Presentation modality of glycoconjugates modulates dendritic cell phenotype†‡
N. A.
Hotaling
a,
D. M.
Ratner
b,
R. D.
Cummings
c and
J. E.
Babensee
*a
aWallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: Julia.Babensee@bme.gatech.edu
bDepartment of Bioengineering, University of Washington, Seattle, WA 98195, USA
cDepartment of Biochemistry, Emory University, Atlanta, GA 30322, USA
First published on 4th July 2014
Abstract
The comparative dendritic cell (DC) response to glycoconjugates presented in soluble, phagocytosable, or non-phagocytosable display modalities is poorly understood. This is particularly problematic, as the probing of immobilized glycans presented on the surface of microarrays is a common screen for potential candidates for glycan-based therapeutics. However, the assumption that carbohydrate–protein interactions on a flat surface can be translatable to development of efficacious therapies, such as vaccines, which are delivered in soluble or phagocytosable particles, has not been validated. Thus, a preliminary investigation was performed in which mannose or glucose was conjugated to cationized bovine serum albumin and presented to DCs in soluble, phagocytosable, or non-phagocytosable display modalities. The functional DC response to the glycoconjugates was assessed via a high throughput assay. Dendritic cell phenotypic outcomes were placed into a multivariate, general linear model (GLM) and shown to be statistically different amongst display modalities when comparing similar surface areas. The GLM showed that glycoconjugates that were adsorbed to wells were the most pro-inflammatory while soluble conjugates were the least. DC interactions with mannose conjugates were found to be calcium dependent and could be inhibited via anti-DC-SIGN antibodies. The results of this study aim to resolve conflicts in reports from multiple laboratories showing differential DC profiles in response to similar, if not identical, ligands delivered via different modalities. Additionally, this study begins to bridge the gap between microarray binding data and functional cell responses by highlighting the phenotypes induced from adsorbed glycoconjugates as compared to those in solution or displayed on microparticles.
Introduction
Soluble and phagocytosable particle-based glycan presentation to antigen presenting cells (APCs) has been previously explored; however, direct comparative data between these versus non-phagocytosable display of glycoconjugates has not yet been performed.1,2 Qi et al.3 examined the differential effect of β-glucan in particulate (nanoparticle) or soluble form on dendritic cell (DC) phenotype. They found that β-glucan particles, derived from yeast, activated DCs and macrophages via Dectin-1 stimulation and that β-glucan delivered in its soluble form caused no increase in activation marker expression.3 However, β-glucan particles are inherently heterogeneous in molecular weight, size, glycan structural composition, and frequently have variability in protein composition. Thus, direct relationships between cell response and ligand factors were difficult to conclude from this study. Another study comparing particulate and soluble presented carbohydrates was performed by Le Cabec et al.4 who showed that mannose receptor (MR) expressed in transfected Chinese hamster ovary (CHO) cells mediated endocytosis of mannosylated glycoproteins in solution but did not support phagocytosis of three of its known particulate ligands: zymosan, Mycobacterium kansasii, and mannosylated latex beads. Furthermore, a differential cell phenotype was observed when identical ligands were presented in soluble or particulate modalities. However, CHO cells were transduced to express human MR; therefore strict conclusions between these cells and primary human APCs is difficult. These previous studies highlight the need for well-controlled comparative studies of DC phenotypic responses to glycoconjugates depending on the method of glycan display.Previous studies have shown that modulation of DC phenotype via phagocytosable nano- and micro-particles bearing carbohydrates is possible.5–9 These studies reported that increased ligand density as well as sugar structure play an important role in DC phenotype modulation. Thus, it has been postulated that glycans presented on particles are recognized by DCs with functional phenotypic effects. However, it has not been demonstrated if this effect is similar when identical ligands are presented via a non-phagocytosable modality. This has relevance to the fields of glycobiology, immunology, and biomaterials because interrogating immobilized glycans presented on the surface of microarrays is a common screen for potential candidates for glycan-based therapeutics. Thus, if the cell response to these surface immobilized glycans is not similar to that of soluble or phagocytosable display of glycans then further study and platform development is needed to address these separate modalities of glycan display. The non-phagocytosable display of glycan to DCs has been interrogated by van Vliet et al. who showed that non-phagocytosable N-acetylgalactosamine (GalNAc) drastically altered DC mobility as compared to other sugars.1 However, no surface marker or cytokine expression was measured for these DCs and thus their overall phenotype, due to these surface displayed glycans, is unknown. Taken cumulatively these studies establish that the mode of glycan presentation can alter DCs phenotype and thus a direct comparison between display modalities using a well-controlled system and defined ligands is needed to quantify these effects.
A platform of bovine serum albumin (BSA) cationized with ethylenediamine (EDA) and modified with the monosaccharides mannose or glucose was chosen for the studies herein. BSA was chosen because Oyelaran et al.25 showed that it was capable of scaling in density to a physiologically relevant density (>20 sugars/protein) that drastically enhanced binding by the carbohydrate-specific protein, C-type lectin receptors (CLRs). Additionally, many studies have shown that BSA is a non-activating support from which to deliver molecules to DCs.36,37 Furthermore, display of glycans in a relatively physiological setting (conjugated to a protein backbone instead of synthetic polymer) was seen as advantageous, as glycoproteins are commonly encountered by DCs in vivo. For example, DCs encounter glycoproteins in circulation because glycoproteins are estimated to represent approximately 50% of all human serum proteins.38
To date, no direct studies have been performed that compare how the charge of a glycoconjugate alters DC phenotype. Historically, cationic vaccine conjugates have been found to enhance the immunogenicity of the vaccine.39–43 Additionally, a recent study by Hotaling et al.10 showed that highly cationized carrier proteins produced highly active glycoconjugates for modification of DC phenotype.10 Glucose was selected as a negative control because no known CLR on DCs can bind to the monosaccharide glucose. Mannose was chosen because mannose-binding CLRs on DCs are extensively studied and well characterized. Also, DCs constantly encounter mannose-rich glycans in different modalities: soluble glycoproteins (plasma glycoproteins),44 particulate bound glycoproteins (bacterial and viral surfaces),45,46 and non-phagocytosable glycoproteins (endothelium and parasites).47,48 The outcome from stimulation by each of these modalities ranges from tolerogenic to pro-inflammatory.
Experimental
Overall experimental approach
In-house prepared glycoconjugates were produced as per the methodology in Hotaling et al.10 Briefly, thiol-oligoethylene glycol (SH-OEG2) functionalized mannose or glucose (Sussex Research; Ottawa, Canada) or OEG3-SH were reduced in tris(2-carboxyethyl)phosphine hydrochloride (TCEP) reducing gel (Pierce; Rockford, IL) in sealed spin cups (Pierce; Rockford, IL) for one hour in degassed buffer 1 (0.1 M EDTA, 0.15 M NaCl, 0.1 M NaH2PO4) at room temperature (RT). Glycans were then added to 1 mg ml−1 maleimide functionalized BSA (Pierce; Rockford, IL) in a 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sugar:carrier molar ratio. Argon gas was passed over the solution and the tubes were sealed with paraffin and allowed to react for 16 hours at RT. After conjugation the glycoconjugates were purified with 10 K Membrane Centrifugal Filter Units (Millipore; Darmstadt, Germany) using 9 rounds of 1:10 buffer exchanges against distilled, endotoxin free, water. Glycoconjugates were then cationized using a stock 1 mg ml−1 glycoprotein solution and adding 1.8 M EDA (pH 4.5) in a 1:1 volume ratio to the glycoprotein. To these solutions 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) was added to a 7.5 mM concentration. The resultant solution was allowed to react for two hours at RT while being shaken at 900 RPM. After conjugation the glycoconjugates were purified as discussed above. The overall experimental approach is shown in Fig. 1 including glycan modification of the maleimide functionalized BSA, cationization of the BSA with 0.9 M EDA, the subsequent adsorption of the glycoconjugates onto 384-well plates or microbeads, and finally assessment of DC phenotype to conjugates that were either soluble, bead adsorbed (BA), or adsorbed to wells (AW) of a 384-well plate.
:1 sugar:carrier molar ratio. Argon gas was passed over the solution and the tubes were sealed with paraffin and allowed to react for 16 hours at RT. After conjugation the glycoconjugates were purified with 10 K Membrane Centrifugal Filter Units (Millipore; Darmstadt, Germany) using 9 rounds of 1:10 buffer exchanges against distilled, endotoxin free, water. Glycoconjugates were then cationized using a stock 1 mg ml−1 glycoprotein solution and adding 1.8 M EDA (pH 4.5) in a 1:1 volume ratio to the glycoprotein. To these solutions 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) was added to a 7.5 mM concentration. The resultant solution was allowed to react for two hours at RT while being shaken at 900 RPM. After conjugation the glycoconjugates were purified as discussed above. The overall experimental approach is shown in Fig. 1 including glycan modification of the maleimide functionalized BSA, cationization of the BSA with 0.9 M EDA, the subsequent adsorption of the glycoconjugates onto 384-well plates or microbeads, and finally assessment of DC phenotype to conjugates that were either soluble, bead adsorbed (BA), or adsorbed to wells (AW) of a 384-well plate.
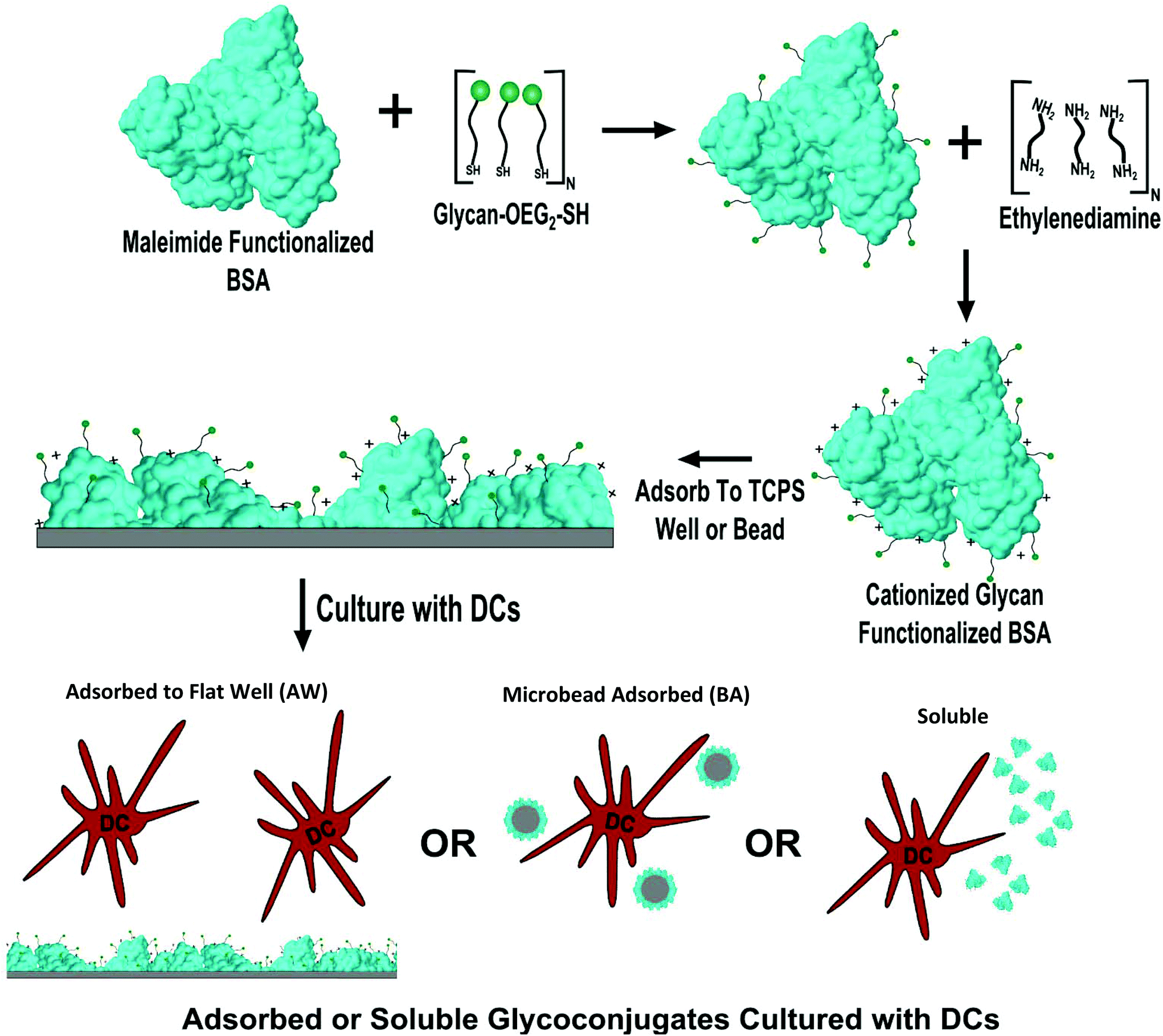 | ||
| Fig. 1 Overall methodology for display of glycoconjugates to DCs. First, maleimide functionalized bovine serum albumin functionalized with an OEG-thiol or glycan–OEG-thiol. After functionalization with glycans/OEG conjugates were aminated via ethylenediamine to increase their isoelectric point. Conjugates were then adsorbed to TCPS of 384-well plates, adsorbed to 1 μm beads or delivered in a soluble display modality to DCs and cultured for 24 hours. | ||
Materials and methods
Preparation and assessment of ζ-potential, mass and endotoxin content of glycoconjugates
All mass spectra and ζ-potential measurements were performed in an identical manner to that of Hotaling et al.10 Briefly, mass spectra of the glycoconjugates were determined using Matrix Assisted Laser Desorption Ionization (MALDI) Mass Spectrometry. The glycoconjugates were first dissolved in ultrapure, endotoxin free water, and then spotted in a 1:1 vol ratio with diammonium hydrogen citrate (DHC) onto a MALDI plate. A linear positive detection method was used for the conjugates. Mass profiles were then exported, plotted and the mean of each mass peak was determined. No significant crosslinking of maleimide conjugates was seen in the spectra. A representative spectra of the conjugates can be seen in Fig. S3 in the ESI.† To determine the isoelectric point (pI) glycoconjugates were diluted to 500 ng ml−1 in ultrapure endotoxin free water and then each conjugate was divided between five different cuvettes (PCS1115, Malvern). The pH in each cuvette was then adjusted to 3.0, 5.0, 7.0, 9.0, or 11.0 using 1 M sterile NaOH or 1 M sterile HCl. Using a Malvern Zetasizer Nano (Malvern, Malvern, Worcestershire, UK.) the ζ-potential and hydrodynamic radius of each solution was then determined. The pH vs. the ζ-potential was then plotted and the subsequent pI of each conjugate was then determined via interpolation of the least squares regression to which point the ζ-potential equaled 0.
The endotoxin contents of the glycoconjugates at a concentration of 100 μg ml−1 (5× the coating concentration used) were measured using an endotoxin assessment kit and the manufacturers recommended protocol (QCL-1000 LAL assay, Lonza). The endotoxin content of all glycoconjugates was determined to be less than 0.2 EU mL−1, which is well below the FDA limit of 0.5 EU mL−1. Furthermore, all mannose conjugates were below the detection limit of the assay for endotoxin content.
Binding assay of recombinant human C-type lectin receptors (CLRs) to glycoconjugates presented in an AW modality
384 well tissue culture polystyrene (TCPS) plates were coated with 20 μg ml−1 of the glycoconjugates overnight at RT (AW modality). During this incubation, recombinant human Dectin-1 (rhDectin-1) or recombinant human Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin-Fc chimeras (rh-DC-SIGN-Fc) (R&D Systems; Minneapolis, MN) were biotinylated individually according to the manufacturer's direction using the ChromaLink™ Biotin Protein Labeling Kit (Solulink Inc, San Diego, CA). Briefly, biotin-PEG3-bis(arylhydrazine)succinimidyl ester dissolved in dimethylformamide at 5 mg ml−1 was added to rhDectin-1 or rhDC-SIGN-Fc in a 10:1 biotin to protein molar ratio and allowed to react for two hours at RT mixing at 900 RPM. Subsequently, proteins were purified via provided Zeba spin columns (Pierce; Rockford, IL) and then diluted to 15 μg ml−1 with lectin buffer (0.1 mM MgCl2, 0.1 mM CaCl2, and 1× phosphate buffered saline (PBS)) (Sigma; St. Louis, Missouri). The extent of biotinylation was then confirmed via included standards and UV fluorescence at 354 nm.
After the completion of the overnight incubation, all wells with glycoconjugates were washed and blocked for two hours at 37 °C with block solution (1× PBS, 5 mg ml−1 biotin-free BSA, 1 mM MnCl2, 1 mM CaCl2 and 0.1 wt% TWEEN20). After blocking, the plates were then washed 5 times with the wash solution 3 (0.5 mg ml−1 BSA in 0.1× PBS, 0.1 mM MnCl2, 0.1 mM CaCl2, and 0.01 wt% TWEEN 20). Next, a 15 μg ml−1 rhDectin1-biotin or 15 μg ml−1 rhDC-SIGN-FC-biotin (diluted in lectin buffer) was incubated with the adsorbed conjugates for three hours at 37 °C or overnight at 4 °C. The plates were then washed 5 times with wash solution 3 and 40 μl of a streptavidin-(horseradish peroxidase) (BD Pharmingen; San Jose, CA) solution diluted 100× with lectin buffer from stock was added to each well and allowed to incubate in the well for 1 hour at RT. The plate was then washed 5× more with the wash solution 3 and a TMB (3,3′,5,5′-tetramethylbenzidine-peroxide) substrate (BD Pharmingen; San Jose, CA) was added and the plates were allowed to develop for 10 minutes. Sulfuric acid (1.0 N) was then added to stop the reaction and the absorbance at 450 nm was determined.
Dendritic cell isolation and culture
Culture and high throughput (HTP) assessment of DC phenotype upon treatment with glycoconjugates presented on various modalities of display was performed as previously presented.11 Briefly, human blood was collected from healthy, consented donors and heparinized (333 U ml−1 blood) (Abraxis Pharmaceutical Products, Schaumburg, IL), in accordance with protocol H10011 of the Georgia Institute of Technology Institutional Review Board. Dendritic cells were derived from human peripheral blood mononuclear cells (PBMCs). PBMCs were isolated by differential centrifugation using lymphocyte separation medium (Cellgro MediaTech, Herndon, VA). After the lysis of residual erythrocytes with red blood cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), the PBMCs were washed with D-PBS and then PBMCs were plated at a concentration of 5 × 106 cells ml−1 in DC medium. After 2 hours of incubation for the selection of adherent monocytes, the dishes were washed and the remaining adherent monocytes were incubated with DC media, supplemented with 1000 U ml−1 recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) and 800 U ml−1 recombinant human interleukin 4 (rhIL-4) (PeproTech, Rocky Hill, NJ), for 5 days to induce the differentiation of monocytes into iDCs.Glycoconjugate presentation to DCs in three modalities
For all experiments where glycan conjugates were presented in an AW modality a 384 well tissue culture polystyrene (TCPS) plate was coated with 20 μg ml−1 of the BSA glycoconjugates dissolved in PBS overnight at RT. All the wells were subsequently washed with complete DC medium [RPMI-1640 (Invitrogen; Grand Island, NY), 10% heat-inactivated fetal bovine serum (FBS) (Cellgro MediaTech; Herndon, VA) and 100 U ml−1 of penicillin/streptomycin (Cellgro MediaTech)] five times and blocked for two hours at 37 °C with 5 mg ml−1 biotin-free human serum albumin (HSA) in 0.1 M NaHCO3. After blocking, the plates were then washed 5× with complete DC media and 40 μl of cells at 7.5 × 105 cells ml−1 (3.0 × 104 cells per well) were added to each well and allowed to incubate for 24 hours.For all soluble conjugate treatments wells of a 384-well plate were pre-coated with complete DC medium overnight. The wells were then washed with complete DC medium five times and blocked for two hours at 37 °C with 5 mg ml−1 biotin-free HSA in 0.1 M NaHCO3. Glycoconjugates were dissolved in complete DC medium (at concentrations starting at 100 μg ml−1 with 1:10 dilutions down to 10 ng ml−1) and used to resuspend DCs, which were then immediately added to the pre-blocked wells of the 384-well plate for a 24 hour incubation. The phenotype of the DCs was then assessed using the HTP methodology discussed below. To ensure that soluble conjugates were not able to adsorb to wells a binding assay similar to that performed above was performed on the wells after blocking with complete DC medium and adding the soluble glycoconjugates. After a 24 hour incubation it was found that due to the diverse glycosylation profile of the proteins in FBS large background noise was generated from the adsorbed proteins even if soluble conjugates were not added (data not shown). To ensure that this non-specific glycoprotein adsorption was not affecting DC maturation DCs were cultured on medium pre-treated wells and they showed no activation and thus the background signal induced from FBS glycoproteins was considered not important to DC activation.
For all experiments where DCs were treated with glycoconjugates that were bead adsorbed (BA), wells of the 384-well plate were pre-coated with complete DC medium overnight. The wells were then washed with complete DC medium five times and blocked for two hours at 37 °C with 5 mg ml−1 biotin-free HSA in 0.1 M NaHCO3. Polystyrene beads (1 μm; Fisher Scientific, Waltham, MA) were coated with 20 μg ml−1 of the BSA glycoconjugates dissolved in PBS overnight at RT. Beads were then centrifuged at 10 K RCF for 3 minutes, supernatants removed, and beads resuspended in complete DC medium for dispersion of beads by vortexing. This process was repeated twice more to wash beads. Beads were then resuspended to their original volume in complete DC medium and added to DCs in the blocked wells of the 384 well plate at bead numbers corresponding to 0.2×, 1.0×, 5.0× and 25.0× the surface area of a well and incubated for 24 hours. The phenotype of the DCs was then assessed using the HTP methodology discussed below.
Phenotypic assessment of DCs
Forty microliters at 7.5 × 105 cells ml−1 (3.0 × 104 cells per well) of DCs in complete medium were placed in treatment wells on day 5 after isolation. After 24 hours all treated DCs and controls were transferred via multi-channel pipette to a black 384-well filter plate (Pall Life Sciences; Port Washington, NY), and the supernatants were immediately collected into a 384-well plate through the filters by stacking the filter plate on top of the collection plate and centrifuging at 300 RCF for 4 min. While spinning down the filter plate, wells of the TCPS plate with glycoconjugates adsorbed to them were incubated with Non-Enzymatic Cell Disassociation Solution (CDS; Sigma; St. Louis, Missouri). The CDS-treated cells were then lightly pipetted up and down and transferred to the black filter plate after its first spin-down. The CDS was removed by stacking the filter plate on top of a new collection plate and centrifuging at 400 RCF for 4 min. To the retained cells 50 μl of 0.05% formaldehyde solution was added and the cells were allowed to fix for 40 minutes at RT while being shaken at 600 RPM. The formaldehyde solution was then removed via centrifugation at 400 RCF for 4 minutes. The cells retained in the wells were assessed for phenotype by immunostaining using antibodies anti-CD86-PE (Clone BU63; Ancell), anti-DC-SIGN-FITC (Clone 120507; R&D Systems; Minneapolis, MN), and anti-ILT3-AF647 (Clone ZM4.1, Biolegend; San Diego, CA). IgG1-PE (clone MOPC31C; Ancell; Bayport, MN) IgG2B-FITC (clone 133303; R&D Systems; Minneapolis, MN); and IgG1κ-AF647 (MOPC-21, Biolegend) isotype-stained DCs were used for background fluorescence subtraction in separate treatment for control wells. CD86 is a costimulatory molecule that is up-regulated upon pro-inflammatory DC maturation,11 DC-SIGN is an endocytic receptor that is slightly down-regulated upon pro-inflammatory maturation,11 and ILT3 is a member of the immunoglobulin superfamily which signals via the immunoreceptor tyrosine-based inhibitory motifs and is up-regulated upon anti-inflammatory DC maturation.12 After 30 minutes of staining the cells were washed three times. The geometric mean fluorescent intensities (gMFIs) were then calculated for each fluorophore (excitation/emission wavelengths: 535/590 PE, 485/535 FITC, and 650/668 AF647) with a Tecan Infinite F500 microplate reader, and the ratio of respective gMFIs were determined as CD86/DC-SIGN, a cell number independent metric named “inflammatory maturation factor” (IMF), and ILT3/CD86, a cell number independent metric named “tolerogenic maturation factor” (TMF) was used to represent DC phenotypic outcomes. The extent of DC maturation was compared to untreated DCs (iDCs) for the negative reference control, lipopolysaccharide (LPS) (1 mg ml−1; E. coli 055:B5; Sigma; St. Louis, Missouri)-treated DCs (mDCs) for the IMF control, and recombinant human interleukin 10 (rhIL10) and recombinant human interferon α (rhIFNα) (R&D Systems; Minneapolis, MN) at 3500 units ml−1 and 35000 units ml−1 respectively for the TMF control (tDC).
Assessment of DC uptake of fluorescent glycoconjugates
To assess uptake of glycoconjugates presented as AW, or as soluble glycoconjugates, the conjugates were fluorescently modified with Alexa-fluor-488-TFP Ester (AF488, Invitrogen according to manufacturer's directions). Briefly, cationized glycan functionalized glycoconjugates were incubated with AF488, 5 mg ml−1 in sterile PBS, at a 10:1 AF488 to protein molar ratio (1 hour, RT). After conjugation, the glycoconjugates were purified using 10 kDa molecular weight cut-off Membrane Centrifugal Filter Unit (Millipore) using 9 rounds of 1:10 buffer exchanges against distilled, endotoxin free, water and stored in the dark. When delivered to cells in a soluble form, all wells were pre-coated with complete DC medium overnight prior to addition of cells or soluble conjugates.
Assessment of DC uptake of soluble, WA, BA, or fluorescent glycoconjugates
All studies where ethylenediaminetetraacetic acid (EDTA) or blocking antibodies were used to block CLR receptors, cells were treated with either 10 mM EDTA, 10 μg ml−1 of mouse anti-human Dentin-1 (clone 259931, R&D Systems; Minneapolis, MN), 10 μg ml−1 of mouse anti-human DC-SIGN (clone 120507, R&D Systems; Minneapolis, MN) or 10 μg ml−1 of mouse anti-human IgG2B (Clone 20116, R&D Systems; Minneapolis, MN) for 30 minutes at 37 °C before exposure to soluble, WA, or fluorescent BA conjugates (1 μm Purple high intensity, Exc./Emm. 590 nm/630 nm, Spherotech; Lake Forest, IL). Similarly, for the negative control, cells were incubated at 4 °C for 30 minutes prior to exposure to the glycoconjugates in each modality and then maintained at 4 °C for four hours in the presence of the fluorescent conjugates or coated fluorescent microbeads. The 4 °C treatment is a common non-specific inhibitor of DC phagocytosis and thus was seen as a negative control and non-specific inhibitor for DC phagocytosis. EDTA is a common inhibitor of CLR activity in DCs because it chelates calcium and prevents these calcium dependent receptors from forming a functional binding pocket. However, EDTA also has broad effects on DC behavior.13 Thus, two blocking antibodies specific for common, well-characterized CLRs, Dectin 1 and DC-SIGN, were chosen for the bead phagocytosis assays to show specific inhibitory ability of DC interaction with conjugates. Cells were then transferred, with media still containing EDTA or antibody (where applicable) to the wells with the fluorescent BA glycoconjugates in a 1:10 cell to bead ratio and the subsequent phagocytosis was assessed after four hours.
For assessment of phagocytosis of soluble, AW or BA conjugates cell suspensions were pipetted up and down vigorously 3 times and then transferred to 1.5 ml eppendorf tubes. Cells were then spun down at 300 RCF for 10 minutes and the resultant supernatant was removed. Cells were washed with PBS, fixed with 1% formaldehyde for 30 minutes, washed again with PBS, and incubated with 0.1% trypsin for 1 minute. Cells were then washed three times with PBS, and phagocytosis was quantified via flow cytometry (BD LSR II Flow Cytometer, BD Biosciences; San Jose, CA).
Viability/cytotoxicity and apoptosis assessment of glycoconjugates
Cytotoxicity associated with DC responses to glycoconjugate treatment was assessed via live/dead staining. The amount of cell apoptosis was of interest due to possibility that cells were impermeable to ethidium homodimer but still in the process of apoptosis. To assess apoptosis DCs were stained with Annexin V-FITC (BD Biosciences; San Jose, CA) and the extent of binding to surface phosphatidylserine was measured. No treatments showed a significantly altered viability compared to untreated cells, and no treatment showed a statistical increase in Annexin V binding except for 100 μg ml−1 β-glucan, which showed a statistically significant increase in Annexin V from untreated cells (Data shown in Fig. S1†). Dead cell controls were freeze–thawed two times prior to placement into wells.Statistical analysis
To observe any significant differences between all sample groups in pairs, a pairwise repeated measures two-way ANOVA followed by Tukey's post test was performed using SAS software (Cary, NC), and the p-value equal to or less than 0.05 was considered significant. Significance of general linear statistical model parameters discussed in this report and seen in Model 1a and b and Model 2 was determined by T value in reference to the referent group discussed in the methods below.Statistical modeling
Table S1 in the ESI† lists the quantitative parameters that were collected and separates them by variable classification: Continuous and categorical/nominal. Table S1A† contains IMF and TMF continuous variables. Table S1B† contains the nominal variables: ligand, modality, and donor. There were 20 total donors for this analysis.Model 1a and b show the general linear models that have IMF (a) or TMF (b) as outcome variables and are a function of the ligand conjugated to BSA, modality of display, and the donor. The models in Model 1a and b isolated and compared the effect of presentation modality on DC IMF (Model 1a) and TMF (Model 1b) when controlling for ligand and donor variations. The null hypothesis was that modality of presentation did not play a role in IMF or TMF and thus that this variable would not have a significant T value when compared to the referent group discussed below.
| IMF = β1 + β2 × donor + β3 × ligand + β4 × modality | (1a) |
| TMF = β1 + β2 × donor + β3 × ligand + β4 × modality | (1b) |
For the comparative model used, the R2 was calculated to determine how well the model fits the data. The R2 value of Model 1a was 0.874 and thus the model was seen as a reasonably good model for the data. The R2 value of Model 1b was 0.723 and thus the model was seen as a reasonably good model for the data. Furthermore, IMF data has historically been shown to be approximately of a normal distribution and the variance of the data remains constant across all samples thus the linear model used herein was further deemed as a valid analysis method.11
Model 2 shows a GLM that has IMF as an outcome variable and is a function of donor used (donor), ligand linked to BSA (ligand), modality of presentation (modality), and the interaction between modality and ligand (ligand × modality). Model 2 isolated and compared the pairwise comparisons between all pairs of ligand on BSA and modality of display of that ligand. The null hypothesis was that no ligand-modality combination would be different from each other. To make this comparison, after the ANOVA was performed, all ligand-modality combinations were compared using Tukey's Post-test. More discussion of the models, their references, and variables can be found in the ESI† of this report.
| IMF = β1 + β2 × donor + β3 × ligand + β4 × modality + β5 × ligand × modality | (2) |
Results
Characterization of glycoconjugates
Table 1 shows the isoelectric point (pI) and hydrodynamic radius for each conjugate. The mean pI of each conjugate was approximately equal for all conjugates and was close to a pI of 10. Hydrodynamic radii of the conjugates were found to decrease with functionalization of ligand from a radius of approximately 5 nm for highly cationized bovine serum albumin (HBSA) to approximately 3 nm for HBSA linked to OEG3 (HBSA–OEG). Approximately 25 glycans/BSA were immobilized on each conjugate with OEG having an average of approximately 4.4 OEG linkers per BSA as determined by MALDI.| Name | Ligands | Isoelectric point | Hydrodynamic radius (nm) |
|---|---|---|---|
| BSA | |||
| HBSA | 0.00 | 10.08 | 5.12 |
| HBSA–OEG | 4.41 | 9.85 | 2.97 |
| HBSA–Glc | 26.20 | 10.10 | 3.62 |
| HBSA–Man | 23.50 | 9.63 | 2.99 |
Recombinant human C-type lectin receptors (rhCLRs) are able to bind to glycoconjugates presented in an AW modality
A binding assay using recombinant human CLRs was performed for all glycoconjugates presented in an AW modality in order to confirm that they had bioavailable glycans for DC–CLR interaction. Fig. 2A shows that rhDC-SIGN-Fc was able to bind to all of the adsorbed conjugates and that the adsorbed HBSA–Man conjugates were able to bind more rhDC-SIGN-Fc than mannan, the positive control. Fig. 2B shows little binding of rhDectin-1 to any of the conjugates as no signal above the detection limit of the assay was seen for binding to HBSA–Glc, HBSA–Man, or mannan. The positive controls (Mannan for DC-SIGN and β-glucan for the Dectin-1) both showed high binding of DC-SIGN and Dectin-1, respectively.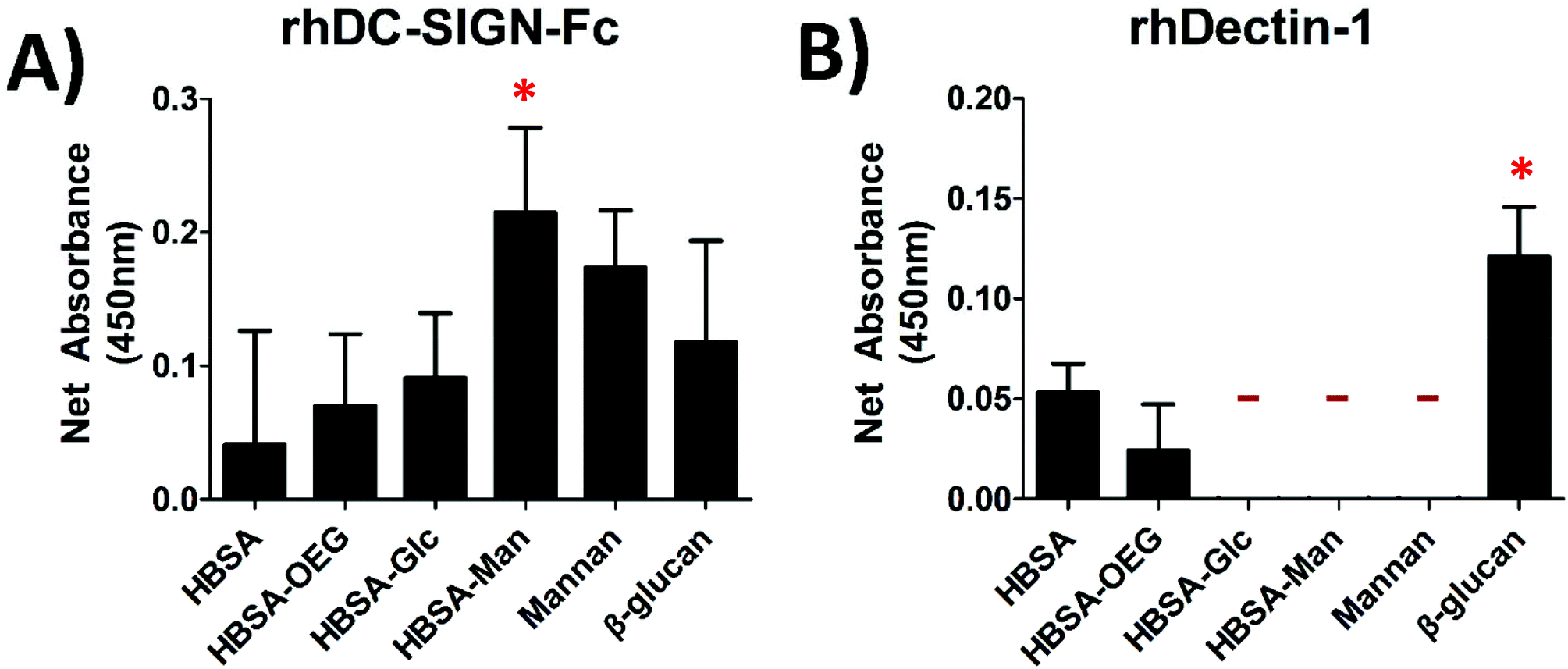 | ||
| Fig. 2 Binding assay for glycan recognition by recombinant human CLRs, rhDC-SIGN-Fc and rhDectin-1, showed CLR specificity for the conjugates. (A) Biotinylated rhDC-SIGN-Fc was incubated with adsorbed conjugates and the subsequent mean absorbance for each conjugate was measured. (B) Shows biotinylated rhDectin-1 incubated with adsorbed conjugates. All signals are background subtracted from untreated wells. N = 4 Trials, 3 wells/trial. “-” indicates below detection limit of the assay. * indicates statistical difference from HBSA. | ||
Dendritic cell response to glycoconjugates was different amongst the three display modalities for DC “inflammatory maturation factor” (IMF)
Assessment of DC responses to glycoconjugates presented in three different modalities shows that the highest level of DC Inflammatory Maturation Factor (IMF) response (Fig. 3E) was found when glycoconjugates were presented in an AW modality. Presentation of glycoconjugates in the soluble form (Fig. 3A) resulted in the lowest effect on DC IMF. Presentation of glycoconjugates in a BA modality resulted in an intermediate level of induced IMF expression by DCs (Fig. 3C). Fig. 3A shows that when control ligands LPS, mannan, or β-glucan in a BA modality (at any bead to well surface area ratio) all caused a statistically significant increase in DC IMF. LPS adsorbed to beads resulted in the highest level of DC activation, with significant increases in IMF for all bead to well surface area ratios. Mannan or β-glucan adsorbed to beads showed a significant increase in DC IMF for 25×, and 5× bead to well surface area ratios, respectively. No other treatments were different from untreated DCs (iDC) except the positive control (mDC). However, of note is the increased trend in DC IMF levels for 5× and 25× bead to well surface area ratios for both HBSA–Glc conjugates and HBSA–Man conjugates. No significant increase in DC TMF was observed for any treatment, except for the positive control of tDC.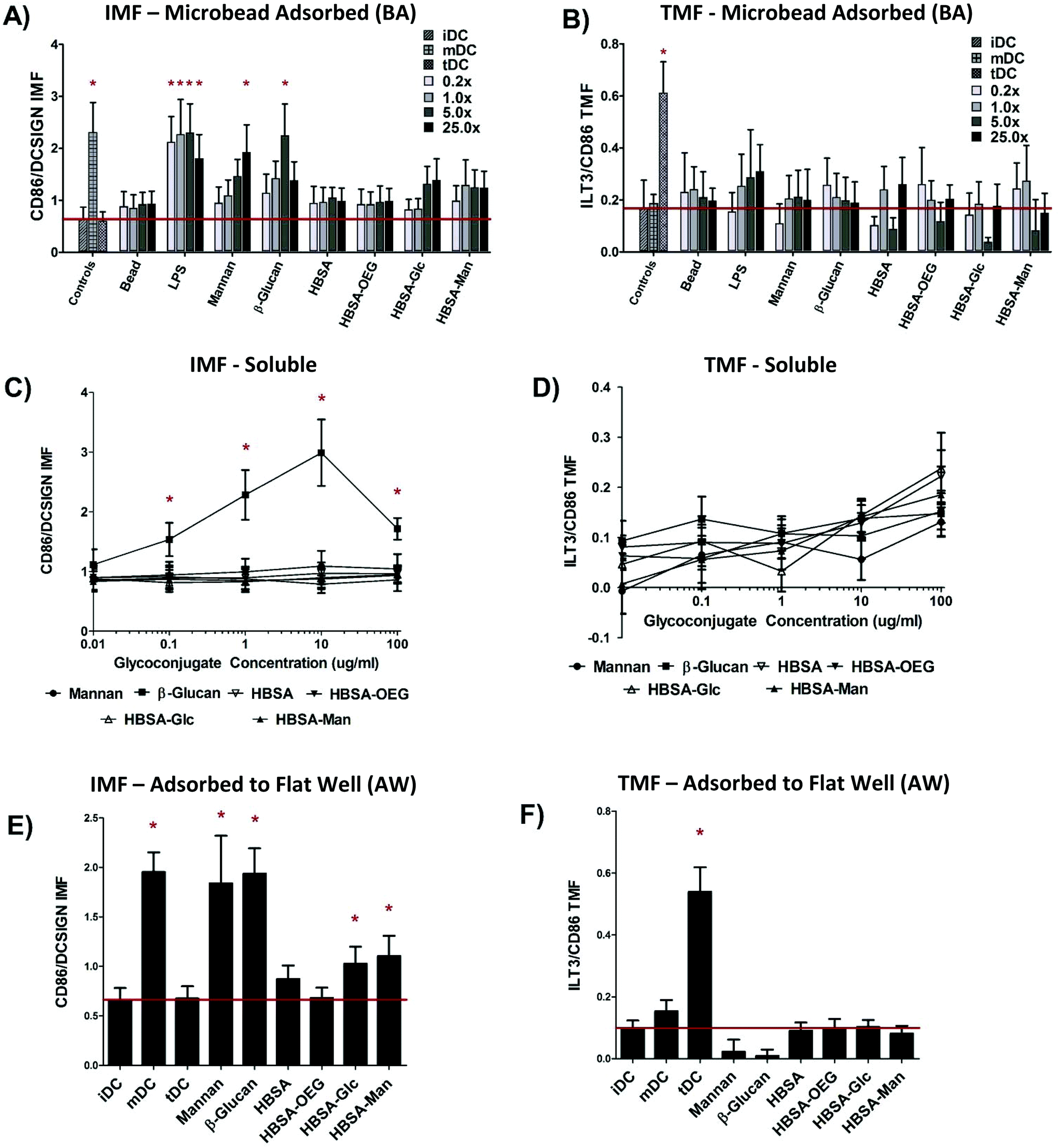 | ||
| Fig. 3 DC Response to glycoconjugates presented in three modalities of display. (A), (C), and (E) show the IMF response of DCs to the conjugates and (B), (D), and (F) show the TMF response from DCs. (A) and (B) show the DC response to glycoconjugates when in a BA modality in 0.2×, 1.0×, 5.0× and 25.0× the surface area of a well. Bead surface area was scaled by increasing bead number until the desired ratio was reached. (C) and (D) indicate DC response to soluble conjugates across five orders of magnitude of concentration. (E) and (F) show DC response to conjugates in an AW modality. For (A–D) N = 6 donors, (E) and (F) N = 12 donors. Error bars represent standard error, red line indicates mean iDC response, * indicates statistical difference from iDC. | ||
The trend of increasing DC IMF level with increasing bead to well surface area ratio was then analyzed using a GLM in which donor and treatment were controlled for and bead to well surface area ratios were compared. Using Tukey's Post-test, all pair-wise comparisons between bead to well surface area ratios were performed and the results are shown in ESI Table S2.† Levels of DC IMF were significantly different for both 5× and 25× bead to well surface area ratios as compared to the levels for 0.2× or 1.0×. However, DC IMF levels were not statistically different for 0.2× as compared to 1.0× or when comparing 5× to 25× bead to well surface area ratios. No significant increase in DC TMF for any BA conjugate was seen except for the TMF positive control, tDC.
No concentration of soluble glycoconjugate caused any significant increase in DC IMF (Fig. 3C) or TMF (Fig. 3D). However, when control ligand β-glucan was delivered to DCs at concentrations between 100 ng ml−1 and 100 μg ml−1, DC IMF levels increased significantly over that of untreated cells. Interestingly, β-glucan treatment at 100 μg ml−1 did not cause the highest level of IMF expression in treated DCs. To test if this was due to cell death, DC apoptosis was assessed (fluorescent intensity of Annexin V-FITC) and a statistically significant increase in apoptosis of DCs at this β-glucan concentration was found and can be seen in Fig. S1.†
Fig. 3E shows that when control ligands Mannan or β-glucan were presented in an AW modality to DCs, IMF levels increased significantly over that of untreated iDCs. Similarly, DCs treated with LPS, the positive IMF control, also showed statistically higher levels of IMF as compared to iDCs. Interestingly, HBSA–Glc or HBSA–Man presented in an AW modality resulted in significantly higher levels of IMF as compared to that of iDCs. Fig. 3F shows that no significant increase in DC levels of TMF were observed upon DC treatment with any of the conjugates presented in an AW modality, except for the tDC control.
Glycoconjugate display modalities are significantly different for induced DC IMF levels when statistically modeled
Two sets of statistical models were constructed to determine whether DC phenotype differed amongst modalities of display when comparing identical conjugates. Model 1a and b assessed whether DC IMF or TMF response to modality of display was different, controlling for ligand of glycoconjugate and donor. Model 2 assessed which specific conjugates and display modality combinations were statistically different for DC IMF levels.The results of Model 1a show that DC IMF levels were statistically different for all modalities when controlling for ligand and donor; with all probabilities being lower than P < 0.0042 (Table 2). The results of Model 1b showed that for DC TMF levels no modalities were statistically different from each other.
| 1 μg ml−1 soluble | 1 μm bead | Well adsorbed | |
|---|---|---|---|
| 1 μg ml−1 soluble | <0.0001 | 0.0042 | |
| 1 μm bead | <0.0001 | <0.0001 | |
| Well adsorbed | 0.0042 | <0.0001 |
Based off of the results from Model 1a and b, a model assessing the interaction between ligand and modality was desired to determine which ligand/modality combinations resulted in significantly different DC IMF levels from each other. The key statistically significant result from Model 2 was that the level of IMF for DCs treated with HBSA–Man in an AW modality was statistically different from that for DCs treated with soluble HBSA–Man or soluble HBSA–Glc. An analysis of DC TMF using an interaction variable was not performed due to results from Model 1b showing no significant change in DC TMF for any modality of display.
Dendritic cell internalization of glycoconjugates in an AW, BA, or soluble modality was inhibited by blocking antibodies or EDTA treatment
Lectin mediated DC phagocytosis of glycoconjugate coated 1 μm beads was specifically inhibited using antibody blocking assays (Fig. 4). Phagocytosis of mannose glycoconjugate coated beads was significantly blocked by DC pretreatment with antibodies specific for the CLRs: DC-SIGN or Dectin-1. Phagocytosis was also inhibited by treatment with 10 mM EDTA or DC incubation at 4 °C (no statistical differences between EDTA and 4 °C treatment) (Fig. 4D). Dendritic cell uptake of uncoated or β-glucan coated beads was not affected by antibody blocking or EDTA treatment; however, phagocytosis of mannan coated beads was inhibited by EDTA (shown in ESI Fig. S2†). DC phagocytosis of adsorbed HBSA (Fig. 4A), HBSA–OEG (Fig. 4B) or HBSA–Glc was only significantly inhibited by 4 °C incubation of DCs.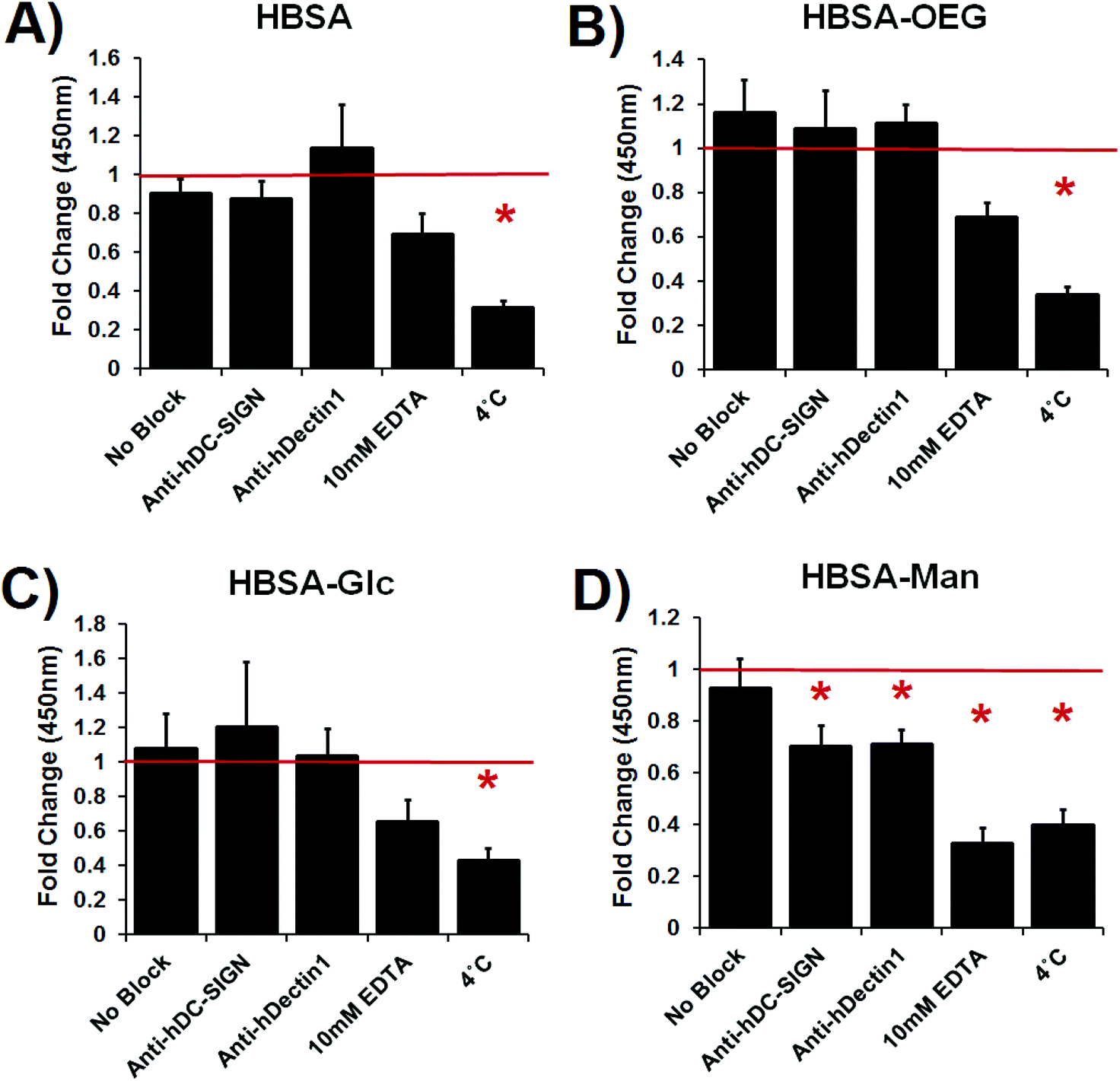 | ||
| Fig. 4 Quantification of phagocytosed 1 μm fluorescent beads with adsorbed conjugates in the presence of CLR blocking antibodies, EDTA, or 4 °C treatment. Data is fold change over isotype control treated DCs. N = 4 donors. Error bars represent standard error, red line indicates mean isotype control treated cells’ internalization of beads fluorescence, * indicates statistical difference from isotype control treated cells. | ||
The extent of internalization of glycoconjugates by DCs was assessed by quantifying uptake of fluorescently-labeled conjugates presented in an AW (Fig. 5A) or soluble (Fig. 5B) modality. In Fig. 5A, DC internalization of fluorescent HBSA–Man conjugates presented in an AW modality was significantly inhibited as compared to isotype antibody treated DCs when the DCs were pre-treated with anti-DC-SIGN, 10 mM EDTA or treated at 4 °C. Dendritic cell internalization of all other adsorbed fluorescent conjugates was only inhibited by the negative control treatment of 4 °C.
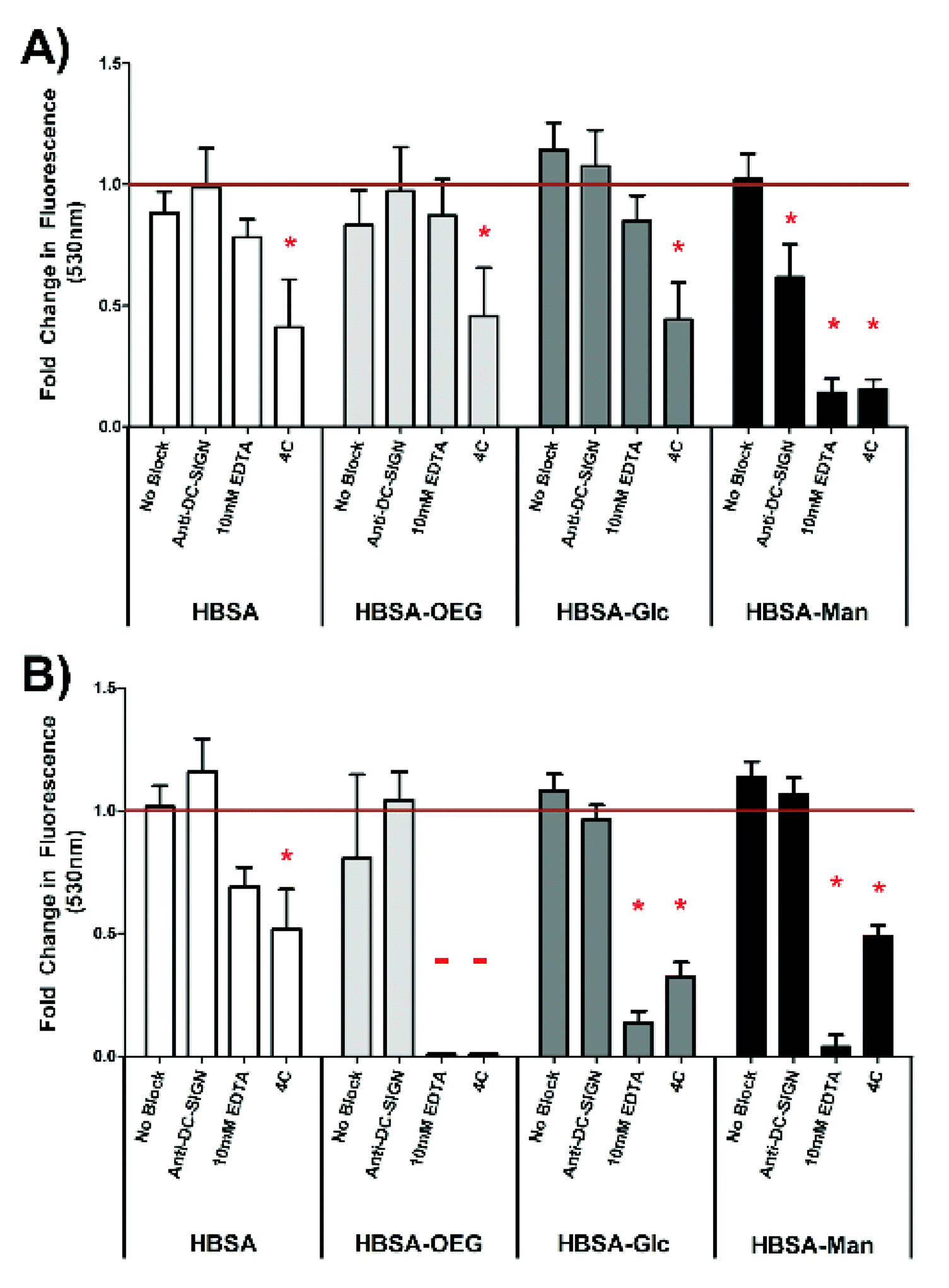 | ||
| Fig. 5 Quantification of internalized fluorescent conjugates presented in an AW modality (A) and 5 μg ml−1 soluble conjugates (B) in the presence of no blocking agent, CLR blocking antibody, EDTA, or 4 °C treatment. Net fluorescence measured by subtracting signal from DCs treated with non-fluorescent equivalent conjugate. Data is shown as fold change for DCs treated with the isotype control for the respective blocking anti-body. N = 3 donors. Error bars represent standard error, red line indicates mean isotype control treated cells’ internalization of beads fluorescence, * indicates statistical difference from isotype control treated cells. | ||
In Fig. 5B, internalization of soluble fluorescently-labeled HBSA–Man or HBSA–Glc conjugates by DCs was significantly inhibited, as compared to isotype antibody treated DCs, when the DCs were pre-treated with 10 mM EDTA or treated at 4 °C. Dendritic cell internalization of soluble fluorescently-labeled HBSA–OEG conjugates, when treated with 10 mM EDTA or at 4 °C, was below the detection limit of the assay. Fluorescently-labeled soluble HBSA conjugate internalization by DCs was only significantly inhibited by the negative control 4 °C treatment. Interestingly, when comparing the internalization of conjugates from an AW modality (Fig. 5A) to soluble conjugates (Fig. 5B), anti-DC-SIGN treatment significantly inhibited internalization of fluorescent conjugates in an AW modality but not soluble conjugates. Furthermore, EDTA shows a much greater inhibitory role in DC internalization of all soluble (other than fluorescent HBSA) conjugates than it does in conjugates presented in an AW modality, indicating a functional requirement for Ca2+ in internalization.
Discussion
A major finding from our studies is that DCs show a distinct response to identical glycoconjugates displayed in phagocytosable vs. non-phagocytosable forms. Differences between DC responses to the glycan presentation in the three modalities were confirmed using a GLM, Model 1a. Additionally, when comparing the raw DC IMF averages amongst modalities for both mannose- or glucose-containing glycoconjugates, DC activation IMF levels were highest for glycoconjugates in an AW modality, intermediate when conjugates were in a BA modality (even for high surface area ratios), and lowest for soluble conjugates delivered to DCs at the relatively high concentration of 100 μg ml−1. Surface area was chosen as the normalizing factor between modalities, rather than bead number or ratio of beads to cells, because it was desired that the results from the BA modality (Fig. 3A and B) be directly compared to those of an AW modality (Fig. 3E and F). Additionally, the difference in DC response to the three modalities, particularly the lower IMF effect of bead or soluble delivery of glycoconjugates was not due to an inability of the DCs to recognize conjugates. This was indicated in Fig. 4 and 5 which showed that DCs were able to recognize and internalize glycoconjugates in a BA modality or delivered at a large range of soluble concentrations. Furthermore, the implication that DC-SIGN is mediating, in part, DC interaction with glycoconjugates is supported by the recombinant human CLR binding assay results shown in Fig. 2.Dendritic cells are capable of interacting with both phagocytosable and soluble presentations of glycoconjugates as supported by both Fig. 3 and 4. However, direct comparisons between modalities could be impossible because glycan display density on flat well surfaces could be significantly different than that of the soluble or phagocytosable modalities. Because CLRs are multivalent receptors, this potential difference could enhance DC interaction with conjugates from a given modality. Determining quantitative estimates of glycan surface interaction with DCs is extremely technically challenging when looking at microbead presentation, and not possible when delivered in solution. Thus, large ranges in concentration and bead number were tested to increase the contact area of glycoconjugate per cell to mitigate this concern. Additionally, the same base material was used (polystyrene) for both the bead and microwell surface display. The glycoconjugate surface density between these two display modalities are predicted to be similar, as steric hindrance of protein adsorption on such relatively large beads (1 μm versus a maximum hydrodynamic radius of 5.2 nm for the conjugates) has been shown to not affect protein adsorption.14,15
DCs did not respond to soluble mannan at any of the concentrations tested. This result is contrary to observations in previous studies wherein soluble mannan was an immune agonist for DCs.6–8 However, in these previous studies, mannan was combined with particulates based on liposomes linked through membrane lipids, such as cholesterol6 or palmitoyl-mannan7 to exert the mannan effect. Therefore, the synergistic effect of the lipids particulate/surface along with the mannan could explain the activation reported in these studies. Indeed, in an elegant study by Wattendorf et al.5 human DCs were treated with phagocytosable PS microbeads functionalized with poly-L-lysine which then had mannan passively adsorbed onto the surface. This presentation was hypothesized to leave the mannan free in solution, and not adsorbed to the bead surface, and no DC maturation was found.
Statistical modeling allowed for the comparison between modality of presentation of conjugates controlling for different ligands and for repeated measures of donors. Model 1a and b showed a quantitative comparison of the different modalities seen in Fig. 2. The result that DC response to HBSA–Man in an AW modality was different from their response to soluble glycoconjugates was anticipated from a cursory comparison of the statistics shown in Fig. 3. This result was further strengthened when considering the antibody-blocking results in Fig. 4. Fig. 4D indicates that for beads coated with HBSA–Man, phagocytosis is mediated through lectin interaction and can be partially inhibited by blocking DC-SIGN. Additionally, Model 2 used the interaction variable between ligand and modality to assess effects of differences between modality/ligand combinations on DC IMF levels. The results of the modeling indicated that the IMF level for DCs treated with HBSA–Man presented in an AW modality was statistically different from soluble delivery of HBSA–Man and soluble HBSA–Glc. In this model, and in Model 1a and b, mannan and β-glucan were not included. Mannan and β-glucan were excluded because they showed clear differences in DC response across modalities. When they were included in the model the large differences from mannan and β-glucan were summed with the differences between glycoconjugates. Thus, each modality was seen as more significantly different when including mannan and β-glucan. Because these ligands were poorly defined, the differential DC response could have been due to factors other than modality and thus they were removed from the model to eliminate confounding effects.
Mannose presenting glycoconjugates are frequently used as agonists for vaccines and to increase DC recognition and uptake of particles.5,16,17 Thus, the lack of induction of tolerogenic DCs as measured through the TMF metric was expected. Similarly, no known effect of the monosaccharide glucose has been shown on DC phenotype. Thus, we expected to observe that no treatments or modalities were significant for the tolerogenic reporter. However, the tolerogenic response of the DCs to the glycoconjugates and controls was tested in all cases because CLRs on DCs are also known to be instrumental in promoting tolerance and maintaining immune cell homeostasis.18–21 The assessment of tolerogenic phenotypes is especially important for DC CLR ligation. It has been shown that identical CLR stimulation can promote tolerance or pro-inflammatory responses from DCs depending on the ligand bound and costimulatory molecules present.22–24 Thus, any methodology that assesses activation and pro-inflammatory responses from DCs using CLRs must also evaluate tolerogenic responses from DCs in order to ensure that both phenotypic outcomes are assessed.
Mass spectra of the conjugates showed that glycan moieties scaled in density with increased molar ratios of glycan. The scaling agrees well with results shown by Oyelaran et al.25 However, it was found that OEG conjugates did not appear to scale with increased molar ratios. The OEG conjugates were created at the same time with identical conditions to those of the other glycoconjugates. It was therefore concluded that the OEG linker mass was small enough that the number of functionalizations per BSA were within the experimental error of the MALDI mass spectra. While no direct evidence of this has been shown in the literature, the average standard deviation of the mass profile for each of the OEG conjugates was 11774 ± 6180 Da. With an average standard deviation of over 6 kDa and a maximum weight of OEG ligands reaching approximately 3 kDa it would be no surprise that the OEG linker weight could be lost in the noise of the mass profile.
The lectin binding assays shown in Fig. 2 showed that rhDC-SIGN-Fc was able to bind to HBSA–Man but neither rhDC-SIGN-F nor rhDectin-1 were able to bind HBSA–Glc. The magnitude of rhDC-SIGN binding to the HBSA–Man conjugates was seen to be greater in magnitude than that of mannan, the positive control; however, this difference was not statistically significant and thus was seen as a confirmation that ligand density was high enough for these conjugates to cause functional binding of rhDC-SIGN-Fc. Additionally, the higher mean fluorescence of the HBSA–Man conjugates from that of wells treated with HBSA, HBSA–OEG, or HBSA–Glc indicated that rhDC-SIGN-Fc was able to bind to the glycoconjugates with relatively high specificity. Therefore, from Fig. 2A it was inferred that HBSA–Man conjugates could be bound by CLRs found on DCs and that the activation of the DCs shown in Fig. 2 could be partially mediated through this receptor. The Glc conjugates did not show any binding affinity above background for the recombinant Dectin-1 receptor while the positive control, β-glucan, showed high binding affinity. Thus, no known CLR on DCs was found to bind to the Glc conjugates (in recombinant form) which indicates that the activation seen in Fig. 3 was not mediated by Dectin-1 and therefore unlikely to have occurred through any lectin mediated process on DCs. This was further confirmed by the EDTA inhibition studies seen in Fig. 4.
When combining the cell response data from Fig. 3E and the antibody blocking data of Fig. 4, it is clear that the DC responses to the HBSA–Man glycoconjugates was at least in part due to DC CLR interaction. However, from Fig. 4 it is clear that the recombinant human CLRs tested do not mediate HBSA–Glc activation of DCs shown in Fig. 3E. This indicates that the DC response to the HBSA–Glc glycans is independent of DC CLR activation due to the fact that no other lectin known on DCs, other than Dectin-1, can bind β-D-glucose.26 Thus, a different mechanism for DC response to the HBSA–Glc conjugates must be at play for the observed activation of the DCs. Other groups have shown that Complement receptor-3 (CR3), lactosylceramides, and scavenger receptors27 can all bind β-D-glucose and cause DC activation, and thus presumably such receptors are mediating the increase in DC IMF for adsorbed HBSA–Glc conjugates.
Phagocytosis of beads was chosen as a means to assess DC interaction with the glycoconjugate for two reasons. First, mannose-binding CLRs on DCs are known to be phagocytic receptors, thus bead internalization can serve as a functional reporter. Second, using the HTP reporter of IMF or TMF could not be performed after 24 hours in the presence of high concentrations of CLR-blocking antibodies due to extraneous activation of the DCs. All antibody (Ab) treatments of DCs were at 10 μg ml−1 of Ab, including isotype, significantly activated DCs after 24 hours. 2.0 μg ml−1 of Ab was assayed but showed no functional blocking of the receptors in terms of phagocytosis at four hours and no influence on IMF or TMF at 24 hours (Data not shown). Thus, isotype antibody control treated DCs were chosen as the reference group in these studies to overcome concerns over non-specific activation, and thus phenotype modulation of DCs, due to Ab treatment. Finally, mannan was not inhibited by either of the CLR blocking antibodies. This result was not surprising given that many other CLRs on DCs are capable of binding and recognizing mannan.28
When comparing the results of Fig. 4 to that of Fig. 5, a nearly identical trend of inhibition of phagocytosis or internalization is observed between conjugates in a BA and AW modality. Interestingly, inhibition trends were different for soluble conjugates. Fig. 5B shows that anti-DC-SIGN did not inhibit soluble conjugate internalization. To our knowledge this is the first report of this phenomenon. However, other researchers have shown differential DC phenotypes when CLR ligands were presented in soluble and particulate form1–3 and macropinocytosis has been shown to mediate mannosylated protein internalization by DCs.29,30 Furthermore, the finding that anti-DC-SIGN does not inhibit internalization of fluorescent conjugates suggests a possible mechanism through which the differential activation profile noted by Wattendorf et al.5 between mannan conjugates could be mediated. Also of importance from Fig. 5 was that, when looking at the signal generated from either modality, it is clear that DCs internalize the conjugates in both soluble and AW modalities. Further, when looking at raw non-normalized signals we observed that DCs internalize over two orders of magnitude more glycan in soluble form than from an AW modality (data not shown). Anti-Dectin 1 was not included in the experiment shown in Fig. 5 because Dectin 1 is not a phagocytic receptor,31 thus it was concluded that it would not alter DC phagocytosis over that of isotype anti-bodies. The results show that the internalization of conjugates from the AW modality must not be the sole process necessary for the increase in DC IMF seen in Fig. 3.
In this study a HTP cellular methodology was used to assess DC phenotype. This assay was adapted for use with adsorbed glycoconjugates from a HTP methodology that has been published previously.11 The IMF reporter was shown to be an excellent indicator of overall DC phenotype11,32,33 as validated by flow cytometry and multiplex cytokine secretion. Cellular adhesion or migration was not used in this study as an indicator of DC phenotype because these outputs have been shown to be poor indicators of DC phenotype.34 Also, the end result of the DC maturation process, whether its pro or anti-inflammatory, CD4 or CD8 stimulating, etc. is independent of adhesion. Thus, adhesion is not an ideal reporter for DC activation.35 High throughput methodologies are necessary when assessing cell response to glycoconjugates because of the extremely limited supply of homogenous, purified, functionalized glycans that are capable of being obtained through biological or synthetic means. While the conjugates used in this study utilized only monosaccharides. The validated assay can now be extended to assess DC phenotype to an array of more complex glycans and glycoconjugates.
This study leaves many questions unanswered which require further investigation. First, given that different modalities of display are capable of producing differential DC phenotypes, what molecular signaling mechanism(s) is involved in the differential response? Possible explanations include mechanical interaction, length of time of interaction with CLRs on the cell surface, ability of CLRs to co-localize for extended periods of time on the cell surface, and/or generation of a “frustrated phagocytosis” state in which DCs release reactive oxygen species and matrix metalloproteinases that leads to activation of surrounding DCs. Finally, the results from Fig. 5 show that conjugates in an AW modality are being internalized by DCs. Whether this internalization is necessary for DC activation and to what extent the internalization of conjugates plays in the phenotype modulation of DCs is an important and unanswered question.
Conclusions
This study established that glycoconjugate presentation modality affects DC phenotype. This is of importance for the glycobiology and biomaterial science fields because it challenges the precepts that glycan structure, density, and context are the only factors of importance for recognition and response from DCs. This study also helps to resolve conflicts in reports from multiple laboratories showing differential DC profiles in response to similar, if not identical, ligands delivered in different modalities. Additionally, this study begins to bridge the gap between microarray binding data and functional cell responses, by highlighting the different phenotypes induced from conjugates in an AW modality as compared to those presented in solution or BA modality. Finally, this study uncovered the importance of non-phagocytosable display of glycans in promoting a pro-inflammatory DC phenotype. Non-phagocytosable display of glycans to DCs has generally received little attention and it is expected that this report will increase its exploration and attention. Use of glycans for implant coatings or as adjuvants for combination products for any other purpose than to increase phagocytosis or targeting of APCs is still relatively unexplored. The studies performed here indicate that not only are glycans able to enhance phagocytosis, they can also serve as immunomodulators in their own right, especially when delivered in a modality that DCs are able to recognize. It is expected that with further optimization of molecular factors, glycoconjugates have the potential to be engineered for the next generation of biomaterials to tune the immune response to any desired outcome.Acknowledgements
We would like to acknowledge Dr Xuezheng Song from Emory University for his invaluable expertise with glycan isolation and functionalization. Also we would like to thank David Smith for the generous contribution of AEAB, mass spectrometer, and lab space. We also wish to acknowledge the Consortium for Functional Glycomics and NIH Grants GM62116, GM103694, P41GM103694 and GM098791 for contributing glycans and other resources. We would like to acknowledge support through the NIH Cell and Tissue Engineering Doctoral Training Grant and through the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454, research supported by NIH 1RO1EB004633-01A1, 1R21EB012339-01A1.Notes and references
- S. J. Van Vliet, L. C. Paessens, V. C. M. Broks-van den Berg, T. B. H. Geijtenbeek and Y. van Kooyk, J. Immunol., 2008, 181, 3148–3155 CrossRef CAS.
- N. Gaudart, P. Ekpo, K. Pattanapanyasat, Y. van Kooyk and A. Engering, FEMS Immunol. Med. Microbiol., 2008, 53, 359–367 CrossRef CAS PubMed.
- C. Qi, Y. Cai, L. Gunn, C. Ding, B. Li, G. Kloecker, K. Qian, J. Vasilakos, S. Saijo, Y. Iwakura, J. R. Yannelli and J. Yan, Blood, 2011, 117, 6825–6836 CrossRef CAS PubMed.
- V. Le Cabec, L. J. Emorine, I. Toesca, C. Cougoule and I. Maridonneau-Parini, J. Leukocyte Biol., 2005, 77, 934–943 CrossRef CAS PubMed.
- U. Wattendorf, G. Coullerez, J. Vörös, M. Textor and H. P. Merkle, Langmuir, 2008, 24, 11790–11802 CrossRef CAS PubMed.
- Z. Cui, S.-J. Han and L. Huang, Pharm. Res., 2004, 21, 1018–1025 CrossRef CAS.
- S. Jain and S. P. Vyas, J. Liposome Res., 2006, 16, 331–345 CrossRef CAS PubMed.
- V. Apostolopoulos, G. A. Pietersz, A. Tsibanis, A. Tsikkinis, H. Drakaki, B. E. Loveland, S. J. Piddlesden, M. Plebanski, D. S. Pouniotis, M. N. Alexis, I. F. McKenzie and S. Vassilaros, Breast Cancer Res., 2006, 8, R27–R27 CrossRef PubMed.
- B. W. Greatrex, S. J. Brodie, R. H. Furneaux, S. M. Hook, W. T. McBurney, G. F. Painter, T. Rades and P. M. Rendle, Tetrahedron, 2009, 65, 2939–2950 CrossRef CAS PubMed.
- N. A. Hotaling, R. D. Cummings, D. M. Ratner and J. E. Babensee, Biomaterials, 2014, 35, 5862–5874 CrossRef CAS PubMed.
- P. M. Kou and J. E. Babensee, Acta Biomater., 2010, 6, 2621–2630 CrossRef CAS PubMed.
- J. S. Manavalan, P. C. Rossi, G. Vlad, F. Piazza, A. Yarilina, R. Cortesini, D. Mancini and N. Suciu-Foca, Transpl. Immunol., 2003, 11, 245–258 CrossRef CAS.
- T. R. Johnson, J. S. McLellan and B. S. Graham, J. Virol., 2012, 86, 1339–1347 CrossRef CAS PubMed.
- N. Singh, A. Karim, F. Bates, M. Tirrell and K. Furusawa, Macromolecules, 1994, 27, 2586–2594 CrossRef CAS.
- G. Pellicane, D. Costa and C. Caccamo, J. Phys.: Condens. Matter, 2003, 15, 375 CrossRef CAS.
- X. Zhou, B. Liu, X. Yu, X. Zha, X. Zhang, Y. Chen, X. Wang, Y. Jin, Y. Wu, Y. Chen, Y. Shan, Y. Chen, J. Liu, W. Kong and J. Shen, J. Controlled Release, 2007, 121, 200–207 CrossRef CAS PubMed.
- S. Hamdy, A. Haddadi, A. Shayeganpour, J. Samuel and A. Lavasanifar, Pharm. Res., 2011, 28, 2288–2301 CrossRef CAS PubMed.
- Y.-P. Li, V. Latger-Canard, L. Marchal, N. Li, J.-P. Ou-Yang and J.-F. Stoltz, Biomed. Mater. Eng., 2006, 16, S163–S170 CAS.
- S. J. Van Vliet, J. den Dunnen, S. I. Gringhuis, T. B. Geijtenbeek and Y. van Kooyk, Curr. Opin. Immunol., 2007, 19, 435–440 CrossRef CAS PubMed.
- Y. Van Kooyk, Biochem. Soc. Trans., 2008, 36, 1478–1481 CrossRef CAS PubMed.
- L. M. Graham and G. D. Brown, Cytokine, 2009, 48, 148–155 CrossRef PubMed.
- T. K. Dam and C. F. Brewer, Glycobiology, 2010, 20, 270–279 CrossRef CAS PubMed.
- G. A. Rabinovich and M. A. Toscano, Nat. Rev. Immunol., 2009, 9, 338–352 CrossRef CAS PubMed.
- G. A. Rabinovich and J. M. Ilarregui, Immunol. Rev., 2009, 230, 144–159 CrossRef CAS PubMed.
- O. Oyelaran, Q. Li, D. Farnsworth and J. C. Gildersleeve, J. Proteome Res., 2009, 8, 3529–3538 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2009, 9, 465–479 CrossRef CAS PubMed.
- S. H. Albeituni and J. Yan, Anticancer Agents Med. Chem., 2013, 13, 689–698 CrossRef CAS.
- L. East and C. M. Isacke, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 364–386 CrossRef CAS.
- V. Maréchal, M.-C. Prevost, C. Petit, E. Perret, J.-M. Heard and O. Schwartz, J. Virol., 2001, 75, 11166–11177 CrossRef PubMed.
- F. Sallusto, M. Cella, C. Danieli and A. Lanzavecchia, J. Exp. Med., 1995, 182, 389–400 CrossRef CAS.
- G. D. Brown, Nat. Rev. Immunol., 2006, 6, 33–43 CrossRef CAS PubMed.
- P. M. Kou, N. Pallassana, R. Bowden, B. Cunningham, A. Joy, J. Kohn and J. E. Babensee, Biomaterials, 2012, 33, 1699–1713 CrossRef CAS PubMed.
- P. M. Kou, Z. Schwartz, B. D. Boyan and J. E. Babensee, Acta Biomater., 2011, 7, 1354–1363 CrossRef CAS PubMed.
- S. Burns, S. J. Hardy, J. Buddle, K. L. Yong, G. E. Jones and A. J. Thrasher, Cell Motil. Cytoskeleton, 2004, 57, 118–132 CrossRef PubMed.
- A. P. Acharya, N. V. Dolgova, M. J. Clare-Salzler and B. G. Keselowsky, Biomaterials, 2008, 29, 4736–4750 CrossRef CAS PubMed.
- I. Van Die, V. Vliet, S. J. A. K. Nyame, R. D. Cummings, C. M. C. Bank, B. Appelmelk, T. B. H. Geijtenbeek and Y. Van Kooyk, Glycobiology, 2003, 13, 471–478 CrossRef CAS PubMed.
- S.-C. Hsu, C.-H. Chen, S.-H. Tsai, H. Kawasaki, C.-H. Hung, Y.-T. Chu, H.-W. Chang, Y. Zhou, J. Fu, B. Plunkett, S.-N. Su, S. Vieths, R. T. Lee, Y. C. Lee and S.-K. Huang, J. Biol. Chem., 2010, 285, 7903–7910 CrossRef CAS PubMed.
- Z. Yang and W. S. Hancock, J. Chromatogr., A, 2004, 1053, 79–88 CrossRef CAS PubMed.
- B. Sayin, S. Somavarapu, X. W. Li, M. Thanou, D. Sesardic, H. O. Alpar and S. Senel, Int. J. Pharm., 2008, 363, 139–148 CrossRef CAS PubMed.
- H. Chen, P. Li, Y. Yin, X. Cai, Z. Huang, J. Chen, L. Dong and J. Zhang, Biomaterials, 2010, 31, 8172–8180 CrossRef CAS PubMed.
- J. M. Martínez Gómez, N. Csaba, S. Fischer, A. Sichelstiel, T. M. Kündig, B. Gander and P. Johansen, J. Controlled Release, 2008, 130, 161–167 CrossRef PubMed.
- L. Thiele, B. Rothen-Rutishauser, S. Jilek, H. Wunderli-Allenspach, H. P. Merkle and E. Walter, J. Controlled Release, 2001, 76, 59–71 CrossRef CAS.
- C. Wischke, H.-H. Borchert, J. Zimmermann, I. Siebenbrodt and D. R. Lorenzen, J. Controlled Release, 2006, 114, 359–368 CrossRef CAS PubMed.
- S. J. Lee, S. Evers, D. Roeder, A. F. Parlow, J. Risteli, L. Risteli, Y. C. Lee, T. Feizi, H. Langen and M. C. Nussenzweig, Science, 2002, 295, 1898–1901 CrossRef CAS PubMed.
- S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS PubMed.
- J. Lodowska, D. Wolny, M. Jaworska-Kik, S. Kurkiewicz, Z. Dzierżewicz and L. Węglarz, Sci. World J., 2012, 2012, 647352 Search PubMed.
- D. A. Harn, J. McDonald, O. Atochina and A. A. Da'dara, Immunol. Rev., 2009, 230, 247–257 CrossRef CAS PubMed.
- J. J. García-Vallejo, E. van Liempt, P. da Costa Martins, C. Beckers, B. van het Hof, S. I. Gringhuis, J.-J. Zwaginga, W. van Dijk, T. B. H. Geijtenbeek, Y. van Kooyk and I. van Die, Mol. Immunol., 2008, 45, 2359–2369 CrossRef PubMed.
Footnotes |
| † This paper is dedicated to Professor Michael V. Sefton in honour of his 65th Birthday and in recognition of his mentorship, service and leadership to the Biomaterials community. |
| ‡ Electronic supplementary information (ESI) available: The supplemental contains a more in-depth description of statistics and statistical models used. Three tables (Tables S1a, S1b and S2). Tables S1a and S1b describe variables used in the modeling of this report. Table S2 shows comparisons between all surface area ratios compared. Finally the supplemental contains two ESI figures. ESI Fig. S1 shows cell apoptosis in response to the conjugates. Fig. S2 shows cell phagocytosis of control coated beads. Fig. S3 shows representative MALDI spectra of the conjugates. See DOI: 10.1039/c4bm00138a |
| This journal is © The Royal Society of Chemistry 2014 |