(b), Wang et al.20 suggested that the phase change-induced (PCI) flow increased significantly at high loads as the operating temperature increased to 90 °C due to an increase in the term d(Psat/T)/dT and the temperature gradient ∇T. They also found that the capillary-driven (CD) flow decreased significantly and even reversed due to the reduced content of liquid water within the CCL. The above results are supported by those of Xu et al.104 and García-Salaberri et al.10 In comparison, the enhanced PCI flow was stronger than the reverse CD flow, which promoted the expulsion of water in the CCL at elevated temperatures,20,105 resulting in an insufficient environmental humidity faced by the ionomer phase. This further decreased the water content in the ionomer and made proton conduction difficult. Moreover, the increased net water drag coefficient at elevated temperatures suggests a more severe water shortage for the ionomer phase on the anode side (Fig. 5(f)),93,97,106 especially in the (H) regime. When the output voltage dropped from 0.75 V to 0.27 V, the membrane dehydration effect caused by temperature increase was enhanced by 13%. Even if the water production increases under a high current density, this water production could not alleviate the dehydration of the ionomer phase at elevated temperatures. Therefore, relying on the back-diffusion of cathode-generated water to alleviate dehydration on the anode side has a limited effectiveness. As shown in Fig. 5(e), Hou et al.88 found that the water generation rate decreased at elevated temperatures, as measured using a pH meter, especially in the (H) regime. Kellegoz et al.107 also indicated that the active reaction area decreased when the cell temperature increased to 85 °C, which decreased the output performance. This was attributed to the presence of liquid water on the surface of the Pt catalyst at lower temperatures, which also provided active sites for the reaction.108 Elevated temperature promotes the vapourization of liquid water, which reduces the active area and subsequently decreases the ORR rate and water production rate.
Other operating conditions, such as gas pressure, stoichiometric ratio, and RH, also influence the transport processes of reactant gas and water (Fig. 4(c)). As demonstrated in Fig. 5(c), an increase in the gas back pressure promotes the regulation of gas flow conditions within the cell, which slows the rate of decrease in the oxygen molar concentration and mitigates the effect of concentration-differential polarization.91 Akitomo et al.92 used X-ray imaging to indicate that when the feed gas was pressurised, the accumulation of liquid water increased (Fig. 5(d)), leading to a decrease in the ohmic overpotential. Therefore, pressurizing reactant gases improves their supply by directly increasing their partial pressures and also suppresses the evaporation of liquid water, thereby alleviating the loss of water within the ionomer phase. However, higher gas pressures are not always better, as they may pose challenges to the durability and dynamic response characteristics of the components. There must be a minimum gas pressure in the flow field, which depends on various factors, including current density, the structure of the MEA, and the design of the flow field. Increasing the gas RH alleviates the evaporation of liquid water within the MEA by externally supplying additional water vapour, but it also reduces the vapour concentration gradient between the MEA interior and the external environment, thereby mitigating vapour outflow. Altogether, these two aspects help relieve water loss from the ionomer phase and can restore the original electrochemically active surface area (Fig. 5(e)).88 As depicted in Fig. 5(f), at a constant flow rate of H2 at the anode, αNMD decreases upon decreasing the stoichiometric ratio of air (γair), but the effect of γair on αNMD is relatively small. However, αNMD can be significantly decreased by decreasing the RH of hydrogen supplied to the anode. Decreasing the anode RH can lower αNMD at elevated temperatures. Notably, Lu et al.21 indicated that increasing the inlet pressure also increased the αNMD.
The reaction gas crossover also changes at elevated temperatures, as shown in Fig. 4(a). Butori et al.109 reported that the H2 permeability increased with temperature and RH, but lower H2 crossover was measured above 100 °C and under humid conditions due to low H2 partial pressure. Consequently, hydrogen crossover is more pronounced in the (M) regime and may lead to a reduction in the open-circuit voltage (OCV) of the fuel cell, which explains the observed decrease in OCV at elevated temperatures.
At elevated temperatures, the vapourization of liquid water is the fundamental driving force behind hindered reactant gas transport, reduced ionomer water content, decreased electrochemically active surface area, and increased H2 crossover. Therefore, elucidating the phase transition mechanism of water within the MEA and suppressing this process is necessary for achieving reliable water–gas management for operation at elevated temperatures. However, the phase transition of water within pores is also heavily influenced by capillary forces. Jatukaran et al.110 observed that in pores smaller than 10 nm, the liquid evaporation pressure was much lower than that in larger pores, suggesting that the phase transition of water at the micro- and nanoscale may significantly differ from that at the macroscopic scale. It is imperative to thoroughly understand the phase transition between liquid water and water vapour in nanopores.
3.2. Charged species conduction
Proton conduction mainly occurs within the ionomer phase of the PEM and CL, which is closely associated with the internal water content, as illustrated in Fig. 6(a).111 Proton conduction is significantly influenced by water transport, as discussed above. Table 4 summarises the charged species (proton and electron) conduction properties at different temperatures. Yin et al.96 reported that as the cell operating temperature increased from 80 °C to 95 °C, the cell ohmic resistance, which is dominated by proton conduction, increased by 14.7% at 100% RH and by 25.1% at 40% RH. Yin et al.111 indicated a sharp drop (∼63.4% compared to 80 °C) in the proton conductivity when the temperature increased to 130 °C (Fig. 6(b)). These results show that elevated temperatures can lead to water loss in the ionomer, causing it to dry out and significantly impair proton conductivity, especially for the (H) regime at a low RH. In contrast, Lufrano et al.15 reported that the proton conductivity of a PEM increased with the temperature, even at 120 °C, as shown in Fig. 6(c). This increase was primarily attributed to the constant and sufficiently high RH environment faced by the PEM, and severe dehydration processes did not occur, which is supported by Butori et al.109 and Eskandari et al.113 The differences in proton conductivity between low temperatures and elevated temperatures can be attributed to the differences in thermally-activated properties such as proton mobility or diffusivity at a constant RH.113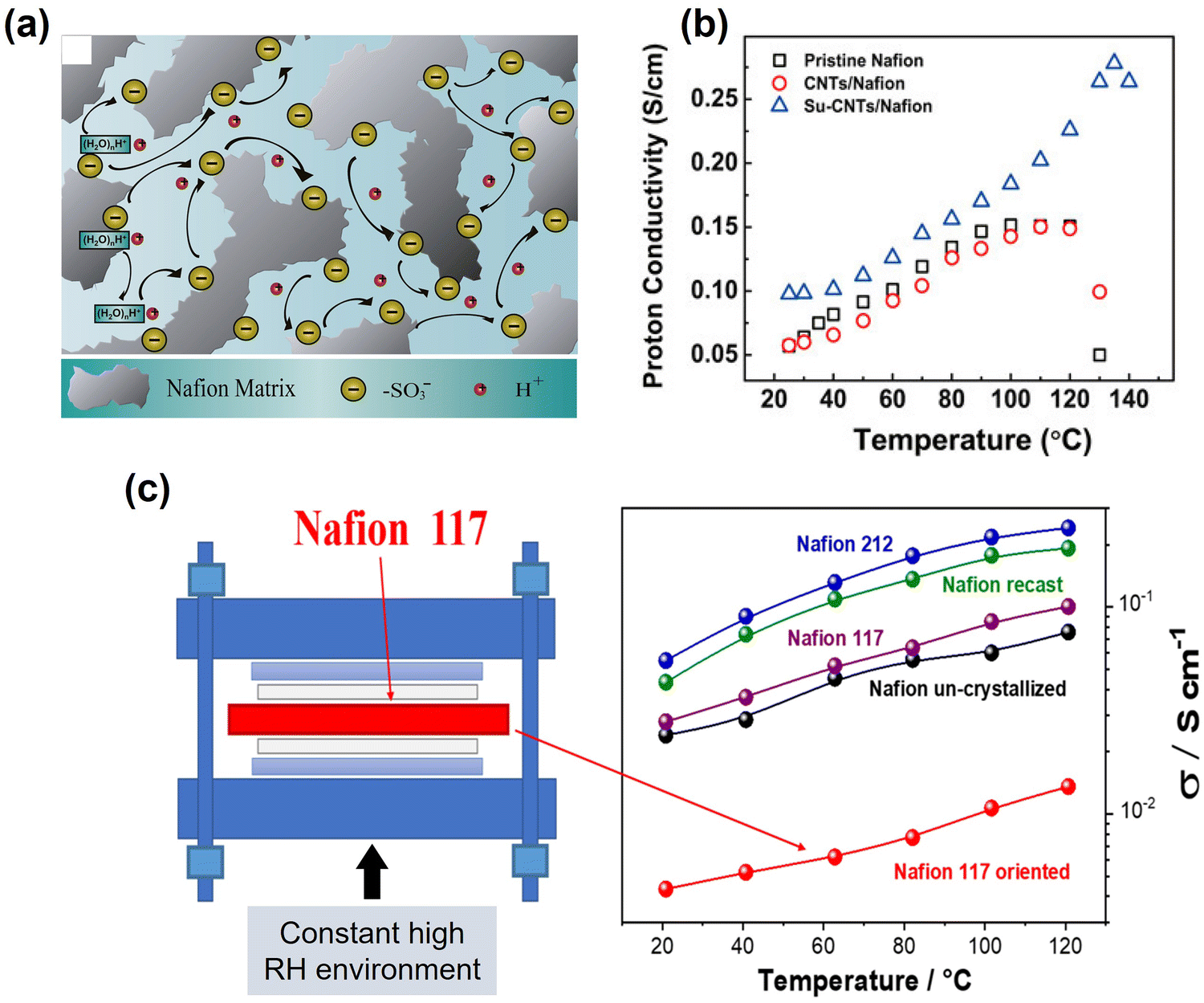 | ||
| Fig. 6 (a) Schematic illustration of proton transportation in water-hydrated pristine Nafion membranes.111 (b) Effect of operating temperatures on the proton conductivity.111 (c) Proton conductivities of Nafion recast, Nafion 212, Nafion un-crystallized, and Nafion 117 as received and Nafion 117 oriented membranes as a function of temperature (20–120 °C) at 90% RH.15 | ||
| Serial number | Operation conditions (anode/cathode) | Items | Values | Ref. |
|---|---|---|---|---|
| 1 | — | Cell high-frequency resistance (HFR) | 13.382 Ω @ 80 °C | 91 |
| 15.441 Ω @ 100 °C | ||||
| 18.382 Ω @ 120 °C | ||||
| 2 | — | Cell ionic resistance | At 0.8 A cm−2: | 10 |
| 77.98 mΩ cm2 @ 70 °C | ||||
| 135.78 mΩ cm2 @ 90 °C | ||||
| 3 | RH: 0.4/0.4, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5 | Cell ohmic resistance | At 1.8 A cm−2 | 96 |
| 82.770 mΩ cm2 @ 80 °C | ||||
| 87.345 mΩ cm2 @ 85 °C | ||||
| 94.238 mΩ cm2 @ 90 °C | ||||
| 103.470 mΩ cm2 @ 95 °C | ||||
| 4 | — | Cell ionic resistance | At 0.8 A cm−2 | 97 |
| 32.83 mΩ cm2 @ 80 °C | ||||
| 70.21 mΩ cm2 @ 90 °C | ||||
| 5 | RH: 1.0/1.0, back pressure (psig): 30/30, flow rate (SLPM): 0.3/1.0; | Cell ohmic transfer resistance | At 0.91 A cm−2: | 13 |
| 0.102 Ω cm2 @ 80 °C | ||||
| 0.095 Ω cm2 @ 100 °C | ||||
| 0.115 Ω cm2 @ 120 °C | ||||
| 6 | RH: 0.4/0.4, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5; | Cell ohmic resistance | At 1.8 A cm−2: | 96 |
| 83.24 mΩ cm2 @ 80 °C | ||||
| 87.55 mΩ cm2 @ 85 °C | ||||
| 94.52 mΩ cm2 @ 90 °C | ||||
| 104.13 mΩ cm2 @ 95 °C | ||||
| 7 | RH: 1.0/1.0, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5; | Cell ohmic resistance | At 1.8 A cm−2: | 96 |
| 64.15 mΩ cm2 @ 80 °C | ||||
| 67.30 mΩ cm2 @ 85 °C | ||||
| 70.20 mΩ cm2 @ 90 °C | ||||
| 73.59 mΩ cm2 @ 95 °C | ||||
| 8 | Fully hydrated before measurement and without external humidification during measurement | Membrane proton conductivity | 0.081 S m−1 @ 40 °C | 111 |
| 0.134 S m−1 @ 80 °C | ||||
| 0.151 S m−1 @ 120 °C | ||||
| 0.049 S m−1 @ 130 °C | ||||
| 9 | RH: constant at 0.9 | Membrane proton conductivity | Nafion 117: | 15 |
| In-plane | ||||
| 40.0 ± 0.8 mS cm−1 @ 40 °C | ||||
| 76.5 ± 1.7 mS cm−1 @ 80 °C | ||||
| 118.4 ± 1.7 mS cm−1 @ 120 °C | ||||
| Through-plane: | ||||
| 36.7 ± 1.2 mS cm−1 @ 40 °C | ||||
| 63.6 ± 1.3 mS cm−1 @ 80 °C | ||||
| 100.6 ± 1.6 mS cm−1 @ 120 °C | ||||
| Nafion 212: | ||||
| In-plane: | ||||
| 74.1 ± 1.5 mS cm−1 @ 40 °C | ||||
| 157.0 ± 2.1 mS cm−1 @ 80 °C | ||||
| 220.0 ± 2.1 mS cm−1 @ 120 °C | ||||
| Through-plane: | ||||
| 76.1 ± 1.6 mS cm−1 @ 40 °C | ||||
| 153.1 ± 2.1 mS cm−1 @ 80 °C | ||||
| 213.1 ± 2.2 mS cm−1 @ 120 °C | ||||
| 10 | Flow rate (ml min−1): 200/200, gas type: humidified nitrogen/humidified hydrogen | Electron conductivity | Carbon paper: | 112 |
| 827.78 S m−1 @ 70 °C | ||||
| 780.56 S m−1 @ 80 °C | ||||
| 788.89 S m−1 @ 90 °C | ||||
| MPL: | ||||
| 86.11 S m−1 @ 70 °C | ||||
| 80.56 S m−1 @ 80 °C | ||||
| 80.55 S m−1 @ 90 °C | ||||
| CL: | ||||
| 16.67 S m−1 @ 70 °C | ||||
| 19.44 S m−1 @ 80 °C | ||||
| 22.22 S m−1 @ 90 °C |
Therefore, maintaining a sufficiently high RH around the ionomer phase is essential. As explained above, increasing the gas pressure and inlet gas RH helps ensure a stable and sufficient humidity within both the CL and PEM to satisfy their constant hydration requirements. In addition, developing novel ionomer materials with enhanced water retention is a critical step in overcoming the limitations of elevated temperatures. Notably, the water absorption and desorption processes of nano-ionomer films within the CL remain poorly understood, with current models relying on empirical equations based on the absorption and desorption observed in the PEM. To alleviate ionomer dehydration in the CL at elevated temperatures, it is essential to first elucidate the phase transition kinetics of water in membranes within the CL and investigate the molecular structures responsible for absorption and desorption. The deformation of ionomer films induced by elevated temperatures also affects the continuity of the proton conduction paths within the CL, while the underlying mechanisms remain unclear.
Limited research has explored changes in electron conduction at elevated temperatures. As depicted in Fig. 4(e), electron conduction is affected by elevated temperature environments mainly within the GDL and CL due to the temperature dependence of electron movement and the altered electron conduction pathways. For the former, Fujita et al.114 reported that the electronic resistance of carbon nanotubes generally decreased, suggesting a thermally activated process. It can be inferred that elevated temperatures activate and enhance electron transport within the MEA components. For the latter, Taş et al.115 proposed that the increased electronic conductivity of GDB at higher temperatures was due to enhanced contact between neighbouring carbon fibres resulting from thermal expansion. Additionally, the electron conduction pathways in the CL were affected by the ionomer thermal expansion at elevated temperatures, leading to carbon particle reorientation or migration. This disrupted the conductive network and altered the electronic conductivity of the CL by inducing component deformation.17–19 Sun et al.112 investigated variations in the electronic conductivity of GDB, MPL, and CL at different temperatures and found that the conductivity of the GDB and MPL decreased from 827.78 S m−1 to 788.89 S m−1 and 86.11 S m−1 to 80.55 S m−1, respectively, as the temperature increased from 70 °C to 90 °C, while the conductivity of CL increased from 16.67 S m−1 to 22.22 S m−1. Consequently, the impact of elevated temperatures on the electronic conductivity of MEA components remains controversial, while this effect is negligible compared with the restricted proton conductivity at elevated temperatures.
3.3. Load responses
 | ||
| Fig. 7 (a) Experimental results of cell voltage, HFR, and current density distribution during the time period of voltage drop when cell operates at elevated temperatures.11 (b) Correlations between different forms of voltage or energy and temperature based on data. The dashed lines represent the cases of water in the liquid state.116 (c) Measured OCVs as a function of temperature.117 (d) Under the conditions of 43% RH, 80/100/120 °C, and 200/250/300 kPa back pressures, respectively, the area resistance values at 0.65 V.118 (e) Polarization and HFR curves of cells composed of Gore M788.12 and Nafion212 at 30%/43% RH and temperatures of 80 °C, 100 °C, and 120 °C.118 | ||
| Serial number | Operation conditions (anode/cathode) | Items | Ratio of elevated temperatures to low temperatures | Relevant processes | Ref. |
|---|---|---|---|---|---|
| 1 | Back pressure (abs, kPa): 200/200, RH: 0.7/0.7 | OCV | 120 °C to 80 °C: 0.94 | Increased hydrogen crossover through PEM | 16 |
| 2 | Back pressure (abs, kPa): 100/100, stoichiometric ratio: very high, RH: 0.8/0.8 | OCV | 100 °C to 60 °C: 0.94 | 92 | |
| 3 | Back pressure (abs, kPa): 200/200; stoichiometric ratio: very high, RH: 0.8/0.8 | OCV | 100 °C to 60 °C: 0.95 | 92 | |
| 4 | Back pressure (abs, kPa): 300/300; stoichiometric ratio: very high, RH: 0.8/0.8 | OCV | 100 °C to 60 °C: 0.94 | 92 | |
| 5 | Back pressure (atm): 3.0/3.0, RH: 1.0/1.0 | OCV | 120 °C to 60 °C: 0.95 | 117 | |
| 6 | Back pressure (atm): 3.0/3.0, RH: 1.0/1.0 | OCV | 120 °C to 60 °C: 0.94 | 117 | |
| 7 | Back pressure (kPa): 130/120, stoichiometric ratio: 1.7/3, RH: 0.5/0.5 | Cell voltage | 90 °C to 60 °C: | 1. Enhanced the phase transition from liquid water to water vapor in the MEA; | 20 |
| 0.95 @ 0.31 A cm−2 | 2. Reduced water content of the ionomer in the PEM and CL | ||||
| 0.93 @ 0.95 A cm−2 | |||||
| 0.82 @ 2 A cm−2 | |||||
| 8 | Back pressure (abs, bar): 1.0/1.0, RH: 0.5/0.5; | Cell voltage | 90 °C to 70 °C: | 103 | |
| 0.97 @ 0.2 A cm−2 | |||||
| 0.92 @ 0.8 A cm−2 | |||||
| 0.60 @ 1.4 A cm−2 | |||||
| 9 | Back pressure (kPa): 100/80, stoichiometric ratio: 1.5/2.5, RH: 0.5/0.5; | Cell voltage | 95 °C to 75 °C: | 21 | |
| 0.97 @ 0.3 A cm−2 | |||||
| 0.96 @ 1 A cm−2 | |||||
| 0.87 @ 2 A cm−2 | |||||
| 10 | Back pressure (abs, kPa): 200/200, RH: 0.43/0.43; | Cell voltage | 120 °C to 80 °C: | 118 | |
| 0.95 @ 0.5 A cm−2 | |||||
| 0.90 @ 1.3 A cm−2 | |||||
| 0.69 @ 2 A cm−2 | |||||
| 11 | Back pressure (abs, kPa): 250/250, RH: 0.43/0.43; | Cell voltage | 120 °C to 80 °C: | 118 | |
| 0.97 @ 0.5 A cm−2 | |||||
| 0.93 @ 1.3 A cm−2 | |||||
| 0.83 @ 2 A cm−2 | |||||
| 12 | Back pressure (abs, kPa): 300/300, RH: 0.43/0.43; | Cell voltage | 120 °C to 80 °C: | 118 | |
| 0.97 @ 0.5 A cm−2 | |||||
| 0.92 @ 1.3 A cm−2 | |||||
| 0.87 @ 2 A cm−2 | |||||
| 13 | Back pressure (abs, kPa): 300/300, RH: 0.3/0.3; | Cell voltage | 120 °C to 80 °C: | 118 | |
| 0.99 @ 0.2 A cm−2 | |||||
| 0.78 @ 1 A cm−2 | |||||
| 14 | RH: 0.4/0.4, back pressure (bar): 1.5/1.5, flow rate (SLPM): 0.7/2.5; | Cell voltage | 95 °C to 80 °C: | 1. Enhanced electrode reaction kinetics in the CL | 96 |
| 1.02 @ 0.3 A cm−2 | 2. Enhanced gas diffusivity in the MEA | ||||
| 1.09 @ 2.1 A cm−2 |
(2) Increased activation overpotential: elevated temperatures can activate the catalyst, thereby improving electrode reaction kinetics and enhancing the electrochemical reaction rate.16,96,119 Conversely, elevated temperatures can lead to the evaporation of the water film covering the Pt catalyst, thereby reducing the ECSA,103 especially in the (H) regime. As presented in Table 5, the ratio of cell voltage at a low current density at elevated temperatures to that at low temperatures is approximately 0.95–0.97.
(3) Increased ohmic overpotential: elevated temperatures induce ionomer dehydration, which increases the proton conduction resistance,20,96,119–121 especially in the (H) regime, but proton conduction is not impeded at a sufficient humidity. As presented in Table 5, the ratio of cell voltage at a moderate current density and elevated temperatures to that at low temperatures is approximately 0.78–0.96.
(4) Increased concentration overpotential: elevated temperatures may accelerate the gas transport while decreasing the gas partial pressure and solubility in the transport medium, especially in the (H) regime. As presented in Table 5, the ratio of cell voltage at a high current density at elevated temperatures to that at low temperatures is approximately 0.6–0.87.
Increasing the gas pressure and gas RH may help mitigate the performance decline at elevated temperatures. Applying a gas backpressure at elevated temperatures is beneficial in reducing Rmt, as presented in Fig. 7(d). Notably, the self-humidification in the thinner membrane is stronger. As shown in Fig. 7(e), when the temperature is kept at 100 °C and the humidity is reduced from 43% to 30%, the HFR of the Nafion212-based cell at a high current density increases by about 70% while that of the Gore12-based cell only increases by about 30%. Therefore, the stable operation of PEMFCs at elevated temperatures should also take membrane thickness into account.
Overall, water phase transitions within the MEA significantly impact the steady-state performance at elevated temperatures. While PEMFC water management has become more straightforward, the stronger escape tendency of water vapour and stronger coupled transport interactions between water vapour and reaction gases compared with liquid water make it difficult to ensure stable operation of PEMFCs at elevated temperatures.
| Dynamic response rate = ΔV × (t1 + t2) | (1) |
| Serial number | Operation conditions (anode/cathode) | Items | Ratio of elevated temperatures to low temperatures | Relevant processes | Ref. |
|---|---|---|---|---|---|
| 1 | Back pressure (kPa): 130/120, stoichiometric ratio: 1.7/3, RH: 0.5/0.5; | Voltage overshoot | 90 °C to 60 °C: | Ionomer dehydration | 20 |
| 9.52 @ 10 to 1000 mA cm−2 | |||||
| 6.80 @ 1800 to 1000 mA cm−2 | |||||
| 0.77 @ 1000 to 10 mA cm−2; | |||||
| 2 | Back pressure (kPa): 100/80, stoichiometric ratio: 1.5/2.5, RH: 0.5/0.5 | Voltage overshoot | 95 °C to 75 °C: | 1. Ionomer dehydration | 21 |
| 0.90, 1.10, 1.04, 1.22 @ 0 to 0.2 A cm−2 | 2. Inadequate oxygen supply | ||||
| 1.06, 1.09, 1.18, 1.22 @ 0 to 0.4 A cm−2 | |||||
| 1.11, 0.76, 1.24, 1.29 @ 0 to 0.7 A cm−2 | |||||
| 1.43 @ 0 to 1.0 A cm−2 | |||||
| 1.31 @ 0.7 to 1.0 A cm−2 | |||||
| 1.44 @ 1.0 to 1.4 A cm−2 | |||||
| 1.27 @ 1.4 to 1.6 A cm−2; | |||||
| 3 | Back pressure (kPa): 250/230, stoichiometric ratio: 1.5/2.5, RH: 0.5/0.5 | Voltage overshoot | 95 °C to 75 °C | 1. Improved membrane hydration | 21 |
| 0.82, 1.00, 0.95, 1.00 @ 0 to 0.2 A cm−2 | 2. Increased partial pressure of reactants | ||||
| 0.92, 1.00, 1.08, 1.00 @ 0 to 0.4 A cm−2 | |||||
| 0.91, 0.91, 0.91, 0.88 @ 0 to 0.7 A cm−2 | |||||
| 0.83@ 0 to 1.0 A cm−2 | |||||
| 0.87 @ 0.7 to 1.0 A cm−2 | |||||
| 0.90 @ 1.0 to 1.4 A cm−2 | |||||
| 0.80 @ 1.4 to 1.6 A cm−2; | |||||
| 4 | Back pressure (kPa): 100/80, stoichiometric ratio: 1.5/2.5, RH: 0.5/0.5; | Dynamic response rate | 95 °C to 75 °C: | 1. Ionomer dehydration | 21 |
| 2.26, 1.00, 0.73, 0.86 @ 0 to 0.2 A cm−2 | 2. Inadequate oxygen supply | ||||
| 1.35, 1.00, 1.43, 0.88 @ 0 to 0.4 A cm−2 | |||||
| 0.57, 0.35, 0.91, 2.06 @ 0 to 0.7 A cm−2 | |||||
| 1.67 @ 0 to 1.0 A cm−2 | |||||
| 2.14 @ 0.7 to 1.0 A cm−2 | |||||
| 1.78 @ 1.0 to 1.4 A cm−2 | |||||
| 1.10 @ 1.4 to 1.6 A cm−2 | |||||
| 5 | Back pressure (kPa): 250/230, stoichiometric ratio: 1.5/2.5, RH: 0.5/0.5; | Dynamic response rate | 95 °C to 75 °C: | 21 | |
| 1.60, 0.84, 0.60, 0.72@ 0 to 0.2 A cm−2 | 1. Improved membrane hydration | ||||
| 0.57, 0.45, 0.78, 0.67 @ 0 to 0.4 A cm−2 | 2. Increased partial pressure of reactants | ||||
| 0.38, 0.45, 0.41, 0.67 @ 0 to 0.7 A cm−2 | |||||
| 0.46 @ 0 to 1.0 A cm−2 | |||||
| 0.93 @ 0.7 to 1.0 A cm−2 | |||||
| 0.79 @ 1.0 to 1.4 A cm−2 | |||||
| 0.73 @ 1.4 to 1.6 A cm−2 |
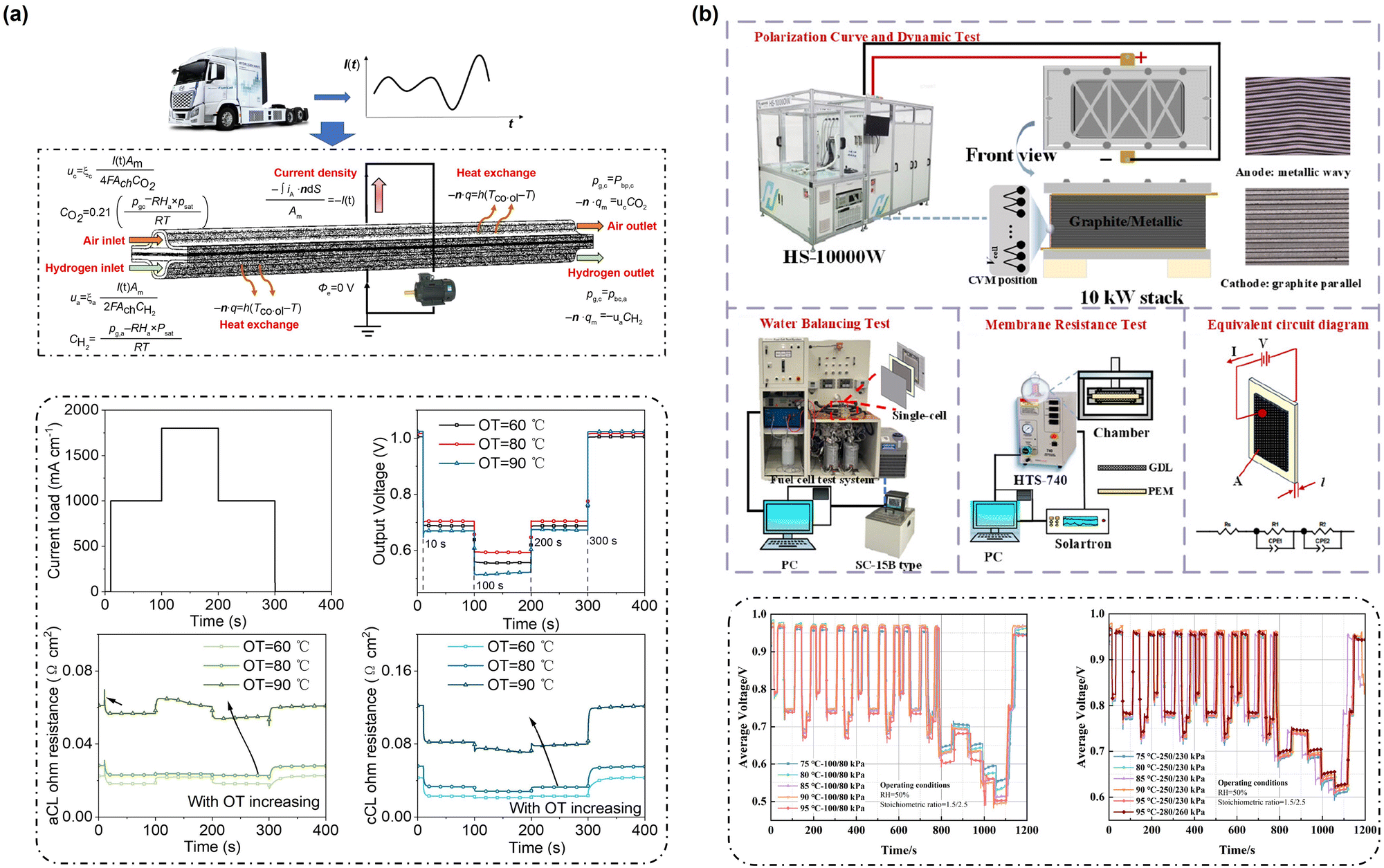 | ||
| Fig. 8 (a) Structure schematics of model and boundary condition settings and current load dynamic profile and dynamic responses of output voltage, ACL average proton ohmic resistance, and CCL average proton ohmic resistance at different OTs.20 (b) Experimental system schematic diagram including the polarization curve test, water balancing test, membrane electric resistance test, and equivalent circuit diagram and dynamic response results at different operating temperatures and inlet pressures during the loading process.21 | ||
Increasing the inlet gas pressure reduces the voltage overshoot and dynamic response rate due to an increased partial pressure of reactants, which creates a more abundant supply of reactants and helps slow voltage undershoots.21,122 Increasing the operating pressure induces the condensation of water vapour, alleviating the ionomer dehydration and the reduction in output performance.20 Overall, similar to the steady-state response characteristics, the phase transitions of water at elevated temperatures also need to be taken seriously during dynamic load. Notably, increasing the reactant gas pressure can optimise the dynamic response characteristics of PEMFCs. However, the interaction between elevated temperatures and variable local factors (e.g., temperature, water content, reactant concentration, and charge) during vehicle operation can considerably impair fuel cell components, particularly the PEM. A well-rounded strategy that optimally balances performance and durability is still lacking. It is crucial to comprehensively understand the sensitivity of the dynamic response characteristics of PEMFCs at elevated temperatures to various operational parameters, but such studies are limited. The main goal of such research is to ensure optimal integration and compatibility of multiple components within a PEMFC, identify refined operational strategies and material designs, and ultimately achieve a stable internal state with minimal fluctuations.
4. Challenges in material and structure durability and their impacts on performance loss
To better understand how elevated temperatures affect MEA durability, MEA failure modes at elevated temperatures are discussed in this section in terms of individual components.4.1. Catalyst layer durability
The CL of PEMFCs is a multi-component/multifunctional medium that consists of the tailored assembly of electrochemical catalyst and ionomer phase. Elevated temperatures can directly degrade the catalyst layer or synergistically exacerbate other degradation factors, leading to a reduction in the performance of its components through complex mechanisms. Table 7 reviews the stability of the catalyst layer at different temperatures. Consequently, it is imperative to examine the impact of elevated temperatures on the degradation of the Pt catalyst, carbon support, and ionomer phase.| Serial number | Durability cases and corresponding conditions (anode/cathode) | Items | Values | Ref. |
|---|---|---|---|---|
| 1 | Durability test under a constant current density of 0.4 A cm−2: operating temperature: 100 °C, back pressure (kPa) 150, gas type: H2/air, RH: 0.25, duration: 480 h; | ECSA loss | 15.6% | 123 |
| 2 | Thermal sintering test: temperature: 100 °C, gaseous atmosphere: 3% H2/He, sample: Pt/Vulcan catalyst (40 wt% Pt); | Pt apparent mean size increase | 60.0% | 124 |
| Pt surface area loss | 4.9% | |||
| 3 | Voltage cycling: square wave potential cycle: 10 s at 0.4 V, 10 s at 0.95 V (20 s per cycle), RH: 1.0, gas: 0.5 SLPM 4% H2/0.5 SLPM N2, pressure: atmospheric pressure, cell temperature: 80 °C and 90 °C; | ECSA loss | 80 °C: | 125 |
| 42.2% @ 10000 cycles |
||||
| 90 °C: | ||||
| 50.0% @ 10000 cycles; |
||||
| Mass activity loss | 80 °C: | |||
| 33.9% @ 10000 cycles |
||||
| 90 °C: | ||||
| 64.1% @ 10000 cycles; |
||||
| 4 | Voltage cycling: voltage range: 0.1–0.96 V, scan rate 10 mV s−1, duration: 300 cycles, temperature: 60 °C, 80 °C, 100 °C, 120 °C; | Pt particle growth | 60 °C: | 126 |
| 22.2% @ 300 cycles | ||||
| 80 °C: | ||||
| 49.4% @ 300 cycles | ||||
| 100 °C: | ||||
| 85.2% @ 300 cycles | ||||
| 120 °C: | ||||
| 117.3% @ 300 cycles | ||||
| 5 | Voltage cycling: voltage range: 0.7–0.9 V, time of one cycle: 60 s, duration: 5000 cycles, RH: 0.25, temperature: 80 °C and 120 °C | Pt surface area loss | 8.7% @ 80 °C | 127 |
| 44.4% @ 120 °C | ||||
| 6 | Thermally induced growth test: atmosphere: N2, duration: 155 h, temperature: 80 °C and 160 °C; | Pt particle growth | 7.14% @ 80 °C | 128 |
| 78.57% @ 160 °C; | ||||
| 7 | Thermal corrosion test: atmosphere: air, duration: 3000 h, sample: Pt/BP2000 catalyst with different Pt loadings (40 wt%, 60 wt%, and 80 wt%), temperature: 125 °C; | Maximum fraction of carbon consumed | 17.78% @ 40 wt% | 129 |
| 33.43% @ 60 wt% | ||||
| 40.36% @ 80 wt%; | ||||
| 8 | Thermal treatment: atmosphere: air, sample: Pt (40 wt%)/XC 72 catalyst temperature: 150 °C | Carbon weight loss | 2.6% | 130 |
| 9 | Durability test under a constant current density of 1.6 A cm−2: operating temperature: 65 °C and 90 °C, gas type: H2/O2, dew point temperature: 65 °C, stoichiometries: 1.5/2.5, duration: 100 h | ECSA loss | 9.59% @ 65 °C | 131 |
| 13.93% @ 90 °C | ||||
| 10 | Durability test under a constant potential of 0.9 V: operating temperature: 75 °C, 80 °C and 90 °C | Carbon mass loss rate | 5.71 μg h−1 @ 75 °C | 4 |
| 7.51 μg h−1 @ 80 °C | ||||
| 12.41 μg h−1 @ 90 °C | ||||
| 11 | On-road heavy-duty transportation: operating temperature of 92 °C, duration: 1000 h; | Pt mass loss | 12.3% | 132 |
| Pt particle growth | 3.92% | |||
| Ionomer degradation | Main-chain: 6.5% | |||
| Side-chain: 14.2%; | ||||
| ECSA loss | 44.75% | |||
| 12 | NEDC near-water boiling temperature: operating temperature: 95 °C, inlet pressure (kPa): 280/260, duration: 100 h; | Pt particle growth | Anode: | 133 |
| 76.8–93.9% | ||||
| Cathode: | ||||
| 83.7–117.8% | ||||
| Catalyst layer thickness | Anode: | |||
| 3.87–37.8% | ||||
| Cathode: | ||||
| 6.78–26.8% | ||||
| ECSA loss | 11.6–82.4% | |||
| 13 | A combined OCV hold and RH cycling testing: gas: H2/air (1000/1000 sccm), pressure: ambient, duration: ∼200 h, temperature: 95 °C; | Pt particle growth | Anode: | 134 |
| 47.6–85.7% | ||||
| Cathode: | ||||
| 195.2–180.9% | ||||
| 14 | Thermal annealing: sample: Nafion film, gas atmosphere: dry N2, duration: 1 h, temperature: 120 °C; | Ionomer proton conductivity loss | 20.91–27.6% | 135 |
| 15 | Thermal annealing: sample: Nafion film, gas atmosphere: vacuum, duration: 1 h, temperature: 110 °C and 146 °C; | Ionomer proton conductivity loss | 81% @ 110 °C | 136 |
| 94% @ 146 °C | ||||
| 16 | Thermal treatment: sample: MEA, gas atmosphere: vacuum, temperature and duration: 90 °C for 10 min, 140 °C for 10 min, 140 °C for 30 min | Decrease of oxygen transport resistance in the CCL | 1.6% @ 90 °C for 10 min | 137 |
| 7.4% @ 140 °C for 10 min | ||||
| 16.0% @ 140 °C for 30 min; | ||||
| 17 | Thermal annealing: sample: Nafion film, gas atmosphere: vacuum, duration: 24 h, temperature: 60 °C and 140 °C | Ionomer Young's modulus increment | 120.4% @ 60 °C | 24 |
| 214.1% @ 140 °C |
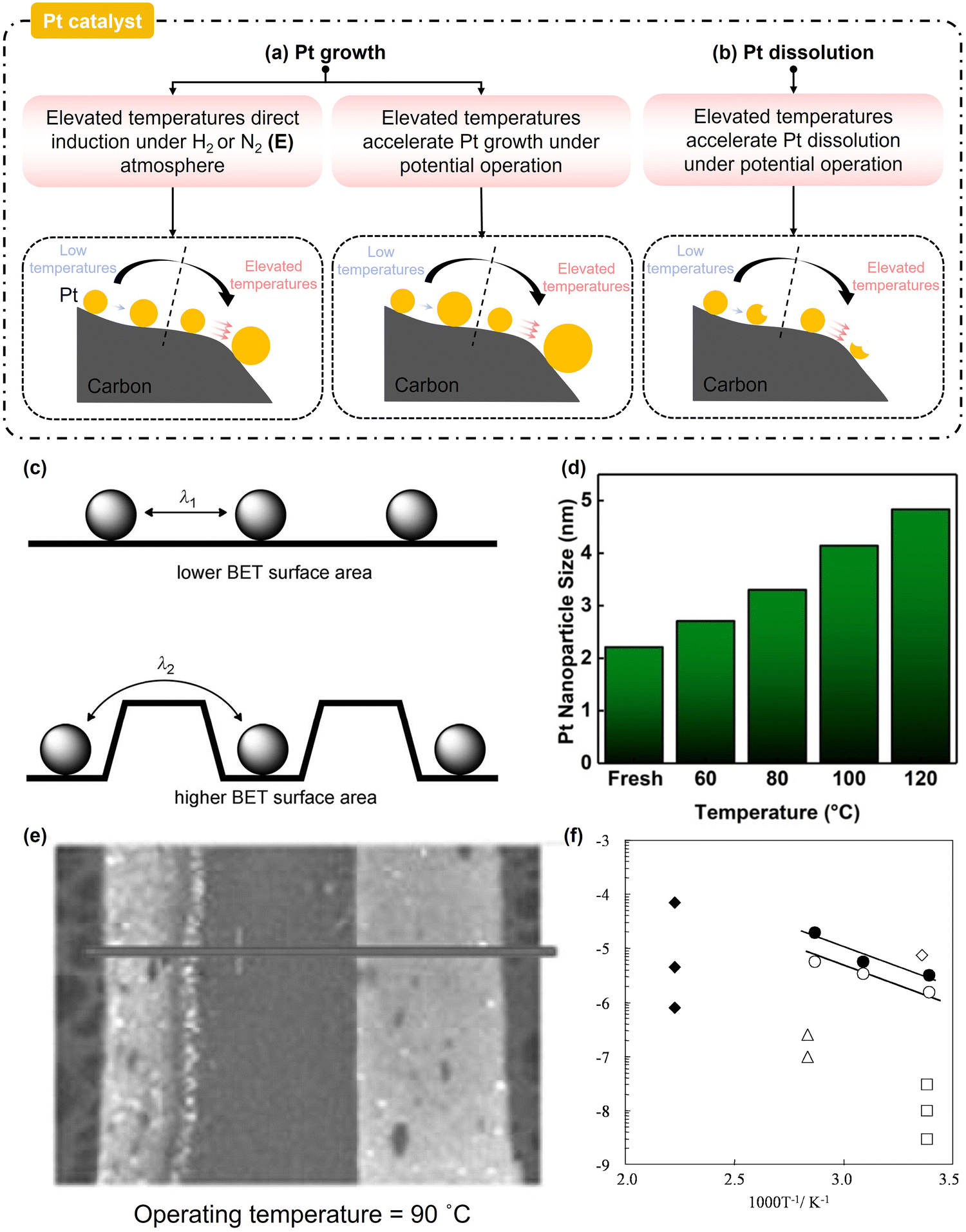 | ||
| Fig. 9 Schemic diagram of Pt catalyst degradation at elevated temperatures: (a) Pt growth and (b) Pt dissolution. (c) Illustration of Pt particle growth as a function of the distance between the nanoparticles.128 (d) Alterations in the Pt catalyst size as the cell temperature increases during voltage cycling (0.1–0.96 V).4 (e) Denser Pt microscopy images at the MEA cross-section.125 (f) Logarithm of Pt solubility as a function of inverse temperature.138 | ||
As illustrated in Fig. 9(a), elevated temperatures can directly induce Pt catalyst growth, but this direct induction mechanism is significantly modulated by specific elevated temperature levels and the surrounding gas atmosphere. Sellin et al.124 reported that increasing temperatures directly induced an increase in the Pt catalyst growth in the entire elevated temperatures range and a H2-containing atmosphere. The reducing effect of H2 gas removed surface oxides on Pt catalysts, which disrupted the protective oxide layer and supported the subsequent crystallite reconstruction and grain boundary diffusion. Sellin et al.124 reported an approximately 4.9% loss of the Pt active surface area and a 60.0% increase in the Pt apparent mean size after thermal sintering at 100 °C. Hasché et al.128 demonstrated significant Pt catalyst growth (∼78.57%) under a N2 atmosphere at 160 °C for 155 h because the Pt catalyst crystallite growth was controlled by the interparticle distance, as indicated in Fig. 9(c). A decrease in the interparticle distance resulted in a shorter time for the Pt catalyst particle diffusion/migration and coalescence. Therefore, the direct elevated temperature induced growth of Pt under a N2 atmosphere was only observed in the (E) regime. However, this direct induction was more challenging under an air atmosphere due to the protective effect of the Pt oxide layer. Shao et al.141 reported no noticeable platinum particle sintering during long-term (3000 h) exposure to dry air at 125–195 °C. Overall, the direct elevated temperature induced growth of the Pt catalyst on the anode side in the PEMFC-matched elevated temperature range is acceptable, while it can be considered negligible on the cathode side.
Catalyst durability studies have also indicated that elevated temperatures can accelerate the Pt catalyst growth during potential cycling (Fig. 9(a)).4,126,142 The influence of elevated temperatures on the accelerated growth of Pt catalysts under voltage cycling appears to increase linearly (Fig. 9(d)), suggesting that Pt catalyst growth is accelerated within the entire elevated temperature range. Elevated temperatures do not trigger explosive Pt growth, even though the underlying acceleration mechanism remains poorly understood. Bi et al.143 reported that a low RH stimulates Pt growth at 60 °C. To our knowledge, the coupled effect of elevated temperatures and RH on Pt catalyst growth during operation has not been studied.
In terms of Pt dissolution at elevated temperatures, Fig. 9(b) shows that elevated temperature has an accelerating effect, exacerbating Pt catalyst dissolution under electrochemical potential operations within the entire elevated temperature range.125,144–146 Yang et al.125 observed a denser Pt band in the membrane after potential cycling at 90 °C (Fig. 9(e)). Borup et al.138 indicated that the Pt dissolution reaction was endothermic, causing the Pt solubility to increase at elevated temperatures, following the Arrhenius relationship (Fig. 9(f)). Huang et al.147 suggested that this was attributed to the impact of the operational temperature on the collective surface tension of Pt, which increased both the net rate of Pt decomposition and the net rate of particulate dissolution. Yue et al.132 reported approximately 12.3% Pt mass loss after 1000 h on-road heavy-duty transportation with a coolant temperature of 92 °C. At low temperatures, a high RH promotes Pt catalyst dissolution due to accelerated Pt ion transport in the ionomer with larger and more abundant water channel networks.143 Similar to Pt growth, the coupling effect of elevated temperature and RH levels on Pt dissolution during potential cycling has not yet been studied. Notably, Pt redeposition equally affects the available amount of Pt catalyst. For instance, at low temperatures, Đukić et al.148 reported that the rates of Pt catalyst dissolution and redeposition both increased with the temperature at a constant potential window, while the overall extent of Pt loss remained minimal. Nevertheless, it is essential to investigate the effect of elevated temperatures on Pt catalyst redeposition (e.g., degree and location) to explicitly understand the overall Pt dissolution.
 | ||
| Fig. 10 Schemic diagram of carbon support corrosion at elevated temperatures: (a) chemical corrosion and (b) electrochemical corrosion. (c) TGA data for 46% Pt/C measured at heating rates of 0.1, 0.25, 1, 2.5, and 10 °C min−1.153 (d) Decline in carbon content against the cell potential at varying temperatures.4 | ||
As depicted in Fig. 10(a), under a H2/O2 atmosphere, elevated temperature promotes carbon corrosion, leading to the corrosion of the part of the carbon support in contact with Pt catalysts, which occurs within the (H) and (E) regimes but not at lower temperatures. Andersen et al.149 found that the degradation of the support without a Pt catalyst was minimal (only ∼1% weight loss) even at temperatures of up to 200 °C, while degradation of the carbon support with Pt was significantly higher, indicating that carbon corrosion was catalysed by Pt. Stevens et al.129 reported that 40.36% of carbon mass was consumed after 3000 h of thermal corrosion at 125 °C when the Pt catalyst loading was 80 wt%. Sellin et al.130 indicated that only 2.6% of the carbon support (Vulcan XC-72) was corroded after thermal treatment at 150 °C, implying that the degree of thermal corrosion resistance of the carbon support itself is equally critical and that the Vulcan XC-72 support has good thermal corrosion resistance. Guterman et al.151 suggested that narrowing the size dispersion of Pt and of support particles and improving the uniformity of Pt distribution increased the exposure of platinum to carbon and resulted in more uniform carbon oxidation. Under an O2 atmosphere, the product of carbon corrosion is either CO or CO2.94,130,152 Sellin et al.130 demonstrated that the carbon support could also be directly corroded at 423 K under a 3% H2/Ar flow, which generated methane or other hydrocarbons. Notably, the rate of change in the thermal environment faced by the carbon support also affects the degree of carbon corrosion. As depicted in Fig. 10(c), Baturina et al.153 conducted thermogravimetric analysis (TGA) at heating rates from 0.1 to 10 °C min−1 and showed that as the heating rate decreased, the percent mass loss increased. This indicated that Pt catalysed the oxidation of carbon during isothermal aging. Therefore, it can be concluded that the contribution of instantaneous thermal overshoot induced within the CL during dynamic processes is not a significant contributor to the thermal corrosion of carbon supports.
In terms of how elevated temperatures accelerate electrochemical corrosion, Fig. 10(b) shows that elevated temperatures further intensify corrosion at pre-existing electrochemical corrosion sites in carbon, thereby exacerbating the extent of electrochemical corrosion over the entire elevated temperature range (Fig. 10(d)). Lochner et al.4 reported that the carbon mass loss rate reached 5.71 μg h−1 at 75 °C and 12.41 μg h−1 at 90 °C during durability tests under a constant potential of 0.9 V. Zhang et al.,94 Roen et al.,154 and Lim et al.152 supported the above results, and Stevens et al.155 suggested that H2O played a significant role in carbon corrosion. Therefore, high RH levels and enhanced water production at a high current density are unsuitable for ensuring that the carbon support remains stable at elevated temperatures, as shown in Fig. 10(a) and (b). Moreover, there has not been a comparative analysis at elevated temperatures between chemical corrosion and electrochemical-induced corrosion of carbon supports.
 | ||
| Fig. 11 Schemic diagram of ionomer degradation at elevated temperatures: (a) elevated temperature direct induction and (b) joint impacts with other factors. | ||
Nevertheless, elevated temperatures alter the properties of ionomers, including decreasing the thermal expansion coefficient (Fig. 12(a)),19 increasing Young's modulus (Fig. 12(b)),24 decreasing the proton conductivity (Fig. 12(c)),136,158 increasing the surface hydrophobicity (Fig. 12(d)),136,158,161 and improving the oxygen transmission capacity (Fig. 12(e)).137 However, the underlying mechanisms between the evolution of phases, clusters, and molecular structures within ionomers and elevated temperatures are not fully understood, making it difficult to understand changes in ionomer properties. Most existing studies on directly-induced ionomer degradation at elevated temperatures have been conducted under vacuum or dry inert gases, but since the actual ionomer phase within the MEA typically exists in a humidified environment, the role of RH in this degradation process needs to be better understood.
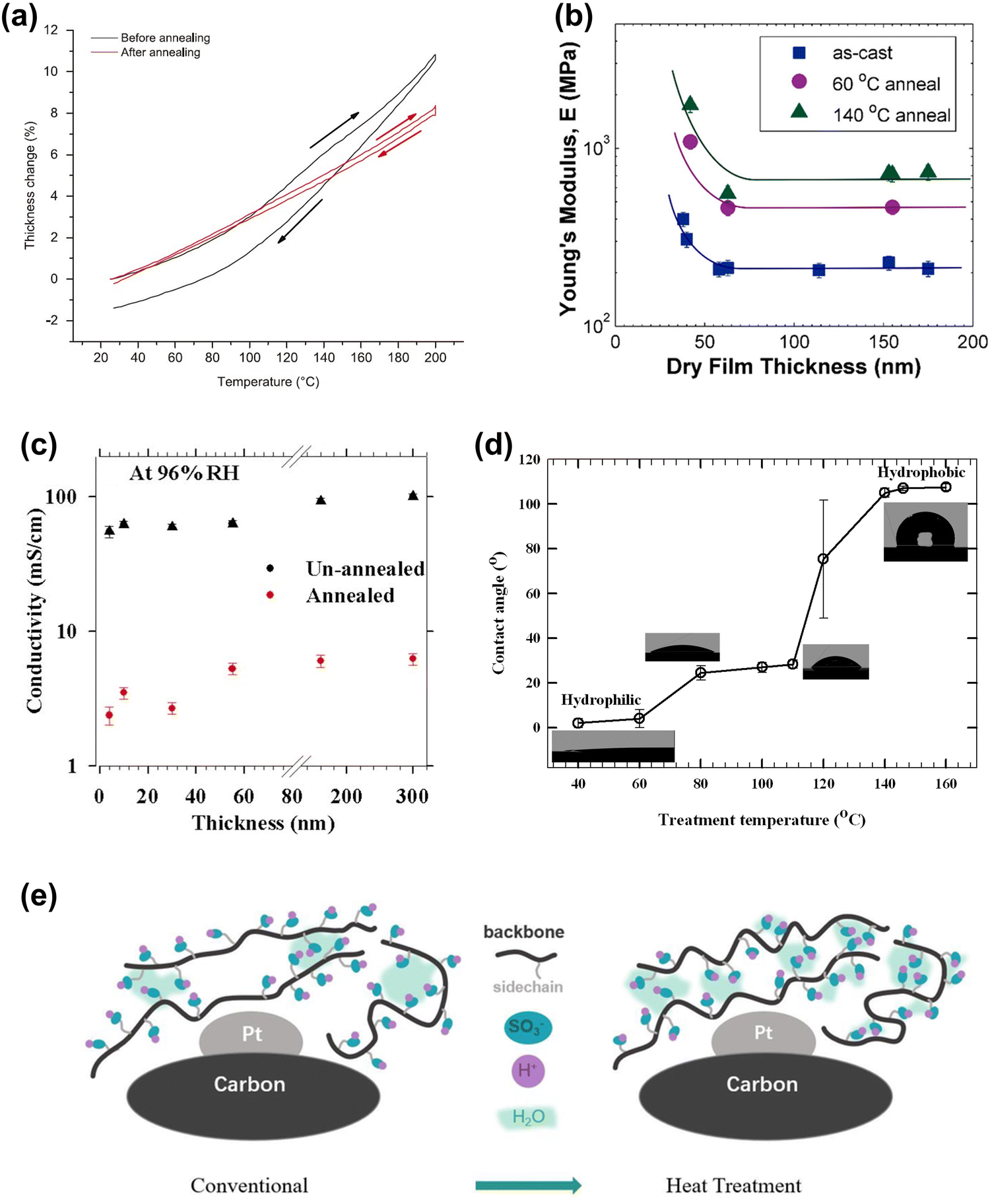 | ||
| Fig. 12 (a) Thickness change of unannealed and annealed Nafion films (50 nm) on SiO2 as a function of temperature.19 (b) Young's modulus at 30% RH: as cast, 60 °C annealing, and 140 °C annealing.24 (c) Proton conductivity of unannealed and annealed Nafion thin films at 96% RH and 60 °C.158 (d) Surface wettability of annealed ultrathin (10 nm) Nafion film.136 (e) Mechanism of enhanced local transport after heat treatment.137 | ||
Mechanical fatigue and chemical degradation processes are affected over the entire elevated temperature range. The mechanical degradation of ionomers is primarily caused by their frequent swelling and shrinkage during cyclic hydrothermal stress. Chang et al.162 suggested that plastic strain within an ionomer could accumulate during cyclic changes in RH. Rong et al.163 reported that the deformation of the Nafion ionomer at elevated temperatures during RH cycling was larger than that at lower temperatures, which was corroborated by Tang et al.164 Therefore, a greater change in plastic strain occurs and propagates within the ionomer phase at elevated temperatures due to larger deformation, which accelerates mechanical degradation of the ionomer, as illustrated in Fig. 11(b). The accumulation of plastic strain may result in the internal breakdown of the ionomer when reaching the breakdown strain, which may significantly impair the structural stability of the CL. The mechanisms of ionomer chemical degradation in PEMFCs involve the formation of hydroxyl and hydroperoxyl radicals (OH˙ and OOH˙) caused by hydrogen peroxide (H2O2) formation on the catalyst surface via the 2e− ORR. Sethuraman et al.165 reported the rates of H2O2 formation in the anode side of a PEMFC at different temperatures and showed that increasing the cell temperature from 25 °C to 95 °C significantly enhanced the rates of H2O2 formation, and the rate of H2O2 formation on the cathode side was the same.165 The anode and cathode H2O2 formation rates cannot be directly correlated with the ionomer degradation rates because there are several intermediate reactions between H2O2 formation and actual ionomer chemical degradation. However, it can be inferred that more significant ionomer chemical degradation may occur due to more severe free radical attack at higher concentrations of H2O2 generated at elevated temperatures,166 as depicted in Fig. 11(b). The temperature-dependence of free-radical attack on ionomers remains unclear.
However, direct experimental observations or modelling calculations on the ionomer mechanical fatigue and ionomer chemical degradation at elevated temperatures have not been conducted. Additionally, thin, photo-sensitive, and low-density organic ionomer films are challenging to characterise and image, and ionomers in real CLs may exhibit non-uniform distribution, discontinuities, and variable thicknesses. The molecular chain configurations within an ionomer, spanning both in-plane and through-plane orientations, are strongly dependent on its thickness.90,136 Domain changes in the ionomer phase, humidity fluctuations, and free radical attacks on ionomer molecular chains may occur simultaneously during PEMFC operation. The synergistic mechanisms underlying these processes and their effects on PEMFC performance are not yet fully characterised, and these uncertainties hinder a comprehensive understanding of ionomer degradation within the real CL at elevated temperatures.
 | ||
| Fig. 13 Structure collapse features in the CL at elevated temperatures. (a) Diagram of collapse characteristics of CL structures at the micrometer and nanometer scales. (b) Cracking on the CL surface.167 (c) Decrease in the thickness.168 (d) Photographs of the MEA cathode and reduction rate of CL thickness in each area after 100 h operation at 1600 mA cm−2 at 65 °C and 90 °C.131 (e) Pore structure evolution.169 (f) TPB structure evolution.162 | ||
Existing studies have provided some insight into the structural evolution of the CL at the microscale, but have mainly focused on experimental phenomena. At the microscale, macroscopic features such as surface cracking and thickness decreases are observed. Han et al.167 conducted two in situ fatigue experiments on a catalyst-coated membrane (CCM) at different operating temperatures and revealed that the CL surface exposed to 95 °C exhibited wider and deeper cracks than those formed at 85 °C, as shown in Fig. 13(b). They emphasised the significant contribution of the glass transition temperature of CCM (∼90 °C) on the CL surface cracking. An increase in the deformation of the PEM during the hydrothermal cycling, especially when the temperature exceeded the glass transition temperature, led to more severe cracking within the CL.164,171 However, the structural stability of the CL was also significantly affected by elevated temperatures, particularly for ionomers. As the binder in the CL, an ionomer helps preserve the CL's surface integrity.172 Therefore, further investigations into the evolution of ionomer adhesion properties on Pt/C catalyst agglomerates and their structural strength at elevated temperatures are warranted. More severe cracking can result in a range of detrimental effects, including an increased risk of PEM perforation,173 water flooding within the CL,173 high free-radical concentrations,173 enhanced brittleness of the CL structure,172 and the loss of certain agglomerates.174 Investigating the deteriorated cracking mechanisms within the CL at elevated temperatures can provide valuable insights for improving the performance and extending the service life of PEMFCs.
Studies have shown that the thickness reduction of the CCL increases at elevated temperatures,142,168,175 as displayed in Fig. 13(c) and (d). Kneer et al.168 and Trogisch et al.142 suggested that the observed reduction in CL thickness was negligible and primarily attributed to the compression effect of the MEA, but elevated temperatures may still substantially accelerate carbon corrosion rates, as discussed above. As shown in Fig. 13(b), Zhao et al.131 reported that compared to 60 °C, the thickness of the CCL decreased by 9.3% after durability tests at 90 °C. The RH level also significantly affects the reduction of CL thickness. In region 9, because there was no water flooding during operation at 90 °C, the thickness reduction of the CCL at 90 °C was less than operation at 60 °C.131 Therefore, as the structural backbone of the CL, progressive carbon corrosion exacerbates thickness reduction, leading to increased catalyst loss and significant performance deterioration. Kneer et al.168 also indicated the increased tortuosity of an aged MEA at elevated temperatures compared with lower temperatures. However, the effects and underlying mechanisms of elevated temperature-induced carbon corrosion on CL thickness and structure in the through-plane direction are unexplored. Additionally, the evolution of ionomer mechanical properties at elevated temperatures and their impact on CL thickness and structure in the through-plane direction warrant further investigation.
Nanoscale features include pore-based mass transport networks and three-phase boundary structures critical for electrochemical reactions. Among the microstructural properties of the CL, the pore size distribution and porosity are particularly critical because they govern the diffusion of reactant gases to active reaction sites and the efficient removal of generated water from the CL. Typically, two types of pores exist within the CL: (i) primary pores, with sizes smaller than approximately 20 nm, and (ii) secondary pores, ranging from 30 nm to 150 nm. Primary pores are located between carbon particles within agglomerates, whereas secondary pores represent the void spaces between agglomerates and are generally larger than primary pores, as depicted in Fig. 13(e). Lu et al.133 tested the durability of a PEMFC stack near the boiling point of water for 100 h and observed an increase in the pore size of the MEA in the range of 10–100 nm. As posited by Kneer et al.,168 the aged samples exhibited a shift in their pore size distribution towards larger pores, while the overall porosity of the cathode remained unchanged. The observed shift was interpreted as an onset of carbon corrosion that resulted in the collapse of the smallest pores and an increase in the number of larger pores. Yue et al.132 demonstrated that the primary pores shrank while the secondary pores expanded after 1000 h of on-road heavy-duty transportation with a coolant temperature of 92 °C, which was attributed to ionomer migration. Elevated temperatures accelerated carbon corrosion and also impacted the properties of ionomers, altering the pore structure within the CL.
Although Kneer et al.168 suggested no obvious differences in the CL pore structure in the cells cycled at 90 °C versus those cycled at 70 °C, their conclusions were based on 3D reconstructions of the CL obtained using focused ion beam scanning electron microscopy, where the identified pores were generally considered secondary pores.132 The evolution of primary pores, however, remains poorly understood, and additional research is needed to clarify the mechanistic relationships between elevated temperatures, compositional changes, and the pore structure evolution.
The TPB structure consists of a Pt catalyst, ionomer/water films, and pore spaces and serves as the critical site for electrochemical reactions, as depicted in Fig. 13(f). Optimizing the TPB structure significantly enhances the power density and catalyst utilization,176 and distortions in the TPB structure induced during operation can severely compromise the cell performance and durability. Current research on the evolution of the TPB structure primarily emphasises interfacial delamination between the ionomer and Pt/C agglomerates.162,163,177–179 Cohesive zone models have demonstrated that the cyclic expansion and contraction of ionomers during hygrothermal cycling cause the partial detachment of ionomers from Pt/C agglomerates, leading to cracks in the CL and a reduction in the ECSA.162,163,177–180 Changes in the water content of ionomers at lower temperatures are considered the primary driver of interfacial delamination.162 Notably, Chang et al.162 reported that low-temperature cycling alone does not significantly induce interfacial delamination. In contrast, the extent of interfacial delamination is governed by the interplay between an ionomer's expansion–contraction and its adhesive properties, both of which are impacted by elevated temperatures. Moreover, Yue et al.132 showed that the contact area between Pt catalysts and ionomers decreased by 37.3% due to ionomer migration at elevated temperatures, indicating significant changes in the TPB structure. However, the detailed distortion mechanisms of the TPB structure at elevated temperatures are unexplored. Notably, the role of humidity in the evolution of pore structure and the TPB structure at elevated temperatures significantly affects the degree of ionomer deformation.
4.2. Proton exchange membrane durability
The PEM plays a crucial role in transporting protons from the anode to the cathode, insulating negative electrons, and separating the reactants, hydrogen, and oxygen. Therefore, the membrane must be an efficient conductor of protons and an effective insulator of electrons and must exhibit both mechanical and chemical robustness. The most commonly employed membranes in PEM fuel cells are PFSA membranes with perfluoroether long side chains with sulfonic acid end groups.138 Recently, alternative PFSA membranes with short side chains have garnered significant attention for investigation.181 Among these, the Nafion membrane is the most widely used due to its long-term stability in both oxidative and reductive environments, relatively low fabrication cost, and ease of processing. Nevertheless, the failure modes of PEMs, including perforation, irreversible reduction in proton conductivity, cracking, etc., at elevated temperatures, emphasize the critical need to investigate and enhance their stability in such environments. Table 8 summarizes the stability of PEMs at different temperatures.| Serial number | Durability cases and corresponding conditions (anode/cathode) | Items | Values | Ref. |
|---|---|---|---|---|
| 1 | NEDC near-water boiling temperature: operating temperature: 95 °C, inlet pressure (kPa): 280/260, duration: 100 h; | PEM thickness decrease | 1.9–8.4% | 133 |
| 2 | A combined mechanical/chemical accelerated stress test: operating temperature: 90 °C, gas type: H2/air, back pressure: no duration: M – 8 for 1512 h, M – 12 for 1488 h | Hydrogen crossover increase | M – 8: 348.4% @ 0 A cm−2 | 182 |
| M – 12: 398.7% @ 0 A cm−2 | ||||
| 3 | Modelling of PEM chemical degradation | PEM mass loss rate | Ratio of 95 °C to 80 °C: 4.83 | 183 |
| 4 | Modelling of PEM chemical degradation | Hydrogen crossover flux | Ratio of 90 °C to 70 °C: 1.53 | 184 |
| 5 | Thermal annealing: sample: short side chain (SSC) membrane, gas atmosphere: vacuum, duration: 1 h, temperature: 140 °C; | PEM water uptake decrease | 8.1–11.1% | 185 |
| PEM conductivity decrease | 3.9–4.7% | |||
| Glass transition temperature | Increase from 134 °C to 143 °C @ 3 M membrane | |||
| increase from 131 °C to 139 °C @ aquivion membrane | ||||
| 6 | Thermal annealing: sample: aquivion 870 membrane, duration: 72 h, temperature: 140 °C | Glass transition temperature | Increase from 124 °C to 131 °C @ aquivion 870 membrane | 186 |
| 7 | Modelling of PEM chemical degradation | Fluoride emission rate | Ratio of 90 °C to 80 °C: 4.9 | 187 |
 | ||
| Fig. 14 (a) TGA curve of the Nafion 117 membrane.188 (b) Instantaneous elastic response of Nafion N1110 as a function of water activity from 23 to 90 °C.189 (c) Typical dynamic mechanical analysis (DMA) profiles of Nafion and 3 M PFSA membranes.190 (d) Differential scanning calorimetry (DSC) data of the Nafion 117 membrane as a function of water content.191 (e) Evolution of proton channels in the direction perpendicular to the membrane surface during the heat treatment process.192 (f) Characterization results for N105 samples annealed at 140 °C, dry conditions and 140 °C, RH = 100%.193 | ||
In the absence of exposure duration to elevated temperatures, as illustrated in Fig. 14(a), TGA-MS can be used to determine the thermal decomposition process of PFSA ionomers at different temperatures. Typically, this occurs in three stages:4,127,188,194–196 (i) loss of membranous water (100–200 ± 20 °C), which culminates in the rupture of H3O+ and the protonation of SO3− to SO3H, as well as the partial decomposition of SO3H → SO2 + OH˙ near 200 °C; (ii) cleavage of the C–S bond (280 ± 30 °C to 400 ± 20 °C) leading to sulfonate-group degradation; and (iii) the final decomposition of the perfluorinated matrix (400 ± 20 °C to 600 ± 40 °C). This demonstrates that in the elevated temperature range required for MEAs, only the loss of water occurs within the PEM, inducing phase transitions in clusters/ion channels without damaging its molecular structure. Rodgers et al.197 confirmed this conclusion and suggested that the Nafion ionomer was thermally stable up to ∼150 °C due to the presence of strong C–F bonds and a shielding effect of the electronegative fluorine atoms on the PTFE-like backbone.
Nevertheless, Majsztrik et al.189 investigated the elastic modulus of Nafion N1110 as a function of water activity from 23–90 °C and suggested that for any given level of hydration (water activity), the elastic modulus always decreased with temperature, as indicated in Fig. 14(b). This was because the internal structure of the PEM evolved, resulting in a decrease in its strength, which became apparent in the (M) regime. In practice, the internal molecular chain structure and domain structure of a PEM typically undergo a thermal transition process as the temperature increases,198 which is captured by dynamic mechanical analysis (DMA).90 A typical DMA curve of the PEM is demonstrated in Fig. 14(c), in which the characteristic thermal transition temperatures are noted.90 The PFSA membrane exhibits three thermal transitions corresponding to different relaxation behaviours: (i) an α relaxation at approximately Tα = 90–120 °C for acid-forming membranes; (ii) a broader β relaxation between Tβ = −40 °C and 20 °C; and (iii) a γ relaxation approximately Tγ = −120 °C to −90 °C. Regarding the three relaxation mechanisms, Kusoglu et al.90 reported that (i) Tα was a dynamic ionic-cluster transition associated with the onset of long-range mobility of the chains due to destabilization of the electrostatic network; (ii) Tβ was the glass-transition temperature of the PFSA matrix attributed to the onset of segmental main-chain motion, facilitated by side-chains; (iii) the low-temperature transition Tγ was related to the local movement of the PTFE backbone.90 When the temperature exceeded Tα, the weakening of the electrostatic interactions within the ionic aggregates triggered the onset of long-range mobility of both the main and side chains, resulting in a significant increase in the chain mobility.199
Due to the glass transition of the membrane, its mechanical properties were greatly altered. The mechanical behavior of a material, i.e., its ability to withstand deformation without rupturing, can be evaluated using its stress–strain curves. Numerous studies have demonstrated that the stress–strain curves of a membrane undergo a significant decline at elevated temperatures due to a reduction in both the elastic modulus and the yield stress, as well as a reduction in ultimate tensile strength and an increase in the strain at break. Marrony et al.200 indicated that the ultimate tensile strength decreased by 54.1% when the temperature increased from 80 °C to 120 °C due to a decrease in intermolecular forces and the presence of amorphous domains.90,164,201 Therefore, PEMs tend to soften in elevated temperature environments, especially close to or exceeding their glass transition temperatures. Notably, the membrane water content significantly influences the magnitude of the glass transition temperature. As depicted in Fig. 14(d), the glass transition temperature decreased from 132 °C to 108 °C when the membrane water content increased from 0% to 24.0%.191 Therefore, the fragility of PEMs containing water needs to be given special consideration.
Considering the effect of the exposure duration on elevated temperature environments, the influence of elevated temperature environments on the PEM is typically referred to as thermal treatment or thermal annealing. It primarily focuses on its influence on the internal domain structure and the evolution of its operational characteristics (proton conductivity, mechanical properties, etc.), as illustrated in Fig. 14(e) and (f).
Studies have shown that thermal treatment of PEMs often decreases the proton conductivity and enhances the mechanical strength, particularly when the thermal treatment temperature falls within the (H) and (E) regimes.185,186,191–193,202–205 This is attributed to increased crystallinity in the hydrophobic domains and changes in the ionic domain structure. An increase in crystallinity enhances the glass transition temperature, tensile strength, etc. Shin et al.185 reported an increase in the glass transition temperature from 134 °C to 143 °C for a 3 M membrane after vacuum thermal treatment for 1 h at 140 °C. Factors leading to an increase in crystallinity are multifaceted, including increased chain mobility, increased long-range order, and the melting and re-crystallization of imperfect PTFE-like crystallites at elevated temperatures.186,191,202 However, Jeon et al.206 suggested that the crystallization of Nafion 212 and Aquivion E87-05S membranes decreased after thermal treatment at 90–140 °C. As demonstrated in the seminal work conducted by Hensley et al.,203 annealing had a marked effect on the crystallinity of films, with a decline observed in the crystallinity of thick films concomitant with an increase in the crystallinity of thin films, as determined by wide-angle X-ray measurements. The heat treatment duration in Jeon's study was much longer than that in the other four studies, which may have re-damaged the already-formed crystalline regions. Regardless, the mechanical properties of the membrane exhibited fluctuations as the exposure time increased at elevated temperatures, which pose a significant challenge to the reliable sealing and stable operation of PEMFCs.
As illustrated in Fig. 14(e), the through-plane conductivity of a PEM significantly decreased after treatment at 120 °C for 30 h. This reduction was caused by the reorientation of proton transport pathways near the membrane surface from perpendicular to parallel to the surface as the annealing temperature approached the glass transition temperature of the membranes, thereby impeding proton conduction.192 Shin et al.185 also suggested a proton conductivity loss in the range of 3.9–4.7% after vacuum annealing for 1 h at 140 °C. Notably, Matos et al.193 reported that the properties of a PEM were significantly less affected when annealing was carried out at high RH, as illustrated in Fig. 14(f). Despite that, the internal evolution of PEMs at elevated temperatures remains unclear, and low-RH conditions pose a significant challenge to the stable operation of PEMs at elevated temperatures. Furthermore, the extent of phase separation in the hydrophobic matrix and the hydrophilic region increases due to the thermal treatment based on the small-angle X-ray scattering measurements.185 This can increase the risk and degree of hydrogen or oxygen crossover through the PEM, thereby reducing the OCV of the PEMFC and inducing chemical degradation.
Elevated temperatures can accelerate the creep deformation of PEMs due to increased molecular mobility, especially when the temperature is close to or exceeds the glass transition temperature,138,208 that is in the (H) and (E) regimes. Excessive creep deformation forms wrinkles and induces membrane thinning. Lu et al.133 observed a 1.9–8.4% PEM thickness decrease after a 95 °C durability test. In addition to creep behaviour, elevated temperature can accelerate membrane fatigue, including but not limited to pinholes and cracks. Alavijeh et al.171 conducted two accelerated mechanical stress tests on the CCMs at cell temperatures of 80 °C and 95 °C and reported that larger and more frequent cracks were observed within the sample at 95 °C. This was attributed to the increased fatigue amplitude and reduced mechanical strength of the membrane at elevated temperatures, suggesting that the MEAs at 95 °C underwent more severe mechanical failure, as shown in Fig. 15(a). Han et al.167 conducted in situ hygrothermal cycling durability tests on a PEM with a reinforced layer of e-PTFE above its glass transition temperature and suggested that the cross-linking degree between molecular chains inside the membrane was weakened after exceeding the glass transition temperature. This resulted in a significant decrease in the mechanical properties of the PEM after durability tests. More severe silver lines and earlier fractures appeared during tensile tests.
 | ||
| Fig. 15 (a) SEM images of fresh and degraded CCMs from the AMST-1 and AMST-2 tests.171 (b) Contour plot of effects of temperature and humidity on the Mid. membrane degradation rate.207 (c) Evolution of cumulative fluoride loss at different operating temperatures.184 | ||
Some studies have conducted modelling of ex situ and in situ fatigue behaviour.209,210 The application of constitutive models and failure criteria to describe materials remains a fundamental challenge in understanding fatigue mechanisms and accurately predicting material lifetimes at elevated temperatures. To achieve high-fidelity computational simulations of the fatigue process, it is essential to incorporate material models that capture the nonlinear viscoelastic and/or viscoplastic effects, particularly those that consider the influence of elevated temperatures. Particular attention should be given to the (H) and (E) regimes, as membrane failure during operation will be significantly accelerated under such conditions. At low temperatures, RH fluctuations and pressure differences between the anode and cathode sides are the main driving forces for membrane fatigue failure. At elevated temperatures, the contribution of RH and pressure difference variation characteristics to membrane mechanical failure requires further investigation to understand membrane degradation during dynamic operation of PEMFCs at elevated temperatures. Regardless, more severe membrane failures significantly increase the crossover leak rate, risking the stable operation of PEMFCs.208
As illustrated in Fig. 15(b), Zhao et al.207 investigated the impact of operating temperature on the durability of membranes using the OCV accelerated stress tests (AST). Their findings revealed a direct proportionality between the membrane degradation rate and the operating temperature. The study identified an optimal operational region, which was determined without changing the membrane materials, suggesting that operation at >90 °C was not feasible. A decrease in RH levels further accelerated the chemical degradation of the PEM. Failure of the Nafion XL membrane was observed only after ∼250 h of the steady-state OCV AST at 30% RH and 90 °C.211 Therefore, elevated temperature dramatically affected the membrane chemical degradation, especially in a low-RH environment. Modelling calculations also confirmed that elevated temperatures accelerate the chemical degradation of the membrane, as evidenced by a significant increase in PEM mass loss and the fluoride emission rate. Chen et al.183 reported that increasing the operating temperature from 80 °C to 90 °C resulted in an approximately 4.83-fold increase in PEM mass loss. Frensch et al. observed a 4.9-fold increase in the fluoride emission rate upon raising the operating temperature from 80 °C to 90 °C. Similarly, as shown in Fig. 15(c), Zheng et al.184 found a dramatic increase in the fluoride ion release rate and hydrogen crossover flux (1.53-fold) at 90 °C compared with 25 °C, indicating the severe chemical degradation degree at elevated temperatures. The PEM chemical degradation at low temperatures is induced by free-radical attack. As discussed in Section 4.1.3, the rates of H2O2 formation significantly increase at elevated temperatures, offering a significant number of sources for free radical generation.165 Chen et al.183 also supported the above conclusion and indicated that the chemical degradation of the PEM was accelerated due to an accelerated chemical reaction in the cell at elevated temperatures, exhibiting an exponential relationship with temperature (60 °C ≤ T ≤ 95 °C). Furthermore, trace metal ions resulting from the corrosion of bipolar or end plates, as well as the leaching of less-noble metals from an alloy catalyst, may also accumulate within the membrane. These ions can catalyse radical attacks previously mentioned and accelerate chemical degradation of the membrane, but there are several intermediate reactions between H2O2 formation and actual ionomer chemical degradation. The effect of elevated temperatures on these intermediate reactions, including free radical formation, subsequent attack of free radicals on membrane polymer chains, and leaching of metal ions, is unclear. Notably, the mechanism between mechanical failure and chemical degradation of membranes has been investigated,212–214 but the role of elevated temperatures is poorly understood. The interplay between elevated temperatures and mechanical and chemical degradation needs further investigation to provide a theoretical basis for extending the lifetime of PEMFCs.
4.3. Gas diffusion layer durability
The primary function of the GDL within a PEMFC is twofold. First, it must facilitate the transport of reactant gases from the flow channels to the CL and also facilitate the drainage of water produced at the CL.217,218 The GDL must also serve as an efficient electron conductor to collect the current. Various types of GDLs are under development, but they are typically comprised of porous carbon fibres and carbon particles,127 and a typical GDL consists of a GDB and a MPL. The primary function of the GDB is the distribution of reactant gases, in conjunction with the current collection. The MPL, which is composed of carbon powder and hydrophobic materials such as PTFE, is responsible for managing water flow. Consequently, it can be concluded that mechanical strength, conductivity, and hydrophobicity are fundamental properties of the GDL.Several mechanisms can cause degradation of the GDL, including chemical degradation, erosion resulting from gas flow, dissolution by water, freezing/thawing effects, and mechanical compression. There is limited research into the impact of elevated temperature on GDL degradation.127,133,215,216,219Table 9 shows the stability of the GDL at elevated temperatures. TGA has shown that the decomposition or volatilization of the GDL occurs only when the temperature reaches 500–650 °C due to thermal degradation of fluorinated polymers. This temperature is much higher than the elevated temperature range required for PEMFCs, indicating that the GDL materials exhibit strong resistance to thermal decomposition. Nevertheless, elevated temperature environments still have a considerable detrimental effect on the operational performance of the GDL, primarily due to thermal softening133,215 and thermal oxidation.127,133,216 The main degradation characteristics include carbon fibre fracture,133,215 reduction in the porosity,212 filler loss,133 thickness reduction,133 PTFE loss,133,216 and more cracking in the MPL.133
| Durability cases and the corresponding conditions | Items | Values | Ref. |
|---|---|---|---|
| NEDC near-water boiling temperature: operating temperature: 95 °C, inlet pressure (kPa) (anode/cathode = 280/260), duration: 100 h; | GDL thickness decreases | Anode: 1.5–10.7% | 133 |
| Cathode: 7.7–23.2%; | |||
| GDB graphitization degree decreases | Anode: 286.3% | ||
| Cathode: 493.2%; | |||
| MPL graphitization degree decreases | Anode: 2.0% | ||
| Cathode: 9.4%; |
Thermal softening describes the phenomenon in which there is a decrease in the storage elastic modulus of binder resins (phenolic resin, epoxy resin, cellulose acetate, etc.) at elevated temperatures,220–224 leading to a decrease in the compressive stiffness of the GDB layer. This also demonstrates the marked temperature dependence on the mechanical stiffness of PTFE at low temperatures. Indeed, the mechanical stiffness of pure PTFE sharply decreases as the temperature increases from −150 °C to 150 °C.225 This temperature-dependent behaviour of these adhesive resins and PTFE is the main cause of the reduction in compressive stiffness of the GDL at elevated temperatures. This softening effect results in more extensive and severe carbon fiber breakage within the GDB,215 as depicted in Fig. 16(a), accompanied by a reduction in the porosity.215 The localised fracture of carbon fibres may lead to the erosion of fillers within the GDB due to water and gas flushing,133 leading to the localised appearance of large pores, as illustrated in Fig. 16(b). The reduction in the GDL thickness (anode side: 1.5–10.7%, and cathode side: 7.7–23.2%) after a durability test at 95 °C may have been induced by the thermal softening,133 as shown in Fig. 16(c). However, the mechanism by which thermal softening affects filler loss and thickness reduction remains unclear. Regardless, the compressive resistance and mass transfer capability of the GDL are compromised at elevated temperatures, amplifying the differences in electrochemical reactions beneath the flow channels and ribs and exacerbating the in-plane non-uniformity of the reaction.
 | ||
| Fig. 16 Durability issues for the GDL at elevated temperatures. (a) SEM images of the uncompressed GDL and compressed GDL at 25 °C, 60 °C, and 90 °C.215 (b) Changes in pinhole distribution on the GDL surface,133 (c) thickness features of the GDL,133 and (d) changes in elemental content of GDL surfaces before and after elevated temperature testing.133 (e) Molecular structure change during the thermal oxidation process of PTFE.216 (f) Surface morphologies of MPL before and after elevated temperature testing.133 | ||
Thermal oxidation refers to the degradation of GDL materials resulting from their reaction with oxygen at elevated temperatures. Qu et al.216 reported that under an air atmosphere at 120 °C, PTFE undergoes thermal oxidation, resulting in a looser surface structure. This loosening was attributed to the degradation of molecular chains during thermal-oxidative aging, with the escape of small molecules, which weakened interactions between chains. The thermal oxidative aging mechanism is shown in Fig. 16(e). As thermal oxidation progresses, PTFE is gradually oxidised from its end groups, which releases volatile small molecules such as CF2O and decreases the PTFE content. PTFE loss was also observed after a 100 h, 95 °C New European Driving Cycle (NEDC) durability test, after which the loss of PTFE in the cathode was more pronounced than in the anode,133 as shown in Fig. 16(d). However, the contribution of thermal oxidation to PTFE loss at elevated temperatures is unclear, as PTFE may also detach due to erosion caused by the reactive flow.226 Additionally, carbon particles and the surfaces of carbon fibres can also undergo oxidation.127,133 Lu et al.133 reported significant GDB graphitization degree decreases of 286.3% at the anode side and 493.2% at the cathode side after a durability test at 95 °C. Similarly, the MPL graphitization degree also decreased, but not significantly, as presented in Table 9. Nevertheless, there are a few studies on the oxidation of these materials at elevated temperatures.
Overall, the reduction in the PTFE content or the oxidation of the GDL diminishes the hydrophobicity of the GDL,133 obstructing the liquid water drainage and increasing the risk of flooding. Lu et al.133 reported increased cracking in the MPL surface after a 100-hour, 95 °C NEDC durability test, as shown in Fig. 16(f). This was attributed to the water and gas transport, but the underlying mechanism was unclear. The more pronounced cracking, coupled with the hydrophilization of the surface, exacerbates the risk of water flooding. The degradation mechanisms of the mass transport properties of GDL at elevated temperatures warrant further investigation. Given the distinct compositions of the GDB and MPL, the degradation of the two components at elevated temperatures should not be generalised.
4.4. Impacts on performance loss
The degradation of MEA components induced by elevated temperatures results in substantial PEMFC performance loss. Fig. 17 presents the performance degradation characteristics of PEMFCs after various durability tests at elevated temperatures, including on-road heavy-duty driving conditions (Fig. 17(a)), NEDC cycling (Fig. 17(b)), in situ hygrothermal fatigue testing (Fig. 17(c)), constant operation at a high current density (Fig. 17(d)), voltage cycling (Fig. 17(e)), and temperature cycling (Fig. 17(f)). PEMFCs experience more severe performance degradation at elevated temperatures due to the harsher deterioration of the MEA. For instance, Ruiu et al.22 demonstrated that short-term temperature cycling between 90 °C and 120 °C caused more pronounced degradation in MEA components compared with long-term stable operation under automotive-relevant conditions at 80 °C. This resulted in a fivefold increase in the rate of irreversible performance loss of the PEMFC. Tables 10 and 11 summarise the performance degradation rates of PEMFCs at different temperatures and the dominant degradation mechanisms of the major MEA components at elevated temperatures, respectively. Other operating conditions also have varying impacts on the service life of the MEA at elevated temperatures. For instance, as shown in Case 10 of Table 10, when the operating temperature is set to 95 °C, the lifetime under 30% RH is only 82 h, which is significantly shorter than 168 h observed at 60% RH.207 The degradation of different MEA components has a profound impact on the performance loss. It is well-established that the output performance loss of PEMFCs is governed by different overpotential increments, including the leakage loss overpotential ηloss, the charge-transfer overpotential (ηact), the ohmic overpotential (ηohm), and the concentration overpotential (ηcon).230 Of these, ηloss is mainly affected by gas permeation and electronic short-circuiting. As illustrated in Fig. 18(a), elevated temperature can increase ηloss due to the PEM degradation due to increased gas crossover flow caused by enhanced phase separation, more severe cracking, pinholes, etc. Furthermore, increased Pt growth and dissolution, more severe carbon support chemical and electrochemical corrosion, and more severe distortion of the ionomer and the TPB structure at elevated temperatures are the main causes that further decrease the ECSA, leading to a higher ηact, as shown in Fig. 18(b). Additionally, the increased crystallinity of the hydrophobic region and decrease in the hydrophilic region in the ionomer phase within the CL and PEM at elevated temperatures further inhibit proton transport, stimulating an increased ηohm, as shown in Fig. 18(c). Moreover, the increased gas transport pathways induced by the increased Pt catalyst dissolution, more severe carbon support corrosion, the distortion of ionomer and pore structure in the CL, and reduced porosity of the GDL lead to an increased ηcon, as shown in Fig. 18(d). | ||
| Fig. 17 (a) Time percentages of operating current and power during the 1000 h on-road heavy-duty transportation with a coolant temperature of 92 °C and voltage reduction ratio at each current density.132 (b) NEDC durability protocol and polarization curves of the stack before and after the durability testing with a coolant temperature of 95 °C for 100 h.133 (c) In situ accelerated fatigue conditions of RH cycling: accelerated mechanical fatigue tests at the operating temperature of 85 °C and 95 °C, the OCV trends, and voltage decay rate after 500 h of RH cycling.167 (d) Polarization curves before and after 100 h of continuous operation at different temperatures.131 (e) Polarization curves after voltage cycling at different operating temperatures.147 (f) Stack temperature, power and voltage observed during 20 thermal cycles at 70 A.22 | ||
| Serial number | Durability cases and corresponding conditions (anode/cathode) | Items | Values | Degradation causes | Ref. |
|---|---|---|---|---|---|
| 1 | On-road heavy-duty transportation: operating temperature of 92 °C, duration: 1000 h; | Cell voltage loss | 7.34% @ 0 A cm−2 | 1. Pt catalyst growth | 132 |
| 47.69% @ 1.8 A cm−2 | 2. Pt catalyst loss | ||||
| 3. Carbon support corrosion | |||||
| 4. Ionomer migration and chemical degradation | |||||
| 2 | In situ hydrothermal cycling: operating temperature: 85 °C and 95 °C, inlet pressure (kPa): 80/60, gas type: H2/N2, dew point temperature: 90 °C, stoichiometric ratio: 1.5/2.5, duration: 500 h | Cell voltage loss | 85 °C | 1. Cracks within the CCM | 167 |
| 4.38% @ 0 A cm−2 | 2. Delamination between the CL and PEM | ||||
| 23.74% @ 2 A cm−2 | |||||
| 95 °C: | |||||
| 5.65% @ 0 A cm−2 | |||||
| 25.17% @ 2 A cm−2 | |||||
| 3 | NEDC near-water boiling temperature: operating temperature: 95 °C, inlet pressure (kPa): 280/260, duration: 100 h | Cell voltage loss | MEA#1: | 1. Polymer melting | 133 |
| 3.34% @ 0 A cm−2 | 2. Carbon support corrosion | ||||
| 1.07% @ 1.0 A cm−2 | 3. Aggregation and growth of Pt nanoparticles | ||||
| 10.85% @ 2.0 A cm−2 | 4. MEA structural breakdown | ||||
| MEA#2: | |||||
| 13.89% @ 0 A cm−2 | |||||
| 56.51% @ 0.4 A cm−2 | |||||
| MEA#3: | |||||
| 11.26% @ 0 A cm−2 | |||||
| 49.13% @ 0.4 A cm−2 | |||||
| MEA#4: | |||||
| 0.27% @ 0 A cm−2 | |||||
| 0.19% @ 1.0 A cm−2 | |||||
| 19.36% @ 2.0 A cm−2 | |||||
| MEA#5: | |||||
| 3.12% @ 0 A cm−2 | |||||
| 8.61% @ 1.0 A cm−2 | |||||
| 58.33% @ 2.0 A cm−2 | |||||
| 4 | A combined mechanical/chemical accelerated stress test: operating temperature: 90 °C, gas type: H2/air, back pressure: no duration: M – 8 for 1512 h, M – 12 for 1488 h; | Cell voltage loss | M – 8: | Cracks or pinholes within the PEM | 182 |
| 12.48% @ 0 A cm−2 | |||||
| M – 12: | |||||
| 8.39% @ 0 A cm−2 | |||||
| 5 | Load-cycle: operating temperature: 120 °C, gas type: H2/O2, stoichiometric ratio: 1/1.5, RH: 40%, range of potential: 0.6–1 V, PEM type: Nafion®, duration: 7 h; | Cell voltage loss | 16.57 μV h−1 @ 0.5 A cm−2 | 1. Hydrogen crossover increase through the PEM | 227 |
| 2. Pt catalyst growth | |||||
| 6 | Load-cycle: operating temperature: 120 °C, gas type: H2/O2, stoichiometric ratio: 1/1.5, RH: 40%, range of potential: 0.6–1 V, PEM type: AquivionTM, duration: 50 h | Cell voltage loss | 2.22 μV h−1 @ 0.5 A cm−2 | ||
| 7 | Voltage cycle: sample A: square wave (2 s), 0.4–0.95 V, 100% RH, 70 °C, 460 h, sample B: square wave (2 s), 0.6–0.95 V, 100% RH, 90 °C, 400 h; | Cell voltage loss | Sample A: | 1. Pt catalyst growth | 168 |
| 0.10% @ 0 A cm−2 | 2. Redistribution of Pt in the MEA | ||||
| 1.84% @ 1.5 A cm−2 | 3. Carbon support corrosion | ||||
| 6.11% @ 2.5 A cm−2 | |||||
| Sample B: | |||||
| 2.79% @ 0 A cm−2 | |||||
| 13.00% @ 1.5 A cm−2 | |||||
| 30.00% @ 2.5 A cm−2 | |||||
| 8 | Hydrothermal cycle under OCV: RH: 100%, operating temperature: 50 °C, 70 °C, and 90 °C duration: 100, 200, 500, and 1000 h | Cell voltage loss | 100 h, 0.6 A cm−2 | 1. Destroyed Nafion structure | 228 |
| 8.6% @ 50 °C | 2. Carbon support | ||||
| 4.7% @ 70 °C | 3. Catalyst aggregation | ||||
| 23.2% @ 90 °C; | |||||
| 200 h, 0.6 A cm−2: | |||||
| 14.5% @ 50 °C | |||||
| 7.7% @ 70 °C, | |||||
| 29.9% @ 90 °C; | |||||
| 500 h, 0.6 A cm−2: | |||||
| 23.3% @ 50 °C | |||||
| 14.7% @ 70 °C | |||||
| 38.8% @ 90 °C; | |||||
| 1000 h, 0.6 A cm−2: | |||||
| 32.2% @ 50 °C | |||||
| 21.9% @ 70 °C | |||||
| 48.1% @ 90 °C; | |||||
| 9 | Aging testing: operating temperature: 70 °C, 80 °C, 90 °C, 100 °C, and 120 °C; PEM types: sulfonated polyether therketone (SPEEK), Nafion® 212, Aquivion™ E790-03S, Aquivion™ E790-03S+ (Gen 2), Aquivion™ E790-03S TT | Lifetime | SPEEK | 1. PEM thinning | 200 |
| >2655 h @ 70 °C | 2. Delamination between the CL and PEM | ||||
| <1558 h @ 80 °C | 3. Pt catalyst dissolution | ||||
| Nafion® 212: | 4. CL cracking | ||||
| >2505 h @ 80 °C | |||||
| 685 h @ 90 °C; | |||||
| Aquivion™ E790-03S: | |||||
| >269 h @ 90 °C | |||||
| <107 h @ 100 °C | |||||
| Aquivion™ E790-03S+ (Gen 2): | |||||
| <200 h @ 90 °C | |||||
| <270 h @ 120 °C; | |||||
| Aquivion™ E790-03S TT: | |||||
| >269 h @ 100 °C | |||||
| >240 h @ 120 °C | |||||
| 10 | OCV hold accelerated stress test: back pressure (bar): 3/2.9, RH: 30% and 60%, operating temperature: 64 °C, 80 °C, and 95 °C | Lifetime | 1100 h @ 64 °C and 60% RH | PEM chemical degradation | 207 |
| 339 h @ 80 °C and 60% RH | |||||
| 168 h @ 95 °C and 60% RH | |||||
| 215 h @ 80 °C and 30% RH | |||||
| 82 h @ 95 °C and 30% RH | |||||
| 11 | In situ accelerated aging procedure: operating temperature: 64 °C, 80 °C, and 95 °C; PEM types: Nafion® 212, Nafion® 115 | Lifetime | Nafion® 212: | PEM degradation | 229 |
| >2820 h @ ≤80 °C | |||||
| <769 h @ 90 °C; | |||||
| Nafion® 115 | |||||
| >2820 h @ ≤90 °C | |||||
| 12 | Thermal cycling test: dynamic temperature cycles between 90 and 120 °C, cell numbers: 30, active cell area: 142 cm2, gas type: H2/air, stoichiometric ratio: 1.5/2.5 | Cell voltage loss | 714 μV h−1 cell−1 @ 70 A | 1. PEM mechanical degradation | 22 |
| 2. PEM chemical degradation | |||||
| 3. CL structural damage | |||||
| 3. CL ionomer degradation | |||||
| 4. Delamination between the CL and PEM |
| Degradation types | Degradation features | Degradation causes | |
|---|---|---|---|
| Pt catalyst degradation | Growth | Direct induction | (i) Entire elevated temperature range, reduced H2 atmosphere, the removal of surface oxides on Pt catalysts, and subsequent crystallite reconstruction and grain boundary diffusion |
| (ii) N2 atmosphere, 160 °C, Pt oxide reduction by the support | |||
| Acceleration assistance | Indeterminate | ||
| Dissolution | Acceleration assistance | (i) Endothermic Pt dissolution reaction | |
| (ii) Impact on the Pt catalyst collective surface tension | |||
| Carbon support corrosion | Chemical corrosion | Direct induction | (i) Environment containing oxygen or hydrogen (H2/O2) |
| (ii) Enhanced the reaction rate between the carbon matrix and oxygen or hydrogen species | |||
| (iii) Catalysis of the Pt catalyst | |||
| (iv) High RH accelerates | |||
| Ionomer degradation | Electrochemical corrosion | Acceleration assistance | (i) Intensified the carbon electrochemical corrosion sites |
| (ii) High RH accelerates | |||
| Ionomer nanoscale structure evolution | Direct induction | (i) Increased crystallinity of the hydrophobic region | |
| (ii) Shrank the hydrophilic region | |||
| (iii) Increased extent of phase separation | |||
| Mechanical fatigue | Acceleration assistance | Increased plastic strain induced by increased ionomer deformation change | |
| Chemical degradation | Acceleration assistance | Enhanced hydrogen peroxide formation rate | |
| CL structural collapse | Surface cracking | Acceleration assistance | (i) Increased in PEM deformation |
| (ii) Altered CL structural stability | |||
| Thickness decrease | Indeterminate | Compression effect, carbon corrosion, or other factors | |
| Pore structure collapse | Indeterminate | Ionomer migration or other factors | |
| TPB distortion | Indeterminate | Ionomer migration or other factors | |
| PEM degradation | Phase transitions in ion channels | Direct induction | Loss of water |
| Glass transition | Direct induction | Destabilization of the electrostatic network | |
| Internal domain structure evolution | Direct induction | (i) Altered crystallinity of the hydrophobic region | |
| (ii) Alerted proton conduction pathway | |||
| (ii) Increased extent of phase separation | |||
| Chemical degradation | Acceleration assistance | Enhanced hydrogen peroxide formation rate | |
| Creep deformation | Acceleration assistance | Increased molecular mobility especially exceeding the glass transition temperature | |
| Mechanical fatigue | Acceleration assistance | (i) Increased fatigue amplitude | |
| (ii) Reduced mechanical strength of the membrane | |||
| GDL degradation | Thermal softening | Direct induction | (i) Reduced storage elastic modulus of binder resins |
| (ii) Decreased mechanical stiffness of PTFE | |||
| Thermal oxidation | Direct induction | Oxidation of PTFE, carbon particles, and carbon fibre | |
| MPL cracking | Indeterminate | Water and gas transport, or other factors | |
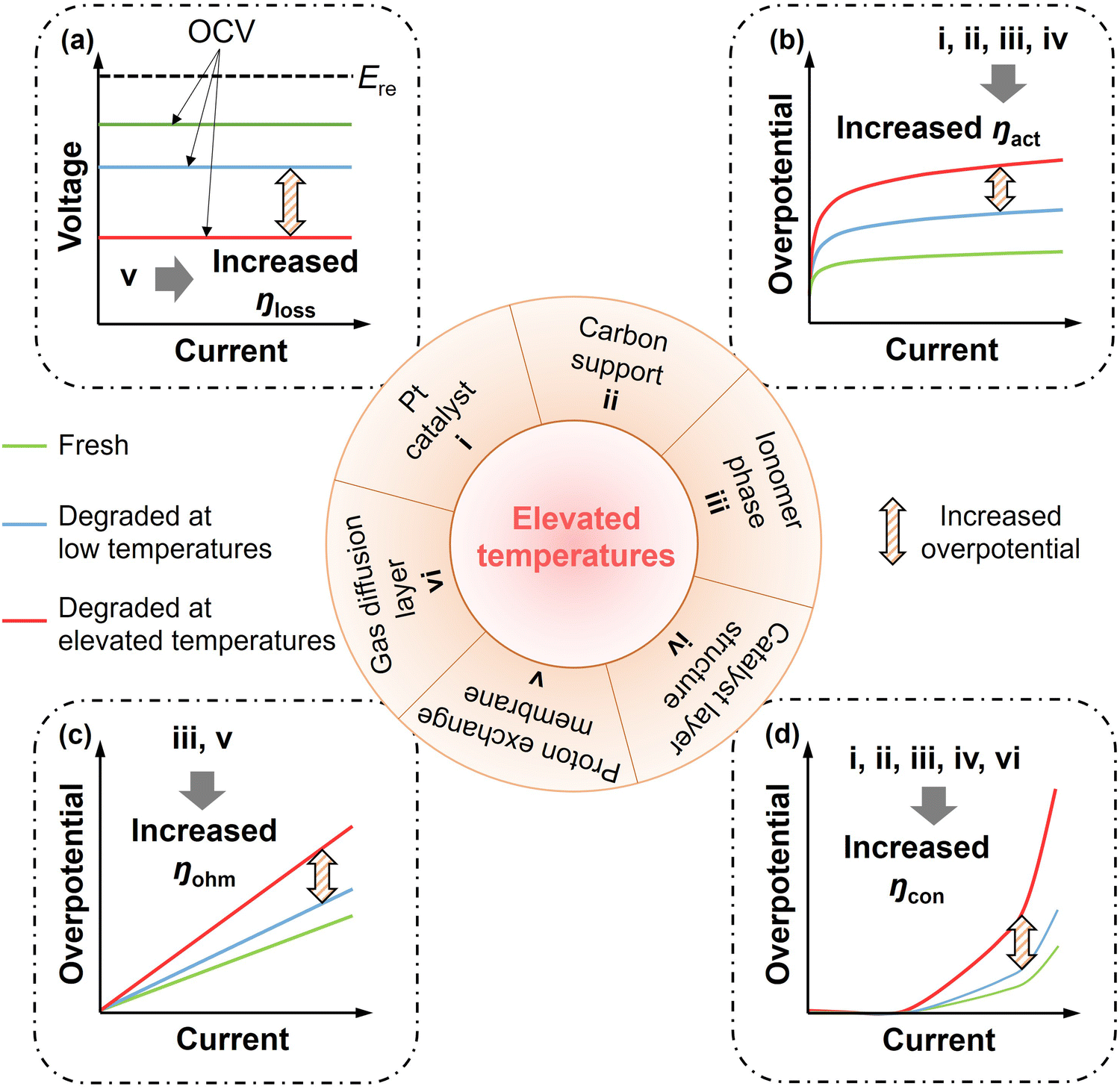 | ||
| Fig. 18 Impacts of MEA component durability on performance loss of PEMFCs after the durability test at different temperatures. | ||
Given the significant impact of elevated temperatures on the degradation of MEA components, it is crucial to recognise and address the associated performance losses in PEMFCs. Establishing a quantifiable relationship between component degradation and performance decline is necessary to optimise the PEMFC durability at elevated temperatures. Because these degradation processes are exacerbated at elevated temperatures, maintaining optimal operating conditions and developing materials with enhanced thermal stability are essential for improving the long-term performance and reliability of a PEMFC.
5. Progress in overcoming the elevated temperature limitations
5.1. Development of PEMs with high proton conductivity at elevated temperatures
As previously discussed, when a MEA operates at elevated temperatures, particularly in the superheated (H) regime, the ionomer phase undergoes significant dehydration, thereby severely hindering proton transport. Concurrently, the substantial expulsion of water vapour markedly reduces the internal RH level within the MEA, resulting in a pronounced decline in the overall performance of the PEMFC. To overcome these performance losses associated with elevated temperature operation, considerable efforts have been devoted to developing new proton conductors in low RH and elevated temperature environments. To date, research progress in this field can be broadly classified into three approaches: (1) composite PFSA membranes with water-retentive fillers; (2) composite PFSA membranes doped with fillers with a high sulfonate concentration; and (3) modified PFSA membranes with additional proton conduction pathways.For route (1), researchers have introduced inorganic fillers such as SiO2, TiO2, and CeO2 into electrolyte membranes using conventional casting and solvent evapouration methods. By enhancing the water retention of the membrane at elevated temperatures, these fillers effectively improve the proton conductivity of the PEM in elevated temperature environments.235–238 However, the overall enhancement in proton conductivity is limited due to the disruption of continuous proton transport pathways and the agglomeration of inorganic fillers within the polymer matrix. To address the above issues, Nam et al.231 uniformly patterned mesoporous TiO2 microporous plates (PTMPs) and embedded them onto the anode-facing surface of the Nafion membrane by employing microporous templates and an ionomer spraying technique, as shown in Fig. 19(a). This innovative approach protected the proton conduction pathways by precisely positioning TiO2 microporous plates on the membrane surface while eliminating agglomeration typically associated with the introduction of hygroscopic inorganic fillers. As a result, the power density was more than 35.2% higher than that of the commercial Nafion 211 membrane at 120 °C and 35% RH, with a 24.5% reduction in the ohmic resistance. The hygroscopic mesoporous PTMPs embedded within the membrane attracted water molecules at elevated temperatures (>100 °C), helping mitigate the reduction in proton conductivity caused by membrane dehydration. Highly proton-conductive nanocomposite membranes were synthesised by incorporating surface-modified silica nanoparticles bearing different acidic functional groups (phosphonate or sulfonate ester groups) in the Nafion matrix,232 as shown in Fig. 19(b). The short oligomeric corona containing phosphonate or sulfonate functional groups facilitated the formation of non-aggregated, discrete nanoparticles that synergistically interacted with the polymer matrix to form a membrane with significantly enhanced proton conductivity. The sulfonate-based nanocomposite exhibited a higher proton conductivity of 0.045 S cm−1 at 130 °C and 30% RH. Nicotera et al.239 also integrated a Nafion matrix with TiO2 nanoparticles directly grown on the surface of multi-walled carbon nanotubes (MWCNTs) to fabricate a Nafion/MWCNTs–TiO2 composite membrane. This synthesis approach enabled the nanoscale dispersion of TiO2 within the host matrix, thereby contributing to superior water-retention capability. The elongated carbon nanotubes provided additional pathways for proton conduction, and the assembled H2/air single cell (with an active electrode area of 5 cm2) achieved a power output of 307.7 mW cm−2 when operated at 120 °C and 30% RH.
 | ||
| Fig. 19 (a) Schematic illustrations of the fabrication process of PTMPs embedded in the Nafion® membrane.231 (b) Schematic illustration of the synthesis of functionalized nanoparticles and the final nanocomposite membranes.232 (c) Proton conduction mechanism of pristine Nafion and HBM Nafion at different humidities.233 (d) Schematic illustration of enhanced proton transportation in Su-CNTs/Nafion membranes with proton highways along Su-CNTs.12 (e) Schematic diagram of the synthesis process and proton conduction for the PFSNPA membrane.234 | ||
For route (2), doping polymers with high sulfonic acid group density or functionalised carbon nanomaterials can be used to form a dense network of proton transport sites and bridging pathways within a membrane. For instance, Xu et al.233 synthesised a hyperbranched polyamide (HBM) featuring a high concentration of sulfonic acid groups and a nanoparticle morphology and incorporated it into a Nafion matrix without structural damage, as depicted in Fig. 19(c). The HBM was designed to imitate ion clusters and accelerate proton transport at elevated temperatures. The carboxyl groups of the HBM formed strong hydrogen bonds with the sulfonate groups of the Nafion matrix, thereby enhancing its structural compatibility and proton transport. As a result, the composite membrane exhibited a proton conductivity of 0.047 S cm−1 at 110 °C and 60% RH, which was 1.9 times higher than that of pristine Nafion. Furthermore, the corresponding power density increased by 30%, demonstrating the feasibility of this approach for extending the operating temperature range of PFSA-based fuel cells. Yin et al.12 prepared a composite membrane by incorporating sulfonated carbon nanotubes (Su-CNTs) into a Nafion matrix, as illustrated in Fig. 19(d). This significantly changed the membrane's microstructure, leading to the formation of numerous proton transport pathways, particularly under elevated temperature and low RH conditions. The carbon nanotubes bridged isolated hydrated clusters, thereby greatly enhancing proton transport efficiency, giving the Su-CNTs/Nafion composite membrane a proton conductivity of ∼0.03 S cm−1 at 130 °C and 50% RH, whereas that of the pristine Nafion remained below 0.01 S cm−1 under the same conditions.
For route (3), Liao et al.234 designed a novel PEM by precisely engineering the side-chain chemical structure of perfluorosulfonyl fluoride by introducing sulfonyl imide and phosphonic acid functional groups, as illustrated in Fig. 19(e). The sulfonyl imide groups exhibited super-acidic properties, while the phosphonic acid groups served as both proton donors and acceptors, and these two functional groups synergistically established unique and additional proton transport channels. Strong interactions between the acidic groups and water molecules enhanced water retention and molecular diffusion, thereby significantly improving the proton conductivity. The resulting perfluorosulfonimide-phosphonic acid (PFSNPA) membrane demonstrated a conductivity of 263 mS cm−1 at 120 °C, and in H2/air fuel cell tests, it delivered a power density of 1076.1 mW cm−2 at 120 °C and 40% RH.
5.2. Optimization of operational strategies
To address the significant reduction in reactant partial pressures at elevated temperatures, along with the pronounced dehydration of the ionomer phase within the MEA, particularly in low-RH environments, researchers have enhanced the performance of PEMFCs at elevated temperatures by solely optimizing operating strategies, without needing to develop new materials. As shown in Fig. 20(a), Wang et al.20 reported that increasing the operating pressure enhances the output voltage of PEMFCs by 50–150 mV, and this improvement was attributed to the higher oxygen concentration and improved membrane hydration. They found that a higher pressure reduced the heat generation, resulting in a 0.6–2 °C decrease in the temperature of the CCL, which also facilitated the accumulation and transport of water vapour and liquid water via CD flow. A higher operating pressure also helped minimise the mismatch in response rates between the ORR and the transport of oxygen and protons. Especially under higher loads, the Damköhler numbers of both oxygen and hydrogen decrease by up to 40% and 54%, respectively. Yang et al.8 also indicated that increases in the operating temperature must be accompanied by an increase in the operating pressure. As illustrated in Fig. 20(b), Lu et al.21 demonstrated that to mitigate the impact of elevated temperatures on the saturated vapour pressure, increasing the inlet pressure of the fuel cell stack enhances both the steady-state and dynamic performance. Nevertheless, increasing the pressure is not a perfect solution, with Lu et al.21 demonstrating that αNMD also increased with both temperature and pressure, making it challenging to mitigate ionomer dehydration on the anode side. Although increasing the cathode-side humidity can enhance back-diffusion and help alleviate excessive membrane drying at the anode, it simultaneously reduces the oxygen partial pressure. Furthermore, dynamic durability often deteriorates under a high pressure, resulting in limited performance improvements, and the associated trade-offs, particularly the potential reduction in fuel cell lifespan, have not been adequately addressed. Shao et al.105 developed an optimised sequence for current loading and temperature elevation during PEMFC operation at elevated temperatures, as illustrated in Fig. 20(c). By first increasing the current before raising the temperature, they utilised the self-humidification effect on the cathode side, which enabled a more uniform in-plane distribution of both current density and membrane hydration, even in dry environments. | ||
| Fig. 20 (a) Schematic of operating pressure (OP) impacts on local mass transfer at higher loads: lower OP and higher OP.20 (b) Schematic of the PEMFC performance improvement at elevated temperatures by increasing the inlet pressure.21 (c) Schematic of the performance loss and recovery mechanism, where the ‘undesirable’ and ‘desirable’ water distributions are shown on the left and right respectively.105 | ||
5.3. Design of materials with enhanced durability at elevated temperatures
At elevated temperatures, catalyst growth and dissolution, carbon support corrosion, and the pronounced degradation of ionomers near or above their glass transition temperature collectively underscore the need to develop MEA components with enhanced thermal stability. As shown in Fig. 21(a), the development of stable core–shell spatially confined structures, the enhancement of interactions between Pt nanoparticles and carbon supports, and the use of thermally corrosion-resistant graphitised carbon supports can improve the thermal stability of Pt/C catalysts.9,244 Ji et al.240 leveraged the corrosion resistance of TiO2 and the strong metal–support interactions between Pt nanoparticles and TiO2 nanofibers, in combination with the high electrochemical conductivity of carbon nanofibers (CNFs), to synthesise a composite electrode consisting of CNT-wrapped Pt/TiO2 nanofibers. This design mitigated Pt dissolution and exhibited remarkable durability even at 120 °C and 40% RH. Zhang et al.245 also exploited the thermal stability of TiO2 and introduced a nanolayer composed of TiO2 nanoparticles between the Pt catalyst and the carbon support, which protected the carbon from corrosion and established strong metal–support interactions between Pt and TiO2. After an AST at 150 °C, the maximum power density of the Pt/C@ TiO2-MEA decreased by only 8.6%, which was significantly lower than the 23.9% reduction observed for the conventional Pt/C-MEA. Therefore, TiO2 may serve as an alternative support material at elevated temperatures due to its stability, but its inherently low electrical conductivity remains a critical limitation. One potential strategy to overcome this issue is to form composites with highly conductive materials. | ||
| Fig. 21 (a) Mitigation strategies for catalyst and support degradation at elevated temperatures.9,240 (b) Open framework ionomer with enhanced durability at elevated temperatures.241 (c) Humidity-induced stress of the as-received Nafion 211 membrane and annealed Nafion 211 membranes.202 (d) Three-dimensional structure scheme of the SSC-PFSA membrane with high durability at elevated temperatures.242 (e) Improved durability of short-side-chain perfluorinated polymer electrolyte membranes by the annealing process.185 (f) High-temperature stability of hydrocarbon-based Pemion® proton exchange membranes.243 | ||
Research on optimizing the thermal stability of ionomer materials in the CL is limited, but Yang et al.241 developed a novel α-aminoketone-linked covalent-organic framework (COF) ionomer and then interweaved it with Nafion to function as “breathable” proton conductors. As depicted in Fig. 21(b), this innovative proton conductor retained 54% of its rated current density after 30k AST cycles at 105 °C, outperforming the 44% retention rate of Nafion ionomers. Further research is needed to enhance the thermal stability of ionomers within the CL because they play a pivotal role in facilitating electrochemical reactions and governing mass and proton transport throughout the CL.
To enhance the thermal stability of PEMs, existing research has primarily focused on three optimization strategies: (1) increasing the PEM crystallinity through thermal treatment; (2) employing short side-chain PEMs; and (3) incorporating thermally stable aromatic rings into the polymer backbone. While these approaches differ, they all help increase the glass-transition temperature of the membrane. Li et al.202 reported that annealing Nafion membranes in the range of 120–160 °C reduced the stress induced by humidity variations, as shown in Fig. 21(c). In addition, short side chain (SSC) PFSA membranes exhibited superior thermal stability compared with Nafion membranes due to their higher glass-transition temperatures.206 As depicted in Fig. 21(d), Guan et al.242 optimised the structure of SSC-PFSA membranes to form an internal “stream-reservoir” morphology, in which the hydrophobic domains served as physical crosslinking centers to maintain a high mechanical strength. As a result, the SSC-PFSA membranes are considered ideal candidates for high-power-density and heavy-duty fuel cells that typically operate in elevated temperature and low-humidity environments. Moreover, Shin et al.185 fabricated physically-reinforced SSC-PFSA membranes via annealing and incorporating porous substrates, which demonstrated significantly improved durability, as shown in Fig. 21(e). Specifically, a 3M 729 reinforced composite membrane annealed at 200 °C withstood 16600 cycles, markedly outperforming its non-annealed counterpart. Furthermore, Mirfarsi et al.243 indicated that the Pemion membrane, a commercially available mechanically reinforced sulfo-phenylated polyphenylene-based PEM, exhibited superior thermo-mechanical stability compared with a reinforced PFSA membrane in a temperature range of 30–120 °C and RH range of 10–90% RH, as illustrated in Fig. 21(f). The Young's modulus and strain hardening of the Pemion membrane remained largely insensitive to temperature, whereas the reinforced PFSA membrane exhibited substantial degradation in its mechanical properties above 90 °C. The Pemion membrane maintained an acceptable yield resistance across the entire test range, while the reinforced PFSA underwent spontaneous yielding above 110 °C under relatively small mechanical stress (e.g., 1 MPa). The pronounced stability of the Pemion membrane was primarily attributed to its rigid-rod polyphenylene backbone, which reduced its segmental mobility, resulting in a high glass transition temperature of 161 °C.
5.4. Diagnosis technique advancements
To gain deeper insights into the structural evolution of MEA during degradation, considerable efforts have recently been devoted to developing advanced characterization techniques. Key areas of progress include non-destructive spectroscopic analysis methods, novel electron microscopy, electrochemical analysis techniques based on impedance–relaxation time correlations, and non-destructive magnetic alignment-assisted imaging approaches.In terms of non-destructive spectroscopic analysis methods, Janssen et al.246 combined in situ small-angle and wide-angle X-ray scattering (SAXS and WAXS) to establish a correlation between the structural parameters of the high-activity skeletal PtCo catalyst and its degradation during start-up/shutdown (SU/SD) cycling, as shown in Fig. 22(a). This integrated approach enabled the detailed in situ monitoring of structural changes within each crystalline phase and revealed two primary degradation mechanisms of the catalyst under SU/SD conditions: Co dissolution and crystallite growth. Bogar et al.252 developed a SAXS-based analytical method coupled with electrochemical characterization to resolve the microstructural features of all functional layers within a MEA, including the ionomer, catalyst support, Pt nanoparticles, and the GDL. The Voigt peak was used to represent the ionomer peak and the matrix inflection point associated with the PEM. The Vulcan carbon support was modeled using the DAB model, while the CL was characterised by a population of Schulz-distributed spheres that aggregated into a mass fractal network that corresponded to Pt nanoparticle clusters. The GDL was represented using a power-law function to account for its larger structural features. Chen et al.253 employed synchrotron-based X-ray fluorescence (XRF) and X-ray absorption spectroscopy (XAS) to investigate the catalyst migration mechanism. A carefully designed MEA cross-sectional exposure method was developed by optimizing a PTFE gasket and minimizing interference through an integrated X-ray window. This configuration enabled the direct and in-depth observation of the internal of a fuel cell, facilitating in situ characterization. Interestingly, Li et al.254 designed a fuel cell compatible with operando XAS to investigate the structural evolution of manganese spinel oxide electrocatalysts in anion-exchange membrane fuel cells (AEMFCs). Notably, ionomer membranes may undergo beam-induced damage caused by X-rays, neutrons, or electron beams, thereby leading to morphological or chemical changes. Yu et al.247 demonstrated a novel approach for distinguishing ionomer phases from carbon supports in a PEMFC by using carbon signals from scanning transmission electron microscopy coupled with electron energy loss spectroscopy (STEM-EELS), as presented in Fig. 22(b). This method enabled the high-spatial-resolution mapping of both ionomer distributions and carbon supports. Although electron beam irradiation may damage samples, the study minimised such damage by carefully controlling the electron dose, even though it was not entirely eliminated. As shown in Fig. 22(c), Chabot et al.248 employed small-angle neutron scattering (SANS) to investigate the nanoscale structural features of fuel cell electrodes, including the size distribution of Pt nanoparticles, the morphology of 2–3 nm thick ionomer films coating the catalyst surface, and the specific locations of water uptake within an electrode. This work highlighted the unique capability of SANS for resolving multiphase interfacial structures and water distribution in complex electrode architectures.
 | ||
| Fig. 22 (a) In situ SAXS & WAXS techniques monitoring the morphological changes of catalysts.246 (b) Identification of ionomers in PEMFCs by EELS.247 (c) Structural information details of the three-phase boundary accessible from the SANS profile of MEA.248 (d) Schematic illustration of identical location transmission electron microscopy.249 (e) DRT technology decoupling of polarization process of PEMFCs.250 (f) Schematics of a designed magnetic array system and a working diagram of a magnetic array.251 | ||
In terms of novel electron microscopy techniques, identical-location transmission electron microscopy (IL-TEM) is a powerful technique for tracking structural evolution. Shokhen et al.249 employed IL-TEM imaging based on ionic liquids to monitor the atomic-scale degradation of the top surface of a CL in an actual PEMFC. As shown in Fig. 22(d), this method was used to precisely track the same location over multiple imaging sessions, providing direct insight into nanoscale structural changes during degradation. Strandberg et al. combined identical-location scanning electron microscopy (IL-SEM) and IL-TEM to investigate the degradation of the cathode Pt/C catalyst layer in an actual operating PEMFC. Under SU/SD conditions, IL-SEM was used to observe the initiation and propagation of cracks within the electrode layer. Nanoscale IL-TEM imaging also showed an ∼20% reduction in the average diameter of the primary carbon support particles, accompanied by an ∼63% increase in platinum nanoparticle size due to their agglomeration and growth. These morphological changes corresponded to a 65% loss in the ECSA. The study identified the corrosion of structurally vulnerable regions of the carbon support as the key factor leading to catalyst layer collapse. Soleymani et al.255 developed an epoxy-free ultramicrotomy technique that, when combined with TEM, enabled the first direct visualization of an ionomer network, carbon particle morphology, and Pt nanoparticle distribution. This method also provided a detailed observation of structural changes during degradation.
The distribution of relaxation time (DRT) analysis based on electrochemical impedance spectroscopy allows for the in situ and non-destructive tracking of the evolution of characteristic peaks associated with different reactions and transport processes within a MEA. This provides insights into the degradation mechanisms of the internal components,250,256 as shown in Fig. 22(e). Interestingly, Sun et al.251 developed a magnetic array imaging system based on magnetic sensing principles to detect variations in external magnetic fields induced by internal current changes to visualise the heterogeneity of degradation in PEMFCs, as presented in Fig. 22(f). This technique was non-destructive, easy to implement, and did not require any modification of the tested structures or materials. It has particular advantages for characterizing spatial heterogeneities in structural degradation within fuel cell stacks, but its accuracy depends on eliminating interference from magnetic contaminants adhered to the PEMFC.
5.5. Exploration of temperature effects on the MEA of anion exchange membrane water electrolyzers
Anion-exchange membrane water electrolyzers (AEMWEs) use the anion-exchange membrane (AEM) to conduct OH− ions and separate hydrogen and oxygen, as illustrated in Fig. 23(a). Their structural configuration is similar to that of PEMFCs, with both systems based on MEAs, but the materials differ. Given the significant influence of elevated temperatures on a PEMFC's performance, this section briefly explores how temperature affects the efficiency and stability of MEA operation in an AEMWE. Table 12 summarises the performance and durability data of AEMWEs at different temperatures. | ||
| Fig. 23 (a) Schematic diagram of the AEMWE.257 (b) Polarization curves for AEMWE on the CCM configuration at different temperatures.258 (c) Stability evaluations at 500 mA cm−2 in 2 M KOH at 60 °C and 80 °C and 1H NMR spectra of the Aemion membrane before and after long-term stability evaluation in the electrolysis cell.259 | ||
| Serial number | MEA configurations | Items | Values | Ref. |
|---|---|---|---|---|
| 1 | FAA3-50 membrane, IrO2 (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed); | Performance | At 1.8 V | 258 |
| 0.6 A cm−2 @ 60 °C | ||||
| 0.9 A cm−2 @ 70 °C | ||||
| 1.05 A cm−2 @ 80 °C | ||||
| At 2 V: | ||||
| 1.2 A cm−2 @ 60 °C | ||||
| 1.7 A cm−2 @ 70 °C | ||||
| 2.1 A cm−2 @ 80 °C | ||||
| 2 | FAA3-50 membrane, IrO2 (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed); | Performance | At 1.8 V | 260 |
| 0.73 A cm−2 @ 50 °C | ||||
| 1.15 A cm−2 @ 70 °C | ||||
| At 2 V | ||||
| 1.42 A cm−2 @ 50 °C, 1.83 A cm−2 @ 70 °C | ||||
| 3 | FAA-3-PK-75 membrane, IrO2 (anode catalyst), Pt/C (cathode catalyst), 0.5 M KOH (feed); | Performance | At 1.8 V | 261 |
| 0.46 A cm−2 @ 50 °C | ||||
| 0.607 A cm−2 @ 60 °C | ||||
| 0.625 A cm−2 @ 70 °C | ||||
| 0.87 A cm−2 @ 80 °C | ||||
| 0.983 A cm−2 @ 90 °C | ||||
| 4 | Fumatech FAA-3-50 membrane, IrO2 (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed) | Performance | At 2 V | 262 |
| 3.5 A cm−2 @ 80 °C | ||||
| Durability | 50 h | |||
| 5 | Aemion membrane, NiFe2O4 (anode catalyst), RANEY® nickel (cathode catalyst), 2 M KOH (feed) | Durability | 0.34 mV h−1 @ 60 °C | 259 |
| 0.44 mV h−1 @ 80 °C | ||||
| 6 | A201 membrane, IrO2 (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed) | Performance | At 1.8 V | 263 |
| 0.399 A cm−2 @ 50 °C; | ||||
| Durability | 500 h | |||
| 7 | AF1-HNN8-50 membrane, NiS2/Ni3S4 (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed) | Performance | At 2.0 V | 264 |
| 1.8 A @ 60 °C | ||||
| Durability | 120 mV kh−1 @ 60 °C | |||
| 8 | PBPA membrane, NiFe2O4-X (anode catalyst), Pt/C (cathode catalyst), 1 M KOH (feed) | Performance | At 1.77 V | 265 |
| 2.0 A @ 80 °C | ||||
| Durability | 240 mV kh−1 @ 80 °C |
In AEMWE systems, the operating temperature is typically limited to below 100 °C and in the range of 50–90 °C. Temperature is a critical operational parameter, where a higher temperature leads to a lower thermodynamic equilibrium potential, enhanced electrochemical reaction kinetics, and improved ionic conductivity, all of which further improve the output performance, as shown in Table 12. Lim et al.261 systematically investigated the influence of temperature within the 50–90 °C range and reported that under a constant overpotential of 591 mV, the current density increased by approximately 50 mA cm−2 for every 10 °C increase. This enhancement was attributed to improved ionic conductivity in the membrane, ionomer, and electrolyte, as well as accelerated electrochemical kinetics. As shown in Fig. 22(b), raising the cell temperature to 90 °C consistently resulted in higher current densities due to improved oxygen and hydrogen evolution reaction kinetics and elevated ionic conductivity.258 However, further increasing the temperature to 100 °C caused the performance to decline due to an increase in series resistance (RS) from 0.09 Ω cm2 at 90 °C to 0.10 Ω cm2 at 100 °C and a rise in charge-transfer resistance (Rct) from 0.095 Ω cm2 to 0.105 Ω cm2. These increases indicate a deterioration of the interface between the PEM and the CL due to dehydration at 100 °C. Therefore, there is an optimal operating temperature range for AEMWEs, and avoiding operation near the evapouration temperature of water is crucial to maintaining their performance and interfacial stability.
Temperature also significantly impacts the stability of MEAs, especially AEMs, as presented in Table 12. Lin et al.266 evaluated the stability of AEMs by immersing membrane samples in aqueous KOH solutions at 60 °C and 80 °C and monitoring the hydroxide ion conductivity over time. After 480 h, the hydroxide conductivity decreased by 6.7% at 60 °C and by 11.1% at 80 °C, indicating that higher temperatures accelerated membrane degradation. Furthermore, as shown in Fig. 22(c), the performance of Aemion cells significantly declined over time, and the degradation rate at 80 °C (0.44 mV h−1) was higher than that at 60 °C (0.34 mV h−1). 1H NMR analysis of membranes before and after degradation revealed detectable structural changes, further confirming that higher temperatures accelerated polymer degradation, leading to faster performance losses.
6. Conclusions and perspectives
Elevated temperatures within MEAs have become a significant focus of research to advance PEMFCs, but current methodologies face significant challenges in ensuring the stable, long-term operation of PEMFCs at elevated temperatures. These limitations include difficulties in mass transfer, charge conduction, electrochemical reactions, component degradation, and water and gas management within the MEA. This review provided a comprehensive overview of the challenges associated with ensuring stable MEAs at elevated temperatures and highlighted key gaps and potential future directions (Fig. 24). | ||
| Fig. 24 Approaches to optimizing MEA performance and longevity at elevated temperatures. | ||
(1) Study on the correlation between the microscale and macroscale thermal environments. At different elevated temperature levels, the transport and degradation of MEA components exhibit different characteristics. A fundamental prerequisite for clarifying these mechanisms is to accurately characterise the thermal environment experienced by each functional layer and its internal nanoscale constituents. The CL, whose nanoscale structure is comprised of a Pt catalyst, carbon support, and ionomer, is highly sensitive to local thermal conditions that directly affect electrochemical reaction kinetics, mass transport pathways, and material durability. However, existing temperature sensing techniques, such as thin-film microsensors,267 only provide localised average temperatures at the CL surface and cannot resolve heterogeneous thermal fields within the nanostructured interior. To address this, systematic investigations into the multi-scale heat transfer mechanisms within MEAs are needed. Specifically, an understanding of the thermal conduction across functional layers and at interfaces between nanoscale materials is essential. Establishing a quantitative correlation between microscopic thermal environments and macroscopic operating temperatures will enable the precise determination of the actual elevated temperatures experienced by MEA components in various operating scenarios.
(2) Development of ionomers with high proton conductivity and anti-dehydration optimization. At elevated temperatures, particularly in the super-boiling regime (H), the ionomer undergoes substantial dehydration, which significantly hinders proton transport, especially on the anode side. To address this, it is essential to elucidate the equilibrium mechanisms governing the interconversion between liquid water, membranous water, and water vapour. Special attention should be given to identifying the key structural features that dictate dehydration behaviour in nanoscale ionomer films. It is also necessary to establish quantitative correlations with high-frequency resistance to enable the real-time assessment of an ionomer's hydration state. From a material innovation perspective, the design of microporous architectures that promote localised condensation, along with highly water-retentive and multi-channel fast proton-conducting ionomer phases, is a promising approach. In terms of optimizing operation, the utilization of cathode-generated water and enhancing the cathode-anode humidity gradient improve the back-diffusion of water, thereby helping replenish water at the anode. Optimizing the control between current density and temperature rise is critical for mitigating severe ionomer dehydration at elevated temperatures.
(3) Optimization of cathode gas and water vapour management: at elevated temperatures, excess water evapouration is the primary cause of reduced reactant gas partial pressures. On the cathode side, although humidification can alleviate ionomer dehydration, it also lowers the partial pressure of reactant gases. Therefore, it is crucial to identify the minimum flow channel pressure threshold required to ensure an adequate gas supply, but maintaining this threshold often necessitates a high operating pressure for PEMFCs at elevated temperatures, which may compromise durability under dynamic conditions. This highlights the need for advanced pressure control techniques to manage the anode–cathode pressure differences. The development of MEA porous networks with low mass transport resistance and the optimization of flow field designs are essential to ensure rapid and efficient delivery of reactant gases at elevated temperatures.
(4) Study on MEA coupled degradation mechanisms and mitigation strategies: the degradation of MEA components is markedly accelerated at elevated temperatures, which includes the growth and dissolution of Pt catalysts, the corrosion of carbon supports, and the deterioration of both the ionomer and GDL. Within the CL and PEM, the ionomer phase undergoes significant structural changes in its internal chain and domain structures, which cause increased mechanical fatigue and chemical decomposition. The GDL is similarly susceptible to thermal softening and oxidative degradation. Notably, the coupled degradation mechanisms of Pt catalysts at elevated temperatures and varying electrochemical potentials remain insufficiently understood. While elevated temperatures induced glass transitions have been confirmed in PEMs, the glass transition of ionomers confined within the CL remains poorly characterised due to their nanoscale thickness and heterogeneous morphologies. The coupled chemo-mechanical degradation of ionomers near or above their glass transition temperatures also remains largely unexplored. Furthermore, the implications of increased molecular mobility on the deformation and adhesion properties of ionomers warrant in-depth investigations, as these factors are critical for understanding the structural collapse of the CL, where ionomers serve as functional binders. Humidity and pressure also play pivotal roles in the degradation of both CL and PEM materials and should be systematically examined. For the GDL, the long-term thermal stability of bonding resins, hydrophobic PTFE, and the MPL needs to be thoroughly investigated. Although the MPL shows more pronounced cracking at elevated temperatures, the increased water vapour fraction reduces its ability to uniformly expel liquid water. Removing the MPL from the GDL structure provides an option for the next-generation design of MEA operating at elevated temperatures to simplify the fabrication, reduce ohmic losses, and enhance the overall durability of MEA components. Importantly, the individual contributions of each MEA component to performance degradation at elevated temperatures are not yet clearly delineated, complicating efforts to identify the primary degradation-limiting factor. Establishing a quantitative relationship between component degradation and cell performance loss is essential for guiding the rational design of PEMFCs with enhanced durability during operation at elevated temperatures.
In terms of material innovations, enhancing the catalyst durability at elevated temperatures necessitates the development of thermally stable catalysts with spatial confinement, strong metal–support interactions, and corrosion-resistant supports (graphitised carbon, TiO2, etc.). In parallel, the development of ionomer materials with low segmental mobility (i.e., high glass transition temperatures) is essential for maintaining mechanical integrity and proton conductivity at elevated temperatures. Strategies such as appropriate thermal treatment, short side-chain structures, or the incorporation of aromatic rings into the polymer backbone can enhance the structural stability of ionomers. However, attention must be paid to preserving the proton conduction channels. Regarding the GDL, future efforts should elucidate the underlying degradation mechanisms in elevated temperature environments to inform the development of targeted durability enhancement strategies.
(5) Development of in situ characterization techniques: high-resolution characterization of MEAs, particularly within the CL, is critical to unravel the complex degradation mechanisms associated with the TPB and porous transport structures. However, conventional techniques face several intrinsic limitations. Most techniques are destructive, providing only ex situ post-mortem analysis with limited relevance to real-time degradation dynamics. The nanoscale dimensions of CL components demand exceptionally high spatial resolution, and the carbonaceous nature of both supports and ionomers hampers clear distinction due to low material contrast. Ionomer structures are often vulnerable to damage or changes under high-energy particle beams, which compromises the reliability of observations. These challenges highlight the need to develop in situ and non-destructive multi-dimensional characterization techniques. Promising directions include non-destructive in situ spectroscopic methods, integrating electron microscopy with optimised sample preparation protocols and visualization techniques based on electrical, magnetic, or acoustic principles. Such integrated approaches may provide deeper insights into the dynamic structural evolution and degradation pathways of MEA components under realistic operating conditions.
Author contributions
Caizheng Yue: writing – original draft, investigation, visualization, formal analysis; Weibo Zheng: writing – reviewing and editing, supervision, funding acquisition; Qiuya Wang: investigation, formal analysis; Zhendong Wang: investigation; Bing Li: supervision. Cunman Zhang: supervision; and Pingwen Ming: writing – reviewing and editing, investigation, supervision, funding acquisition.Data availability
Data will be made available upon request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFB2504201) and National Natural Science Foundation of China (no. 52276210 and 52106255).References
- X. Lü, Y. Qu, Y. Wang, C. Qin and G. Liu, Energy Convers. Manage., 2018, 171, 1273–1291 CrossRef.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- M. A. Sadeghi, Z. A. Khan, M. Agnaou, L. Hu, S. Litster, A. Kongkanand, E. Padgett, D. A. Muller, T. Friscic and J. Gostick, Appl. Energy, 2024, 353, 122004 CrossRef CAS.
- T. Lochner, R. M. Kluge, J. Fichtner, H. A. El-Sayed, B. Garlyyev and A. S. Bandarenka, ChemElectroChem, 2020, 7, 3545–3568 CrossRef CAS.
- J. Xu, C. Zhang, Z. Wan, X. Chen, S. H. Chan and Z. Tu, Renewable Sustainable Energy Rev., 2022, 155, 111908 CrossRef.
- E. Jannelli, M. Minutillo and A. Perna, Appl. Energy, 2013, 108, 82–91 CrossRef CAS.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- P. C. C. Yang, S. Srinivasan, J. Benziger and A. B. Bocarsly, J. Power Sources, 2001, 103, 1–9 CrossRef.
- Q. Meyer, C. Yang, Y. Cheng and C. Zhao, Electrochem. Energy Rev., 2023, 6, 16 CrossRef CAS.
- P. A. García-Salaberri and A. Sánchez-Ramos, Appl. Energy, 2024, 367, 123332 CrossRef.
- Y. Shao, L. Xu, Z. Hu, L. Xu, X. Zhang, G. Zhao, J. Li and M. Ouyang, J. Power Sources, 2023, 581, 233520 CrossRef CAS.
- G. Xu, X. Dong, B. Xue, J. Huang, J. Wu and W. Cai, Energies, 2023, 16, 1565 CrossRef CAS.
- J. Z. Yanghua Tang, C. Song, H. Liu, J. Zhang, H. Wang, S. Mackinnon, T. Peckham, J. Li, S. McDermid and P. Kozak, J. Electrochem. Soc., 2006, 153, A2036–A2043 CrossRef.
- T. S. T.-C. Jao, S. Uemura, T. Yoshida and S. Hirai, ECS Trans., 2016, 75, 179–188 CrossRef CAS.
- E. Lufrano, C. Simari, M. L. Di Vona, I. Nicotera and R. Narducci, Polymers, 2021, 13, 359 CrossRef CAS PubMed.
- M. Butori, B. Eriksson, N. Nikolić, C. Lagergren, G. Lindbergh and R. W. Lindström, J. Power Sources, 2023, 563, 232803 CrossRef CAS.
- D. R. P. Morris, S. P. Liu, D. Villegas Gonzalez and J. T. Gostick, ACS Appl. Mater. Interfaces, 2014, 6, 18609–18618 CrossRef CAS PubMed.
- M. Tesfaye, D. I. Kushner, B. D. McCloskey, A. Z. Weber and A. Kusoglu, ACS Macro Lett., 2018, 7, 1237–1242 CrossRef CAS PubMed.
- C. Zhang, M. Davies and K. Karan, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 343–352 CrossRef CAS.
- Q. Wang, J. Lu, W. Zheng, B. Li, J. P. Zheng, G. Cui, L. Hao and P. Ming, Fuel, 2025, 381, 133623 CrossRef CAS.
- Y. Lu, H. Wu, D. Yang, W. Zhuge, P. Ming, Y. Zhang, J. Chen and X. Pan, Energy Fuels, 2024, 38, 7331–7343 CrossRef CAS.
- T. Ruiu, A. M. Dreizler, J. Mitzel and E. Gülzow, J. Power Sources, 2016, 303, 257–266 CrossRef CAS.
- Q. Wang, B. Li, D. Yang, H. Dai, J. P. Zheng, P. Ming and C. Zhang, J. Power Sources, 2021, 492, 229613 CrossRef CAS.
- B. R. Frieberg, K. A. Page, J. R. Graybill, M. L. Walker, C. M. Stafford, G. R. Stafford and C. L. Soles, ACS Appl. Mater. Interfaces, 2016, 8, 33240–33249 CrossRef CAS PubMed.
- J. Ramousse, S. Didierjean, O. Lottin and D. Maillet, Int. J. Therm. Sci., 2008, 47, 1–6 CrossRef.
- K. Suárez-Alcántara, A. Rodríguez-Castellanos, R. Dante and O. Solorza-Feria, J. Power Sources, 2006, 157, 114–120 CrossRef.
- M. V. V. D. B. Sepa, J. Electrochem. Soc., 1987, 134, 845–848 CrossRef.
- Y. S. Hui Xu, H. Russell Kunz and J. M. Fenton, J. Electrochem. Soc., 2005, 152, A1828–A1836 CrossRef.
- H. Yano, J. Inukai, H. Uchida, M. Watanabe, P. K. Babu, T. Kobayashi, J. H. Chung, E. Oldfield and A. Wieckowski, Phys. Chem. Chem. Phys., 2006, 8, 4932 RSC.
- V. Ramani, H. R. Kunz and J. M. Fenton, J. Membr. Sci., 2004, 232, 31–44 CrossRef.
- S. Bose, T. Kuila, T. X. H. Nguyen, N. H. Kim, K.-T. Lau and J. H. Lee, Prog. Polym. Sci., 2011, 36, 813–843 CrossRef CAS.
- R. Haider, Y. Wen, Z.-F. Ma, D. P. Wilkinson, L. Zhang, X. Yuan, S. Song and J. Zhang, Chem. Soc. Rev., 2021, 50, 1138–1187 RSC.
- J. N. Adam and Z. Weber, J. Electrochem. Soc., 2006, 153, A2205–A2214 CrossRef.
- K. P. Peter Berg, J. St. Pierre, J. Stumper and B. Wetton, J. Electrochem. Soc., 2004, 151, A341–A335 CrossRef.
- R. H. Qingfeng Li, J. Oluf Jensen and N. J. Bjerrum, Chem. Mater., 2003, 15, 4896–4915 CrossRef.
- A. Liu, X. Liu, L. Liu, Y. Pu, K. Guo, W. Tan, S. Gao, Y. Luo, S. Yu, R. Si, B. Shan, F. Gao and L. Dong, ACS Catal., 2019, 9, 7759–7768 CrossRef CAS.
- R. H. Qingfeng Li, J.-A. Gao, J. Oluf Jensen and N. J. Bjerrum, J. Electrochem. Soc., 2003, 150, A1599–A1605 CrossRef.
- S. G. Kandlikar and Z. Lu, Appl. Therm. Eng., 2009, 29, 1276–1280 CrossRef CAS.
- N. Alhazmi, M. S. Ismail, D. B. Ingham, K. J. Hughes, L. Ma and M. Pourkashanian, J. Power Sources, 2013, 241, 136–145 CrossRef CAS.
- O. S. Burheim, H. Su, H. H. Hauge, S. Pasupathi and B. G. Pollet, Int. J. Hydrogen Energy, 2014, 39, 9397–9408 CrossRef CAS.
- M. Ahadi, M. Tam, M. S. Saha, J. Stumper and M. Bahrami, J. Power Sources, 2017, 354, 207–214 CrossRef CAS.
- O. S. Burheim and J. G. Pharoah, Curr. Opin. Electrochem., 2017, 5, 36–42 CrossRef CAS.
- B. G. P. Odne and S. Burheim, ECS Trans., 2018, 86, 97–109 Search PubMed.
- J. Zhou, S. Shukla, A. Putz and M. Secanell, Electrochim. Acta, 2018, 268, 366–382 CrossRef CAS.
- A. Goshtasbi, P. García-Salaberri, J. Chen, K. Talukdar, D. G. Sanchez and T. Ersal, J. Electrochem. Soc., 2019, 166, F3154–F3179 CrossRef CAS.
- R. Bock, H. Karoliussen, B. G. Pollet, M. Secanell, F. Seland, D. Stanier and O. S. Burheim, Int. J. Hydrogen Energy, 2020, 45, 1335–1342 CrossRef CAS.
- Q. Chen, G. Zhang, X. Zhang, C. Sun, K. Jiao and Y. Wang, Appl. Energy, 2021, 286, 116496 CrossRef CAS.
- W. Wang, Z. Qu, X. Wang and J. Zhang, Membranes, 2021, 11, 148 CrossRef CAS PubMed.
- Y. Huang, X. Xiao, H. Kang, J. Lv, R. Zeng and J. Shen, Energy Convers. Manage., 2022, 254, 115221 CrossRef CAS.
- L. Yang, N.-N. Nik-Ghazali, M. A. H. Ali, W. T. Chong, Z. Yang and H. Liu, Renewable Sustainable Energy Rev., 2023, 187, 113737 CrossRef CAS.
- A. U. Yakubu, J. Zhao, Q. Jiang, X. Ye, J. Liu, Q. Yu and S. Xiong, Heliyon, 2024, 10, e38556 CrossRef CAS PubMed.
- Z. Zhang, J. Mao, H. Wei, C. Cheng and Z. Liu, Appl. Therm. Eng., 2024, 239, 122083 CrossRef CAS.
- G. Unsworth, N. Zamel and X. Li, Int. J. Hydrogen Energy, 2012, 37, 5161–5169 CrossRef CAS.
- O. S. Burheim, H. Su, S. Pasupathi, J. G. Pharoah and B. G. Pollet, Int. J. Hydrogen Energy, 2013, 38, 8437–8447 CrossRef CAS.
- J. G. P. Odne, S. Burheim, H. Lampert, P. J. S. Vie and S. Kjelstrup, J. Fuel Cell Sci. Technol., 2011, 8, 021013 CrossRef.
- B. H. Robert Bock, M. Onsrud, H. Karoliussen, F. Seland and O. S. Burheim, ECS Trans., 2019, 92, 223–245 CrossRef.
- Z. L. A. Radhakrishnan and S. G. Kandlikar, ECS Trans., 2010, 33, 1163–1176 CrossRef.
- G. Karimi, X. Li and P. Teertstra, Electrochim. Acta, 2010, 55, 1619–1625 CrossRef CAS.
- V. Leduc, G. Sdanghi, R. Bligny, J. Dillet, S. Didierjean, T. Schmitt, M. Hanauer, U. Sauter and G. Maranzana, Electrochim. Acta, 2024, 505, 145001 CrossRef CAS.
- F. Liu, Q. Wang, B. Li, C. Zhang and P. Ming, eTransportation, 2022, 12, 100162 CrossRef.
- I. Nitta, O. Himanen and M. Mikkola, Fuel Cells, 2008, 8, 111–119 CrossRef CAS.
- M. Ahadi, M. Andisheh-Tadbir, M. Tam and M. Bahrami, Int. J. Heat Mass Transfer, 2016, 96, 371–380 CrossRef CAS.
- Y. Wang and M. Gundevia, Int. J. Heat Mass Transfer, 2013, 60, 134–142 CrossRef CAS.
- A. Pfrang, D. Veyret, F. Sieker and G. Tsotridis, Int. J. Hydrogen Energy, 2010, 35, 3751–3757 CrossRef CAS.
- O. Burheim, P. J. S. Vie, J. G. Pharoah and S. Kjelstrup, J. Power Sources, 2010, 195, 249–256 CrossRef CAS.
- O. S. Burheim, G. Ellila, J. D. Fairweather, A. Labouriau, S. Kjelstrup and J. G. Pharoah, J. Power Sources, 2013, 221, 356–365 CrossRef CAS.
- C. Csoklich, M. Sabharwal, T. J. Schmidt and F. N. Büchi, J. Power Sources, 2022, 540, 231539 CrossRef CAS.
- E. Sadeghi, N. Djilali and M. Bahrami, J. Power Sources, 2011, 196, 246–254 CrossRef CAS.
- S. Wang, J. Wang and Y. Zhu, J. Power Sources, 2023, 575, 233179 CrossRef CAS.
- G. Xu, J. M. LaManna, J. T. Clement and M. M. Mench, J. Power Sources, 2014, 256, 212–219 CrossRef CAS.
- N. Zamel and X. Li, Prog. Energy Combust. Sci., 2013, 39, 111–146 CrossRef CAS.
- L. Chen, Y.-F. Wang and W.-Q. Tao, Therm. Sci. Eng. Prog., 2020, 19, 100616 CrossRef.
- H. Sadeghifar, N. Djilali and M. Bahrami, J. Power Sources, 2015, 273, 96–104 CrossRef CAS.
- T.-F. Cao, Y.-T. Mu, J. Ding, H. Lin, Y.-L. He and W.-Q. Tao, Int. J. Heat Mass Transfer, 2015, 87, 544–556 CrossRef.
- O. S. Burheim, ECS Trans., 2017, 80, 509–525 CrossRef CAS.
- Q. Wang, F. Tang, B. Li, H. Dai, J. P. Zheng, C. Zhang and P. Ming, J. Power Sources, 2021, 488, 229419 CrossRef CAS.
- Q. Wang, F. Tang, B. Li, H. Dai, J. P. Zheng, C. Zhang and P. Ming, Appl. Energy, 2022, 308, 118377 CrossRef CAS.
- Q. Wang, F. Tang, X. Li, J. P. Zheng, L. Hao, G. Cui and P. Ming, Chem. Eng. J., 2023, 463, 142286 CrossRef CAS.
- H. Pourrahmani, A. Yavarinasab, M. Siavashi, M. Matian and J. Van herle, Energy Rev., 2022, 1, 100002 CrossRef.
- F. Cai, S. Cai and Z. Tu, Energy Convers. Manage., 2024, 307, 118348 CrossRef CAS.
- Z. Z. Mufan Li, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016, 354, 1414–1419 CrossRef PubMed.
- T. J. Bvumbe, P. Bujlo, I. Tolj, K. Mouton, G. Swart, S. Pasupathi and B. G. Pollet, Hydrogen Fuel Cells, 2016, 1, 1–20 Search PubMed.
- J. N. Qiao, H. Guo, F. Ye and H. Chen, Int. J. Heat Mass Transfer, 2024, 226, 125456 CrossRef CAS.
- W. Yoon and A. Z. Weber, J. Electrochem. Soc., 2011, 158, B1007 CrossRef CAS.
- D. Banham, J. Zou, S. Mukerjee, Z. Liu, D. Yang, Y. Zhang, Y. Peng and A. Dong, J. Power Sources, 2021, 490, 229515 CrossRef CAS.
- S. Liu, S. Yuan, Y. Liang, H. Li, Z. Xu, Q. Xu, J. Yin, S. Shen, X. Yan and J. Zhang, Int. J. Hydrogen Energy, 2023, 48, 4389–4417 CrossRef CAS.
- S. Kreitmeier, P. Lerch, A. Wokaun and F. N. Büchi, J. Electrochem. Soc., 2013, 160, F456–F463 CrossRef CAS.
- K.-H. Hou, C.-H. Lin, M.-D. Ger, S.-W. Shiah and H.-M. Chou, Int. J. Hydrogen Energy, 2012, 37, 3890–3896 CrossRef CAS.
- L. Chen, Y. Chen and W.-Q. Tao, Renewable Sustainable Energy Rev., 2023, 173, 113050 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, Z. Xiao, J. Tan, H. Zhang and J. Yu, Membranes, 2024, 14, 72 CrossRef CAS PubMed.
- F. Akitomo, T. Sasabe, T. Yoshida, H. Naito, K. Kawamura and S. Hirai, J. Power Sources, 2019, 431, 205–209 Search PubMed.
- H. Ito, T. Mimoto, S. Someya and T. Munakata, J. Electrochem. Soc., 2021, 168, 124505 CrossRef CAS.
- J. Zhang, Z. Xie, J. Zhang, Y. Tang, C. Song, T. Navessin, Z. Shi, D. Song, H. Wang, D. P. Wilkinson, Z.-S. Liu and S. Holdcroft, J. Power Sources, 2006, 160, 872–891 CrossRef CAS.
- V. Ionescu, Phys. Scr., 2020, 95, 034006 CrossRef CAS.
- T. Yin, D. Chen, T. Hu, S. Hu, R. Li, T. Wei, Y. Li, Y. Li, X. Xu and P. Pei, Energy Convers. Manage., 2024, 315, 118740 CrossRef CAS.
- J. Shen, C. Zhang, L. Li, S. Liu, H. Liu, B. Chen and C. Du, Energy, 2024, 308, 132851 CrossRef CAS.
- A. Martín-Alcántara, J. Pino and A. Iranzo, Int. J. Hydrogen Energy, 2023, 48, 13987–13999 CrossRef.
- C. Song, Y. Tang, J. L. Zhang, J. Zhang, H. Wang, J. Shen, S. McDermid, J. Li and P. Kozak, Electrochim. Acta, 2007, 52, 2552–2561 CrossRef CAS.
- H. Sun, C. Xie, H. Chen and S. Almheiri, Appl. Energy, 2015, 160, 937–944 CrossRef CAS.
- J. Zhang, Y. Tang, C. Song and J. Zhang, J. Power Sources, 2007, 172, 163–171 CrossRef CAS.
- J. Zhang, Y. Tang, C. Song, Z. Xia, H. Li, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 5315–5321 CrossRef CAS.
- S. Wang, X. Hu, Z. Zhou, Q. Wang, K. Ye, S. Sui, M. Hu and F. Jiang, Sustainable Energy Technol. Assess., 2023, 60, 103529 CrossRef.
- Y. Xu, R. Fan, G. Chang, S. Xu and T. Cai, Energy Convers. Manage., 2021, 248, 114791 CrossRef CAS.
- Y. Shao, L. Xu, L. Xu, X. Zhang, Z. Wang, G. Zhao, Z. Hu, J. Li and M. Ouyang, eTransportation, 2023, 18, 100285 CrossRef.
- J. Zhang, Y. Tang, C. Song, X. Cheng, J. Zhang and H. Wang, Electrochim. Acta, 2007, 52, 5095–5101 CrossRef CAS.
- I. O. M. Kellegoz and M. S. Kilickaya, J. Optoelectron. Adv. Mater., 2008, 10, 369–372 Search PubMed.
- X. Li, F. Tang, Q. Wang, B. Li, H. Dai, G. Chang, C. Zhang, W. Zheng and P. Ming, Int. J. Hydrogen Energy, 2024, 50, 1228–1238 CrossRef CAS.
- M. Butori, B. Eriksson, N. Nikolić, C. Lagergren, G. Lindbergh and R. Wreland Lindström, Int. J. Hydrogen Energy, 2024, 95, 1158–1170 CrossRef.
- A. Jatukaran, J. Zhong, A. H. Persad, Y. Xu, F. Mostowfi and D. Sinton, ACS Appl. Nano Mater., 2018, 1, 1332–1338 CrossRef CAS.
- C. Yin, J. Li, Y. Zhou, H. Zhang, P. Fang and C. He, ACS Appl. Mater. Interfaces, 2018, 10, 14026–14035 CrossRef CAS PubMed.
- R. Sun, Z. Xia, C. Yang, F. Jing, S. Wang and G. Sun, Prog. Nat. Sci.: Mater. Int., 2020, 30, 912–917 CrossRef CAS.
- H. Eskandari, D. K. Paul, A. P. Young and K. Karan, ACS Appl. Mater. Interfaces, 2022, 14, 50762–50772 CrossRef CAS PubMed.
- S. Fujita and A. Suzuki, J. Appl. Phys., 2010, 107, 1 CrossRef.
- M. Taş and G. Elden, Fuel Cells, 2020, 20, 531–539 CrossRef.
- Y. Wang, Y. Pang, H. Xu, A. Martinez and K. S. Chen, Energy Environ. Sci., 2022, 15, 2288–2328 RSC.
- J. Zhang, Y. Tang, C. Song, J. Zhang and H. Wang, J. Power Sources, 2006, 163, 532–537 CrossRef CAS.
- Y. Zhang, Z. Xiao, X. Zhao, J. Wang, Y. Wang and J. Yu, Membranes, 2025, 15, 72 CrossRef CAS PubMed.
- J.-C. Shyu, K.-L. Hsueh and F. Tsau, Energy Convers. Manage., 2011, 52, 3415–3424 CrossRef CAS.
- Q. Yan, H. Toghiani and H. Causey, J. Power Sources, 2006, 161, 492–502 CrossRef CAS.
- Q. Wang, F. Tang, B. Li, H. Dai, J. P. Zheng, C. Zhang and P. Ming, Chem. Eng. J., 2022, 433, 133667 CrossRef CAS.
- H. Chen, Y. Liu, C. Deng and J. Chen, Int. J. Hydrogen Energy, 2023, 48, 1075–1089 CrossRef CAS.
- M. W. H. Xu, Y. Liu, V. Mittal, R. Vieth, H. R. Kunz, L. J. Bonville and J. M. Fenton, ECS Trans., 2006, 3, 561–568 CrossRef.
- C. D. G. Remy Sellin, S. Arrii-Clacens, S. Pronier, J.-M. Clacens, C. Coutanceau and J.-M. Leger, J. Phys. Chem. C, 2009, 113, 21735–21744 CrossRef.
- Z. Yang, S. Ball, D. Condit and M. Gummalla, J. Electrochem. Soc., 2011, 158, B1439 CrossRef CAS.
- R. L. Borup, J. R. Davey, F. H. Garzon, D. L. Wood and M. A. Inbody, J. Power Sources, 2006, 163, 76–81 CrossRef CAS.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- F. Hasché, M. Oezaslan and P. Strasser, ChemPhysChem, 2012, 13, 828–834 CrossRef PubMed.
- D. A. Stevens and J. R. Dahn, Carbon, 2005, 43, 179–188 CrossRef CAS.
- R. Sellin, J.-M. Clacens and C. Coutanceau, Carbon, 2010, 48, 2244–2254 CrossRef CAS.
- J. Zhao, X. Huang, H. Chang, S. Hwa Chan and Z. Tu, Energy Convers. Manage.: X, 2021, 10, 100087 CAS.
- C. Yue, W. Zheng, Y. Lian, J. Kang, S. Chen, X. Dong, B. Li, C. Zhang and P. Ming, J. Energy Storage, 2024, 104, 114426 CrossRef.
- Y. Lu, D. Yang, H. Wu, L. Jia, J. Chen, P. Ming and X. Pan, Renewable Energy, 2024, 234, 121166 CrossRef CAS.
- S. Lee, J. Nam, J. Ahn, S. Yoon, S. C. Jeong, H. Ju and C. H. Lee, Int. J. Hydrogen Energy, 2024, 96, 333–342 CrossRef.
- X. Gao, K. Yamamoto, T. Hirai, T. Uchiyama, N. Ohta, N. Takao, M. Matsumoto, H. Imai, S. Sugawara, K. Shinohara and Y. Uchimoto, Langmuir, 2020, 36, 3871–3878 CrossRef CAS PubMed.
- D. K. Paul and K. Karan, J. Phys. Chem. C, 2014, 118, 1828–1835 CrossRef CAS.
- Y. Liang, Y. Feng, H. Li, M. He, D. Yang, S. Shen, X. Yan, J. Zhang, X. Xu and R. Masrour, presented in part at the International Conference on Optoelectronic Information and Functional Materials (OIFM 2024), 2024.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon and D. Wood, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- J. C. Meier, C. Galeano, I. Katsounaros, J. Witte, H. J. Bongard, A. A. Topalov, C. Baldizzone, S. Mezzavilla, F. Schüth and K. J. J. Mayrhofer, Beilstein J. Nanotechnol., 2014, 5, 44–67 CrossRef PubMed.
- A. P. Soleymani, L. R. Parent and J. Jankovic, Adv. Funct. Mater., 2021, 32, 2105188 CrossRef.
- Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558–566 CrossRef CAS.
- N. Trogisch, D. Babik, A. Orfanidi, G. Jegert, A. Albert and R.-A. Eichel, J. Electrochem. Soc., 2024, 171, 104506 CrossRef CAS.
- W. Bi, Q. Sun, Y. Deng and T. F. Fuller, Electrochim. Acta, 2009, 54, 1826–1833 CrossRef CAS.
- G. Inzelt, B. Berkes and Á. Kriston, Electrochim. Acta, 2010, 55, 4742–4749 CrossRef CAS.
- Z. Zheng, F. Yang, C. Lin, F. Zhu, S. Shen, G. Wei and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 35088–35097 CrossRef CAS PubMed.
- T. F. F. Wu Bi, ECS Trans., 2007, 11, 1235 CrossRef.
- Z. Huang, Z. Zhou, J. Zhao, W.-T. Wu, L. Wei, C. Hu, Y. Yang, Y. Li and Y. Song, Int. J. Hydrogen Energy, 2024, 53, 1107–1122 CrossRef CAS.
- T. Đukić, L. J. Moriau, L. Pavko, M. Kostelec, M. Prokop, F. Ruiz-Zepeda, M. Šala, G. Dražić, M. Gatalo and N. Hodnik, ACS Catal., 2021, 12, 101–115 CrossRef PubMed.
- S. M. Andersen, M. Borghei, P. Lund, Y.-R. Elina, A. Pasanen, E. Kauppinen, V. Ruiz, P. Kauranen and E. M. Skou, Solid State Ionics, 2013, 231, 94–101 CrossRef CAS.
- S. Maass, F. Finsterwalder, G. Frank, R. Hartmann and C. Merten, J. Power Sources, 2008, 176, 444–451 CrossRef CAS.
- V. E. Guterman, S. V. Belenov, V. V. Krikov, L. L. Vysochina, W. Yohannes, N. Y. Tabachkova and E. N. Balakshina, J. Phys. Chem. C, 2014, 118, 23835–23844 CrossRef CAS.
- K. H. Lim, H.-S. Oh, S.-E. Jang, Y.-J. Ko, H.-J. Kim and H. Kim, J. Power Sources, 2009, 193, 575–579 CrossRef CAS.
- S. R. A. Olga, A. Baturina and K. J. Wynne, Chem. Mater., 2006, 18, 1498–1504 CrossRef.
- C. H. P. L. M. Roen and T. D. Jarvi, Electrochem. Solid State Lett., 2004, 7, A19–A22 CrossRef.
- M. T. H. D. A. Stevens, G. M. Haugen and J. R. Dahn, J. Electrochem. Soc., 2005, 152, A2309–A2315 CrossRef.
- M. A. Modestino, A. Kusoglu, A. Hexemer, A. Z. Weber and R. A. Segalman, Macromolecules, 2012, 45, 4681–4688 CrossRef CAS.
- A. Kusoglu, D. Kushner, D. K. Paul, K. Karan, M. A. Hickner and A. Z. Weber, Adv. Funct. Mater., 2014, 24, 4763–4774 CrossRef CAS.
- D. K. Paul, H. K. K. Shim, J. B. Giorgi and K. Karan, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1267–1277 CrossRef CAS.
- A. Alentiev, J. Kostina and G. Bondarenko, Desalination, 2006, 200, 32–33 CrossRef CAS.
- D. Chu, D. Gervasio, M. Razaq and E. Yeager, J. Appl. Electrochem., 1990, 20, 157–162 CrossRef CAS.
- R. P. Dowd, Jr., Y. Li and T. Van Nguyen, J. Appl. Electrochem., 2020, 50, 993–1006 CrossRef.
- Y. Chang, J. Zhao, S. Shahgaldi, Y. Qin, Y. Yin and X. Li, Int. J. Hydrogen Energy, 2020, 45, 29904–29916 CrossRef CAS.
- F. Rong, C. Huang, Z.-S. Liu, D. Song and Q. Wang, J. Power Sources, 2008, 175, 699–711 CrossRef CAS.
- Y. Tang, A. M. Karlsson, M. H. Santare, M. Gilbert, S. Cleghorn and W. B. Johnson, Mater. Sci. Eng., A, 2006, 425, 297–304 CrossRef.
- J. W. W. Vijay, A. Sethuraman, A. T. Haug, S. Motupally and L. V. Protsailo, J. Electrochem. Soc., 2008, 155, B50–B57 CrossRef.
- C. Yue, W. Zheng, S. Chen, B. Li, C. Zhang, F. Tang and P. Ming, Int. J. Hydrogen Energy, 2024, 50, 1515–1525 CrossRef.
- S. Han, W. Zheng, Y. Lu, Y. Lian, B. Li, P. Ming and C. Zhang, Int. J. Hydrogen Energy, 2024, 58, 279–288 CrossRef CAS.
- A. Kneer, J. Jankovic, D. Susac, A. Putz, N. Wagner, M. Sabharwal and M. Secanell, J. Electrochem. Soc., 2018, 165, F3241–F3250 CrossRef CAS.
- A. Malekian, S. Salari, J. Stumper, N. Djilali and M. Bahrami, Int. J. Hydrogen Energy, 2019, 44, 23396–23405 CrossRef CAS.
- S. Kim, J.-H. Kim, J.-G. Oh, K.-L. Jang, B.-H. Jeong, B. K. Hong and T.-S. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 15391–15398 CrossRef CAS PubMed.
- A. S. Alavijeh, R. M. H. Khorasany, Z. Nunn, A. Habisch, M. Lauritzen, E. Rogers, G. G. Wang and E. Kjeang, J. Electrochem. Soc., 2015, 162, F1461–F1469 CrossRef CAS.
- J.-B. Pyo, J. H. Kim and T.-S. Kim, J. Mater. Chem. A, 2020, 8, 24763–24773 RSC.
- S. Kundu, M. W. Fowler, L. C. Simon and S. Grot, J. Power Sources, 2006, 157, 650–656 CrossRef CAS.
- J. Zhao, S. Shahgaldi, X. Li and Z. Liu, J. Electrochem. Soc., 2018, 165, F3337–F3345 CrossRef CAS.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- C. Li, K. Yu, A. Bird, F. Guo, J. Ilavsky, Y. Liu, D. A. Cullen, A. Kusoglu, A. Z. Weber, P. J. Ferreira and J. Xie, Energy Environ. Sci., 2023, 16, 2977–2990 RSC.
- Y. Zhang, X. Li and A. Klinkova, Int. J. Hydrogen Energy, 2021, 46, 11071–11083 CrossRef CAS.
- F. Rong, C. Huang, Z.-S. Liu, D. Song and Q. Wang, J. Power Sources, 2008, 175, 712–723 CrossRef CAS.
- J. Liu, Y. Yin, J. Zhang, T. Zhang, X. Zhang and H. Chen, J. Power Sources, 2021, 512, 230487 CrossRef CAS.
- Y. Chang, J. Liu, R. Li, J. Zhao, Y. Qin, J. Zhang, Y. Yin and X. Li, Energy Convers. Manage., 2019, 189, 24–32 CrossRef CAS.
- A. Stassi, I. Gatto, E. Passalacqua, V. Antonucci, A. S. Arico, L. Merlo, C. Oldani and E. Pagano, J. Power Sources, 2011, 196, 8925–8930 CrossRef CAS.
- Z. Yao, F. Zhou, C. Tu, J. Tan and M. Pan, Int. J. Hydrogen Energy, 2024, 50, 200–208 CrossRef.
- S. Chen, M. Hao, Y. Hu, K. Liu and Y. Li, J. Power Sources, 2024, 599, 234238 CrossRef CAS.
- W. Zheng, L. Xu, Z. Hu, Y. Ding, J. Li and M. Ouyang, J. Power Sources, 2021, 487, 229367 CrossRef CAS.
- S.-H. Shin, P. J. Nur, A. Kodir, D.-H. Kwak, H. Lee, D. Shin and B. Bae, ACS Omega, 2019, 4, 19153–19163 CrossRef CAS PubMed.
- S. Giancola, R. A. B. Arciniegas, A. Fahs, J.-F. Chailan, M. L. Di Vona, P. Knauth and R. Narducci, Membranes, 2019, 9, 134 CrossRef CAS PubMed.
- S. H. Frensch, G. Serre, F. Fouda-Onana, H. C. Jensen, M. L. Christensen, S. S. Araya and S. K. Kær, J. Power Sources, 2019, 420, 54–62 CrossRef CAS.
- M. Feng, R. Qu, Z. Wei, L. Wang, P. Sun and Z. Wang, Sci. Rep., 2015, 5, 9859 CrossRef CAS PubMed.
- A. B. B. Paul, W. Majsztrik and J. B. Benziger, Macromolecules, 2008, 41, 9849–9862 CrossRef.
- S. Shi, A. Z. Weber and A. Kusoglu, J. Membr. Sci., 2016, 516, 123–134 CrossRef CAS.
- H.-Y. Jung and J. W. Kim, Int. J. Hydrogen Energy, 2012, 37, 12580–12585 CrossRef CAS.
- S. W. Osung Kwon and D.-M. Zhu, J. Phys. Chem. B, 2010, 114, 14989–14994 CrossRef PubMed.
- B. R. Matos, M. A. Dresch, E. I. Santiago, L. P. R. Moraes, D. J. Carastan, J. Schoenmaker, I. A. Velasco-Davalos, A. Ruediger, A. C. Tavares and F. C. Fonseca, Electrochim. Acta, 2016, 196, 110–117 CrossRef CAS.
- C. A. Wilkie, J. R. Thomsen and M. L. Mittleman, J. Appl. Polym. Sci., 2003, 42, 901–909 CrossRef.
- M. Pan, C. Pan, C. Li and J. Zhao, Renewable Sustainable Energy Rev., 2021, 141, 110771 CrossRef CAS.
- A. M. Dafalla, L. Wei, B. T. Habte, J. Guo and F. Jiang, Energies, 2022, 15, 9247 CrossRef CAS.
- M. P. Rodgers, L. J. Bonville, H. R. Kunz, D. K. Slattery and J. M. Fenton, Chem. Rev., 2012, 112, 6075–6103 CrossRef CAS PubMed.
- Y. K. Hamdy, F. M. Mohamed, C. S. Kuroda and A. Ohira, Macromol. Chem. Phys., 2011, 212, 708–714 CrossRef.
- K. M. C. Kirt, A. Page and R. B. Moore, Macromolecules, 2005, 38, 6472–6484 CrossRef.
- M. Marrony, D. Beretta, S. Ginocchio, Y. Nedellec, S. Subianto and D. J. Jones, Fuel Cells, 2013, 13, 1146–1154 CrossRef CAS.
- P. Choi, N. H. Jalani, T. M. Thampan and R. Datta, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2183–2200 CrossRef CAS.
- J. Li, X. Yang, H. Tang and M. Pan, J. Membr. Sci., 2010, 361, 38–42 CrossRef CAS.
- J. E. Hensley, J. D. Way, S. F. Dec and K. D. Abney, J. Membr. Sci., 2007, 298, 190–201 CrossRef CAS.
- D. DeBonis, M. Mayer, A. Omosebi and R. S. Besser, Renewable Energy, 2016, 89, 200–206 CrossRef CAS.
- M. Casciola, G. Alberti, M. Sganappa and R. Narducci, J. Power Sources, 2006, 162, 141–145 CrossRef CAS.
- Y. Jeon, H.-k Hwang, J. Park, H. Hwang and Y.-G. Shul, Int. J. Hydrogen Energy, 2014, 39, 11690–11699 CrossRef CAS.
- N. Zhao, Y. Chu, Z. Xie, K. Eggen, F. Girard and Z. Shi, Fuel Cells, 2020, 20, 176–184 CrossRef CAS.
- D. Qiu, L. Peng, X. Lai, M. Ni and W. Lehnert, Renewable Sustainable Energy Rev., 2019, 113, 109289 CrossRef CAS.
- M. N. Silberstein and M. C. Boyce, J. Power Sources, 2011, 196, 3452–3460 CrossRef CAS.
- A. Kusoglu, M. H. Santare and A. M. Karlsson, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1506–1517 CrossRef CAS.
- R. Mukundan, A. M. Baker, A. Kusoglu, P. Beattie, S. Knights, A. Z. Weber and R. L. Borup, J. Electrochem. Soc., 2018, 165, F3085–F3093 CrossRef CAS.
- Y. Singh, F. P. Orfino, M. Dutta and E. Kjeang, J. Electrochem. Soc., 2017, 164, F1331–F1341 CrossRef CAS.
- S. Shi, X. Sun, Q. Lin, J. Chen, Y. Fu, X. Hong, C. Li, X. Guo, G. Chen and X. Chen, Int. J. Hydrogen Energy, 2020, 45, 27653–27664 CrossRef CAS.
- S. Bhattacharya, J. Leung, M. V. Lauritzen and E. Kjeang, Electrochim. Acta, 2020, 352, 136489 CrossRef CAS.
- Y. Chen, C. Jiang and C. Cho, Polymers, 2018, 10, 971 CrossRef PubMed.
- F. Qu, L. Liu, G. Tao, W. Zhan, S. Zhan, Y. Li, C. Li, X. Lv, Z. Shi and H. Duan, Tribol. Int., 2023, 188, 108850 CrossRef CAS.
- Q. Meyer, Y. Zeng and C. Zhao, Adv. Mater., 2019, 31, 1901900 CrossRef CAS PubMed.
- P. Irmscher, D. Qui, H. Janßen, W. Lehnert and D. Stolten, Int. J. Hydrogen Energy, 2019, 44, 23406–23415 CrossRef CAS.
- F. Lapicque, M. Belhadj, C. Bonnet, J. Pauchet and Y. Thomas, J. Power Sources, 2016, 336, 40–53 CrossRef CAS.
- R. Taherian, M. Matboo Ghorbani and S. R. Kiahosseini, J. Electroanal. Chem., 2018, 815, 90–97 CrossRef CAS.
- A. Kausar, I. Rafique and B. Muhammad, Polym.-Plast. Technol. Eng., 2016, 55, 1167–1191 CrossRef CAS.
- M. C. Gutiérrez, M.-A. De Paoli and M. I. Felisberti, Ind. Crop. Prod., 2014, 52, 363–372 CrossRef.
- A. P. G. Agarwal and R. K. Sharma, J. Eng. Sci. Technol., 2014, 9, 590–604 Search PubMed.
- J. Zhou, Z. Yao, Y. Chen, D. Wei, Y. Wu and T. Xu, Polym. Compos., 2013, 34, 1245–1249 CrossRef CAS.
- P. K. Z. Zhang and K. Friedrich, Compos. Sci. Technol., 2002, 62, 1001–1009 CrossRef.
- Y. Lian, W. Zheng, C. Yue, S. Han and P. Ming, Int. J. Heat Mass Transfer, 2024, 223, 125239 CrossRef CAS.
- Y. Jeon, H. Na, H. Hwang, J. Park, H. Hwang and Y.-G. Shul, Int. J. Hydrogen Energy, 2015, 40, 3057–3067 CrossRef CAS.
- H. Lee, S. Jeong, J. Song, M. Kim, C. Chu, Y. Lee, D. Kim and M. Kim, J. Mech. Sci. Technol., 2023, 37, 2095–2108 CrossRef.
- M. Marrony, R. Barrera, S. Quenet, S. Ginocchio, L. Montelatici and A. Aslanides, J. Power Sources, 2008, 182, 469–475 CrossRef CAS.
- P. Ren, P. Pei, Y. Li, Z. Wu, D. Chen and S. Huang, Prog. Energy Combust. Sci., 2020, 80, 100859 CrossRef.
- L. V. Nam, E. Choi, S. Jang and S. M. Kim, Renewable Energy, 2021, 180, 203–212 CrossRef CAS.
- J.-S. Park, M.-S. Shin and C.-S. Kim, Curr. Opin. Electrochem., 2017, 5, 43–55 CrossRef CAS.
- G. Xu, A. Ke, G. Xu, Y. Liu, Y. Zuo, X. Yang, Y. Dong, J. Wang, J. Zheng, J. Li and W. Cai, Int. J. Hydrogen Energy, 2024, 56, 330–337 CrossRef CAS.
- Y. Liao, S. Zhao, G. Liu, H. Li, J. Shuai, L. Wang, B. Liu and H. Tang, Chem. Eng. J., 2024, 488, 150971 CrossRef CAS.
- G. Gnana Kumar, A. R. Kim, K. Suk Nahm and R. Elizabeth, Int. J. Hydrogen Energy, 2009, 34, 9788–9794 CrossRef.
- J. Niu, S. Zhang, Y. Li, X. Li, J. Zhang, S. Lu and Q. He, Polymer, 2023, 273, 125869 CrossRef CAS.
- S. Vengatesan, H. Kim, S. Lee, E. Cho, H. Yongha, I. Oh, S. Hong and T. Lim, Int. J. Hydrogen Energy, 2008, 33, 171–178 CrossRef CAS.
- A. A. Wani, N. Shaari, S. K. Kamarudin, N. F. Raduwan, Y. N. Yusoff, A. M. Khan, S. Yousuf and A. Mohamed Nainar Mohamed, Energy Fuels, 2024, 38, 18169–18193 CrossRef CAS.
- I. Nicotera, L. Coppola and C. Simari, Mater. Renewable Sustainable Energy, 2024, 13, 307–318 CrossRef.
- Y. Ji, Y. i Cho, Y. Jeon, C. Lee, D.-H. Park and Y.-G. Shul, Appl. Catal., B, 2017, 204, 421–429 CrossRef CAS.
- H. X. Jianwei Yang, J. Li, K. Gong, F. Yue, X. Han, K. Wu, P. Shao, Q. Fu, Y. Zhu, W. Xu, X. Huang, J. Xie, F. Wang, W. Yang, T. Zhang, Z. Xu, X. Feng and B. Wang, Science, 2024, 385, 1115–1120 CrossRef PubMed.
- Y. Z. Panpan Guan, M. Zhang, W. Zhong, J. Xu, J. Lei, H. Ding, W. Feng, F. Liu and Y. Zhang, Sci. Adv., 2023, 9, eadh1386 CrossRef PubMed.
- S. H. Mirfarsi, A. Kumar, J. Jeong, M. Adamski, S. McDermid, B. Britton and E. Kjeang, Int. J. Hydrogen Energy, 2024, 50, 1507–1522 CrossRef CAS.
- C. Li, W. Wang, S. Zhu, H. Pan, Q. Xu, P. Shi and Y. Min, Chem. Eng. J., 2023, 466, 143105 CrossRef CAS.
- W. Zhang, Y. Chen, Y. Jin, H. Liu, Q. Ma, Q. Xu and H. Su, Int. J. Hydrogen Energy, 2024, 65, 829–836 CrossRef CAS.
- M. Janssen, J. Drnec, I. Martens, J. Quinson, R. Pittkowski, D. Park, P. Weber, M. Arenz and M. Oezaslan, ChemSusChem, 2024, 17, e202400303 CrossRef CAS PubMed.
- K. Yu, J. L. Hart, J. Xie, M. L. Taheri and P. Ferreira, Nano Energy, 2023, 111, 108393 CrossRef CAS.
- F. Chabot, P. Lionel, L. Guétaz, S. Rosini and A. Morin, J. Electrochem. Soc., 2024, 171, 124506 CrossRef CAS.
- V. Shokhen, L. Strandberg, M. Skoglundh and B. Wickman, J. Mater. Chem. A, 2023, 11, 21029–21035 RSC.
- C. Wang, J. Li, S. Zhang, X. Li, X. Duan, Y. Wu, Q. Zhang, T. Yang and J. Liu, Appl. Energy, 2024, 367, 123377 CrossRef CAS.
- Y. Sun, L. Mao, Z. Hu, X. Zhang and R. Peng, Adv. Sci., 2024, 11, 2403631 CrossRef CAS PubMed.
- M. Bogar, Y. Yakovlev, J. Nováková, A. M. Darabut, M. Kriechbaum, H. Amenitsch, R. Taccani and I. Matolínová, Int. J. Hydrogen Energy, 2024, 58, 1673–1681 CrossRef CAS.
- J. Chen, M. Perez-Page, C. M. A. Parlett, Z. Guo, X. Yang, Z. Zhou, H. Zhai, S. Bartlett, T. S. Miller and S. M. Holmes, Chem. Eng. J., 2024, 487, 150670 CrossRef CAS.
- Q. Li, C. J. Pollock, J. Soto, A. M. Villarino, Z. Shi, M. R. Krumov, D. A. Muller and H. D. Abruña, Nat. Commun., 2025, 16, 3008 CrossRef CAS PubMed.
- A. P. Soleymani, M. Reid and J. Jankovic, Adv. Funct. Mater., 2022, 33, 2209733 CrossRef.
- Y. Ao, Z. Li, S. Laghrouche, D. Depernet, D. Candusso and K. Zhao, J. Power Sources, 2024, 603, 234420 CrossRef CAS.
- C. Liu, Z. Geng, X. Wang, W. Liu, Y. Wang, Q. Xia, W. Li, L. Jin and C. Zhang, J. Energy Chem., 2024, 90, 348–369 CrossRef CAS.
- I. Gatto, A. Caprì, C. Lo Vecchio, S. Zignani, A. Patti and V. Baglio, Int. J. Hydrogen Energy, 2023, 48, 11914–11921 CrossRef CAS.
- M. Rossini, D. Pan, B. Koyutürk, S. Chen, A. Khataee, G. Lindbergh, P. Jannasch and A. Cornell, J. Mater. Chem. A, 2024, 12, 12826–12834 RSC.
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS.
- A. Lim, H.-J. Kim, D. Henkensmeier, S. Jong Yoo, J. Young Kim, S. Young Lee, Y.-E. Sung, J. H. Jang and H. S. Park, J. Ind. Eng. Chem., 2019, 76, 410–418 CrossRef CAS.
- S. Y. Kang, J. E. Park, G. Y. Jang, O.-H. Kim, O. J. Kwon, Y.-H. Cho and Y.-E. Sung, Int. J. Hydrogen Energy, 2022, 47, 9115–9126 CrossRef CAS.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- L. Xia, W. Jiang, H. Hartmann, J. Mayer, W. Lehnert and M. Shviro, ACS Appl. Mater. Interfaces, 2022, 14, 19397–19408 CrossRef CAS PubMed.
- X. Wang, Z. Jiang, Y. Ma, X. Su, X. Zhao, A. Zhu and Q. Zhang, J. Power Sources, 2024, 591, 233819 CrossRef CAS.
- C. X. Lin, X. Q. Wang, L. Li, F. H. Liu, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Power Sources, 2017, 365, 282–292 CrossRef CAS.
- L. Yuan, Q. Wang, F. Tang, B. Li, P. Ming and C. Zhang, Etransportation, 2022, 13, 100178 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |