The hydrophobic effect of nanoparticles composed of amphiphilic poly(γ-glutamic acid) on the degradability of the encapsulated proteins
Fumiaki
Shima
,
Takami
Akagi
and
Mitsuru
Akashi
*
Department of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita 565-0871, Japan. E-mail: akashi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7359; Tel: +81-6-6879-7356
First published on 4th July 2014
Abstract
For the development of safe and effective next-generation vaccine carriers, their physicochemical properties (size, shape, surface charge, and hydrophobic/hydrophilic balance) are crucial to control their interactions (cellular uptake, intracellular degradability of the loaded antigen, and intracellular localization) with immune cells. Recently, the hydrophobicity of carriers affected the cellular uptake and immune response, which demonstrated that hydrophobicity is one of the most important factors to control the behaviors of the loaded antigens and carriers. In this study, we investigated the effect of the hydrophobicity of nanoparticles (NPs) composed of amphiphilic poly(γ-glutamic acid)-graft-phenylalanine ethyl ester (γ-PGA-Phe) with various grafting degrees of hydrophobic side chains on cellular uptake of the encapsulated antigens, their degradability, and their release behavior in the endosomal environment. These NPs could encapsulate proteins, and the degradability of the encapsulated proteins was changed by the hydrophobicity of NPs. On the other hand, the release behavior of the encapsulated proteins was not changed by the hydrophobicity of NPs. These results suggest that the intracellular behaviors of the encapsulated protein could be controlled by the hydrophobicity of NPs, and could result in the manipulation of the antigen-specific immune responses.
Introduction
Recently, purified subunits, recombinant proteins, and synthetic peptides have been paid much attention to develop next-generation vaccines. As these antigens possess low immunogenicity, adjuvants and antigen delivery systems are required for the targeted delivery and controlled release of antigens at the targeted site.1 Polymeric nanoparticles (NPs) composed of biodegradable or biocompatible copolymers have been developed as vaccine carriers for the targeted delivery of antigens. Antigen presenting cells (APCs), especially dendritic cells (DCs) and macrophages, take up these antigen-loaded NPs, and initiate the antigen-specific immune responses.2,3 The internalized antigens are degraded to peptides, followed by the presentation on the surface of the APCs, and then antigen-specific humoral and cellular immunity is induced.4 The intracellular behavior of these antigens is crucial to induce the immune responses. Particularly, the degradability of the antigens drastically affects the induction level of immune responses. Tran and Shen5 have reported that the induction efficiency of antigen-specific cellular immunity was increased when the antigens loaded onto polymeric particles were protected from degradation. Recently, the field of “immunoengineering” has been drawn much attention to develop effective vaccines.6,7 This provides for the design of carriers such as NPs, dendrimers and liposomes prepared from non-cytotoxic or biodegradable polymers to target and manipulate immune responses.8,9 Particularly, there are some reports that the hydrophobicity of particulate polymeric adjuvants and antigens is a key factor for controlling the interactions with cells and initiating the immune responses.10–15 Therefore, we hypothesized that the hydrophobicity of NPs should also affect the intracellular degradability of the encapsulated antigens into NPs, and investigated the effect of hydrophobicity of NPs on the degradability of the encapsulated proteins.In a previous study, we synthesized amphiphilic copolymers composed of poly(γ-glutamic acid) (γ-PGA) as the hydrophilic backbone and L-phenylalanine ethyl ester (Phe) as the hydrophobic segment (γ-PGA-Phe) and prepared NPs (γ-PGA-Phe NPs) to develop the nanoparticle-based vaccines which ensure both safety and efficacy.16 The potent antigen-specific immune response could be induced due to the efficient cellular uptake of the encapsulated antigen by APCs.17 We reported that γ-PGA-Phe NPs themselves stimulated APCs via Toll-like receptor (TLR) 4 expressed on the cell surface, which suggests that these NPs could be a great candidate for adjuvant and antigen delivery systems.18 These γ-PGA-Phe NPs also possessed a membrane disruptive property,19 and could escape from the endosomes after cellular uptake, which resulted in the induction of antigen-specific cellular immunity.20,21 Moreover, we have revealed that the activation potential of DCs and induction of antigen-specific cellular immunity were correlated with the hydrophobicity (grafting degree of Phe) of γ-PGA-Phe NPs, which could manipulate about 5- to 30-fold and 10- to 40-fold more than the conventional vaccine, respectively.22 Although the detailed mechanism about how the hydrophobicity of γ-PGA-Phe NPs affected the antigen behaviors (intracellular degradation, release from NPs) is unclear, in this study, we investigated the effects of the hydrophobicity of γ-PGA-Phe NPs on intracellular degradation of protein-encapsulated γ-PGA-Phe NPs and release behavior of antigens from γ-PGA-Phe NPs in the endosomal environment in vitro.
Materials and methods
Materials
γ-PGA (Mw = 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and dimethyl sulfoxide (DMSO) were purchased from Wako Pure Chemical Industries (Osaka, Japan). L-Phenylalanine ethyl ester (Phe), phosphate-buffered saline (PBS), RPMI 1640 medium, and fetal bovine serum (FBS) were purchased from Sigma-Aldrich (MO, USA). The antibiotics were purchased from Nacalai Tesque Inc. (Kyoto, Japan). Granulocyte macrophage colony stimulating factor (GM-CSF) was purchased from PeproTech (NJ, USA). FITC-labeled ovalbumin (FITC-OVA) and DQ OVA were purchased from Molecular Probes (OR, USA).
:1 to 1.6:1. The synthesized γ-PGA-graft-Phe (γ-PGA-Phe) copolymers were then dialyzed against water for 3 days and freeze-dried for 2 days. The synthesis of γ-PGA-Phe copolymers was determined by FT-IR spectroscopy (Spectrum 100, Perkin Elmer, USA) and 1H NMR (ECS-400, JEOL, Japan). γ-PGA-Phe, whose grafting degrees of Phe were 43, 50, 61, 65, and 71%, was used in this study. γ-PGA-Phe NPs were prepared for scanning electron microscopy (SEM) according to a previous report.22 Briefly, γ-PGA-Phe (10 mg ml−1 in DMSO) was added to the same volume of NaCl solution. To regulate the size of γ-PGA-Phe NPs around 200 nm, the salt concentration was varied from 0.08 to 0.6 M. The resulting solution was then dialyzed against pure water for 2 days, and observed by SEM.
000), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and dimethyl sulfoxide (DMSO) were purchased from Wako Pure Chemical Industries (Osaka, Japan). L-Phenylalanine ethyl ester (Phe), phosphate-buffered saline (PBS), RPMI 1640 medium, and fetal bovine serum (FBS) were purchased from Sigma-Aldrich (MO, USA). The antibiotics were purchased from Nacalai Tesque Inc. (Kyoto, Japan). Granulocyte macrophage colony stimulating factor (GM-CSF) was purchased from PeproTech (NJ, USA). FITC-labeled ovalbumin (FITC-OVA) and DQ OVA were purchased from Molecular Probes (OR, USA).
:1 to 1.6:1. The synthesized γ-PGA-graft-Phe (γ-PGA-Phe) copolymers were then dialyzed against water for 3 days and freeze-dried for 2 days. The synthesis of γ-PGA-Phe copolymers was determined by FT-IR spectroscopy (Spectrum 100, Perkin Elmer, USA) and 1H NMR (ECS-400, JEOL, Japan). γ-PGA-Phe, whose grafting degrees of Phe were 43, 50, 61, 65, and 71%, was used in this study. γ-PGA-Phe NPs were prepared for scanning electron microscopy (SEM) according to a previous report.22 Briefly, γ-PGA-Phe (10 mg ml−1 in DMSO) was added to the same volume of NaCl solution. To regulate the size of γ-PGA-Phe NPs around 200 nm, the salt concentration was varied from 0.08 to 0.6 M. The resulting solution was then dialyzed against pure water for 2 days, and observed by SEM.
To evaluate the effect of hydrophobicity of γ-PGA-Phe NPs on the cellular uptake amount of proteins, FITC-OVA was used. For the preparation of FITC-OVA-encapsulated γ-PGA-Phe NPs (FITC-OVA-NPs) with various grafting degrees of Phe, γ-PGA-Phe (10 mg ml−1 in DMSO) was added to the same volume of FITC-OVA solution (875 μg ml−1). The size of the FITC-OVA-NPs was regulated by changing the salt concentration in the FITC-OVA solution from 0.08 to 0.6 M. After purification by centrifugation, the FITC-OVA-NPs were dispersed into PBS. The FITC-OVA-NPs were then added to the same volume of 4% SDS to dissolve the NPs, and the encapsulated amount of protein was measured by fluorescence intensity. The size distributions of the FITC-OVA-NPs in PBS were measured by the dynamic light scattering (DLS) method using a Zetasizer Nano ZS (Malvern Instruments, UK).
To investigate the effect of hydrophobicity on the intracellular degradation of the encapsulated proteins into the NPs, DQ OVA was used. DQ OVA is an OVA modified with self-quenched fluorescent dyes that exhibits bright-green fluorescence upon proteolytic degradation. DQ OVA-encapsulated γ-PGA-Phe NPs (DQ OVA-NPs) with various grafting degrees of γ-PGA-Phe were prepared by the same method as FITC-OVA-NPs mentioned above. To regulate the size of DQ OVA-NPs, the salt concentration in the DQ OVA solution was varied from 0.1 to 0.63 M. The DQ OVA-NPs were then added to the same volume of 4% SDS to dissolve the NPs, and the protein loading content was measured by the Lowry method.
Results and discussion
Preparation of γ-PGA-Phe NPs and protein-encapsulated γ-PGA-Phe NPs with various hydrophobicity
γ-PGA-Phe copolymers were synthesized by coupling the amino group of Phe with the carboxylic groups in γ-PGA using EDC. The grafting degree of Phe was determined by the integral of peaks measured by 1H NMR spectroscopy. The grafting degree was controlled from 43 to 71% by changing the molar ratio of Phe to γ-PGA from 0.8 to 1.6. These copolymers could form NPs due to their hydrophobic interactions. In this study, the size of γ-PGA-Phe NPs was regulated to around 200 nm by changing the NaCl concentration as it has been reported that the 200 nm sized NPs were efficiently taken up by immune cells compared with smaller sized NPs.26Fig. 1 shows the SEM images of γ-PGA-Phe NPs. The morphology was universally spherical regardless of the grafting degree. γ-PGA-Phe NPs with 43% grafting degree showed a soft and flexible morphology compared with γ-PGA-Phe NPs with higher grafting degrees. This may be due to the decrease in the hydrophobic interaction between γ-PGA-Phe. As γ-PGA-Phe with 43% grafting degree possessed lower amounts of hydrophobic side chains than those with higher grafting degrees, this resulted in weak interaction between copolymers, and thus became flexible. The sizes of γ-PGA-Phe NPs were smaller (about 100 nm) than when measured by the DLS method (about 200 nm, data not shown). These findings suggest that water may have existed inside the NPs and that the NPs contracted when they were dried to remove water for SEM observation. | ||
| Fig. 1 SEM images of γ-PGA-Phe NPs with various grafting degrees (43–71%). The scale bars represent 100 nm. | ||
In order to evaluate the effect of hydrophobicity of γ-PGA-Phe NPs on the cellular uptake of proteins, FITC-OVA-encapsulated γ-PGA-Phe NPs (FITC-OVA-NPs) were prepared. The amount of encapsulated FITC-OVA was about 40 μg per 1 mg NPs regardless of the hydrophobicity of γ-PGA-Phe NPs and the size was about 200 nm (data not shown). To evaluate the effect of hydrophobicity on the intracellular degradation of the encapsulated proteins, DQ OVA was used. DQ OVA is an OVA modified with self-quenched fluorescence dyes that exhibits pH-insensitive bright-green fluorescence upon proteolytic degradation. DQ OVA-encapsulated γ-PGA-Phe NPs (DQ OVA-NPs) were prepared by the same method as FITC-OVA-NPs. The size of DQ OVA-NPs was regulated to about 200 nm by changing the salt concentration from 0.1 to 0.63 M (Fig. 2a). When the grafting degree was low, a higher NaCl concentration was necessary for the preparation of 200 nm DQ OVA-NPs (0.63 M NaCl for γ-PGA-Phe with a 43% grafting degree). On the other hand, a low NaCl concentration was sufficient for the preparation of DQ OVA-NPs with higher grafting degrees (0.1 M NaCl for γ-PGA-Phe with a 71% grafting degree). These may be due to the amphiphilic balance of γ-PGA-Phe and the electrostatic repulsion of the carboxyl groups in γ-PGA-Phe. Since γ-PGA-Phe with lower grafting degrees possessed fewer hydrophobic moieties and a larger number of carboxyl groups, it might require a greater amount of salt to reduce the electrostatic repulsions. On the other hand, γ-PGA-Phe with higher grafting degrees possessed more hydrophobic moieties and a lesser number of carboxyl groups, it might require less salt for attenuating the repulsion. When the feeding amount of DQ OVA was 875 μg ml−1, the amount of encapsulated DQ OVA was about 2 times larger for γ-PGA-Phe NPs (about 40 μg per 1 mg NPs for 65 and 71% grafting degrees) than for γ-PGA-Phe NPs with lower grafting degrees (43–61%). The encapsulation efficiencies, which were calculated from the following equation: encapsulated amount of DQ OVA into NPs/feeding amount of DQ OVA × 100 (%), were about 40% for γ-PGA-Phe NPs (65 and 71%), and about 20% for γ-PGA-Phe NPs (43 to 61%). It is suggested that OVA may be encapsulated into γ-PGA-Phe NPs by hydrogen bonding and hydrophobic interactions between hydrophobic side chains of the γ-PGA-Phe copolymer and hydrophobic moieties of OVA. As the hydrophobicity of γ-PGA-Phe NPs increased, the hydrophobic interactions between γ-PGA-Phe NPs and DQ OVA became stronger, which resulted in the increase in the encapsulated amount. The amount of encapsulated DQ OVA could be controlled to about 20 μg per 1 mg NPs for all γ-PGA-Phe NPs by changing the feeding amount of DQ OVA as shown in Fig. 2b. FITC-OVA-NPs and DQ OVA-NPs with various grafting degrees of Phe showed a negative zeta potential (about −30 mV) due to the ionization of the carboxyl groups of the γ-PGA located near the surface of the NPs (data not shown). We employed these FITC-OVA-NPs and DQ OVA-NPs, whose sizes were 200 nm, for the following experiments.
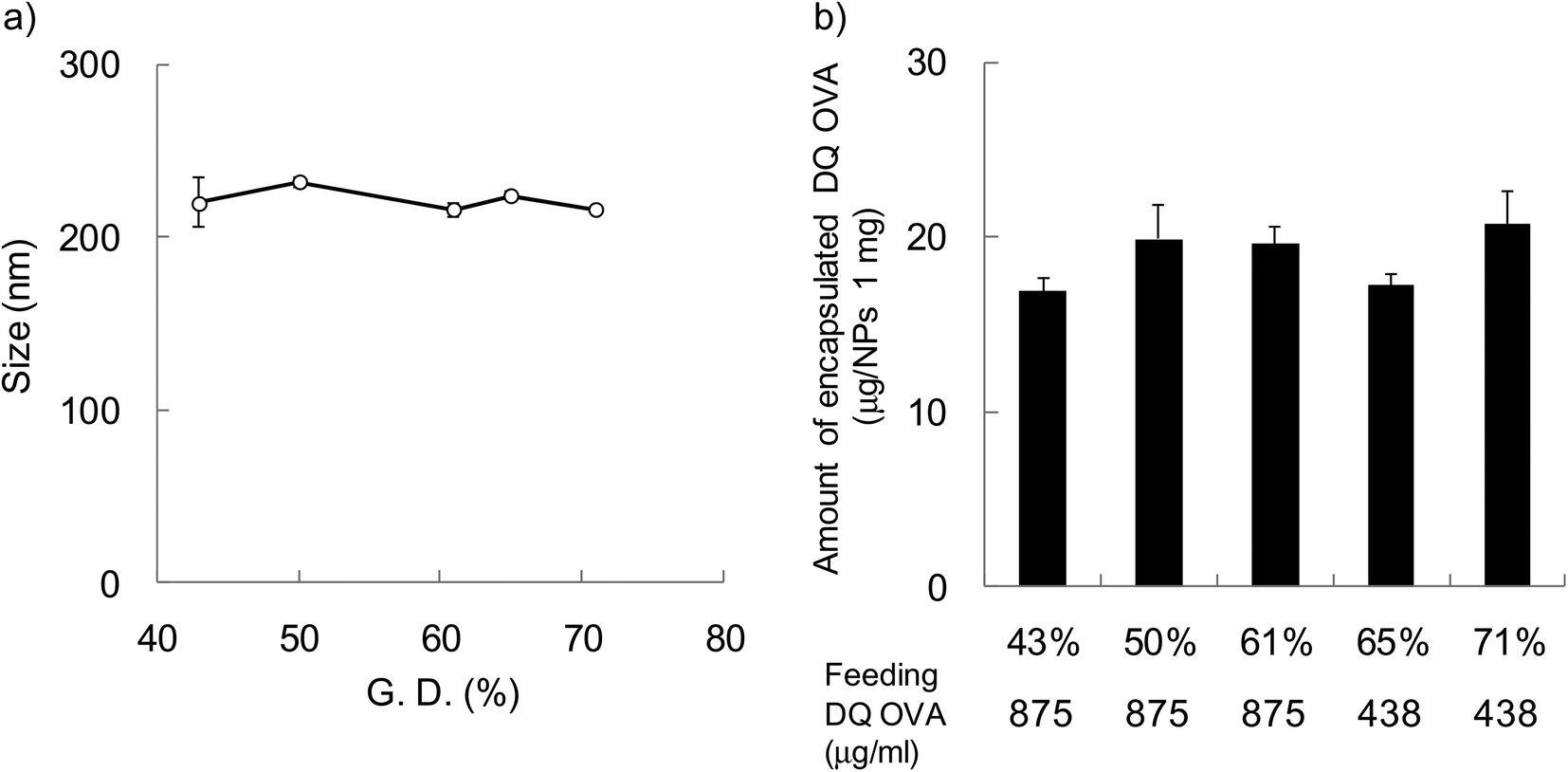 | ||
| Fig. 2 (a) Particle size and (b) encapsulated amount of DQ OVA-NPs prepared at various grafting degrees (43–71%) of γ-PGA-Phe. The particle size in PBS was measured by DLS (n = 3). The amount of encapsulated DQ OVA was measured by the Lowry method (n = 3). γ-PGA-Phe (10 mg ml−1 in DMSO) was added to the same volume of 875 μg ml−1 or 438 μg ml−1 DQ OVA. To regulate the size of the DQ OVA-NPs, the NaCl concentration was varied from 0.1 to 0.63 M. | ||
Uptake of FITC-OVA-NPs by DCs
To evaluate the effect of the hydrophobicity of γ-PGA-Phe NPs on the uptake amount of the encapsulated proteins by DCs, DCs were incubated with various concentrations of FITC-OVA or FITC-OVA-NPs (43–71% grafting degree) for 1 h at 37 °C. To quantify the amount of FITC-OVA taken up by DCs, the fluorescence intensity of the lysates was measured. Fig. 3 shows the hydrophobicity and dose dependency on the uptake of FITC-OVA-NPs by DCs. It demonstrated that FITC-OVA-NPs were efficiently taken up by DCs about 25- to 50-fold more than FITC-OVA alone. The uptake amount of FITC-OVA increased as the feeding amount of FITC-OVA increased. Moreover, there is significant difference in the uptake amount of FITC-OVA when compared with FITC-OVA-NPs (43% grafting degree) and FITC-OVA-NPs (71% grafting degree) (p < 0.05). This may be due to the presence of the hydrophobic side chains near the surface of NPs. Chiu et al. reported that the uptake amount of hydrophobically-modified chitosan NPs increased with an increasing degree of substitution of hydrophobic groups due to the presence of hydrophobic groups on the surface of the NPs, which were then able to interact efficiently with the cell membranes.12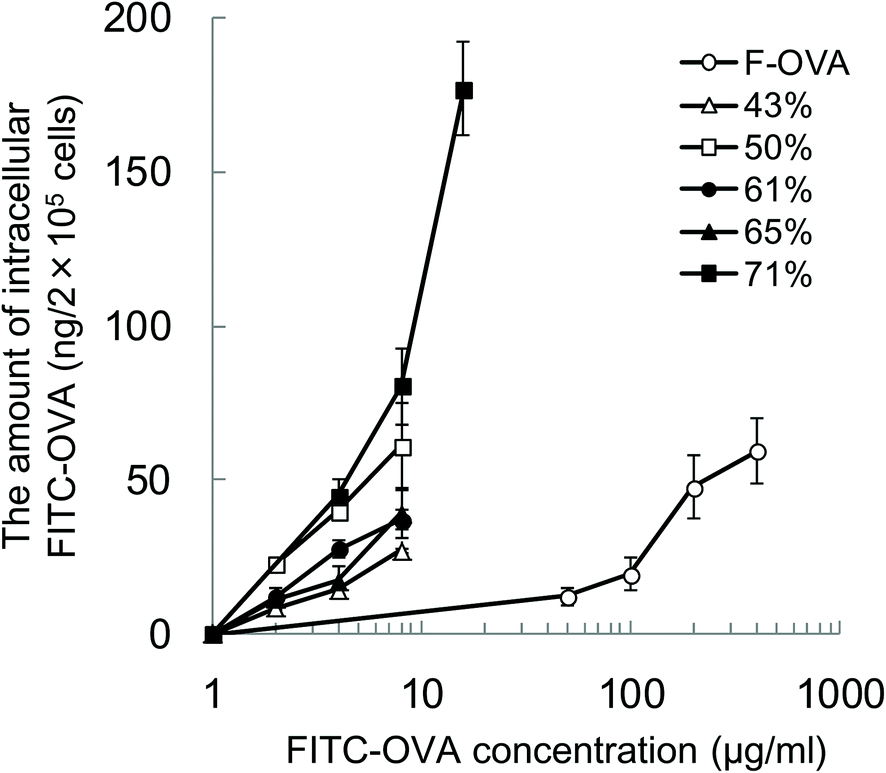 | ||
| Fig. 3 The hydrophobicity and dose dependency on the uptake of FITC-OVA-NPs by DCs. The cells were cultured with the indicated concentrations of FITC-OVA alone, or FITC-OVA-NPs with various grafting degrees (43–71%) for 1 h at 37 °C. To quantify the amount of intracellular FITC-OVA-NPs, the cells were dissolved, and then the fluorescence intensity of lysates was measured. The fluorescence incorporation was calculated as the uptake amount of FITC-OVA per cell numbers (n = 3). | ||
Intracellular degradation of OVA encapsulated into γ-PGA-Phe NPs
The relationship between the hydrophobicity of γ-PGA-Phe NPs and the intracellular degradation of the encapsulated proteins was evaluated. DCs were incubated with DQ OVA-NPs (43–71% grafting degree) or DQ OVA alone for 30 min at 37 °C. Since the hydrophobicity of γ-PGA-Phe NPs influenced the amount of OVA uptake by DCs, DQ OVA-NPs and DQ OVA alone were pulsed to the cells under the conditions where equal amount of DQ OVA were taken up by the cells. After washing with PBS, the cells were incubated in PBS for up to 1.5 h at 37 °C and intracellular degradation was measured by flow cytometry. The uptake of DQ OVA alone by DCs resulted in the early and excessive degradation. As the incubation time increased, the fluorescence from the degraded DQ OVA became more intense. As expected, the degradation of DQ OVA encapsulated into γ-PGA-Phe NPs was attenuated as compared with the free DQ OVA (Fig. 4). The relative degradation of the encapsulated DQ OVA was calculated as mentioned above. The degradation of the encapsulated DQ OVA was attenuated by about 60–80% compared with the free DQ OVA, and the degradability was changed by the hydrophobicity of γ-PGA-Phe NPs. A difference in DQ OVA degradation kinetics was observed between the hydrophobicity of γ-PGA-Phe NPs, γ-PGA-Phe NPs of 50% grafting degree showed the minimum degradation of DQ OVA. On the other hand, γ-PGA-Phe NPs of 71% grafting degree showed the highest degradation among the DQ OVA-NPs, followed by the γ-PGA-Phe NPs of 43%, 65%, and 61%. In a previous study, the cellular uptake and maturation of DCs by OVA-NPs, whose grafting degree was 71%, was highest, while the induction of the antigen-specific cellular immunity by γ-PGA-Phe NPs of 71% decreased as compared with those of 65%.22 According to the results of intracellular DQ OVA degradation, it is suggested that the increased degradation of the encapsulated DQ OVA in γ-PGA-Phe NPs of 71% compared with those of 65% may be one of the factors. These results also showed the same tendency as Tran and Shen reported.5 It has been reported that the antigens taken up by DCs are first degraded to the peptides, and then presented on the surface of the cells.4 The excess degradation of the encapsulated proteins may cause destruction of the peptides which are important for antigen presentation, and may result in the lower degree of induction of the antigen specific immune response. We also evaluated the size effect of γ-PGA-Phe NPs on the degradation of the encapsulated DQ OVA, although there was no significant difference.21 Our results suggest that the intracellular degradation behavior of antigens could be controlled by changing the hydrophobicity of γ-PGA-Phe NPs. | ||
| Fig. 4 Intracellular degradation of DQ OVA-NPs in DC. The cells were incubated with DQ OVA alone, DQ OVA-NPs prepared at various grafting degrees (43–71%) for 30 min at 37 °C. After washing with PBS, the cells were incubated with fresh PBS and the degradation of DQ OVA was evaluated by flow cytometry. The indicated incubation time was total incubation time. DQ OVA and DQ OVA-NPs were pulsed to the cells under the conditions where an equal amount of DQ OVA was taken up by the cells (n = 3). | ||
Release behavior of OVA encapsulated into γ-PGA-Phe NPs in the endosomal environment
To evaluate the relationship between the degradability of the encapsulated proteins and their release from NPs with various hydrophobicities in the endosomal environment, the release behavior was investigated under the condition that mimicked the endosomal environment (early endosomes, pH 6.9 and 20 mM Cl−)25in vitro. In this study, FITC-OVA-NPs (43–71% grafting degree) containing 40 μg of FITC-OVA per 1 mg of NPs were used. FITC-OVA-NPs were incubated in the endosomal environment for determined time periods at 37 °C and the amount of released proteins was measured by fluorescence intensity of the supernatant. As shown in Fig. 5, the release of the encapsulated proteins was not changed by the hydrophobicity of γ-PGA-Phe NPs. The sizes of FITC-OVA-NPs did not change during the incubation period and they did not show aggregation (data not shown). As mentioned above, the degradability of the encapsulated proteins was changed by the hydrophobicity of γ-PGA-Phe NPs. Taken together, it is suggested that the release behavior of proteins from NPs was not a dominant factor for the difference in the degradability, and physical properties of NPs such as polymer density and the position of the encapsulated protein might be crucial. All results in this study are summarized in Fig. 6. As previously reported, the protein-encapsulated γ-PGA-Phe NPs escaped from endosomes after they were taken up.21 Also, the potential of the endosomal escape was changed by the hydrophobicity of γ-PGA-Phe NPs.27 Taken together, the extent of the endosomal escape of the encapsulated proteins should be changed by the hydrophobicity of γ-PGA-Phe NPs. As there are many proteolytic enzymes in endosomes, the degradability of the proteins decreases when they escape from endosomes. Therefore, the change in the degradability of the encapsulated proteins may be due to the difference in the extent of the endosomal escape of the encapsulated proteins. The difference in the distribution of the proteins on/in the NPs may also affect the degradation of the encapsulated proteins. It has been reported that the distribution of the proteins on/in the particles significantly affected their degradability.28 The proteins loaded into the core of the particles were protected against proteolytic degradation while proteins on the surface of the particles were rapidly degraded. According to this report, the distribution of the encapsulated proteins into γ-PGA-Phe NPs with lower grafting degree (43%) and with higher grafting degree (71%) might be close to the surface, and thus the enzymes in the endosomes might easily degrade. On the other hand, the encapsulated proteins into γ-PGA-Phe NPs (50–65%) might be distributed to the inner part of NPs and might be protected against degradation. The hydrophobic/hydrophilic balance of γ-PGA-Phe and the rigidity of NPs might affect the distribution of the proteins. When the hydrophobicity is too low or high, they form fewer or larger hydrophobic domains into the NPs, which might affect the distribution of the proteins. D. F. Moyano et al. reported a direct, quantitative correlation between hydrophobicity of gold NPs with different surface hydrophobicity and immune system activation.13 Y.-L. Chiu et al. reported that the cellular uptake of NPs made of hydrophobically modified chitosan increased as the degree of substitution of hydrophobic groups increased, which was due to the existence of more hydrophobic groups near the surface, and thus could interact with the cell membranes.12 Y. Liu et al. demonstrated that the increased surface hydrophobicity of poly(lactic acid)-based microparticles (MPs) greatly promoted antigen internalization into DCs and elevated cytokine secretion levels.29 They also showed that the interaction between MPs and cell membranes was highly dependent on the MP surface hydrophobicity. Taken together, it is reasonable to think that the surface hydrophobicity of the carriers is a key factor to control the interaction with DCs and the consequent immune responses. In this study, it is suggested that the surface hydrophobicity of γ-PGA-Phe NPs was changed by the grafting degrees of Phe, which resulted in the difference in the interaction with the cell surface, and thus the cellular uptake and subsequent degradation of the encapsulated protein was changed. Although the relationships between the hydrophobicity of γ-PGA-Phe NPs and endosomal escape of the encapsulated proteins and distribution of the proteins remain to be elucidated, our research revealed that the degradability of the encapsulated proteins affected the induction level of the antigen-specific immune response.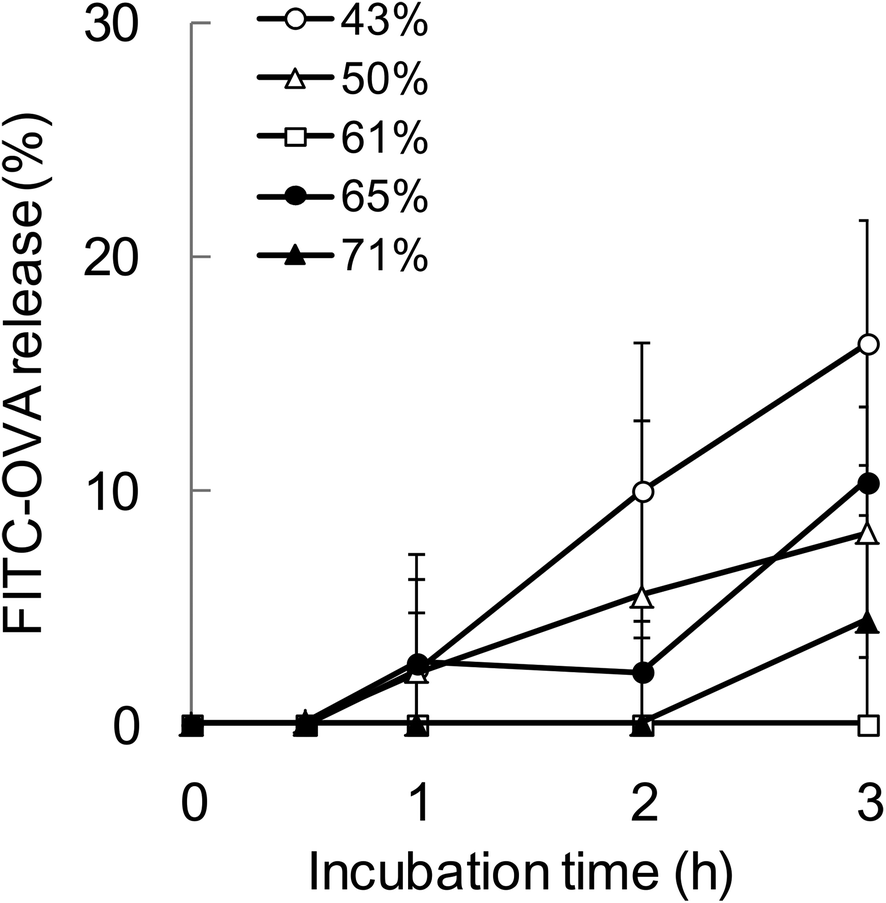 | ||
| Fig. 5 Release behavior of FITC-OVA from γ-PGA-Phe NPs with various grafting degrees (43–71%) in the endosomal environment. The FITC-OVA-NPs were incubated with PBS which mimicked the endosomal environment (pH 6.9) at 37 °C, and the amount of released FITC-OVA was measured by the fluorescence intensity. | ||
 | ||
| Fig. 6 Schematic illustration of the effect of hydrophobicity of γ-PGA-Phe NPs on interactions with dendritic cells. | ||
Conclusions
In this study, we clearly demonstrated that the hydrophobicity of γ-PGA-Phe NP vaccine adjuvant is a key factor to control the protein behaviors in the immune cells. The degradability of the encapsulated proteins was changed by controlling the hydrophobicity of γ-PGA-Phe NPs. On the other hand, the release behavior of the proteins from γ-PGA-Phe NPs was not changed by the hydrophobicity of NPs. As γ-PGA-Phe NPs could easily control their hydrophobicity, they should possess great potential as vaccine adjuvants that could control the degradability of the encapsulated antigens, which should result in the manipulation of the antigen specific immune responses.Acknowledgements
This work was supported by a Research Fellow of the Japan Society for the Promotion of Science, partially by a Grant-in-Aid for Scientific Research (S) from the Ministry of Education, Culture, Sports, Science, and Technology (23225004).References
- L. J. Peek, C. R. Middaugh and C. Berkland, Adv. Drug Delivery Rev., 2008, 60, 915–928 CrossRef CAS PubMed.
- C. Foged, A. Sundblad and L. Hovgaard, Pharm. Res., 2002, 19, 229–238 CrossRef CAS.
- V. Kanchan and A. K. Panda, Biomaterials, 2007, 28, 5344–5357 CrossRef CAS PubMed.
- J. Neefjes, M. L. M. Jongsma, P. Paul and O. Bakke, Nat. Rev. Immunol., 2011, 11, 823–836 CAS.
- K. K. Tran and H. Shen, Biomaterials, 2009, 30, 1356–1362 CrossRef CAS PubMed.
- J. A. Hubbell, S. N. Thomas and M. A. Swartz, Nature, 2009, 462, 449–460 CrossRef CAS PubMed.
- M. A. Swartz, S. Hirosue and J. A. Hubbell, Sci. Transl. Med., 2012, 4, 148rv9 Search PubMed.
- J. J. Moon, B. Huang and D. J. Irvine, Adv. Mater., 2012, 24, 3724–3746 CrossRef CAS PubMed.
- D. T. O'Hagan and N. M. Valiante, Nat. Rev. Drug Discovery, 2003, 2, 727–735 CrossRef PubMed.
- J. Kreuter, E. Liehl, U. Berg, M. Soliva and P. P. Speiser, Vaccine, 1988, 6, 253–256 CrossRef CAS.
- S. Y. Seong and P. Matzinger, Nat. Rev. Immunol., 2004, 4, 469–478 CrossRef CAS PubMed.
- Y. L. Chiu, Y. C. Ho, Y. M. Chen, S. F. Peng, C. J. Ke, K. J. Chen, F. L. Mi and H. W. Sung, J. Controlled Release, 2010, 146, 152–159 CrossRef CAS PubMed.
- D. F. Moyano, M. Goldsmith, D. J. Solfiell, D. L. Milo, O. R. Miranda, D. Peer and V. M. Rotello, J. Am. Chem. Soc., 2012, 134, 3965–3967 CrossRef CAS PubMed.
- N. Murthy, J. R. Robichaud, D. A. Tirrell, P. S. Stayton and A. S. Hoffman, J. Controlled Release, 1999, 61, 137–143 CrossRef CAS.
- R. A. Jones, C. Y. Cheung, F. E. Black, J. K. Zia, P. S. Stayton, A. S. Hoffman and M. R. Wilson, Biochem. J., 2003, 372, 65–75 CrossRef CAS PubMed.
- M. Matsusaki, K. Hiwatari, M. Higashi, T. Kaneko and M. Akashi, Chem. Lett., 2004, 33, 398–399 CrossRef CAS.
- T. Uto, T. Akagi, T. Hamasaki, M. Akashi and M. Baba, Immunol. Lett., 2009, 125, 46–52 CrossRef CAS PubMed.
- T. Uto, T. Akagi, K. Yoshinaga, M. Toyama, M. Akashi and M. Baba, Biomaterials, 2011, 32, 5206–5212 CrossRef CAS PubMed.
- T. Akagi, H. Kim and M. Akashi, J. Biomater. Sci., Polym. Ed., 2010, 21, 315–328 CrossRef CAS PubMed.
- T. Yoshikawa, N. Okada, A. Oda, K. Matsuo, K. Matsuo, Y. Mukai, Y. Yoshioka, T. Akagi, M. Akashi and S. Nakagawa, Biochem. Biophys. Res. Commun., 2008, 366, 408–413 CrossRef CAS PubMed.
- T. Akagi, F. Shima and M. Akashi, Biomaterials, 2011, 32, 4959–4967 CrossRef CAS PubMed.
- F. Shima, T. Akagi, T. Uto and M. Akashi, Biomaterials, 2013, 34, 9709–9716 CrossRef CAS PubMed.
- T. Uto, X. Wang, K. Sato, M. Haraguchi, T. Akagi, M. Akashi and M. Baba, J. Immunol., 2007, 178, 2979–2986 CrossRef CAS.
- K. Y. Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS PubMed.
- N. D. Sonawane, J. R. Thiagarajah and A. S. Verkman, J. Biol. Chem., 2002, 277, 5506–5513 CrossRef CAS PubMed.
- F. Shima, T. Uto, T. Akagi, M. Baba and M. Akashi, Acta Biomater., 2013, 9, 8894–8901 CrossRef CAS PubMed.
- F. Shima, T. Akagi and M. Akashi, J. Biomater. Sci., Polym. Ed., 2014, 25, 203–210 CrossRef CAS PubMed.
- H. Takahata, E. C. Lavelle, A. G. A. Coombes and S. S. Davis, J. Controlled Release, 1998, 50, 237–246 CrossRef CAS.
- Y. Liu, Y. Yin, L. Wang, W. Zhang, X. Chen, X. Yang, J. Xu and G. Ma, J. Mater. Chem. B, 2013, 1, 3888–3896 RSC.
| This journal is © The Royal Society of Chemistry 2014 |