DOI:
10.1039/D5CC00123D
(Feature Article)
Chem. Commun., 2025,
61, 5883-5898
Harnessing radical mediated reactions of thioacids for organic synthesis
Received
8th January 2025
, Accepted 19th March 2025
First published on 20th March 2025
Abstract
Thiyl radical mediated reactions are of burgeoning importance for organic synthesis. This Feature Article focuses specifically on thioacid- and thioacetate-derived thiyl radicals as versatile intermediates for the synthesis of a diverse range of organic compounds under mild conditions with a high degree of chemo-, regio- and diastereoselectivity. We review recent developments in the field, including novel approaches for radical initiation, strategies for the synthesis of a wide range of functional groups, peptide and glycan diversification, protein labelling and radical dethiocarboxylation. We outline our own contributions to the field over several years, including concomitant strategies to furnish native peptide bonds and discuss the future directions of this field.
Introduction
Thioacids, the sulfur analogues of carboxylic acids in the form RCOSH, are of increasing importance in organic synthesis due to their unique reactivity.1 The bond dissociation energy (BDE) of the thioacid S–H bond is found to be in the same range as that of alkanethiols (≈87 kcal mol−1), enabling homolytic reactivity comparable to thiol-derived thiyl radicals.2 Facile homolytic cleavage of the thioacid S–H bond is often achieved via ultraviolet (UV) or visible light irradiation, or by thermal radical initiation to chemoselectively form thiyl radicals. Herein, reactions where thioacid-derived radicals function as key reactive intermediates for a diverse range of organic transformations are reviewed. The aim of the review is to highlight the considerable synthetic potential of thioacid-derived radicals for the synthesis of a range of valuable functionalities including amides, thioesters and thiolactones amongst others and to outline their broad application for organic synthesis.
Amide bond formation via diacyl disulfides
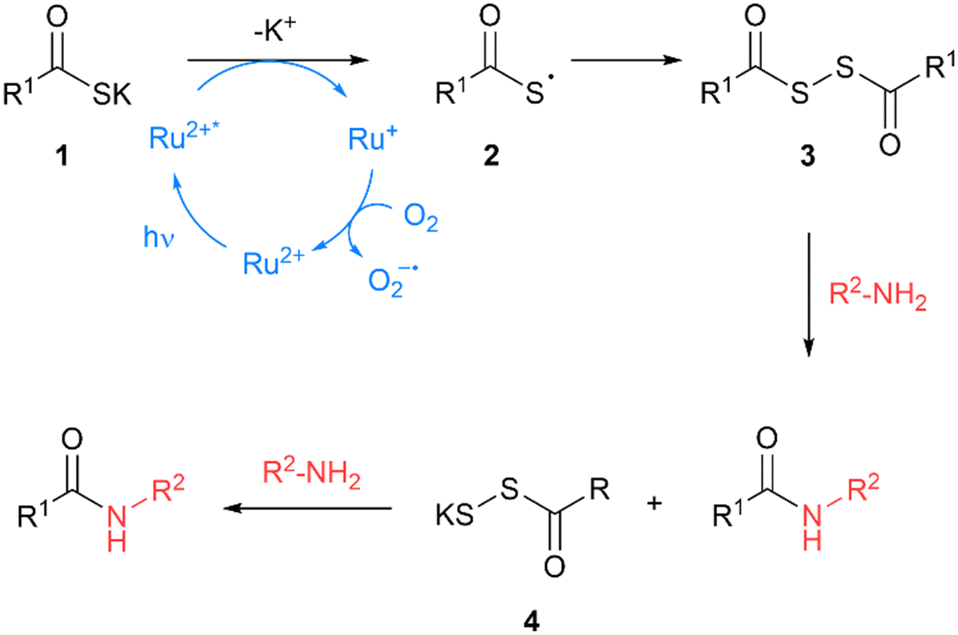
The formation of amide bonds is one of the most widely utilised and researched reactions in organic chemistry due to the ubiquity of this linkage in pharmaceuticals, biomolecules, and natural products.3,4 In recent years, there has been a drive to develop alternatives to the traditional coupling reagent-based amide formation methods in order to improve the atom economy, selectivity and moisture sensitivity of the reaction.5,6 To address such challenges, Tan and co-workers developed a novel amide bond-forming reaction between thioacids/potassium thioacetate and amines, facilitated by visible light photoredox catalysis.7 Mechanistic studies by the authors demonstrated that the reaction is initiated upon visible-light photoexcitation of the Ru2+ photocatalyst, Ru(bpy)3Cl2, leading to the formation of excited Ru2+* (Fig. 1). This excited species is reduced to Ru1+ by single-electron transfer (SET) with an electron rich thioacetate 1 to form thioacid thiyl radical 2. The active Ru2+ catalyst is regenerated by atmospheric oxygen to complete the catalytic cycle. The thioacid thiyl radical 2 undergoes diradical coupling with another thiyl radical to form a key diacyl disulfide intermediate 3, which undergoes subsequent aminolysis to form the amide. The perthioacetate by-product 4 also undergoes direct aminolysis with another equivalent of the amine to form an amide. Compared to traditional coupling reagent-based amide bond-forming strategies, this process is highly atom efficient, generating only sulfur salts as the by-product.
 |
| | Fig. 1 Proposed mechanism of amide bond formation using potassium thioacetate and Ru(bpy)3Cl2.7 | |
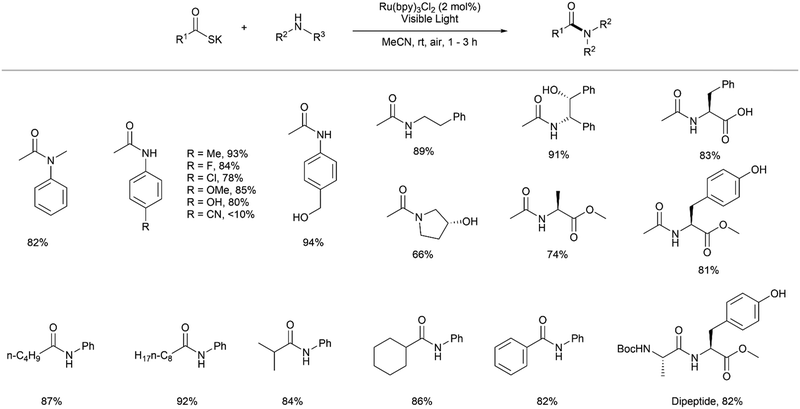
Investigating the scope of the reaction, Tan and co-workers first reacted primary and secondary anilines with potassium thioacetate using 2 mol% of the photocatalyst Ru(bpy)3Cl2 in acetonitrile with visible light irradiation using a 45 W household bulb for 1–3 h.7 High yields of the corresponding aromatic amides were obtained for a range of anilines substituted with both electron-donating and electron-withdrawing groups (Fig. 2). Notably, the reaction showed high selectivity for the acylation of the desired amine over phenols or benzylic alcohols. The only amide which was isolated in a poor yield (<10%) contained a highly electron-withdrawing para-nitrile substituent, reducing the reactivity of the amine. Furthermore, the authors reacted a range of aliphatic amines under the same conditions to yield aliphatic amides in moderate to high yield. Some biologically relevant N-acyl derivatives of α-amino acids such as alanine, phenylalanine and tyrosine were also prepared with excellent yields, including with an unprotected C-terminal carboxyl group. Tan and co-workers demonstrated that the reaction proceeded in high yields within 3 h for a variety of long chain aliphatic thioacids, branched and hindered thioacids and aromatic thioacids. The authors even demonstrated a peptide coupling reaction using these conditions through reaction of thioalanine with a methyl ester of tyrosine to form the Ala-Tyr dipeptide in 82% yield, paving the way for the application of this chemistry for peptide synthesis.
 |
| | Fig. 2 Selected examples of amides formed by the coupling of potassium thioacetates and amines.7 | |
Subsequently, Biswas and co-workers reported the amide bond-forming reaction between thioacids and amines at room temperature in water, using CdS nanoparticles as a photocatalyst upon irradiation with a household 30 W compact fluorescent light (CFL) bulb.8 The scope of the reaction was demonstrated through reaction of thiobenzoic acid and thioacetic acid with a range of substituted anilines, as well as alkyl amines in good to excellent yields (Fig. 3). Furthermore, the reaction of thiobenzoic acid with aniline was conducted on multigram scale under the standard conditions to furnish N-phenylbenzamide in 88% yield, demonstrating the scalability of the process. The authors established that the CdS nanoparticles could be recycled at least 6 times without significant loss of catalytic activity by recovering the catalyst through simple centrifugation after the reaction. After this point, agglomeration of the nanoparticles during the reaction began to slowly diminish catalytic activity. To further demonstrate the ‘sustainable’ credentials of the reaction, the authors conducted the reaction of thiobenzoic acid and aniline under moderate sunlight (44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lux) and an excellent conversion of 90% was obtained after 3 h. Following extensive mechanistic studies, a mechanism was proposed whereby surface binding of the thioacetates onto the CdS nanoparticles resulted in the generation of thioacid thiyl radicals by SET upon visible light irradiation. The thioacid thiyl radicals then readily couple to form diacyl disulfides which are aminolysed to the corresponding amide.
000 lux) and an excellent conversion of 90% was obtained after 3 h. Following extensive mechanistic studies, a mechanism was proposed whereby surface binding of the thioacetates onto the CdS nanoparticles resulted in the generation of thioacid thiyl radicals by SET upon visible light irradiation. The thioacid thiyl radicals then readily couple to form diacyl disulfides which are aminolysed to the corresponding amide.
 |
| | Fig. 3 Selected scope of the coupling of thioacids and amines to form amide bonds using CdS nanoparticles.8 | |
In pursuit of the development of improved ‘green’, environmentally benign conditions, Ramón and co-workers conducted photocatalytic amide bond formation between thioacids and amines in deep eutectic solvents (DESs).9 The authors used 2 mol% of Ru(bpy)3Cl2 as a photocatalyst with blue light-emitting diode (LED) (450 nm) irradiation. After optimisation, a DES system consisting of 1:2 choline chloride:urea was found to give the best results, which is attributed in part to the basic nature of the mixture which could deprotonate the thioacid to facilitate the reaction. The reaction of thioacetic acid with a wide range of anilines substituted with electron donating and withdrawing groups gave good to excellent yields in 1 h under the optimised conditions (selected examples illustrated in Fig. 4). Additionally, a range of aromatic and alkyl thioacids underwent reaction in good to excellent yields. Interestingly, terminal alkenes which are known to trap thioacid thiyl radicals, were well tolerated under these conditions as evidenced by the compatibility of undec-10-enoic thioacid with the reaction. To test the reusability of the DES solvent system, the organic compounds were extracted with ethyl acetate after reaction and the DES and photocatalyst which remained were reused after being vacuum dried. The DES could be recycled for up to 3 cycles without significant loss of reaction yield. Following detailed mechanistic studies, the key intermediate was once again determined to be a diacyl disulfide formed by diradical coupling of two thioacid radicals, which undergoes direct aminolysis to furnish the amide.
 |
| | Fig. 4 Selected scope of the coupling of thioacids and amines in a DES solvent system to form amide bonds.9 | |
Li and co-workers studied the coupling of thioacids and amines utilising the photoredox catalyst, 9-mesityl-10-methylacridinium tetrafluoroborate (Mes-Acr-MeBF4) 5, under blue LED irradiation (450 nm).10 Under the optimised conditions, 2 mol% of the photoredox catalyst in acetonitrile, a range of substituted anilines and various alkyl and aryl thioacids were reacted to furnish amide products in good to excellent yields. The authors also studied the coupling of α-amino thioacids and showed that commonly used α-amino protecting groups, including Fmoc, Cbz and Boc were well tolerated (Fig. 5). The coupling of Gly, Ala, Pro, Met, Thr and Phe derivatives with various anilines proceeded in good to excellent yields with good functional group tolerances. Heterocycles, secondary alcohols and sulfides were unaffected by the reaction conditions. The potential of the reaction to access biologically relevant substrates was demonstrated through synthesis of the drugs moclobemide, melatonin and acetazolamide in excellent yield, without the requirement for any protecting group manipulations due to the high-selectivity of the amide bond-forming reaction.
 |
| | Fig. 5 Selected scope of amides formed by coupling of thioacids and amines using the photoredox catalyst Mes-Acr-MeBF45.10 | |
Recently, Li and coworkers reported a facile bioconjugation strategy which exploits the chemoselectivity of the photochemical thioacid mediated amide formation reaction to acylate lysine (Lys) residues.11 After optimisation of reaction conditions in a high-throughput microfluidic robotic system, it was found that riboflavin tetraacetate (RFTA) served as a suitable photoredox catalyst under blue light irradiation (450 nm). The reaction is postulated to proceed through the formation of thioacid thiyl radicals which dimerise to form a highly reactive diacyl disulfide intermediate, followed by aminolysis by Lys residues to yield the amide. Firstly, the authors reacted a diverse range of peptides including drugs such as somatostatin and melanotan with various thioacids under the optimised conditions to yield acylated Lys derivatives with good to excellent conversions, with concomitant acylation of the N-terminal amine. Furthermore, the reaction was also tested on proteins such as carbonic anhydrase, trypsin, myoglobin, both an antibody and nanobody (CD39Nb) against CD38, and trastuzumab along with thioacids bearing azide or biotin functionalities which can be used as handles for further modification (selected example in Fig. 6). High labelling efficiency of up to 90% was observed with reaction times as low as one minute for these isolated proteins. To investigate the compatibility of the protocol with the labelling of Lys residues in a complex mixture, the reaction was conducted on cell lysate using a biotin bearing thioacid and Lys-labelled proteins were successfully isolated. Other nucleophilic residues such as cysteine, serine, arginine and tyrosine accounted for only 6.8% of the labelled sites, demonstrating the good selectivity of the method for labelling Lys residues.
 |
| | Fig. 6 Selected example of the photoredox Lys labelling of proteins using thioacids.11 | |
N-Acylation of sulfoximines via diacyl disulfide
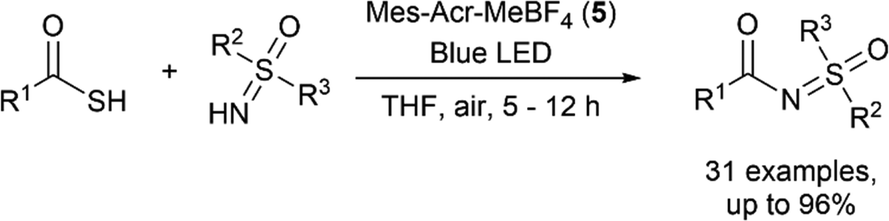
Song and co-workers used a similar thioacid mediated strategy to form C–N bonds in the N-acylation of sulfoximines (Fig. 7).12 Using the photosensitiser Mes-Acr-MeBF4 (5) and blue LED irradiation in air, a wide range of sulfoximines were acylated with various alkyl and aryl thioacids in yields of up to 96% with good functional group tolerance. The proposed mechanism involves formation of a thioacid radical which undergoes diradical coupling to form a highly electrophilic diacyl disulfide intermediate. Subsequent aminolysis of this intermediate by a sulfoximine generates the N-acyl sulfoximine.
 |
| | Fig. 7
N-Acylation of sulfoximines using thioacids.12 | |
Thioester bond formation via diacyl disulfide
In a related approach to the aforementioned reactions, Shah and co-workers reported the synthesis of thioesters through coupling of thioacids and thiols under blue LED irradiation in air.13 The proposed mechanism, developed via detailed mechanistic studies, involves deprotonation of the thioacid 6 by a base to form the thiocarboxylate 7, which, upon irradiation by blue light, forms an excited thiocarboxylate species 8 (Fig. 8). This intermediate undergoes SET with atmospheric O2 to form a thioacid radical 9 and a superoxide radical (O2˙−). The superoxide radical (O2˙−) abstracts a proton from another molecule of thioacid and is eventually converted to hydrogen peroxide (H2O2). The thioacid radical 9 undergoes diradical coupling to form a diacyl disulfide intermediate 10 which is thiolysed to form thioester 11 and a dihydrosulfide intermediate 12. The intermediate 12 can undergo further reaction with another thiol to form a thioester, analogous to that observed in amide bond forming reactions.
 |
| | Fig. 8 Mechanism proposed by Shah and co-workers for thioester bond formation between thioacids and thiols under photochemical conditions.13 | |
A diverse array of aryl thiols displaying both electron donating and withdrawing substituents underwent thioester formation with thiobenzoic acid in good to excellent yields and in a reaction time of 6 h (selected examples illustrated in Fig. 9).13 In addition, heterocyclic thiols like 2-methyl-3-furanthiol and thiophene-2-thiol reacted in good yield. Aliphatic thiols were also compatible with the conditions but displayed generally lower yields compared to the aromatic thiols. Various thioacids were also screened including aromatic thioacids with electron donating and withdrawing substituents, aliphatic thioacids including those derived from amino acids (Phe, Val and Leu), and drugs such as (S)-naproxen and gabapentin.
 |
| | Fig. 9 Scope of thioester bond formation.13 | |
Thioester bond formation by C(sp3) activation
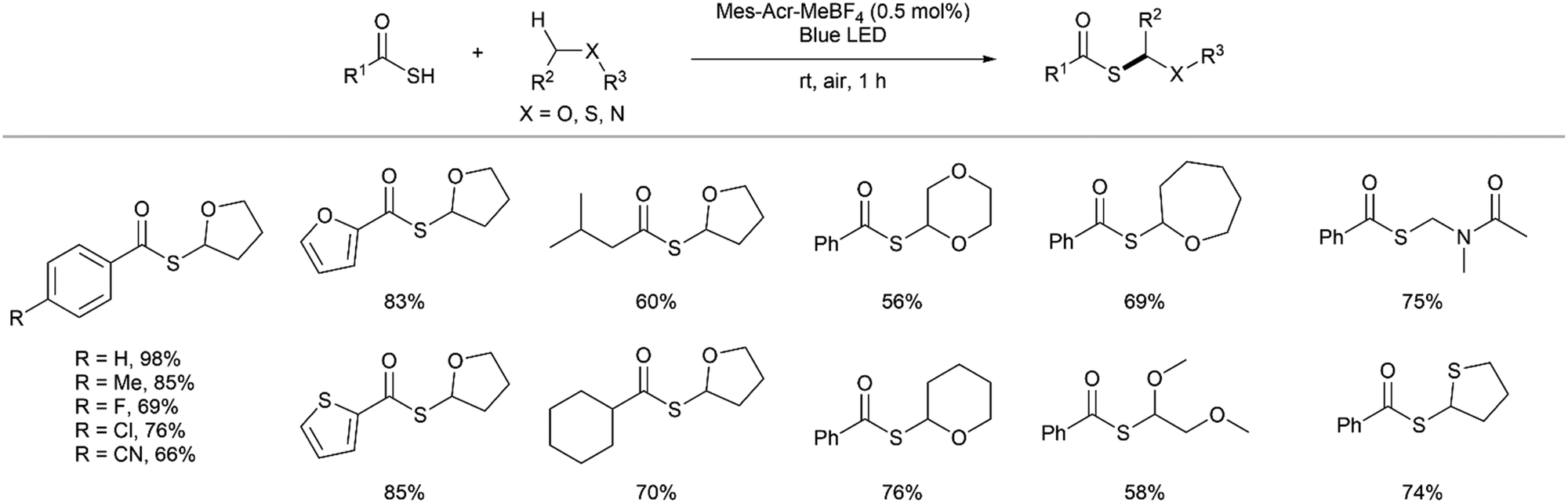
Song and co-workers reported the first synthesis of thioesters via radical-mediated cross-coupling of thioacids with C(sp3)–H bonds adjacent to heteroatoms under visible light.14 Following optimisation, 0.5 mol% 9-mesityl-10-methylacridinium tetrafluoroborate (Mes-Acr-MeBF4) 5 was identified to be the optimal photocatalyst. The authors demonstrated that a diverse range of aryl and alkyl thioacids could undergo cross-coupling with tetrahydrofuran (THF) at the position adjacent to the oxygen, to furnish thioesters in good to excellent yields (Fig. 10). Furthermore, various unactivated C(sp3) compounds including cyclic ethers such as 1,4-dioxane, tetrahydropyran and oxepane, as well as linear ethers furnished thioesters in good yields. Dimethylformamide (DMF) and tetrahydrothiophene were also compatible and both underwent coupling at C(sp3)–H bonds adjacent to the heteroatom. The authors further utilised the thioesters formed by this reaction in a one-pot Liebeskind–Srogl cross-coupling with various aryl boronic acids to form ketones.
 |
| | Fig. 10 Selected scope for the synthesis of thioesters by coupling of thioacids to C(sp3)–H bonds.14 | |
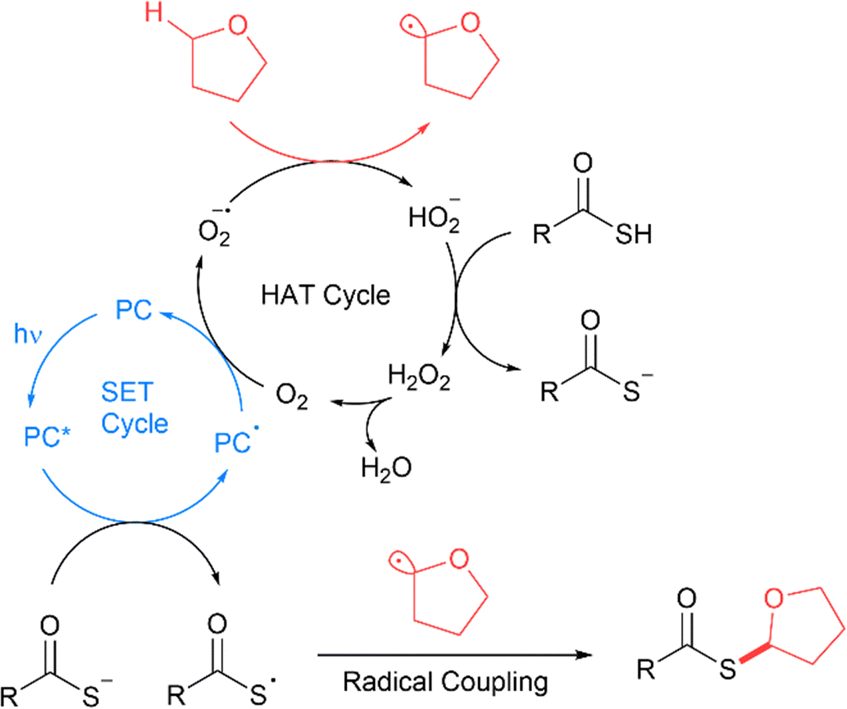
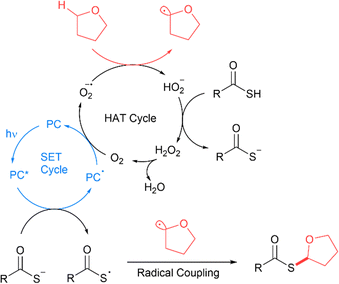
The proposed radical relay mechanism involves an initial SET cycle with Mes-Acr-MeBF45 to form the thioacid radical, interfaced to a hydrogen atom transfer (HAT) cycle with molecular oxygen and the C(sp3) compound to form the C(sp3) radical (Fig. 11).14 The thioacid radical and C(sp3) radicals then undergo homolytic cross-coupling to furnish the thioester product. The catalytic cycle is replenished by proton transfer between protonated thioacids and HO2− formed in the HAT cycle to form hydrogen peroxide (H2O2) and a thioacetate. The hydrogen peroxide formed decomposes to release oxygen to restart the cycle.
 |
| | Fig. 11 Mechanism of thioester bond formation by coupling of thioacids to C(sp3)–H bonds. PC = photocatalyst.14 | |
Thioester bond formation by C(sp2) activation
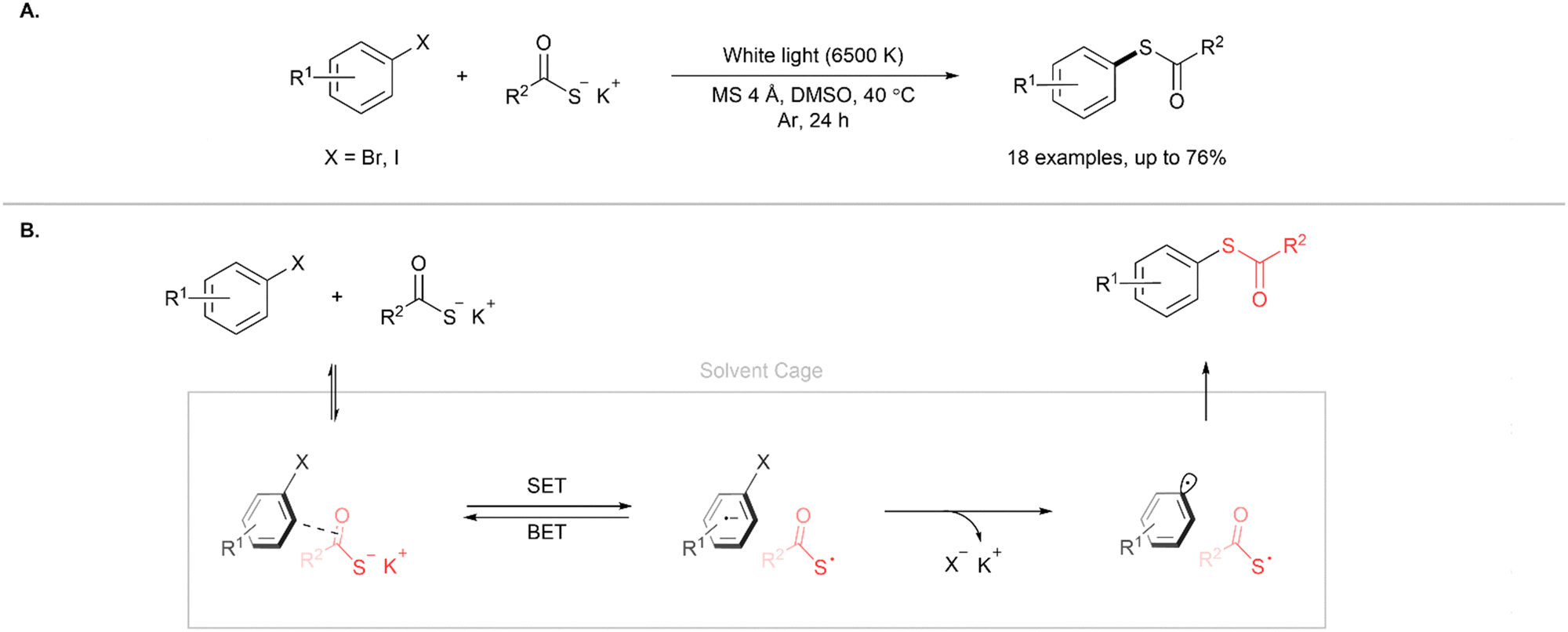
In another approach, Karchava and co-workers reported the formation of C(sp2)–S bonds through the coupling of electron-deficient aryl halides and potassium thiocarboxylates to form aryl thioesters (Fig. 12A).15 Of particular note is that the reaction does not use transition metal or organic photocatalysts. The reaction is initiated by the formation of an electron donor–acceptor (EDA) complex due to anion–π or halogen bonding interactions between the aryl halide and thiocarboxylate (Fig. 12B). The EDA complex serves as a photosensitiser and irradiation with visible light leads to the formation of an S-centred radical-aryl radical pair due to intrasystem single electron transfer (SET). Subsequent rapid fragmentation of the complex by an irreversible carbon–halogen bond scission forms a thioacid radical and an aryl radical. These radicals couple to form the aryl thioester product. The authors also demonstrated that the aryl thioesters formed could serve as versatile intermediates for the synthesis of aryl disulfides, sulfonamides and sulfonyl chlorides from the corresponding aryl halides, thus avoiding the use of transition metals or malodorous reagents commonly associated with these transformations. Lin and co-workers have also reported the blue light photoredox synthesis of thioesters by the coupling of aryl, heteroaryl and vinyl iodides with potassium thioacetates.16 This cross-coupling reaction comprises of a nickel/photoredox dual catalysis system using 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzlPN) as a photosensitiser, NiCl2·glyme as the Ni source and methoxyl-2,2′-dipyridyl as the ligand.
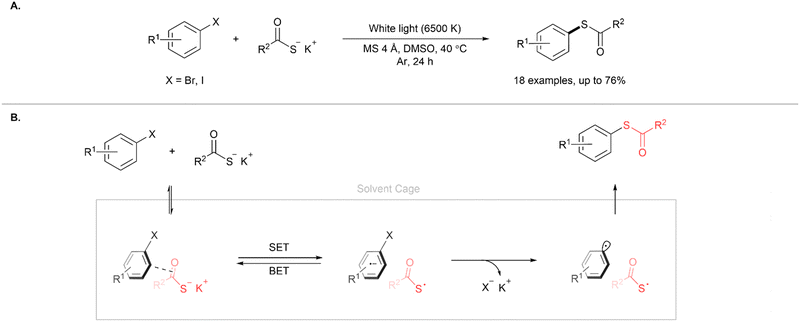 |
| | Fig. 12 (A) Synthesis of aryl thioesters using aryl halides and potassium thioacetates. (B) Proposed mechanism.15 | |
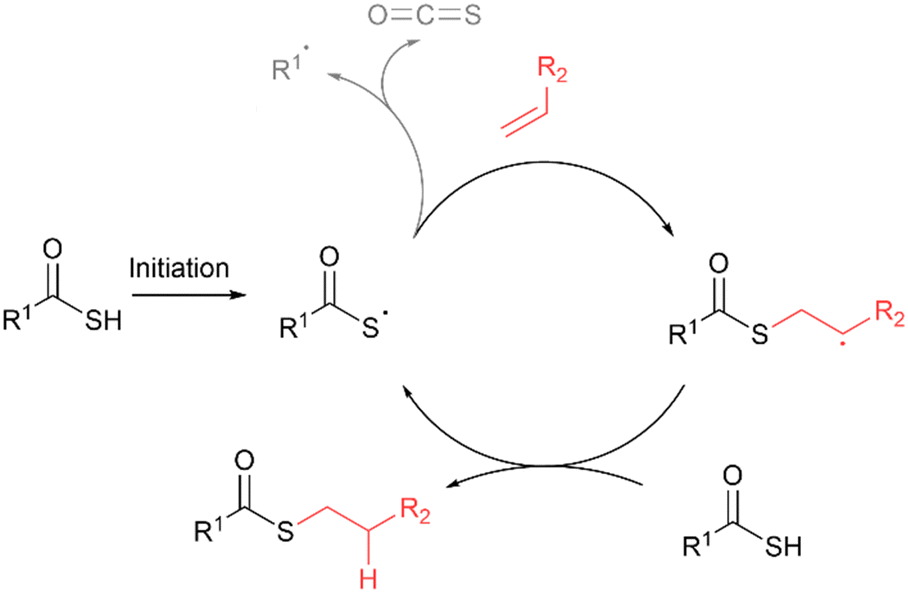
Thioester bond formation via acyl thiol–ene
The addition of thioacid radicals to alkenes via the acyl thiol–ene reaction offers another elegant solution for the chemoselective synthesis of thioesters. Following thioacid radical generation, addition to the alkene proceeds with anti-Markovnikov selectively via formation of the more stable carbon-centered radical intermediate (Fig. 13).17 In certain instances, the thioacid radical can undergo a competing dethiocarboxylation side reaction via the elimination of carbonyl sulfide (COS) to form a carbon-centred radical.
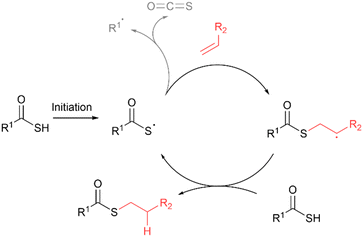 |
| | Fig. 13 General mechanism for the acyl thiol–ene reaction. Side reaction of dethiocarboxylation of the thioacid radical via elimination of carbonyl sulfide (COS) is also shown. | |
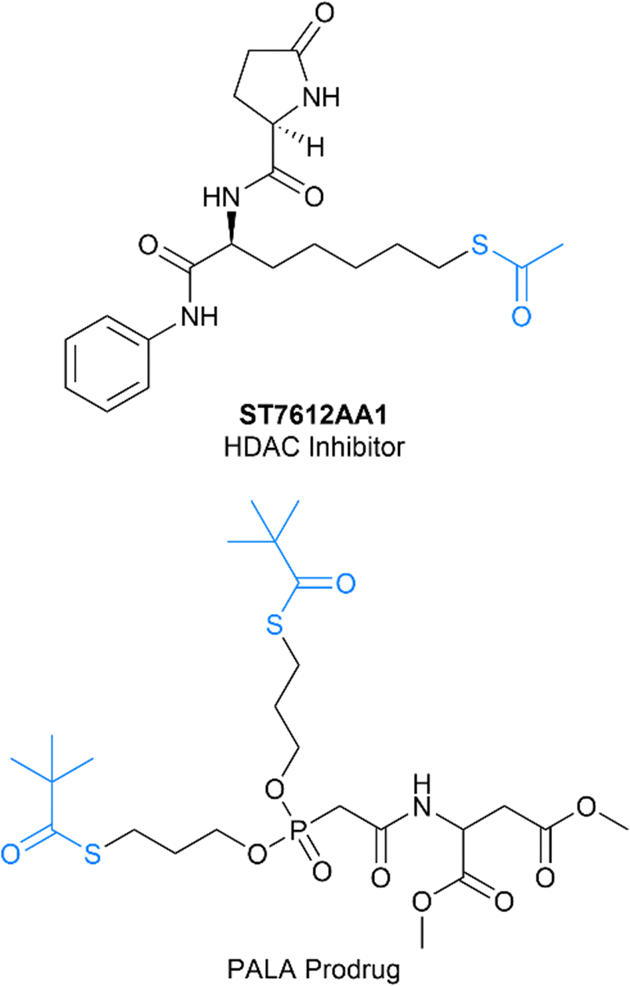
One particularly interesting application of the acyl thiol–ene reaction has been the synthesis of thioester containing prodrugs. In this case, the chemoselectivity of the acyl thiol–ene reaction enables late stage-diversification of drugs modified with an alkene handle to install a thioester, which is cleaved in vivo to release a biologically active thiol group (Fig. 14). This process has been used for the synthesis of thioester prodrugs of histone deacetylase (HDAC) inhibitors for the treatment of latent HIV and cancer,18,19 the pyrimidine antimetabolite N-phosphonoacetyl-L-aspartate (PALA)20 and the antiviral Foscarnet.21
 |
| | Fig. 14 Representative examples of thioester containing prodrugs synthesised using photochemical acyl thiol–ene.19,20 | |
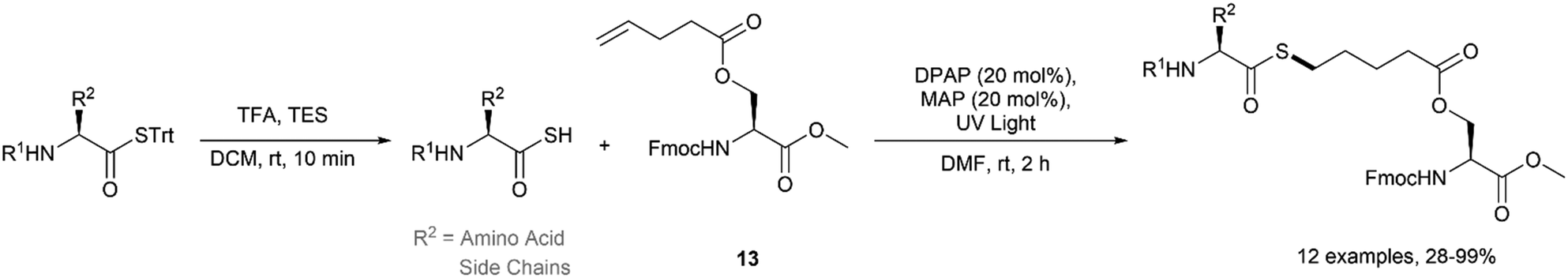
Recently, our group reported the application of the acyl thiol–ene reaction for the synthesis of thioester derivatives of biomolecules.17 In this study, thioacids were prepared by deprotection of the corresponding S-trityl thioester upon treatment with trifluoroacetic acid (TFA) prior to use. A diverse range of amino acid derived thioacids were reacted with serine-derived alkene 13 under UV irradiation (354 nm) using 20 mol% 2,2-dimethoxy-2-phenylacetophenone (DPAP) as the photoinitiator and 20 mol% 4-methoxyacetophenone (MAP) as a photosensitiser in DMF (Fig. 15). It was necessary to use at least 3 equivalents of the thioacid to force quantitative conversion to the thioester during optimisation due to the competing dethiocarboxylation of the thioacid radical depleting the active species. The reaction was found to be compatible with Fmoc, Boc and tBu protecting groups which are frequently used in peptide chemistry.
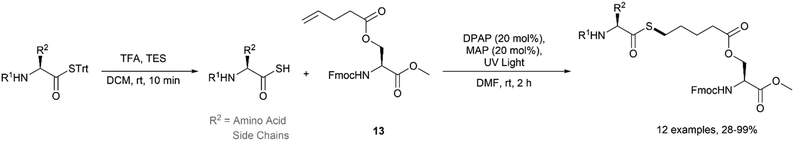 |
| | Fig. 15 Acyl-thiol–ene functionalisation of auxiliary-functionalised serine. | |
In the same work, we studied peptide thioesters capable of undergoing S,N acyl transfer to furnish native peptides.17 For instance, Gly thioacid 14 was reacted with β-γ-didehydrovaline dipeptide 15 under UV irradiation (354 nm) using DPAP/MAP photoinitiation in ethyl acetate to form thioester 17 (Fig. 16). Following acid-mediated α-amine deprotection of 17, S,N acyl transfer over a 6-membered transition state formed the thiol-functionalised tripeptide 19 in 73% overall yield. This tripeptide was subjected to desulfurisation using TCEP/VA50 to form the native tripeptide 21. Similarly, Gly thioacid 14 was reacted with vinylglycine dipeptide 16 to form thioester 18. Following α-amine deprotection, this thioester also underwent S,N acyl transfer to furnish the homocysteinyl tripeptide 20 in 66% overall yield. The thiol of tripeptide 20 was methylated with trimethylsilyldiazomethane to form the native tripeptide 22.
 |
| | Fig. 16 Acyl thiol–ene mediated peptide ligation strategy.17 | |
A similar reaction process involving (i) acyl thiol–ene of a thioacid onto an alkene containing auxiliary on an amino sugar, (ii) S,N acyl transfer and (iii) auxiliary removal, was used to form anomeric amides (Fig. 17).17 Peracetylated N-acetylglucosamine (GlcNAc) derivative 23 containing a 4-pentenoic ester at the 6-position and an amino group at the anomeric position was synthesised and subjected to acyl thiol–ene with a variety of thioacids using UV irradiation with DPAP/MAP in DMF for 1 h to form thioester intermediate 24. The reaction mixture was subsequently stirred at room temperature for a further 16 h without UV irradiation to facilitate S,N acyl transfer over a 13-membered transition state, furnishing the anomeric amide. Following removal of the solvent in vacuo, the crude mixture was redissolved in 8:2 MeOH:H2O with triethylamine (TEA) to cleave the 4-pentenoic ester auxiliary at the 6-position to yield the GlcNAc amide derivatives. These examples highlight the potential of acyl thiol–ene to enable chemoselective amide bond formation on fully unprotected sugars in carbohydrate ligation methodologies.
 |
| | Fig. 17 Synthesis of anomeric amides using acyl thiol–ene followed by S-to-N transfer.17 | |
Our group has also reported the use of inexpensive nanocomposites to initiate acyl thiol–ene reactions.22 Compared to photoinitiation using organic or transition metal complex based photoinitiators, these nanocomposites can be easily removed from the reaction mixture by filtration to simplify purification. During optimisation, a variety of nanodiamond, carbon nanoonion and graphene oxide nanomateirals containing Bi2O3 (2 mol%) and WO3 (2 mol%) were screened and all resulted in complete conversion of the model reaction in 1 h. The nanocomposites were prepared in a two-step protocol whereby the carbon nanomaterial and metal oxide were sonicated together in ethyl acetate and then filtered through a nylon syringe filter (1 μm) to remove large particles. Having comparable results, the graphene oxide–Bi2O3 nanocomposite was selected to take forward since both materials required for its synthesis were commercially available and inexpensive. Using the graphene oxide–Bi2O3 catalyst, a selection of thioesters including amino acid and sugar derivatives were prepared by reaction with thioacetic acid in good yields. The catalysts were also studied under blue LED irradiation (405 nm), but while conversions of up to 90% was observed, full conversion could not be achieved even with increased concentration of the catalyst and reaction times.
Our group has also shown that thioacids can undergo radical acyl thiol–ene reaction under atmospheric oxygen-initiated conditions, thus completely avoiding the requirement for photoinitiators or light irradiation (Fig. 18).23 After optimisation, it was found that heating a solution of alkene in chloroform to reflux with 2 equivalents of both the thioacid and TFA gave quantitative conversion. The TFA is thought to aid the reaction by ensuring that the thioacid remains in a protonated state for efficient proton abstraction by triplet oxygen to form the thioacid radical. A wide range of thioacids and alkenes underwent reactions under these conditions, including highly functionalised terminal and internal alkenes as well as alkyl and aryl thioacids. In all examples, complete anti-Markovnikov addition of the thioacid radical to the alkene was observed. Recently, our group has expanded on the atmospheric oxygen-initiated reaction to take advantage of the reduced need for additives.24 Through using a DES solvent system, additive-free acyl thiol–ene could be achieved for a selection of substrates, though this study focused primarily on thiol reactants.
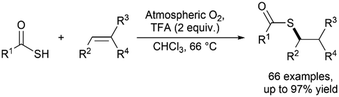 |
| | Fig. 18 Atmospheric oxygen-initiated acyl thiol–ene.23 | |
Liang and co-workers have employed a modified acyl thiol–ene reaction to vinyl thioesters under solvent and catalyst free conditions.25 A series of aryl, heteroaryl and alkyl thioacids readily added onto 1,2-diarylethenes at 100 °C under ambient light in air to form a C(sp2)–S bond and give (Z)-vinyl thioesters in up to 85% yield (Fig. 19A). Mono-aryl substituted alkenes only generated alkyl thioesters resulting from simple acyl thiol–ene addition of the thioacid radical to the alkene. Based on observed experimental data, the authors proposed a mechanism initiated by the formation of a thioacid-olefin complex due to a S–H⋯π non-covalent interaction (Fig. 19B). Upon visible light irradiation, this complex is converted to its excited state. A thioacid-olefin co-oxidation involving molecular oxygen leads to the formation of a thioacid radical and peroxide radical (HO2˙). The thioacid radical undergoes anti-Markovnikov addition to the alkene to form a C–S bond and a carbon-centred radical intermediate. Cross coupling of the carbon radical intermediate with the peroxide radical forms an organic peroxide intermediate which was identified by X-ray crystallography and NMR. Subsequently the peroxide can directly eliminate hydrogen peroxide (H2O2) to form the vinyl thioester. Alternatively, cleavage of the peroxide bond with the assistance of a thioacid forms an alcohol intermediate which can eliminate water to form the vinyl thioester.
 |
| | Fig. 19 (A) Thioacid mediated synthesis of (Z)-vinyl thioesters of 1,2-diarylethenes. (B) Proposed mechanism for the reaction.25 | |
Thiolactone synthesis via acyl thiol–ene/yne
The photochemical acyl thiol–ene reaction can also be performed in an intramolecular fashion to furnish cyclic thioesters, also known as thiolactones. Thiolactones have found broad application across chemical biology,26 medicinal chemistry27 and polymer chemistry.28 The traditional condensation approach to thiolactone synthesis involves a multi-step synthesis to introduce a thiol functionality followed by intramolecular coupling to a carboxylic acid, often in low yield.29 In contrast, the intramolecular radical addition of a thioacid onto an alkene to furnish the thiolactone offers significant advantages in terms of selectivity and reactivity. Recently, our group developed a thiol–ene strategy for the synthesis of thiolactones via cyclisation of γ-alkenyl thioacids using 20 mol% DPAP and 20 mol% MAP with UV irradiation (350 nm).30 A wide range of thiolactones were formed under these conditions in high yields with good functional group tolerance and excellent regioselectivity as almost exclusive 5-exo–trig addition was observed (Fig. 20). The reaction was also highly diastereoselective with dr values of >95:5 observed for substrates with substituents at the β-position relative to the carbonyl. A significantly lower diastereoselectivity of (dr = 60:40) was recorded for substituents at the α-position. The reaction was also highly efficient for the formation of complex polycyclic thiolactones. The only substrate for which 6-endo–trig was favoured was the diene-containing sorbic acid derivative, possibly due to the requirement for a trans/cis isomerisation prior to cyclisation. Computational studies showed that the 5-exo–trig product was both the kinetic and thermodynamic product for γ-alkenyl thioacids, justifying the regioselectivity observed. This is in contrast to the results obtained for radical thiol–ene cyclisations, which exhibit 6-endo–trig selectivity, especially for terminal alkenes, since 6-endo addition furnishes the thermodynamic product.
 |
| | Fig. 20 Synthesis of thiolactones by intramolecular acyl thiol–ene.30 | |
In the same work, we also reported the cyclisation of γ-alknyl thioacids via the acyl thiol–yne reaction to form unsaturated thiolactones.30 In general, yields were significantly lower for acyl thiol–yne cyclisation compared to acyl thiol–ene cyclisation, and selectivity was less robust (Fig. 21). Simple γ-thiolactones 25–27 were formed by 5-exo–dig cyclisation of the thioacid radical with the alkyne in moderate yields and without the formation of other isomers. However, increasing the complexity of the alkyne with dimethyl substituents at the β-position and the use of an internal alkyne resulted in cyclisation by 5-exo–dig and 6-endo–dig to form a mixture of γ- and δ-thiolacones 28a and 28b in an approximately 1:1 ratio. In fact, the alkene of the endo product 28b was reduced under the reaction conditions to form a fully reduced δ-thiolactone. Surprisingly, when one of the methyl groups at the β-position was substituted for a bulky phenyl group, the cyclisation highly favoured 5-exo–dig cyclisation to form the γ-thiolactone 29a over 6-endo–dig cyclisation to form the δ-thiolactone 29b.
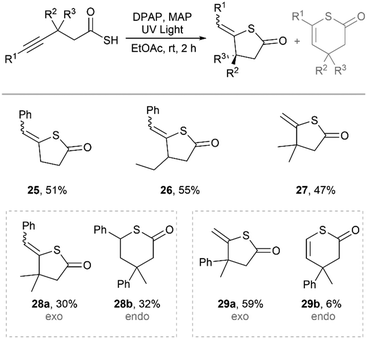 |
| | Fig. 21 Synthesis of thiolactones by intramolecular acyl thiol–yne.30 | |
Thiolactones are also found in several naturally occurring peptides such as the depsipeptides thiocoraline31 and verrucosamide,32 and most notably the autoinducing peptides (AIPs) found in Gram positive bacteria.33 AIPs act as quorum sensing peptides and have a key role in the production of virulence factors.34 Therefore, extensive research is underway to investigate AIP analogues containing thiolactone linkages to treat S. aureus infections.27,35 Current methods for the synthesis of macrocyclic peptide thiolactones have been reviewed recently by Gordon.36 In summary, these methods generally require fully protected peptides and long reaction times, or elevated temperatures to achieve good conversions, though alternative chemistries, including transthioesterification approaches have shown potential.
Our group has recently reported the application of photochemical acyl thiol–ene chemistry for the synthesis of peptide thiolactones, through cyclisation of fully unprotected linear peptides bearing both a thioacid and alkene residue.37 Linear peptide precursors were readily synthesised by Fmoc solid phase peptide synthesis (SPPS) in high yields and purity. Cyclisation under UV irradiation (354 nm) using equimolar DPAP in a solvent mixture of 1:1 MeCN:H2O containing 0.1% TFA for 15 min was used to form a series of structurally diverse macrocyclic peptide thiolactones (Fig. 22). Exclusive anti-Markovnikov addition of the thioacid radical to the terminal alkene was observed. While the isolated yields after semi-preparative HPLC purification were moderate, full conversion was observed by analytical HPLC during the reaction. Thiolactones 32 and 33 were based on the structures of AIP-I and AIP-II respectively, highlighting the potential utility of this method for the synthesis of biologically relevant peptides. In the case of 33, no cyclisation was observed under the standard conditions, likely due to intramolecular hydrogen bonding of the thioacid to the adjacent serine residue preventing proton abstraction to initiate the reaction. However, addition of the chaotropic agent, 6 M guanidine hydrochloride to the solvent mixture resulted in complete consumption of the linear peptide and the corresponding thiolactone product was isolated in a yield of 61%. Importantly, side-chain functional groups including phenols, amines, carboxylic acids, alcohols, sulfides and indoles were all compatible with the protocol.
 |
| | Fig. 22 Synthesis of peptide thiolactones by acyl thiol–ene. # 6 M guanidinium chloride added to reaction mixture. *20% of inseparable co-eluting impurity present.37 | |
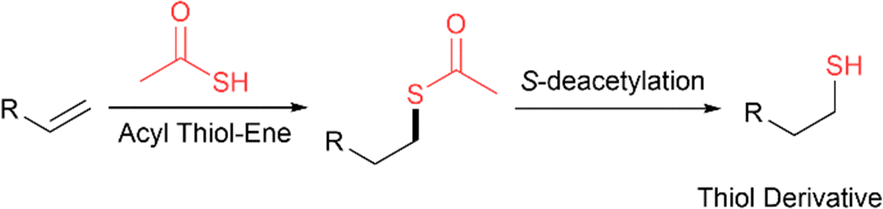
Acyl thiol–ene and S-deacetylation
Another common strategy that takes advantage of the chemoselectivity and fast kinetics of the acyl thiol–ene reaction is the synthesis of thiol derivatives by a sequential two-step process involving radical acyl thiol–ene followed by S-deacetylation (Fig. 23). In this way, thiol derivatives can be easily prepared from the corresponding alkenes which are widely commercially available. Often, a very simple thioacid such as thioacetic acid is used, since the acyl component is removed following hydrothiolation. This strategy has been used extensively in materials chemistry to form thiol-functionalised linkers for the synthesis of gold nanoparticles,38–41 gold surfaces,42–44 polyhedral oligomeric silsesquioxanes,45 carbosilane dendrimers,46,47 rotaxanes48 and fullerenes.49 Additionally, thiol derivatives of sugars,50,51 steroids,52 silylalkanes,53 cyclooctadiene,54 pyridine ligands,55para-terphenylalkanes56 and ferrocene have been prepared using this strategy.57
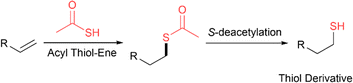 |
| | Fig. 23 General scheme for the synthesis of thiol derivatives from alkenes via acyl thiol–ene followed by S-deacetylation. | |
Our group has reported the use of this strategy to synthesise δ-thiolactones from γ-unsaturated esters (Fig. 24).58 As reported in our previous study, intramolecular acyl thiol–ene cyclisation of γ-alkenyl thioacids strongly favours 5-exo–trig cyclisation to form γ-thiolactones. However, δ-thiolactones can be formed from similar γ-alkenyl ester substrates by first performing an intermolecular anti-Markovnikov acyl thiol–ene addition to the alkene, followed by S-deacetylation to install a thiol at the δ-position. Next, intramolecular Steglich condensation forms the δ-thiolactone. The acyl thiol–ene step was conducted with thioacetic acid under UV irradiation (350 nm) using DPAP/MAP (10 mol%) for 1 h to form δ-thioacetates in good to excellent isolated yields (>80%). The thioacetate and ester groups were hydrolysed concomitantly upon treatment with aqueous NaOH at 90 °C for 45 min to expose the δ-thiol carboxylic acid. The δ-thiols were subjected to Steglich thiolactonisation with EDC·HCl and 4-(dimethylamino)pyridine (DMAP) in dilute conditions for 8–14 h at room temperature to form the desired δ-thiolactones. A wide range of δ-thiolactones were formed, including alkyl, aryl and polycyclic thiolactones in good to excellent yields.
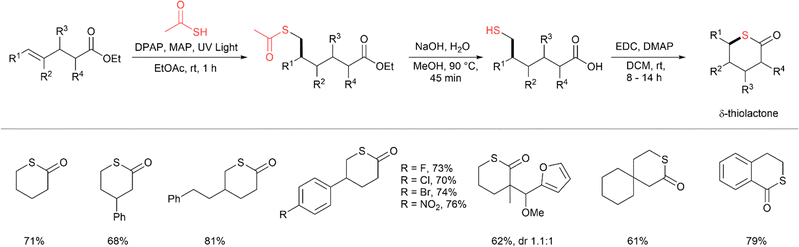 |
| | Fig. 24 Sequential acyl thiol–ene, S-deacetylation and intramolecular condensation for synthesis of δ-thiolactones.58 | |
While the aforementioned studies have yielded products in which the S-atom has been incorporated into the ring, our group has also recently reported the application of thioacid radicals to the initiation of 1,6-diene cyclisation, yielding 5-membered carbocyclic products with an exocyclic sulfur atom (Fig. 25).59 The resulting thioester can then be hydrolysed to yield an exocyclic thiol, for which a variety of chemistries were demonstrated for further manipulation. Notably, photochemical desulfurisation was performed to yield the traceless cyclic product. Computational investigation of the reaction mechanism showed that the lowest energy for the transition states corresponding to 5-exo–trig over 6-endo–trig cyclisation, as well as preference for cis diastereomeric products accounted for by the Beckwith–Houk model.
 |
| | Fig. 25 Acyl thiol–ene followed by S-deacetylation to form carbocycles with an exocyclic sulfur atom.59 | |
Dethiocarboxylation
Thioacid thiyl radicals can also undergo dethiocarboxylation via elimination of carbonyl sulfide (COS) to form C(sp3) radicals. This was first reported by Otaka and co-workers in 2016 for the synthesis of C-terminal N-alkylamide peptides by the dethiocarboxylation of C-terminal thioacids.60 In this instance, the intermediate C(sp3) radical formed after dethiocarboxylation is reduced by hydrogen atom transfer (HAT) to form the N-alkylamide. Otaka and co-workers first synthesised C-terminal peptide thioacids by the hydrothiolysis of a C-terminal thioester formed in situ from N-sulfanylethylanilide (SEAlide) peptides. The thioacid was then subjected to thermal radical initiation using the azo initiator, VA-044, at 37 °C in aqueous phosphate buffer adjusted to pH 7 containing 5 M guanidinium hydrochloride, 9 mM glutathione and 34 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl). Peptides bearing various amino acids at the C-terminal were dethiocarboxylated under these conditions in 2–6 h to form the corresponding N-alkylamides in excellent conversion as determined by HPLC. With Gly at the C-terminal, a significant ratio (26%) of the peptide underwent thioacid hydrolysis to form the C-terminal carboxylic acid rather than dethiocarboxylation. Finally, Otaka and co-workers synthesised the anticancer agent ABT-510 which contains a C-terminal N-ethylamide in an isolated yield of 47% (Fig. 26).
 |
| | Fig. 26 Synthesis of C-terminal peptide N-alkylamide by dethiocarboxylation of a C-terminal thioacid with thermal radical initiation.60 | |
Our group has subsequently reported the dethiocarboxylation of amino acid substrates under photochemical conditions in organic solvent to form N-alkylamines.61 During optimisation of the reaction conditions, it was shown that dethiocarboxylation proceeds efficiently in ethyl acetate with either UV irradiation (354 nm) using 20 mol% DPAP or with blue LED irradiation (440 nm) with 25 mol% Eosin Y. However, blue LED irradiation required a longer reaction time of 1 h compared to 15 min for UV irradiation to reach >95% conversion. Having comparable results, blue LED irradiation was pursued further, due to its milder and safer properties. Thereafter, a series of amino acid thioacids were prepared from the corresponding S-trityl thioesters and dethiocarboxylated to form N-alkylamines (Fig. 27). A wide range of functional groups were compatible, including carbamates, ethers, alcohols, sulfides, carboxylic acids and indoles. Highly electron withdrawing groups such as alkylammonium (R-NH4+) at the α-position to the thioacid resulted in no reaction, possibly due to destabilisation of the intermediate C(sp3) radical. Additionally, the glycine derivative was only detected in trace amounts by NMR, with dethiocarboxylation being disfavoured due to the relatively high energy methyl radical intermediate that must form. This is consistent with our previous studies into the photochemical acyl thiol–ene reaction, as dethiocarboxylation was not detected as a notable side-reaction for thioacids without any substituents at the α-position.37 Finally, we also conducted the dethiocarboxylation reaction with UV irradiation in flow, demonstrating the facile scalability of this process.61
 |
| | Fig. 27 Scope of dethiocarboxylation of amino acid thioacid derivatives under photoinitiation.61 | |
Thioacid to carboxylic acid
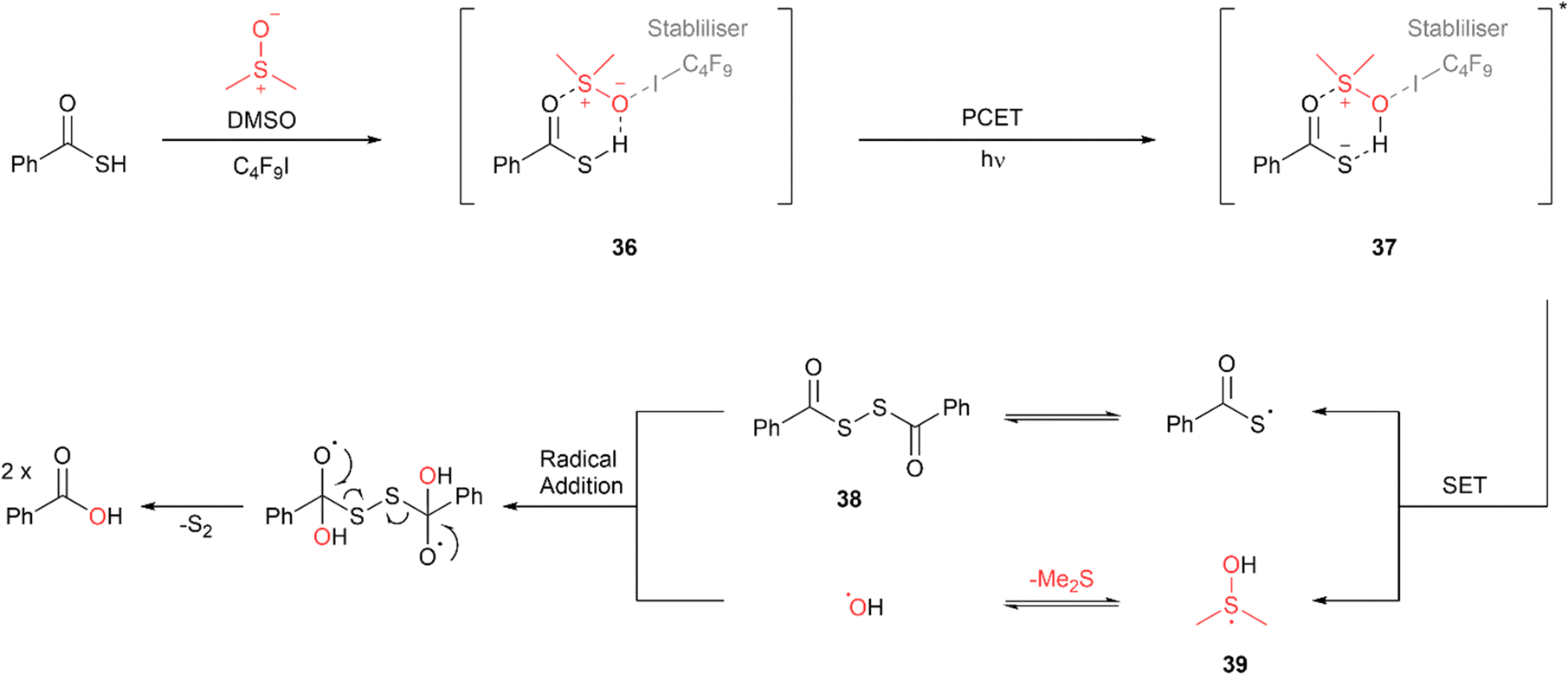
Liang and co-workers have reported a visible-light mediated conversion of thioacids to the corresponding carboxylic acids using dimethyl sulfoxide (DMSO) as both the oxygen donor and solvent.62 While the reaction proceeded with excellent conversion of >90% with CFL bulbs and ambient light, blue LED irradiation (458 nm) led to quantitative conversion in the shortest reaction time. The use of 10 mol% perfluorobutyl iodine (C4F9I), which acts as an intermediate stabiliser, was also required for reaction completion. The proposed mechanism involves formation of adduct 36 due to hydrogen bonding interactions between the thioacid, DMSO and the stabiliser, C4F9I (Fig. 28). Absorption of light results in the transfer of the proton from the weakened S–H bond to the DMSO via proton coupled electron transfer (PCET) to form the excited complex 37. Subsequent SET from the thioacid anion to the DMSO cation forms a thioacid radical which undergoes homocoupling to form diacyl disulfide intermediate 38. The SET process also forms a dimethyl sulfanol radical 39 which collapses with the elimination of dimethyl sulfide to form a hydroxyl radical. This radical adds to diacyl disulfide 38 leading to the formation of the carboxylic acid product and elemental sulfur. A wide range of aryl thioacids with both electron withdrawing and donating substituents underwent reaction with excellent yields with reaction times ranging from 24 to 34 h. Long chain, benzylic and cyclic hydrocarbons also reacted smoothly with excellent yields in 21 to 28 h.
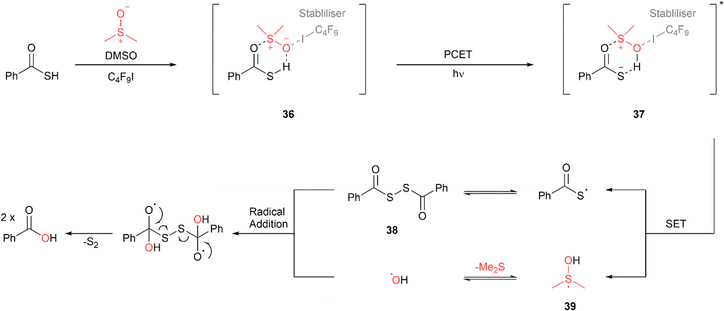 |
| | Fig. 28 Photochemical conversion of thioacids to carboxylic acids via thiyl radical intermediate.62 | |
Conclusions
In an era of intense focus on the development of mild and controllable radical-mediated reactions for the synthesis and further elaboration of molecular architectures, thiyl-radical mediated reactions have found prominence for their ease of formation, high-efficacy and ability to undergo complex reaction pathways under broad reaction conditions and in the presence of a diverse range of unprotected functional groups. Within the context of thiyl radical chemistry, the facile generation of radical intermediates derived from both simple and complex thioacids offers fascinating prospects for thioester synthesis alongside innovative concomitant processes that exploit the unique reactivity of thioesters and related intermediates for further reaction, including amide bond formation and peptide ligation amongst others. Thioacid derived thiyl radicals can be formed under very mild conditions from both the protonated acid and the corresponding thioacetate, permitting operation across a wide pH range. Mild photochemical, thermal and even atmospheric oxygen mediated conditions have been found to be highly effective in generating the desired radical intermediate. Upon formation, a broad range of possible reactive pathways exist and highly sought after functional groups such as amides and thioesters can be generated under mild and chemoselective conditions. In addition, thioacid radicals readily participate in acyl-thiol–ene and thiol–yne coupling reactions in both an intra- and intermolecular fashion to furnish thiolactone and thioester products in high yield. Addition reactions of thioacids radicals to alkenes or alkynes followed by mild deacetylation enables efficient introduction of a reactive thiol handle. The high level of compatibility of thioacid radicals with biomolecules such as peptides, proteins and glycans have enabled the synthesis of native peptide bonds, anomeric amides and even Lys-labelling of proteins in cell lysate. The competing dethiocarboxylation process remains underexplored with few reported examples to date, however, owing to the considerable synthetic importance of mild methods for the chemoselective modification of carboxylic acids via decarboxylation, it is expected that interest in this topic will surge in the coming years. This review highlights the versatility of thioacid-derived thiyl radicals for organic synthesis and the abundance of possibility for further investigation of these useful intermediates. Tantalising prospects exist for the application of acyl thiol–ene reactions as key components of drug discovery efforts, new methods in chemical biology and the development of molecular machines amongst others.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- N. Narendra, V. M. Thimmalapura, B. Hosamani, G. Prabhu, L. R. Kumar and V. V. Sureshbabu, Org. Biomol. Chem., 2018, 16, 3524–3552 RSC.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- M. Lubberink, W. Finnigan and S. L. Flitsch, Green Chem., 2023, 25, 2958–2970 CAS.
- H. Liu, L. Zhao, Y. Yuan, Z. Xu, K. Chen, S. Qiu and H. Tan, ACS Catal., 2016, 6, 1732–1736 CrossRef CAS.
- S. Das, S. Ray, A. B. Ghosh, P. K. Samanta, S. Samanta, B. Adhikary and P. Biswas, Appl. Organomet. Chem., 2018, 32, e4199 Search PubMed.
- D. Procopio, X. Marset, G. Guillena, M. L. Di Gioia and D. J. Ramón, Adv. Synth. Catal., 2024, 366, 870–876 CrossRef CAS.
- W. Song, K. Dong and M. Li, Org. Lett., 2020, 22, 371–375 CAS.
- Z. Hou, C. Wan, H. Jiang, Y. Wang, Y. Xing, J. Wang, Z. Liu, X. Guo, Y. An, W. Han, R. Wang, X. Zhang, F. Yin and Z. Li, Green Chem., 2024, 26, 11238–11248 CAS.
- P. Qiu, X. Duan, M. Li, Y. Zheng and W. Song, Org. Lett., 2022, 24, 2733–2737 CAS.
- M.-U.-S. Bhat, M. A. Ganie, S. Kumar, M. A. Rizvi, S. Raheem and B. A. Shah, J. Org. Chem., 2024, 89, 4607–4618 CAS.
- G. Liu, N. Zheng, X. Duan, X. Sun and W. Song, Green Chem., 2023, 25, 5035–5040 CAS.
- A. A. Volkov, D. I. Bugaenko, A. V. Bogdanov and A. V. Karchava, J. Org. Chem., 2022, 87, 8170–8182 CAS.
- W. Zheng, Y. Xu and L. Lin, ChemPhotoChem, 2021, 6, e202100264 Search PubMed.
- J. T. McLean, P. Milbeo, D. M. Lynch, L. McSweeney and E. M. Scanlan, Eur. J. Org. Chem., 2021, 4148–4160 CAS.
- R. Badia, J. Grau, E. Riveira-Muñoz, E. Ballana, G. Giannini and J. A. Esté, Antiviral Res., 2015, 123, 62–69 CAS.
- G. Giannini, L. Vesci, G. Battistuzzi, D. Vignola, F. M. Milazzo, M. B. Guglielmi, M. Barbarino, M. Santaniello, N. Fantò, M. Mor, S. Rivara, D. Pala, M. Taddei, C. Pisano and W. Cabri, J. Med. Chem., 2014, 57, 8358–8377 CrossRef CAS PubMed.
- V. Gagnard, A. Leydet, V. Le Mellay, M. Aubenque, A. Morère and J.-L. Montero, Eur. J. Med. Chem., 2003, 38, 883–891 CrossRef CAS PubMed.
- V. Gagnard, A. Leydet, A. Morère, J.-L. Montero, I. Lefèbvre, G. Gosselin, C. Pannecouque and E. De Clercq, Bioorg. Med. Chem., 2004, 12, 1393–1402 CrossRef CAS PubMed.
- V. Maffeis, R. O. McCourt, R. Petracca, O. Laethem, A. Camisasca, P. E. Colavita, S. Giordani and E. M. Scanlan, ACS Appl. Nano Mater., 2018, 1, 4120–4126 CrossRef CAS.
- R. O. Mccourt and E. M. Scanlan, Chem. – Eur. J., 2020, 26, 15804–15810 CrossRef CAS PubMed.
- M. D. Nolan, A. Mezzetta, L. Guazzelli and E. M. Scanlan, Green Chem., 2022, 24, 1456–1462 RSC.
- R. Wang, J.-L. Yuan, K.-L. Liang, J.-Y. Hu, Q. Fu and F.-S. Liang, J. Org. Chem., 2024, 89, 9597–9608 CrossRef CAS PubMed.
- S. Aubry, K. Sasaki, L. Eloy, G. Aubert, P. Retailleau, T. Cresteil and D. Crich, Org. Biomol. Chem., 2011, 9, 7134–7143 RSC.
- J. K. Vasquez and H. E. Blackwell, ACS Infect. Dis., 2019, 5, 484–492 CrossRef CAS PubMed.
- N. Illy and E. Mongkhoun, Polym. Chem., 2022, 13, 4592–4614 RSC.
- M. Langlais, O. Coutelier and M. Destarac, ACS Omega, 2018, 3, 17732–17742 CrossRef CAS PubMed.
- R. O. McCourt, F. Dénès, G. Sanchez-Sanz and E. M. Scanlan, Org. Lett., 2018, 20, 2948–2951 CrossRef CAS PubMed.
- F. Romero, F. Espliego, J. Pérez Bas, T. García de Quesada, D. Grávalos, F. De la Calle and J. Luis Fernández-Puentes, J. Antibiot. Res., 1997, 47, 734–737 CrossRef PubMed.
- V. Nair, M. C. Kim, J. A. Golen, A. L. Rheingold, G. A. Castro, P. R. Jensen and W. Fenical, Mar. Drugs, 2020, 18, 549 CrossRef CAS PubMed.
- B. Wang, A. Zhao, R. P. Novick and T. W. Muir, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10679–10684 Search PubMed.
- B. Wang and T. W. Muir, Cell Chem. Biol., 2016, 23, 214–224 CrossRef CAS PubMed.
- K. H. J. West, C. G. Gahan, P. R. Kierski, D. F. Calderon, K. Zhao, C. J. Czuprynski, J. F. McAnulty, D. M. Lynn and H. E. Blackwell, Angew. Chem., Int. Ed., 2022, 61, e202201798 CrossRef CAS PubMed.
- C. P. Gordon, Org. Biomol. Chem., 2020, 18, 379–390 RSC.
- A. Benny and E. M. Scanlan, Chem. Commun., 2024, 60, 7950–7953 RSC.
- S. A. Svarovsky, Z. Szekely and J. J. Barchi, Tetrahedron:Asymmetry, 2005, 16, 587–598 CrossRef CAS.
- C.-C. Lin, Y.-C. Yeh, C.-Y. Yang, G.-F. Chen, Y.-C. Chen, Y.-C. Wu and C.-C. Chen, Chem. Commun., 2003, 2920–2921 RSC.
- P. E. Laibinis, C. D. Bain, R. G. Nuzzo and G. M. Whitesides, J. Phys. Chem., 1995, 99, 7663–7676 CrossRef CAS.
- Z. Chu, Y. Han, P. Král and R. Klajn, Angew. Chem., Int. Ed., 2018, 57, 7023–7027 CrossRef CAS PubMed.
- S.-C. Ng, T. Sun and H. S. O. Chan, Tetrahedron Lett., 2002, 43, 2863–2866 CrossRef CAS.
- K. Motesharei and D. C. Myles, J. Am. Chem. Soc., 1998, 120, 7328–7336 CrossRef CAS.
- H. H. Cao, N. Nakatsuka, S. Deshayes, J. M. Abendroth, H. Yang, P. S. Weiss, A. M. Kasko and A. M. Andrews, Chem. Mater., 2018, 30, 4017–4030 CrossRef CAS PubMed.
- M. Lo Conte, S. Staderini, A. Chambery, N. Berthet, P. Dumy, O. Renaudet, A. Marra and A. Dondoni, Org. Biomol. Chem., 2012, 10, 3269–3277 RSC.
- J. A. Camerano, M. A. Casado, M. A. Ciriano, C. Tejel and L. A. Oro, Dalton Trans., 2005, 3092–3100 RSC.
- K. Matsuoka, H. Oka, T. Koyama, Y. Esumi and D. Terunuma, Tetrahedron Lett., 2001, 42, 3327–3330 CrossRef CAS.
- A. Boccia, V. Lanzilotto, V. Di Castro, R. Zanoni, L. Pescatori, A. Arduini and A. Secchi, Phys. Chem. Chem. Phys., 2011, 13, 4452–4462 RSC.
- E. Zhang, D. Wang, Z. Huang and M. Wang, Chin. J. Chem., 2010, 28, 1690–1696 CrossRef CAS.
- R. Roy, M.-G. Baek and K. Rittenhouse-Olson, J. Am. Chem. Soc., 2001, 123, 1809–1816 CrossRef CAS PubMed.
- K. Igarashi and T. Honma, J. Org. Chem., 1970, 35, 606–610 CAS.
- B. J. Kim, S. Yamada, T. Funada, Y. Kadoma and H. Morita, Bioorg. Med. Chem. Lett., 2000, 10, 357–359 CrossRef CAS PubMed.
- G. A. Gornowicz, J. W. Ryan and J. L. Speier, J. Org. Chem., 1968, 33, 2918–2924 CrossRef CAS.
- J. M. Locke and E. W. Duck, Chem. Commun., 1965, 151a RSC.
- J. A. Krall, P. J. Rutledge and J. E. Baldwin, Tetrahedron, 2005, 61, 137–143 CAS.
- J. Müller, M. Brunnbauer, M. Schmidt, A. Zimmermann and A. Terfort, Synthesis, 2005, 998–1004 Search PubMed.
- M. Herberhold, O. Nuyken and T. Pöhlmann, J. Organomet. Chem., 1995, 501, 13–22 CAS.
- R. O. McCourt and E. M. Scanlan, Org. Lett., 2019, 21, 3460–3464 CAS.
- D. M. Lynch, M. D. Nolan, C. Williams, L. Van Dalden, S. H. Calvert, F. Dénès, C. Trujillo and E. M. Scanlan, J. Org. Chem., 2023, 88(14), 10020–10026 CAS.
- T. Shimizu, R. Miyajima, N. Naruse, K. Yamaoka, K. Aihara, A. Shigenaga and A. Otaka, Chem. Pharm. Bull., 2016, 64, 375–378 CAS.
- A. Benny, L. Di Simo, L. Guazzelli and E. M. Scanlan, Molecules, 2024, 29, 1465 CAS.
- R. Wang, K.-J. Xie, Q. Fu, M. Wu, G.-F. Pan, D.-W. Lou and F.-S. Liang, Org. Lett., 2022, 24, 2020–2024 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Mark D.
Nolan
,
Mark D.
Nolan