
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancing metallomimetic catalysis through structural constraints of cationic PIII species
Deependra
Bawari
 *,
Donia
Toami
and
Roman
Dobrovetsky
*
*,
Donia
Toami
and
Roman
Dobrovetsky
*
School of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel Aviv University, Tel Aviv 69978, Israel. E-mail: rdobrove@tau.ac.il; deependrabawari@gmail.com
First published on 20th March 2025
Abstract
In recent years, the concept of structural constraints on the main-group (MG) centers has emerged as a powerful strategy to enhance their reactivity. Among these, structurally constrained (SC) phosphorus centers have garnered significant attention due to their ability to cycle between two stable oxidation states, P(III) and P(V), making them highly promising for small molecule activation and catalysis. Structural constraints grant phosphorus centers transition metal (TM)-like reactivity, enabling the activation of small molecules by these SC P(III) centers, a reactivity previously inaccessible with conventional phosphines or other phosphorus derivatives. This feature article reviews recent advances in the chemistry of cationic, structurally constrained P(III) (CSCP) compounds, emphasizing their ability to mimic TM behavior in small-molecule activation and catalysis, particularly through the key elementary steps of TM-based catalysis, such as oxidative addition (OA), migratory insertion (MI), ligand metathesis (LM), reductive elimination (RE), etc. The development of these SC cationic P(III) species highlights the interplay between structural constraints and cationic charge, facilitating analogous metallomimetic reactivity in other main-group elements.
1. Introduction
The search for sustainable alternatives to transition-metal (TM) based catalysts has driven scientists to explore main-group (MG) compounds for small-molecule activation and catalysis, making it a vibrant field of research over the past two decades.1–4 Among MG elements, pnictogens stand out for their ability to switch between two stable oxidation states (En ⇌ En+2; E = P, Sb, Bi), a property that enables them to mimic TMs’ chemistry and thus react via similar key elementary steps such as oxidative addition (OA) and reductive elimination (RE).5–12 These properties position pnictogens as emerging players in TM-free redox catalysis.While most neutral pnictogen species exhibit limited reactivity towards small molecules in their common oxidation states, imposing structural constraints on these molecular centers,13–19 particularly phosphorus (P), has unlocked their potential for small-molecule activation.20–24 A common strategy for achieving this involves enclosing pnictogen centres within rigid pincer-type ligands.20–24 This constraint induces a deviation from typical VSEPR geometries, disrupting the local symmetry around the pnictogen atom (Fig. 1). Consequently, the frontier molecular orbitals rehybridize, and the HOMO–LUMO energy gap decreases, which enhances the ambiphilicity (nucleo- and electrophilicity) of these centers and significantly improves their ability to activate strong chemical bonds.20–24
 | ||
| Fig. 1 A qualitative Walsh diagram illustrating the changes in molecular orbital energies and their corresponding shapes as a trigonal pyramidal phosphorus center distorts toward a planar T-shaped geometry. | ||
In the past decade, there has been a steady rise in the development of molecules featuring structurally constrained (SC) phosphorus centers. The earliest examples of structurally constrained phosphorus (SCP) species (I–III) were reported in the early 1980s by the groups of Houalla,25–28 Contreras,29–32 and Baccolini.33–35 While these pioneering studies primarily focused on the synthesis of phosphorus (PIII and PV) derivatives (Fig. 2a), they laid the foundation for exploring the reactivity of modern SCP compounds.
 | ||
| Fig. 2 Known examples of SCPs and their use in the activation of various E–H bonds. | ||
In 1984, Arduengo reported a seminal discovery of an SC, T-shaped phosphorus center (IV) in an ONO-type pincer ligand (Fig. 2b), capable of activating O–H bonds in MeOH via an OA-type reaction at the PIII centre.36,37 After a period of limited progress, Radosevich and co-workers revived interest in SCPs in 2012 by demonstrating the catalytic transfer hydrogenation of azobenzene using IV and H3NBH3 as hydrogen source (Fig. 2b).38
The same group later showed OA-type reactions of the N–H bonds in NH3 and alkyl/arylamines at the PIII centre in IV.39 Subsequently, Radosevich reported an SCP compound (V) in an NNN-type pincer ligand with an aromatic backbone.40 This SCP species activated the E–H bonds (E = N, O) in NH3, alkyl/arylamines, carboxylic acids, and alkyl/aryl alcohols (Fig. 2c). Mechanistic studies revealed that the OA step was preceded by E–H bond cleavage through phosphorus-ligand assisted cooperation (LA), followed by intramolecular σ3-P → σ5-P tautomerism. Interestingly, a later-reported derivative of V, featuring an ethylene-bridged backbone (VI), exhibited a preference for the σ5-P product, despite retaining the same local structure and bonding around the P-center as V.41 This shift was attributed to the restricted rotation of the Caryl–N bond in the σ3-P product, favouring the σ5-P isomer (Fig. 2d). Notably, the resulting PV product, V-B, could undergo reductive elimination of anilines and alcohols at elevated temperatures, highlighting its potential for catalytic applications (Fig. 2c).
Additionally, V successfully activated the B–H and C–F bonds and was subsequently employed in the hydroboration of imines and the non-catalytic, metallomimetic hydrodefluorination of fluoroarenes, clearly underscoring the significant catalytic potential of these species.42,43
Ammonia activation is essential in energy production and synthetic chemistry.44a,b While most TMs do not activate NH3 through formal oxidative addition, but instead form Werner-type complexes,44c,d certain MG compounds have demonstrated the potential to activate the N–H bonds in ammonia.39,45–53 In the case of SCPs, Hirao and Kinjo used an NPN-type pincer ligand with an aliphatic backbone to synthesize a diazadiphosphapentalene (VII), which effectively activated the H2N–H bond via σ bond metathesis (Fig. 3a).54 Aldridge and Goicoechea reported the SCP species, VIII, featuring an ONO-type pincer ligand with an aromatic backbone (Fig. 3c).55VIII readily activated the N–H bond in NH3 through oxidative addition (OA) to the P-center (Fig. 3c). Interestingly, the O–H bonds in H2O were also activated via OA to the P-center, with both O–H bonds reacting when two equivalents of VIII were used (Fig. 3c).
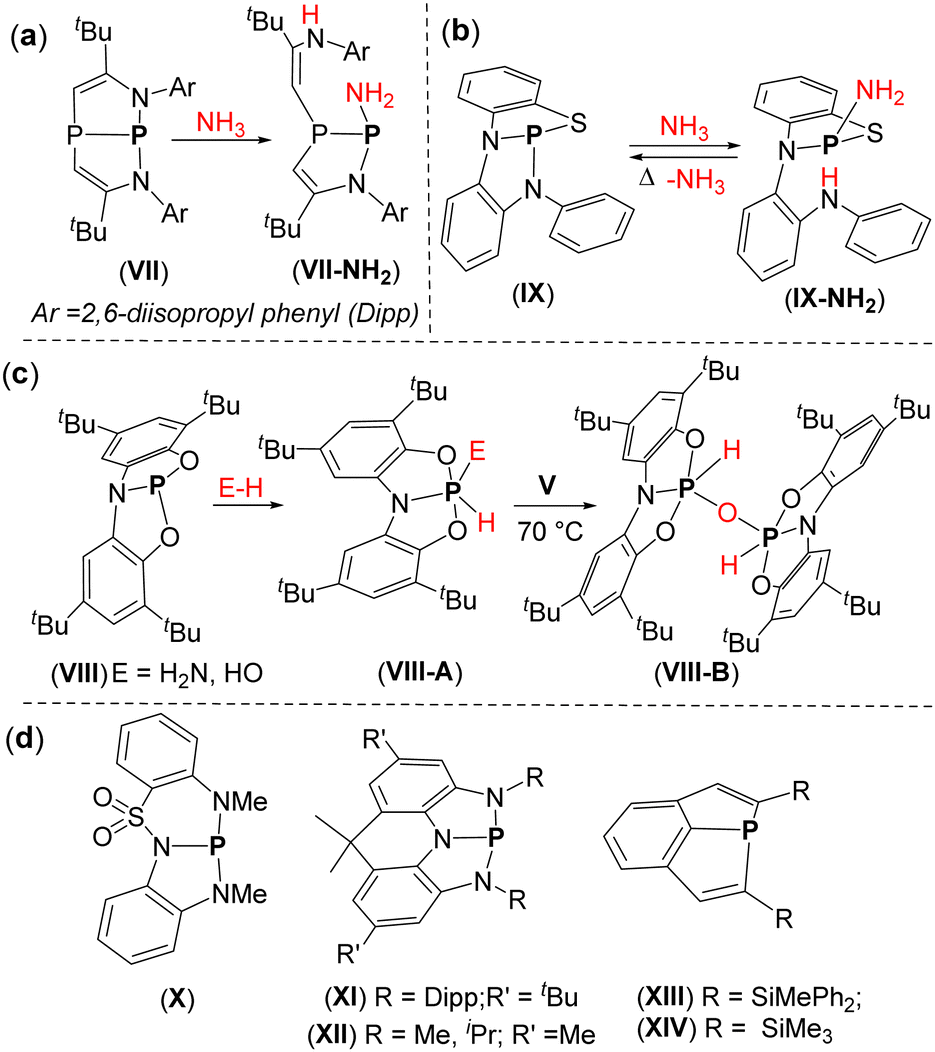 | ||
| Fig. 3 Known examples of SCPs and their use in the activation of various E–H bonds. | ||
Noteworthy, all the SCP systems discussed above activated NH3 irreversibly (Fig. 3a and c) i.e., no reductive elimination of the ammonia regenerating the starting SCP species was observed. The first neutral SCP in an NNS-type ligand (IX) capable of activating NH3 and releasing it upon mild heating was reported by Goicoechea and co-workers in 2021 (Fig. 3b).56 In 2023, Vlugt and co-workers introduced an SCP centre in a non-symmetric NNN-type ligand (X) that could reversibly activate the N–H bond in dimethylamine (Fig. 3d).57 Beyond these examples, recent years have seen the emergence of several other intriguing SCP species, including V (R = iPr, 2-Py, SiMe3)19,40,58 and XI–XIV59–61 each showcasing unique and fascinating chemistry (Fig. 3d).
2. Cationic, structurally constrained PIII species
The field of PIII cations began with the synthesis of phosphenium cations in 1964 by Dimroth and Hoffmann,62 and had been largely dominated by N-heterocyclic phosphenium cations (NHPs).63,64 While NHPs are isolable, akin to N-heterocyclic carbenes (NHCs),65,66 their electronic properties are fundamentally different. Due to their cationic nature, NHPs are weak σ-donors and strong π-acceptors,67,68 making them highly effective ligands for electron-rich TM complexes.69–73 In contrast to ambiphilic carbenes,45,74,75 NHPs exhibit relatively low reactivity toward small molecules. In fact, only a few NHPs have demonstrated the ability to activate bonds in small molecules.76,77 Non-NHP phosphenium cations, particularly dications, have been shown to activate O–H bonds, but exhibit limited reactivity with Si–H and B–H bonds.78,79As mentioned earlier, the use of rigid scaffolds to constrain phosphorus centers has enabled the activation of E–H bonds (E = B, N, O, S, C–X, where X = N, F, Cl, Br, I) in neutral SCP species.36–43,54–61,80 The catalytic applications of neutral SCP compounds, however, have been largely limited to species IV and V. One strategy to further enhance the reactivity of SCP centers is to introduce a cationic charge, preferably localized on the phosphorus atom. This modification is expected to significantly boost reactivity by further lowering the LUMO. Literature precedents demonstrate that the presence of a cationic charge often yields more electrophilic species compared to their neutral counterparts. A notable example is borenium ions, which can catalyze reactions by activating a variety of small molecules.81 This type of enhanced reactivity is well illustrated by recent examples of cationic SCP species (CSCPs) (2, 6, 11, 13, 17, 24, and 34), which demonstrated the ability to activate a broad range of bonds, including O–H, N–H, C–H, C–F, Si–H, and H–H bonds (Fig. 4–13).82–88
 | ||
| Fig. 4 Synthetic scheme for the synthesis of: (a) [1A][OTf] and [1B][OTf]; (b) [2][B(C6F5)4] and the activation of O–H and N–H bonds by 2. | ||
 | ||
| Fig. 5 Synthetic scheme showing: (a) phosphorus mediated rearrangement of an OCO type ligand; (b) Sb-to-P metathesis to prepare CSCP, [6][SbCl4] and [6][Cb]; (c) activation reactions of O–H and N–H bonds by [6][Cb]. | ||
 | ||
| Fig. 6 Synthetic scheme for the synthesis of (a) [11][W]; (b) isomerization between bent structures of 11via a T-shaped planar structure. | ||
 | ||
| Fig. 7 (a) DFT computed mechanism for C–H bond activation in 1-methylindole by 11. Synthetic scheme to show: (b) the C–H activation by 11A–D and (c) and (d) the reductive elimination of C–H bond. | ||
 | ||
| Fig. 8 (a) Synthetic scheme for the synthesis of [13][W]; (b) phosphinophosphination of alkynes, olefins, and carbonyls with 13. | ||
 | ||
| Fig. 9 (a) Synthetic scheme for the synthesis of [17][OTf] and [17][B(C6F5)4]; (b) activation of O–H, N–H bonds and (c) H3NBH3 by [17][OTf]. | ||
 | ||
| Fig. 10 (a) Si–H bond activation by [17][OTf] and [17][B(C6F5)4]; (b) DFT optimized structure of 17-O(H)Me, 17-H-SiEt3 and their bond lengths around P atom; (c) hydrosilylation reaction catalyzed by [17][B(C6F5)4]. | ||
 | ||
| Fig. 11 Synthetic scheme for (a) the synthesis of [24][PF6]; (b) the formation of [25][PF6] by the activation of C–F bond and its further reactions; (c) the hydrodefluorination and C–N bond forming cross-coupling reactions catalyzed by 24; (d) and the scheme for proposed computational mechanism. | ||
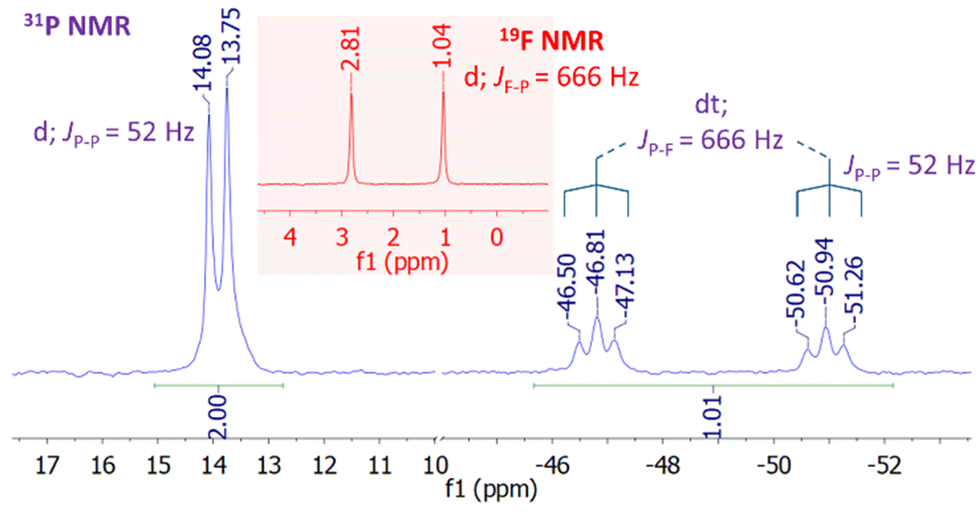 | ||
| Fig. 12 31P and 19F NMR spectra showing the formation of the OA product [25][PF6]. | ||
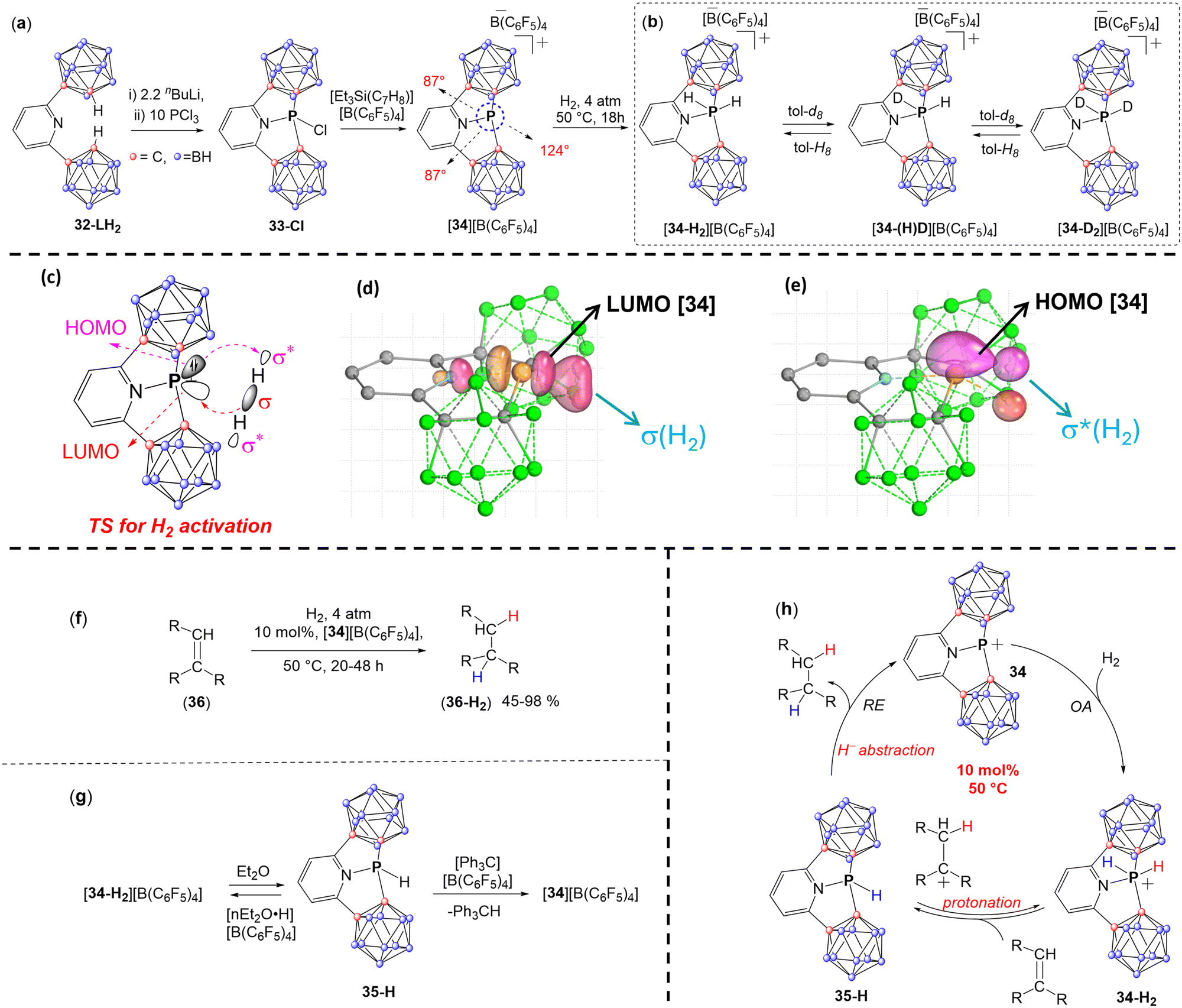 | ||
| Fig. 13 Synthetic scheme for (a) the synthesis of [34][B(C6F5)4]; (b) the activation of dihydrogen by [34][B(C6F5)4] and the exchange reaction between H and D atom between [34-H2][B(C6F5)4] and toluene-d8 and vice-versa; (c) schematic representation to show electrophilically driven activation of the H–H bond by 34.; (d) σ-bond of H2 approaching the P-centre from the direction of the LUMO of 34; (e) followed by the interaction of the HOMO of the P-centre with the LUMO (σ*) of H2; (f) [34][B(C6F5)4] catalyzed hydrogenation reactions of unsaturated C–C species; (g) equilibrium of [34][B(C6F5)4] with 35-H and reaction of 35-H with [Ph3C][B(C6F5)4]; and (h) the proposed catalytic cycle. | ||
Notably, in contrast to neutral SCP species, their cationic analogues have also found application in metallomimetic catalysis. For instance, the OA type reaction of the Si–H bond has been employed in the catalytic hydrosiliylation of aldehydes;86 the OA of C–F bonds has been utilized in catalytic hydrodefluorination and C–N bond cross-coupling reactions;87 and the OA of the H–H bond has been applied in catalytic hydrogenation.88
While the chemistry of neutral SCP species has been periodically reviewed over the years, the field of CSCP species is relatively young and remains less explored. This feature article provides an overview of recent advancements in CSCP chemistry, highlighting key distinctions from neutral SCP compounds in terms of synthetic methods, chemical properties, and potential applications.
Aldridge and Goicoechea reported the reaction of VIII with HOTf or MeOTf (OTf = trifluoromethanesulfonate) leading to PIII cations, 1A and 1B (Fig. 4a).89 The single crystal X-ray diffraction (SCXRD) structures of 1A and 1B revealed a significant distortion around the phosphorus centre and considerable elongation of the N–P bonds (1.92 and 1.95 Å, respectively) compared to the starting SCP compound, VIII (1.757(1) Å). However, the chemistry of these cations (1A and 1B) was not investigated.
In 2018, Dobrovetsky and co-workers reported a cationic phosphenium species, 2, featuring an ONO scaffold (2-L) with pyridine as the central donor.82 The synthesis of 2 involved first the preparation of chlorophosphine (2-Cl) by reacting PCl3 with 2-LH2 (Fig. 4b). Notably, 2-Cl was shown to be solvent dependent, in polar solvents the P–Cl bond dissociated forming the cationic [2][Cl]. An anion exchange reaction with weakly coordinating anion [B(C6F5)4]− resulted in the formation of a ion separated phosphenium cation, [2][B(C6F5)4]. The SCXRD of [2][B(C6F5)4] revealed a significant distortion in local symmetry around the P-centre. Noteworthy, the P–N bond length in 2 was significantly shorter (1.81 Å), in comparison to the P–N bond lengths in 1A and 1B (1.92 and 1.95 Å, respectively).
The preliminary reactivity of 2 with H2O and ROH (R = Me, iPr, tBu, and Ph) was studied. Remarkably, 2 reacted with H2O and ROH (R = Me, iPr, tBu) by the OA-type reactions of the O–H bonds to the P-center producing PV cations, 3a–3d (Fig. 4c). However, no reaction was observed with phenol, even at elevated temperatures. Additionally, 2 reacted with NH3via an OA-type reaction, leading to the formation of a PV compound (3e) which, interestingly, undergoes reductive elimination of NH3 at 70 °C, regenerating 2. The reversible nature of NH3 activation at phosphorus centre was unprecedented at the time and emphasized the novel reactivity derived from the combination of structural constraint and cationic character of the P-centre.
In recent years, dianionic, tridentate pincer-type ligands with NHC central donor have demonstrated significant utility in stabilizing a range of both early and late TM complexes.90–93 However, their application to MG elements has remained relatively underexplored.94–96
Attempts to obtain CSCP species 6 (Fig. 5b) through the reaction of 4-LH3 with PCl3 led to the rearrangement of imidazolinium to oxazolium ring (4A) mediated by the phosphorus centre (Fig. 5a).97 Interestingly when the reaction is carried out in the presence of H2O or CH3CN, different rearrangements lead to the formation of a neutral (4B) through a multi-step process, as illustrated in Fig. 5a.80a All the attempts to deprotonate 4A to obtain the CSCP species 5 failed (Fig. 5a). In contrast, the reaction of 4-LH3 with SbCl3 readily leads to the antimony chloride 4-SbCl (Fig. 5b).83 While the synthesis of 6via the reaction of deprotonated 4-LH3 with PCl3 failed, the use of 4-SbCl as a template for the Sb-to-P metathesis with PCl3 led directly to the desired [6][SbCl4] (Fig. 5b). The SCXRD structure of [6][SbCl4] revealed notable distortion around the phosphorus center (∠O1–P–O2 = 103°; ∠O1–P–C = 89°; ∠O2–P–C = 95°). Both the molecular structure and density functional theory (DFT) computational analysis of 6 suggested that the formal positive charge is divided between the two nitrogen atoms of the imidazolinium ring.
The reactivity of [6][SbCl4] with H2O and amines showed that [SbCl4]− anion is non-innocent in these reactions. Therefore, it was replaced with the carborane anion, [CB11H12]− (Cb) using CsCb, yielding [6][Cb] (Fig. 5b). [6][Cb] successfully activated O–H and N–H bonds in H2O and NH3via formal OA of the O–H or N–H bonds to the P-center leading to 7-H2O and 8-NH3 (Fig. 5c). 7-H2O was not a stable compound under the reaction conditions and rapidly underwent rearrangement of the imidazolinium unit to oxazolium (7′-H2O). Unlike the reversible H2N–H bond activation by 2 (Fig. 4c), 8-NH3 did not exhibit reversible reductive elimination of the NH3 fragment.
More interesting results were obtained in the reaction of [6][Cb] with alcohols and amines. In these cases, the reaction of [6][Cb] with MeOH, iPrNH2, PhOH, and PhNH2 reaches an equilibrium between the LA and OA products. Notably, in the case of PhOH and PhNH2, the LA product undergoes reductive elimination of PhOH and PhNH2 at room temperature, regenerating [6][Cb]. DFT calculations supported the thermoneutral nature of these reactions. It is worth noting that unlike 2, 6 was capable of activation of the PhO–H bond.
While Dobrovetsky and co-workers were unsuccessful in the synthesis of CSCP species 5 (Fig. 5a), Radosevich and Greb recently reported a series of phosphenium cations similar to 5, 11A–D, with pyridine-type donors in NNO scaffolds (Fig. 6a).84 The CSCP 11A–D were synthesized by chloride abstraction from the corresponding chlorophosphines (10-Cl) using NaB(C6F5)4 or LiAl(ORF)4 (Fig. 6a). In contrast to the nitrogen-donor-based cationic PIII species 1 and 2, the SCXRD structure of 11A–D revealed more pronounced distortion, with bond angles ∠N1–P–O = 108°, ∠N1–P–N2 = 86°, and ∠N2–P–O = 94°.
The low-temperature NMR and DFT calculations of 11A–D suggested a rapid dynamic conformational process in solution, occurring via the isomerization of the bent 11A structures. This process involves a metastable, T-shaped planar intermediate (11-IM; ΔG = 4.1 kcal mol−1) (Fig. 6b). Notably, the calculations also revealed that planarization of the bent global minimum positions the frontier orbitals at the P center, whereas in the bent form, these orbitals reside on the ligands. Additionally, the planarization significantly lowers the energy of the LUMO orbitals, resulting in a smaller HOMO–LUMO gap (2.5 eV). It was therefore suggested by the authors that the T-shaped intermediate (11-IM) is the key intermediate in bond activation reactions.
Remarkably 11A activates the C–H bonds in 1-methylindole and phenylacetylene at room temperature, producing OA-type products (Fig. 7b). DFT calculations of 11A with N-methylpyrrole as a model substrate revealed that the C–H bond activation proceeds through T-shaped intermediate 11-IM. Initially, the C–H bond is cleaved between the P- and Npyridine centres through a phosphorus-ligand cooperation mechanism. The resulting species, 11-IM-A, then isomerizes to 11-IM-B, which undergoes σ3-P/σ5-P tautomerization, transferring the hydrogen from nitrogen to the phosphorus centre and leading to the formation of the C–H activated product 12 (Fig. 7a). Notably, the intermediates (11-IM-A) and (11-IM-B) were not observed experimentally, suggesting their rapid conversion to the final OA product.
The more electron-poor CSCP species, 11B and 11D, could activate less reactive C–H bonds in thiophenes and furanes, leading to OA products (Fig. 7b). Remarkably, 11B and 11D also activated the C–H bond in benzene, which overall is very rare in MG chemistry.98 Similarly, 11D could activate the C–H bonds in toluene at ortho, meta, and para positions as well as the C–H bond in alkene (in 1,1-diphenylethylene) (Fig. 7b). 11C was capable of activating the C–H bond in chlorobenzene affording isomeric mixture of the C–H activated products (Fig. 7b).
Most importantly, the OA products herein also undergo reductive elimination and regeneration of the PIII species. For example, heating the OA product of 12B at 60 °C after 1 day leads the regeneration 11B (71%) and bromothiophene (Fig. 7c). This strategy could be effectively utilized for exchange reactions with other C–H bond-containing arenes. For example, the OA products of 11B or 11D with methylthiophene in the presence of 1-methylindole, when kept at rt for one day, yield a more stable indole addition product, with the elimination of methylthiophene. Similarly, the OA product 12D formed from the reaction of benzene with 11D, in the presence of toluene-d8, leads to the formation of the OA product with toluene-d8, while benzene undergoes reductive elimination (Fig. 7d).
Notably, electrophilic, two-coordinated phosphenium ions can also insert PIII centres into the C–H bonds, though their reactivity is generally limited to the activated C–H bonds in metallocenes, xanthene, and cycloheptatriene.78,99,100 This study emphasized the role of structural constraints in cationic PIII species, which enabled the activation of more challenging C–H bonds in a broader range of arenes. Importantly, the reversible RE observed in these reactions suggests promising applications in catalysis.
Recently, Greb and co-workers introduced phosphanylphosphenium 13, which is a modified version of 11, where the pyridine-based donor is replaced by a phosphine unit.85 The central P-centre in 13 is distorted from the C3v symmetry with the bond angles around it ∠P–P–O = 104°, ∠P–P–N = 87° and ∠O–P–N = 94° (in SCXRD) (Fig. 8a). Greb and co-workers show that the structural constraints at the P–P bond in 13 enable it to efficiently engage in the phosphinophosphination (addition of two phosphines to the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds) of various non-activated π-bonded substrates, such as alkynes, alkenes, aldehydes, and ketones (Fig. 8b).
X bonds) of various non-activated π-bonded substrates, such as alkynes, alkenes, aldehydes, and ketones (Fig. 8b).
In 2022, Dobrovetsky and co-workers reported CSCP species 17 in which the P centre is incorporated in an NNN-type scaffold with anionic pyrrole “arms” and a central pyridinyl donor.8617 was synthesized through the chlorophosphine precursor (17-Cl) via the double deprotonation of the bis(pyrrolyl)pyridine ligand (17-LH2) (Fig. 9a). Reacting 17-Cl with AgOTf or [Et3Si(C6H4F2)][B(C6F5)4] led to the facile formation of the corresponding cationic PIII species 17 (Fig. 9a).
The SCXRD structure of 17 clearly shows a strong deviation from a local C3v symmetry that could be easily concluded by different bond angles around the P-centre: ∠N1–P–N2 = 123°, ∠N1–P–N3 and ∠N2–P–N3 = 85° (Fig. 9a). Notably, 17 attributes a shorter bond between the pyridine nitrogen and phosphorus atoms (N3–P 1.752(3)Å) than that of nitrogen of pyrrole ring and phosphorus atom (N1–P 1.781(2), and N2–P 1.786(3) Å).
17 rapidly activates O–H and N–H bonds in MeOH and Et2NH, leading to the formation of LA products as syn and anti-isomers (Fig. 9b). 17 does not react with dihydrogen, however, it does react with H3NBH3 (1 equiv.) producing LA product (19-H2) with isomeric forms (Fig. 9c). Interestingly, 17 reacted with 3 equiv. of H3NBH3, producing formally a phosphinidene (PI product) (19-(BH3)2) in which the P center is coordinated to two BH3 molecules (Fig. 9c). This PI species likely forms via the addition of BH3 to the P-centre in an intermediate (19-BH3), which can also be independently synthesized by reacting 17 with 2 equiv. H3NBH3, with a concomitant liberation of [NH4]+.
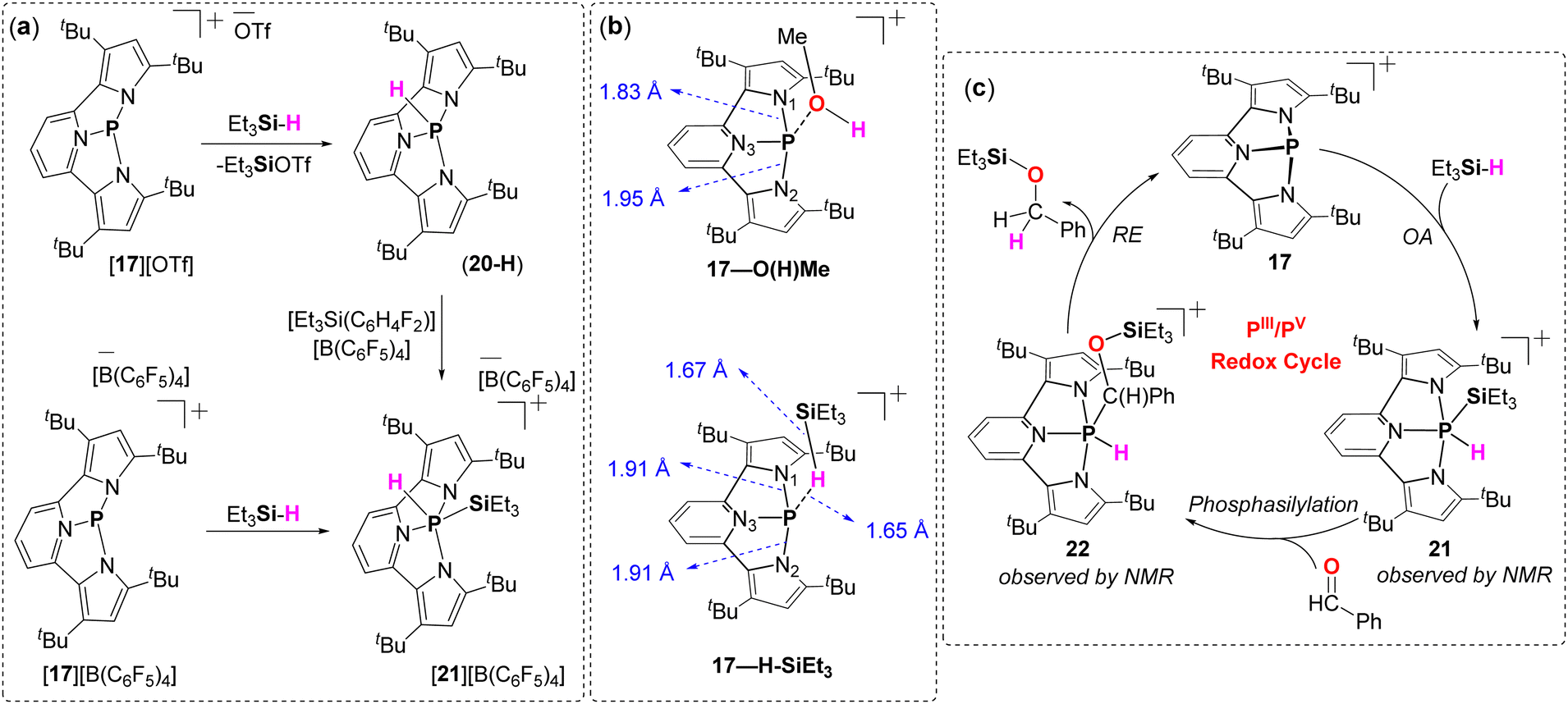
Interestingly, [17][OTf] reacts rapidly with Et3SiH, via a different reaction route compared to its reactions with MeOH, Et2NH, and H3NBH3 that involves the participation of the TfO− anion (Fig. 10a). The reaction proceeds via the formation of a hydro-phosphine (20-H) and Et3SiOTf, ultimately resulting in a complex mixture of unidentified products. The Lewis acidic nature of 17 and the non-innocent behaviour of the TfO− anion likely contribute to this unusual reactivity. Confirmation of this mechanism comes from the reaction of 20-H (obtained from 17-Cl and DIBAL-H) with Me3SiOTf, which shows a similar 31P NMR pattern to the activation of Et3SiH by [17][OTf].
Due to the weaker coordinating nature of the (C6F5)4B− anion compared to TfO− anion, the activation of Et3Si–H bond by [17][B(C6F5)4] leads to the rapid formation of 21via an unprecedented OA-type reaction of the Si–H bond to the PIII center of 17 (Fig. 10a). The formation of this OA product is clearly indicated by the corresponding doublets in the 1H NMR (9.56 ppm) and 31P NMR (−88.7 ppm), with a similar coupling constant (J = 577 Hz). Notably, 21 was unstable, leading to a complex 31P NMR spectrum that displays a mixture of unidentified products similar to the one obtained from the reaction of [17][OTf] with Et3SiH. Similar 31P NMR spectrum was observed when 20-H was reacted with [Et3Si(C6H4F2)][B(C6F5)4].
DFT computation showed that prior to the activation of the E–H bonds (E = N, O Si) there is an adduct formation between the substrate and 17. In the case of MeO–H bond activation, first the P–O type adduct is formed (17-O(H)Me) with the loss of the symmetry of the P-center supporting NNN-ligand (P–N1: 1.83 Å; P–N2: 1.95 Å; P–N3: 1.78 Å). These computations clearly show how 17-O(H)Me consequently leads to LA product 18-OMe by proton transfer to the N2 atom of the ligand (Fig. 10b). In contrast, the adduct 17-H-SiEt3 that is formed prior the OA product 21 has symmetrical NNN-ligand around P atom with slightly elongated P–N1 (1.91 Å) and P–N2 (1.91 Å) bonds and a shorter P–N3 (1.79 Å) bond (Fig. 10b). In addition, longer P–H (1.65 Å) and Si–H (1.67 Å) bonds in 17-H-SiEt3 compared to the P–H bond in 20-H (1.36 Å), and calculated Et3Si–H bond (1.50 Å), further support the weakening of the Si–H bond in 17-H-SiEt3 that eventually lead to OA type reaction. Eventually, in 17-H-SiEt3 the bulky Et3Si-group is positioned far from the pyrrole's basic nitrogen, preferring to bind to the less sterically encumbered lone-pair at the P-center. In contrast, in MeOH and Et2NH activation, the larger MeO- and Et2N-groups bind to P-center, while the pyrrole abstracts a proton, avoiding steric congestion (Fig. 10b).
The most compelling feature of this work is the catalytic hydrosilylation of CO bonds in benzaldehyde, mediated by 17, which emulates the behaviour of the TM complexes by following a similar catalytic cycle (Fig. 10c). In this process, benzaldehyde reacts with Et3SiH in the presence of 5 mol% of [17][B(C6F5)4], resulting in the formation of the corresponding silyl ether, Et3SiO–C(H2)Ph, after 12 hours at 50 °C. The stepwise progress of this reaction revealed first formation of an OA product 21 (31P(δ) = −88.73 ppm), followed by a phosphasilylation producing the migratory insertion intermediate 22 (31P(δ) = −74.54 ppm; 1JP–H = 648 Hz) and finally, a reductive elimination type reaction from PV centre in 22 producing Et3SiO–C(H2)Ph and regeneration of the active catalyst 17 (Fig. 10c).
Carbone-based carbodiphosphorane (CDP)101 scaffolds are increasingly being employed to construct complexes with MG elements.14–16,102–104 Specifically, the hexaphenyl-carbodiphosphoranyl-based CCC pincer-type ligand (23-LH2) is benefited from a strong donor carbon center and the facile deprotonation of the phenyl rings, which together impart an excellent ability to obtain dianionic, tridentate CCC pincer-type ligand that can be used to structurally constrain MG centers. Recent examples highlight the use of 23-LH2 to incorporate neutral beryllium species, as well as cationic antimony and bismuth species.14–16,102–104 In the case of phosphorus, deprotonation of 23-LH2, followed by reaction with PCl3, results in the production of a cationic SC PIII species, [24][Cl].87 An anion exchange with K[PF6] leads to of [24][PF6] (Fig. 11a).
A significant distortion around the P center is induced by the CCC framework in 24, resulting in wider ∠C1–P–C2 (106°) and narrower ∠C1–P–C3 (95°) and ∠C2–P–C3 (96°) bond angles (Fig. 10a). However, the degree of distortion in 24 is less pronounced than that observed in previously reported SCP species (XIII-XIV) featuring the CCC framework (Fig. 3d).
Notably, 24 is inert toward Et2NH, MeOH, and Et3SiH, however, it reacts with electron-poor fluoroarenes at elevated temperatures (80–120 °C) readily leading to the formation of the OA products of the C–F bond to the PIII centre in 24 (Fig. 11b). For instance, the reaction of [24][PF6] with pentafluoropyridine in 1,2-difluorobenzene (oDFB) for 3 h at 80 °C clearly demonstrates the formation of the OA product [25][PF6], as indicated by a doublet of triplets (1JPF = 666 Hz; 2JPP = 52 Hz) at δ = −48.87 ppm in 31P NMR spectrum with a complementary doublet (1JPF = 666 Hz) at δ = 1.92 ppm in 19F NMR (Fig. 12).
Interestingly, heating [25][PF6] to 110 °C for 10 hours results in the formation of a difluoro-species, [26][PF6], likely being mediated through a disproportionation mechanism involving a series of ligand exchange reactions followed by the elimination of perfluoro-4,4′-bipyridine (27) (Fig. 11b). In contrast, heating [25][PF6] in the presence of PhSiH3 at 80 °C for 3 h directly leads to the regeneration of [24][PF6], PhSiF3 and the product of hydrodefluorination (29). This transformation likely proceeds through hydrophosphorane intermediate [28][PF6] followed by a rapid RE of the C–H bond from the P-centre (Fig. 11b). Notably, similar reactivity was observed for the reaction with neutral SCP (V), but this reaction led to the hydrodefluorination products via a stable hydrophosphorane intermediate in the reaction with DIBAL-H.43
In contrast to V, the reactivity of 24 with fluoroarenes, coupled with its inertness toward hydrosilanes allowed the catalytic hydrodefluorination of fluoroarenes using PhSiH3 and 10 mol% [24][PF6] (Fig. 11c). Various fluoroarenes with different functional groups (F3C-, NC-, MeOC(O)-) could be catalytically hydrodefluorinated in good to excellent yields (90–95%). Importantly, the catalytic hydrodefluorination of fluoroarenes by 24 proceeds via a metallomimetic PIII/PV redox cycle.
Remarkably, the reactivity of 24 was also successfully extended to the catalytic C–N bond-forming cross-coupling reaction products (Fig. 11c). The substrate scope includes the reaction of various fluoroarenes with Et3Si–NR2 (R = Et, (H)Ar) in presence of 10 mol% of [24][PF6] in oDFB, at elevated temperatures (80–120 °C).
Both experimental and computational studies suggest that the mechanisms of both hydrodefluorination and C–N bond forming cross-coupling proceed in a metallomimetic fashion through the OA → LM → RE steps (Fig. 11d). Notably, a similar catalytic transformation was later reported with simple phosphines, however, the mechanism was distinctively different.7
Very recently, the Dobrovetsky group reported another example of pyridine based CSCP, [34][B(C6F5)4], using a CNC-type of pincer type ligand (32-LH2) (Fig. 13a).88 The CNC-type ligand consists of a pyridine ring attached to two o-carborane units.105 While the nitrogen atom of the pyridine ring stabilizes the cationic P center, the electron-withdrawing nature of the carborane units enhances the Lewis acidity of the P centre. Additionally, the o-carboranyl units are easily deprotonated at the C–H position, and their rigid structure imposes additional constraint at the P centre. Thus deprotonation of 32-LH2, followed by reaction with PCl3 affords a chlorophosphine 33-Cl (Fig. 13a). Chloride abstraction from 33-Cl using [Et3Si(C7H8)][B(C6F5)4] results in the formation of the cationic PIII species, [34][B(C6F5)4].
34 exhibited a highly distorted geometry around the P center (∠C1–P–C2 = 124°, ∠C1–P–N and ∠C2–P–N = 87°) (Fig. 13a). In contrast to the aforementioned CSCPs, a significant difference in the reactivity was observed for 34, as evidenced by its ability to activate dihydrogen (H2) at a single PIII centre, resulting in the formation of the formal OA product 34-H2 (Fig. 13b). It is important to note that while the activation of H2 by OA-type process has been observed at several low-valent MG centres, such as carbenes,3,45,106 borylenes,3,107–109 and their heavier analogues,110–115 the activation of H2 at a single PIII centre was never reported, which makes the reactivity of H2 with 34 unprecedented and provides new insight into the influence of SC and cationic character of this center. It is also important to note that the activation of H2 between either two phosphorus centers or between phosphorus and other MG center was reported with Frustrated Lewis acid–base pairs being the prime examples of this type of reactivity.
Importantly, all attempts to liberate H2 from [34-H2][B(C6F5)4] failed and the reductive elimination did not occur, which indicated that the H2 activation by 34 is irreversible. Interestingly, when [34-H2][B(C6F5)4] was dissolved in deuterated aromatics (C6D6, tol-d8) at 50 °C, an exchange reaction between H and D atoms occurred at the aromatic ring over the course of 3 h, leading to [34-D2][B(C6F5)4] and C6H6 (or C6H5CD3) (Fig. 13b). Conversely, a similar behaviour was observed for [34-D2][B(C6F5)4] in the presence of toluene leading to the formation of [34-H2][B(C6F5)4] and C6D5CH3.
The DFT computed mechanism of H2 activation by 34 reveals that this process is electrophilically driven, with the H–H σ-bond approaching the cationic P-centre from the direction of the LUMO in the asynchronous transition state (TS) (Fig. 13c–e). This is followed by the interaction of the HOMO of the P-centre with the LUMO (σ*) of the H–H bond (Fig. 13c–e).
As mentioned previously, despite the ability of several low-valent MG compounds such as carbenes, borylenes, and their heavier analogues to activate the H–H bond by OA, the hydrogenation reaction using these MG species was never reported. In contrast, [34][B(C6F5)4] could be further used in hydrogenation of various unsaturated C–C bonds and fused aromatic systems (Fig. 13f). Mechanistic and experimental studies of the hydrogenation process suggested that the first step of this process is OA of H2 to PIII centre in 34 producing 34-H2. Deprotonation of 34-H2 by the doubly-bonded species at next step, produces the corresponding carbocation and 35-H (Fig. 13g and h). The last step is the hydride abstraction from the 35-H by the carbocation producing the hydrogenated products and regenerated catalyst (34) (Fig. 13g and h). This was supported by a sequential deprotonation and hydride abstraction of 34-H2 by first Et2O and second Ph3C+ (Fig. 13g).
3. Conclusions
In this review, we discussed the chemistry of cationic structurally constrained phosphines (CSCPs) and their use in the activation of small molecules and catalysis. While most phosphines do not exhibit significant reactivity toward challenging bonds, the application of the structural constraint (SC) to phosphorus enhances their reactivity and leads to ambiphilic phosphorus centers capable of activating such bonds. Despite significant progress with structurally constrained phosphines, their ability to catalyze reactions remains very limited.Recently, a new class of SCPs—cationic SCPs (CSCPs)—was designed, in which the combination of SC and cationic charge enables these P-compounds to not only activate challenging bonds but also participate in catalysis. The CSCPs highlighted in this review demonstrated reversible activation of O–H and N–H bonds. They were also shown to activate C–F bonds in fluoroarenes through oxidative addition (OA)-type reactions, enabling catalytic hydrodefluorination and C–N bond-forming cross-coupling reactions with elementary steps reminiscent of TM-based catalytic cycles. Remarkably, these species were capable of activating Si–H, C–H, and H–H bonds through OA-type reactions, and participate in phosphino-phosphination reactions of CC double bonds. While the activation of C–H bonds has yet to find applications in catalysis, Si–H and H–H bond activations were successfully employed in catalytic hydrosilylation and hydrogenation reactions, mimicking TM-like behavior.
CSCPs have already proven to be promising species for the activation of challenging bonds. However, their full potential for activating other challenging bonds and their broader applications in catalysis remain to be explored.
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Israeli Science Foundation, Grant 195/22, and the Israel Ministry of Science Technology & Space, Grant 01032376. D. B. thanks Raymond & Beverley Sackler for the Postdoctoral fellowship. D. T. thanks the Ariane de Rothschild Women Doctoral scholarship for outstanding female PhD students.References
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed.
- C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 CrossRef CAS PubMed.
- H. Fujimoto, K. Yasui and M. Tobisu, Bull. Chem. Soc. Jpn., 2023, 96, 872–886 CrossRef CAS.
- S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 2005–2014 CrossRef CAS PubMed.
- P. Šimon, R. Jambor, A. Růžička and L. Dostál, Organometallics, 2013, 32, 239–248 CrossRef.
- C. Ganesamoorthy, C. Wölper, L. Dostál and S. Schulz, J. Organomet. Chem., 2017, 845, 38–43 CrossRef CAS.
- M. Huang, K. Li, Z. Zhang and J. Zhou, J. Am. Chem. Soc., 2024, 146, 20432–20438 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CrossRef CAS PubMed.
- M. K. Mondal, L. Zhang, Z. Feng, S. Tang, R. Feng, Y. Zhao, G. Tan, H. Ruan and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 15829–15833 CrossRef CAS PubMed.
- D. Obi, D. A. Dickie, W. Tiznado, G. Frenking, S. Pan and R. J. GilliardJr, Inorg. Chem., 2022, 61, 19452–19462 CrossRef PubMed.
- A. D. Obi, C.-L. Deng, A. J. Alexis, D. A. Dickie and R. J. Gilliard Jr., Chem. Commun., 2024, 60, 1880–1883 RSC.
- L. Warring, K. S. Westendorff, M. T. Bennett, K. Nam, B. M. Stewart, D. A. Dickie, C. Paolucci, T. B. Gunnoe and R. J. GilliardJr, Angew. Chem., Int. Ed., 2025, 64, e202415070 CrossRef CAS PubMed.
- M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850–7855 CrossRef CAS PubMed.
- T. Hynes, J. D. Masuda and S. S. Chitnis, ChemPlusChem, 2022, e202200244 CrossRef CAS PubMed.
- K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem. – Eur. J., 2019, 25, 16414–16424 CrossRef CAS PubMed.
- S. Kundu, Chem. – Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- A. Maiti, R. Yadav and L. Greb, Structural constraint effects on p-block elements: Recent advances, chapter 8, Adv. Inorg. Chem., 2023, 82, 261–299 CrossRef CAS.
- T. J. Hannaha and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 RSC.
- D. Houalla, F. H. Osman, M. Sanchez and R. Wolf, Tetrahedron Lett., 1977, 35, 3041–3044 CrossRef.
- R. Wolf, Pure Appl. Chem., 1980, 52, 1141–1150 CrossRef CAS.
- C. Bonningue, D. Houalla, M. Sanchez and R. Wolf, J. Chem. Soc., Perkin Trans. 2, 1981, 19 CAS.
- C. Bonningue, D. Houalla and R. Wolf, J. Chem. Soc., Perkin Trans. 2, 1983, 773–776 CAS.
- R. Contreras, A. Murillo, G. Uribe and A. Klaébé, Heterocycles, 1985, 23, 2187–2192 CAS.
- A. Murillo, L. M. Chiquete, P. Joseph-Nathan and R. Contreras, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 53, 87–101 CAS.
- R. Contreras, A. Murillo and P. Joseph-Nathan, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 215–224 CAS.
- C. Camacho-Camacho, F. J. Martínez-Martínez, M. De Jesús Rosales-Hoz and R. Contreras, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 91, 189–203 CAS.
- G. Baccolini, E. Mezzina, P. E. Todesco and E. Foresti, J. Chem. Soc., Chem. Commun., 1988, 304–305 CAS.
- G. Baccolini, E. Mezzina and P. E. Todesco, J. Chem. Soc., Perkin Trans. 1, 1988, 3281–3283 CAS.
- G. Baccolini, C. A. Mosticchio, E. Mezzina, C. Rizzoli and P. Sgarabotto, Heteroat. Chem., 1993, 4, 319–322 CAS.
- S. A. Culley and A. J. Arduengo, J. Am. Chem. Soc., 1984, 106, 1164–1165 CAS.
- A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef CAS.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed.
- S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS PubMed.
- W. Zhao, S. M. McCarthy, T. Yi Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed.
- H. W. Moon, A. Maity and A. T. Radosevich, Organometallics, 2021, 40, 2785–2791 CrossRef CAS.
- Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS PubMed.
- S. Lim and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 16188–16193 CrossRef CAS PubMed.
- (a) O. Elishav, B. M. Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. G. Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed; (b) S. Streiff and F. Jérôme, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC; (c) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed; (d) E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, J. Am. Chem. Soc., 2009, 131, 14234–14236 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 CrossRef CAS PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed.
- A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- D. Wendel, T. Szilvási, D. Henschel, P. J. Altmann, C. Jandl, S. Inoue and B. Rieger, Angew. Chem., Int. Ed., 2018, 57, 14575–14579 CrossRef CAS PubMed.
- F. Dankert, J.-E. Siewert, P. Gupta, F. Weigend and C. Hering-Junghans, Angew. Chem., Int. Ed., 2022, 61, e202207064 CAS.
- A. L. Humphries, G. A. Tellier, M. D. Smith, A. R. Chianese and D. V. Peryshkov, J. Am. Chem. Soc., 2024, 146, 33159–33168 CAS.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS PubMed.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- J. Abbenseth, O. P. E. Townrow and J. M. Goicoechea, Angew. Chem., Int. Ed., 2021, 60, 23625–23629 CrossRef CAS PubMed.
- N. Beims, T. Greven, M. Schmidtmann and J. I. van der Vlugt, Chem. – Eur. J., 2023, 29, e202302463 CrossRef CAS PubMed.
- A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 8114–8118 CrossRef CAS PubMed.
- A. J. King, J. Abbenseth and J. M. Goicoechea, Chem. – Eur. J., 2023, 29(1–12), e202300818 CrossRef CAS PubMed.
- A. Hentschel, A. Brand, P. Wegener and W. Uhl, Angew. Chem., Int. Ed., 2018, 57, 832–835 CrossRef CAS PubMed.
- A. Brand, A. Hentschel, A. Hepp and W. Uhl, Eur. J. Inorg. Chem., 2020, 361–369 CrossRef CAS.
- K. Dimroth and P. Hoffmann, Angew. Chem., Int. Ed. Engl., 1964, 3, 384 Search PubMed.
- D. Gudat, Acc. Chem. Res., 2010, 43, 1307–1316 CrossRef CAS PubMed.
- D. Gudat, Dalton Trans., 2016, 45, 5896–5907 RSC.
- D. Gudat, Eur. J. Inorg. Chem., 1998, 1087–1094 CAS.
- D. Gudat, A. Haghverdi, H. Hupfer and M. Nieger, Chem. – Eur. J., 2000, 6, 3414–3425 CrossRef CAS PubMed.
- H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2007, 46, 10693–10706 CrossRef CAS PubMed.
- A. L. Brazeau, C. A. Caputo, C. D. Martin, N. D. Jones and P. J. Ragogna, Dalton Trans., 2010, 39, 11069–11073 RSC.
- D. Gudat, Coord. Chem. Rev., 1997, 163, 71–106 CrossRef CAS.
- H. Nakazawa, J. Organomet. Chem., 2000, 611, 349–363 CrossRef CAS.
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606–626 CrossRef CAS.
- N. Burford, D. E. Herbert, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2004, 126, 17067–17073 CrossRef CAS PubMed.
- C. A. Caputo, M. C. Jennings, H. M. Tuononen and N. D. Jones, Organometallics, 2009, 28, 990–1000 CrossRef CAS.
- T. W. Hudnall, J. P. Moerdyka and C. W. Bielawski, Chem. Commun., 2010, 46, 4288–4290 RSC.
- U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedeckec and G. Frenking, Chem. Sci., 2010, 1, 697–704 RSC.
- C. A. Caputo, J. T. Price, M. C. Jennings, R. McDonald and N. D. Jones, Dalton Trans., 2008, 3461–3469 RSC.
- C. M. Feil, F. Goerigk, Y. Stöckl, M. Nieger and D. Gudat, Dalton Trans., 2025, 54, 1806–1814 RSC.
- N. Đorđević, R. Ganguly, M. Petković and D. Vidović, Inorg. Chem., 2017, 56, 14671–14681 CrossRef PubMed.
- M. Q. Y. Tay, Y. Lu, R. Ganguly and D. Vidović, Chem. – Eur. J., 2014, 20, 6628–6631 CrossRef CAS PubMed.
- (a) D. Bawari, S. Volodarsky, Y. Ginzburg, K. Jaiswal, P. Joshi and R. Dobrovetsky, Chem. Commun., 2022, 58, 12176–12179 RSC; (b) P. Wang, Q. Zhu, Y. Wang, G. Zeng, J. Zhu and C. Zhu, Chin. Chem. Lett., 2021, 32, 1432–1436 CrossRef CAS.
- X. Tan and H. Wang, Chem. Soc. Rev., 2022, 51, 2583–2600 RSC.
- S. Volodarsky and R. Dobrovetsky, Chem. Commun., 2018, 54, 6931–6934 CAS.
- D. Bawari, I. Malahov and R. Dobrovetsky, Angew. Chem., Int. Ed., 2024,(7 of 9), e202419772 Search PubMed.
- D. Roth, A. T. Radosevich and L. Greb, J. Am. Chem. Soc., 2023, 145, 24184–24190 CAS.
- L. You, D. Roth and L. Greb, Chem. Sci., 2025, 16, 1716–1721 RSC.
- S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61(1–8), e202208401 CAS.
- K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786–3794 CAS.
- D. Bawari, D. Toami, K. Jaiswal and R. Dobrovetsky, Nat. Chem., 2024, 16, 1261–1266 CrossRef CAS PubMed.
- T. P. Robinson, S.-K. Lo, D. De Rosa, S. Aldridge and J. M. Goicoechea, Chem. – Eur. J., 2016, 22, 15712–15724 CrossRef CAS PubMed.
- M. J. Panzner, C. A. Tessier and W. J. Youngs, Pincer complexes of N-heterocyclic carbenes. Potential uses as pharmaceuticals, The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, The Netherlands, 2007, 139–150 Search PubMed.
- D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC.
- R. Taakili and Y. Canac, Molecules, 2020, 25, 2231 CrossRef CAS PubMed.
- I. Jain and P. Malik, Eur. Polym. J., 2021, 150, 110412 CrossRef CAS.
- N. Matsumura, J. Kawano, N. Fukunishi and H. Inoue, J. Am. Chem. Soc., 1995, 117, 3623–3624 CrossRef CAS.
- C. Romain, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2014, 33, 5730–5739 CrossRef CAS.
- J. M. Farrell and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 5214–5217 CrossRef CAS PubMed.
- D. Bawari, S. Volodarsky, Y. Ginzburg, K. Jaiswal, P. Joshi and R. Dobrovetsky, Dalton Trans., 2021, 50, 16478–16482 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- A. H. Cowley, R. A. Kemp and C. A. Stewart, J. Am. Chem. Soc., 1982, 104, 3239–3240 CrossRef CAS.
- A. Jayaraman and B. T. Sterenberg, Organometallics, 2016, 35, 2367–2377 CrossRef CAS.
- F. Ramirez, B. Hansen, N. B. Desai and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 CrossRef CAS.
- M. R. Buchner, S. Pan, C. Poggel, N. Spang, M. Müller, G. Frenking and J. Sundermeyer, Organometallics, 2020, 39, 3224–3231 CrossRef CAS.
- P. Dabringhaus, A. Molino and R. J. GilliardJr, J. Am. Chem. Soc., 2024, 146, 27186–27195 CrossRef CAS PubMed.
- Z. Liu, Z. Wang, H. Mu, Y. Zhou, J. Zhou and Z. Dong, Nat. Commun., 2024, 15, 9849 CrossRef CAS PubMed.
- K. P. Anderson, H. A. Mills, C. Mao, K. O. Kirlikovali, J. C. Axtell, A. L. Rheingold and A. M. Spokoyny, Tetrahedron, 2019, 75, 187–191 CrossRef CAS PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef CAS PubMed.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 823–8261 CrossRef PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CAS.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CAS.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CAS.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, Angew. Chem., Int. Ed., 2023, 62, e202302071 CAS.
| This journal is © The Royal Society of Chemistry 2025 |