
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis and application of aryldihydronaphthalene derivatives
Xia Chen
 a,
Zhaolong He
a,
Shiqiang Xu
*a,
Yu Zou
*a and
Yonghui Zhang
*b
a,
Zhaolong He
a,
Shiqiang Xu
*a,
Yu Zou
*a and
Yonghui Zhang
*b
aInstitute of Pharmaceutical Process, Hubei Province Key Laboratory of Occupational Hazard Identification and Control, School of Medicine, Wuhan University of Science and Technology, Wuhan 430065, China. E-mail: xushiqiang@wust.edu.cn; zouyu@wust.edu.cn
bHubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. E-mail: zhangyh@mails.tjmu.edu.cn
First published on 11th October 2024
Abstract
Aryldihydronaphthalenes (ADHNs) and their derivatives are widely found in many types of natural products, bioactive compounds, and functional materials, and are also important synthetic intermediates in organic chemistry, attracting widespread attention from both organic and pharmaceutical chemists. In the past two decades, the chemical synthesis and biological activity of ADHNs and their derivatives have become two hot spots. This review summarizes the synthetic protocols of ADHN derivatives, introduces some representative examples of the reaction mechanism, and focuses on the research progress of ADHNs in natural product chemistry and chemical biology since 2000.
 Xia Chen | Xia Chen received her PhD degree in 2021 from Huazhong University of Science and Technology, under the supervision of Professor Yonghui Zhang, where she studied medicinal chemistry and natural products chemistry. After two years of post-doctoral study in collaboration with Hubei University, she joined the Medical College, Wuhan University of Science and Technology as a lecturer in 2023. Her research interests include organic synthesis, pharmaceutical chemistry and biological activity studies of natural products and organic molecules. |
 Zhaolong He | Zhaolong He is currently studying pharmaceutical chemistry at Wuhan University of Science and Technology, pursuing a master's degree in pharmaceutical chemistry. His research interests focus on organic synthesis and pharmaceutical chemistry. |
 Shiqiang Xu | Shiqiang Xu received his master's degree in medicinal Chemistry from Tongji Medical College of HUST in 2001. After 6 years working in the R&D department of CR DOUBLE-CRANE (WUHAN) pharmaceutical CO., Ltd, he joined the School of Medicine, Wuhan University of Science and Technology. His main research interests include the design, synthesis and activity study of small molecule compounds against cancer. |
 Yu Zou | Yu Zou received his PhD degree from China Pharmaceutical University in 2017. He started his independent research at Wuhan University of Science and Technology, focusing on the medicinal chemistry and drug discovery from natural products and NO donors. He also worked as a program officer in China Science and Technology Exchange Center, collaborating with several scientific research groups in USA, UK and Italy. |
 Yonghui Zhang | Yonghui Zhang received his doctorate in 2004 from Huazhong University of Science and Technology (HUST), where he studied natural-product chemistry. He joined the faculty of HUST as an associate professor (2005) and was then promoted to professor (2008). He was elected as an associate dean (2010) and then the dean (2014) of the School of Pharmacy. He is the director of the Key Laboratory of Natural Pharmaceutical Chemistry and Resource Evaluation in Hubei Province. His research interest is the discovery of bioactive metabolites from fungi and plants as well as the innovative drugs. |
1. Introduction
Lignans, naturally occurring bioactive chemicals, are always formed by the oxidative dimerization of two phenylpropanoid units.1 Although the molecular skeleton of lignans consists of only two phenylpropane (C6–C3) units, lignans exhibit remarkable structural diversity, primarily attributed to variations in the oxidation levels of their side chains and the substitution patterns on their aromatic rings.2 Aryldihydronaphthalene (ADHN) is an important subtype of the lignan family, which represents a potential drug lead due to the potent activity of its analogues against many diseases. According to the distribution of the double bonds and pendent phenyl rings in the dihydronaphthalene structure, ADHN lignans can be classified into: 1-phenyl-1,2-dihydronaphthalene (1), 1-phenyl-1,4-dihydronaphthalene (2), 2-phenyl-1,2-dihydronaphthalene (3), 2-phenyl-1,4-dihydronaphthalene (4), 3-phenyl-1,2-dihydronaphthalene (5) and 4-phenyl-1,2-dihydronaphthalene (6) (Fig. 1). | ||
| Fig. 1 Six isomers of ADHNs. | ||
ADHNs are not only widely found in natural products, functional materials and bioactive substances,3 but also very important intermediates in organic synthesis, which can be used for the rapid preparation of highly functional and structurally diverse aryltetralins/aryltetrahydronaphthalenes and arylnaphthalenes.4 In view of the significant applications of ADHN derivatives, their synthetic methods have garnered significant attention within the realm of chemistry, and this subclass of lignans stands as one of the most thoroughly studied substructural types of lignans.
Traditionally, the preparation methods of ADHNs include: (1) oxidation by different oxidants: for example, (a) FeCl3/O2 oxidizes two molecules of cinnamic acids or cinnamates to obtain dilactones, which rearrange to form ADHNs under acidic conditions;5 (b) horseradish peroxidase HRP-H2O2 or Cu(NO3)2·3H2O can serve as oxidants to deliver ADHNs;6 (2) intramolecular didehydro-Diels–Alder (DDDA) reactions of styrene-ynes by means of refluxing acetic anhydride;7 (3) aryl tetrahydrofuran reacts with perchloric acid and acetic acid;8 (4) the oxidative coupling of ethyl ferulates generates the 1,4-diphenyl-1,3-butadiene intermediates, which then undergoes intramolecular cyclization catalyzed by aluminum trichloride.9 Although these methods documented in previous literature have provided viable avenues for the synthesis of ADHNs, their further utilization is constrained by stringent reaction conditions, poor functional group tolerance, and low yields. In addition, traditional approaches tend to be narrowly focused on attaining the target compounds, while neglecting the exploration of structural diversity. The aforementioned constraints impede the synthesis of derivatives based on ADHN scaffolds, thereby preventing pharmaceutical chemists from further investigating the structure–activity relationships. As modern synthetic techniques pivot around modular and diverse synthesis, it is imperative to develop efficient and convenient approaches for building the useful ADHN frameworks.
In recent decades, significant research progress has been made in the synthesis of ADHNs. However, comprehensive overviews encompassing the synthetic methodologies and applications of ADHN skeletons remain scarce. This review classifies the approaches for synthesizing ADHN derivatives into two primary categories: intramolecular and intermolecular reactions. Specifically, each reaction mode is further delineated based on its specific type, for instance, intramolecular reactions will encompass intramolecular cyclization, Heck and elimination reactions, etc., whereas intermolecular reactions involve coupling reactions and addition reactions, and so forth. This review will introduce the intramolecular and intermolecular reactions involved in the synthesis of ADHNs, focusing on their applications in the total synthesis of natural products and the construction of bioactive molecules, and summarize their profound implications in the field of biology.
2. Chemical synthesis of ADHN derivatives
2.1 Intramolecular reactions
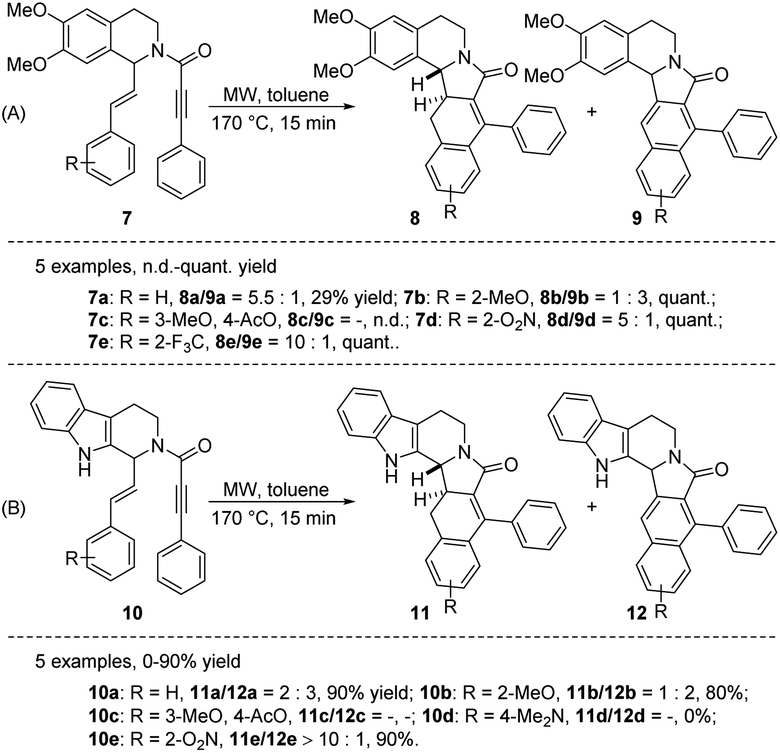
Heating, particularly when combined with microwave irradiation, serves as an efficacious means to facilitate diverse reactions,11 notably the renowned intramolecular dehydrogenative Diels–Alder transformation. In 2010, Ruijter et al. reported microwave-assisted intramolecular Diels–Alder reactions of tetrahydroisoquinolines 7 and tetrahydro-β-carbolines 10 to provide the polycyclic alkaloid-type compounds 8–9 and 11–12, respectively (Scheme 1A and B).12 Unfortunately, the reaction is often accompanied by inseparable byproducts 9 or 12. 10 examples were given in this conversion (7a–e and 10a–e), while the formation of a mixture of regioisomers made accurate assessment of yields and product ratios impossible in this case (7c and 10c). The substrate with dimethylamino group (10d) did not afford any cyclization product 11d or 12d. Although the authors attempted to selectively obtain rearrangement (8 and 11) and oxidation products (9 and 12) in the presence of inert atmosphere or dehydrogenators (e.g., S, Pd/C), respectively, no complete selectivity was observed.
 | ||
| Scheme 1 Synthesis of ADHNs via microwave-assisted intramolecular Diels–Alder reactions of styrene-ynes. | ||
Shortly after, in 2011, Matsubara, Kurahashi and coworkers proposed an example of ADHN 14 by promoting the reaction through heating at 160 °C (Scheme 2).13 The reaction process is simple and can proceed without the addition of Lewis acids, transition metals, or oxidants. In their research, they primarily introduced the dehydrogenative cycloaddition reaction of silyl-substituted dieneynes and demonstrated that the presence of a silyl group attached to the alkyne moiety drives the occurrence of the dehydrogenation reaction.
 | ||
| Scheme 2 Synthesis of ADHNs via heat-promoted dehydrogenative cycloaddition. | ||
In 2014, Brummond and coworkers proposed a solvent-regulated intramolecular dehydro-Diels–Alder (DDA) reaction of styrene precursors 15 to selectively produce ADHN lactones 16 (Scheme 3).4b The styrenyl precursor 15 was subjected to microwave irradiation (MWI) at 180 °C for 20 minutes. This is the first report of entirely selective formation of ADHN lactones through the DDA reaction of styrene-ynes. Interestingly, the solvent exerts a pivotal influence on the product selectivity in the intramolecular DDA reaction. Employing DMF as the reaction solvent results exclusively in the formation of ADHN lactones 16, while PhNO2 selectively yields arylnaphthalene lactones (not shown here). The authors speculated that the hydrogen atom donor properties of DMF may be a factor contributing to the selectivity of the DDA reaction in DMF, while the oxidation capacity of PhNO2 may explain the exclusivity of arylnaphthalene lactones products.
 | ||
| Scheme 3 Synthesis of ADHNs through DMF-regulated intramolecular dehydro-Diels–Alder reactions of styrene-ynes. | ||
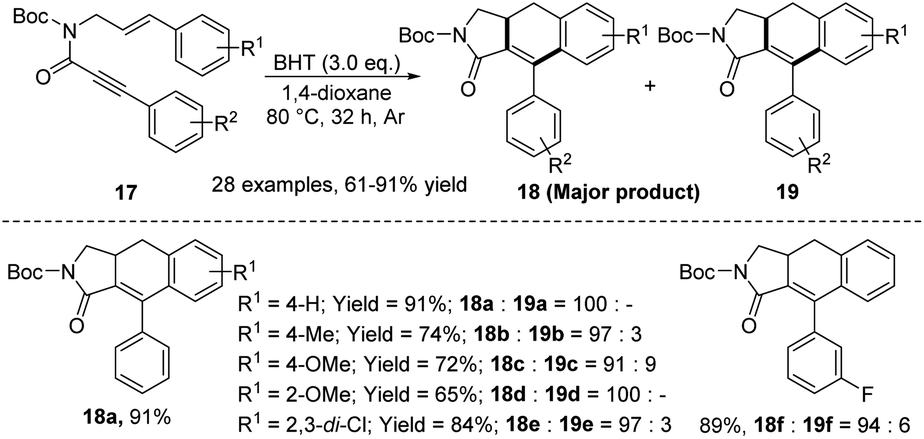
In 2022, our group developed an intramolecular didehydro-Diels–Alder (DDDA) reactions of styrene-ynes 17 (Scheme 4).14 Although the preparation of ADHN skeletons using styrene as a substrate is generally considered a challenging task, this reaction achieved moderate to good yields under relatively mild reaction conditions (80 °C) without any transition metal. This promising result may be explained by the critical propiolamide moiety, which was previously reported by our research group.15 The study showed that the argon atmosphere played an important role in the selective generation of ADHNs, with 2,6-di-tert-butyl-4-methylphenol (BHT) serving as an antioxidant. The synthetic value of this protocol was validated through a gram-scale reaction (1.0 g, 82% yield), showcasing the successful deacylation of the N-bound acyl group and the oxidation of ADHN with oxygen (not shown here).
 | ||
| Scheme 4 Synthesis of ADHNs via intramolecular DDDA reaction of styrene-ynes. | ||
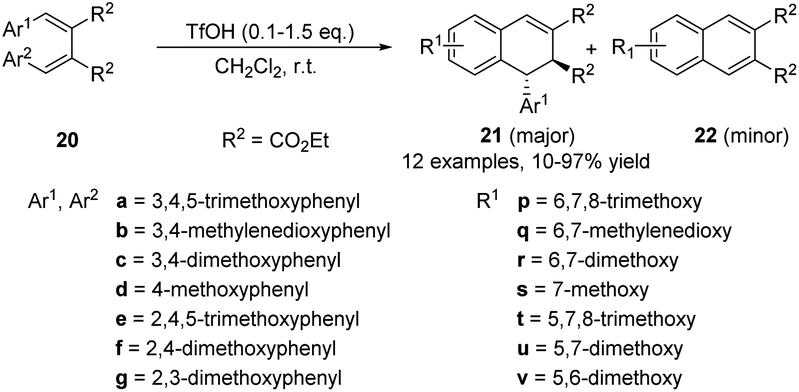
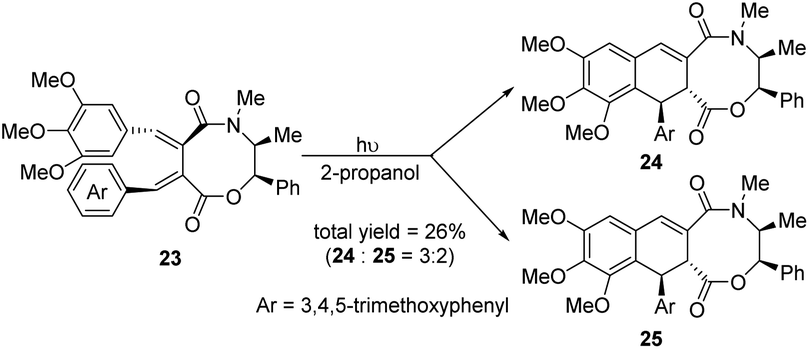
In 2001, Charlton et al. reported an acid-catalyzed cyclization of 2,3-dibenzylidenesuccinates 20 (Scheme 5),16 which has been exploited to prepare the naturally occurring lignans (±)-cagayanin and (±)-galbulin, with overall yields of 18% and 23%, respectively. It was found that the reactivity of the diesters was highly dependent on the position of the alkoxy substituents on the aryl rings. By prolonging the heating time or using excessive triflic acid, the yield of the products 21 was reduced and the dearylized compounds 22 was induced. Since (E,E)-dibenzylidenesuccinates can be easily prepared by the Stobbe condensation, it becomes an attractive substrate for the formation of lignans. Subsequently, in 2004, the same group prepared mandelate ester from the same raw material, 2,3-dibenzylidenesuccinate, and then transferred the (−)-ephedrine cyclic amide ester 23 to obtain optically active ADHNs 24 and 25 through an asymmetric photocyclization reaction (Scheme 6).17
 | ||
| Scheme 5 Synthesis of ADHNs via acid-catalyzed cyclization of diethyl E,E-dibenzylidenesuccinates. | ||
 | ||
| Scheme 6 Synthesis of ADHNs via photochemical closure of styrene-enes. | ||
In 2010, Nishii et al. reported the ring-expansion of phenyl hydroxy methyl cyclopropane carboxylates 26 and its diaryl analogs 28 mediated by Lewis acids, resulting in the corresponding 1,2-dihydronaphthalene-3-carboxylic acid esters 27 and 29, respectively (Scheme 7A and B).18 Various Lewis acids such as TiCl4, SnCl4, TBDMSOTf, BF3·Et2O, Yb(OTf)3 and Sc(OTf)3 were screened in the conversion. The results showed that Yb(OTf)3 and Sc(OTf)3 were the most optimal choices among the lanthanide triflates with the highest yields (90% and 95%). EDC (1,2-dichloroethane) was identified as the best solvent. The authors cited several examples (27a–e and 29a–d) to demonstrate the substrate scope, with yields ranging from 60% to 99%.
 | ||
| Scheme 7 Synthesis of ADHNs via Lewis acid-mediated ring-expansion reaction of cyclopropane carboxylates. | ||
Subsequently, in 2017, the same group reported the asymmetric homo-Nazarov-type cyclization of racemic donor–acceptor (D–A) cyclopropanes 30 mediated by TiCl4 to synthesize poly-substituted ADHNs 31 and 31′ (Scheme 8). The OH groups of the products 31 are in equilibrium with their corresponding keto-isomers 31′ and is subsequently converted to the corresponding triflates. Notably, the stoichiometric amount of TiCl4 significantly improved the yield, and the optimal result (92%) was achieved using 1.5 equivalents of TiCl4 at 83 °C. The practicability of the reaction was verified through further transformations of the above mentioned triflates, such as Suzuki–Miyaura coupling and Sonogashira coupling.
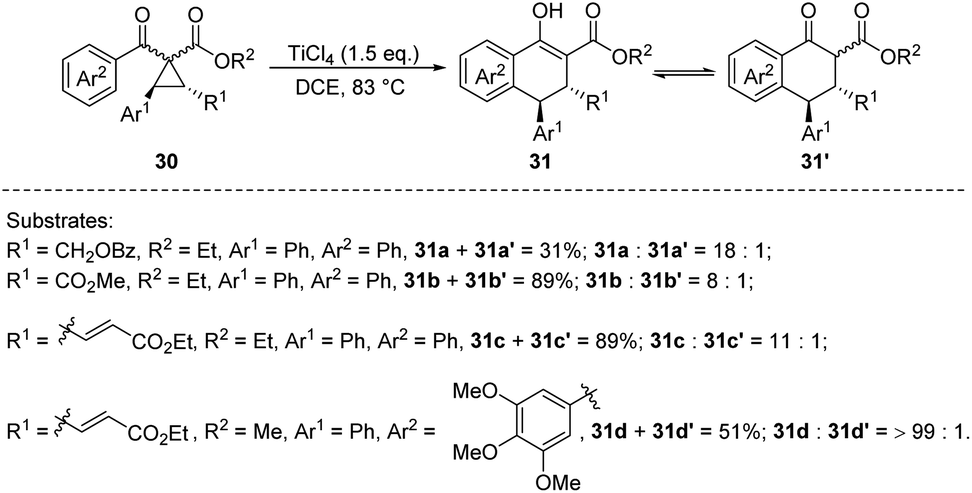 | ||
| Scheme 8 Synthesis of ADHNs via TiCl4-mediated ring-opening cyclization of D–A cyclopropanes. | ||
In 2014, Li et al. showed the construction of ADHN skeletons via the acid-catalyzed cyclization of 32, providing the target ADHNs 33 (similar to the structure of 21, Scheme 5) accompanied with a small amount of dearylated products 34 (Scheme 9).19 Differed from Charlton's work (Scheme 5), this work used compound 32 as the substrate, and it was treated with either perchloric acid in acetic acid at room temperature or p-toluenesulfonic acid in toluene under reflux. As a possible mechanism, they suggest that the tetrahydrofuran ring in 32 is opened under acidic conditions to form a quinone methide intermediate, which then undergoes an intramolecular nucleophilic attack to provide the five-membered spiro structure. During the subsequent dienone-phenol rearrangement of the spiro intermediate, the oxygen-substituted side-chain migrates and H2O is eliminated, leading to the formation of ADHN 33.
 | ||
| Scheme 9 Synthesis of ADHNs via acid-catalyzed cyclization of tetrahydrofuran. | ||
In 2022, the group of Zhang and Chen disclosed a method for the cyclization of 35 promoted by boron trifluoride diethyl etherate (Scheme 10).20 In this work, the authors initially optimized the conditions for preparing 36 from benzhydrol 35. Both Brønsted acids and Lewis acids were evaluated, and boron trifluoride diethyl etherate was proved to be the best one. The relatively low temperature was conducive to production, −40 °C being better than 0 °C, −30 °C and −50 °C. The reaction exhibited good functional group tolerance and good to excellent yields. The applicability of the reaction was further validated by a scaled-up reaction (2.1 mmol).
 | ||
| Scheme 10 Synthesis of ADHNs via Brønsted acid-catalyzed cyclization of benzhydrol. | ||
The cascade cyclization reaction strategy has attracted considerable attention in view of the convenience in the synthesis of polysubstituted polycyclic compounds.21 The traditional protocols mainly focused on acetic anhydride or base-mediated cyclization, while the high loadings of acetic anhydride7a,d or base7d result in toxic byproducts, limiting their applications. Recently, some progress has been made in base-catalyzed cyclization reactions. In 2010, Li et al. demonstrated the intramolecular cascade reactions of aryl-substituted enynes 37 catalyzed by P4-t-Bu, resulting in the synthesis of 9-aryl-3a,4-dihydronaphtho[2,3-c]furan-1(3H)-ones 38 (Scheme 11). In this reaction, a number of bases such as Cs2CO3, K3PO4, NaOH, NaOEt, DABCO, DBU and K2CO3 were examined, while all of them were less effective than 10 mol% P4-t-Bu. Solvents such as toluene, tetrahydrofuran, and N,N-dimethylformamide were also tested, and none of them succeed to provide a higher yield than DCE. The optimal reaction temperature for this transformation was 100 °C. Noteworthy, the electronic properties of aryl allyl groups had little influence on the conversion. The authors suggested that an intramolecular Diels–Alder cyclization mechanism might be involved in this process.
 | ||
| Scheme 11 Synthesis of ADHNs via P4-t-Bu-mediated intramolecular Diels–Alder reactions of aryl-substituted enynes. | ||
Subsequently, in 2011, the same group reported a silver fluoride-promoted intramolecular cascade reaction of aryl-substituted 1,6-enynes 39 to afford ADHN lignan lactone 40 (Scheme 12).22 The reaction was carried out at 100 °C under an argon atmosphere, using a mixture of AgF/K2CO3 as the base and CH2ClCH2Cl as the optimal solvent. When the alkynyl allyl ester moiety is simultaneously linked with electron-donating groups and electron-withdrawing groups, the corresponding high-yield products can be obtained. Unfortunately, however, enyne ethers 39d failed to complete this conversion (40d, 0% yield). The author suggested that the reason why silver ions can promote the reaction is probably due to their ability to coordinate with triple bonds, instantly activating them, and thus facilitating the reaction. Among silver salts, AgF likely exhibits the best effect due to its strong alkaline nature, while other silver salts are neutral or weakly acidic. Transition metal catalysts are undoubtedly one of the most fundamental and useful tools for organic chemists, playing a pivotal role in enriching reaction pathways and significantly broadening the range of substrates. They are widely used to form carbocyclic and heterocyclic frameworks as precursors for building a variety of organic molecules.23 In 2013, Shia et al. developed a general approach to construct ADHNs 42 via the palladium(0)/copper(I)-catalyzed radical cyclization of 41 (Scheme 13).24 Although aryl 1-cyanoalk-5-ynyl ketones 41 and aryl iodides were used as the starting materials, the Pd(0) catalyzed Sonogashira coupling reaction generated the crucial intermediate 1-cyanoalk-5-ynyl ketone, which underwent a [4 + 2] intramolecular cyclization in the reaction. Air was vital for the radical cyclization of the intermediate, facilitating the production of the target products 42. Under the optimal conditions, both electron-withdrawing and electron-donating groups were tolerable, yielding nearly equivalent product amounts.
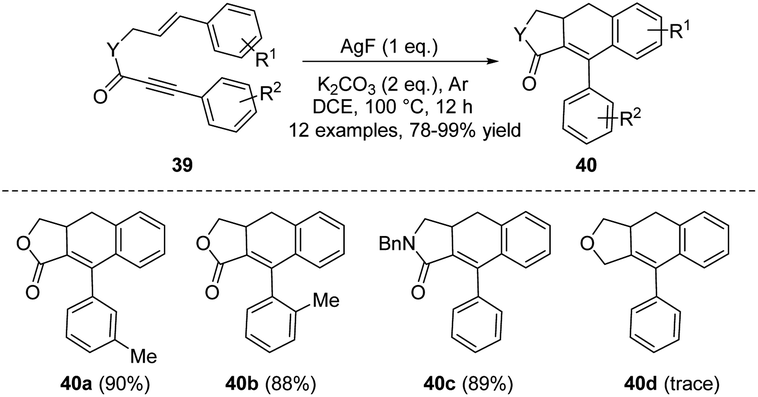 | ||
| Scheme 12 Synthesis of ADHNs via silver fluoride-promoted intramolecular cascade reactions of aryl-substituted 1,6-enynes. | ||
 | ||
| Scheme 13 Synthesis of ADHNs via Pd(0)/Cu(I) catalyzed tandem annulation of aryl 1-cyanoalk-5-ynyl ketones. | ||
Shortly after, in 2014, the same group reported the oxidative cyclization of aryl 1-cyanoalk-5-ynyl ketone 43 catalyzed by manganese(III) (Scheme 14).25 Unfortunately, this protocol primarily focused on the acyl tandem cyclization to aryl 1-cyanoalk-5-ynyl ketones, and the ADHN product was presented (44). Compared with the traditional styrenyl DDA reaction, this reaction is characterized by mild reaction conditions. Furthermore, operational simplicity, high product yields and the potential synthetic elaboration shed light on its further synthetic value.
 | ||
| Scheme 14 Synthesis of ADHNs via manganese(III)-catalyzed oxidative cyclization of aryl 1-cyanoalk-5-ynyl ketone. | ||
In 2018, Kang, Ahn and coworkers established an Fe(II)/Fe(III)-catalyzed microwave assisted intramolecular DDDA reaction of styrene-ynes 45 (Scheme 15).26 Despite their primary focus being on the synthesis of arylnaphthalenes 47, during their initial experiments to identify the optimal conditions, they discovered that varying types of iron, solvents, and iron catalyst loadings significantly influenced the ratio of the ADHNs 46 and arylnaphthalenes 47. Mechanistic investigations revealed that an Fe(III) or Fe(III) catalyst favored the dehydrogenation process, leading to the production of arylnaphthalene products.
 | ||
| Scheme 15 Synthesis of ADHNs via Fe(II)/Fe(III)-catalyzed and microwave assisted intramolecular DDDA reactions of styrene-ynes. | ||
In 2012, the Shia group described the intramolecular radical cascade reaction of α-cyano-aryl-capped alkynyl aryl alkyl ketones 48 based on radical catalytic process to offer a variety of highly functionalized ADHNs 49 (Scheme 16).27 The reaction employed tert-butyl hydroperoxide (TBHP) as the oxidant and tetrabutylammonium iodide (TBAI) as a substitute for transition metal catalysts, rendering it a metal-free and highly efficient process. Unfortunately, this protocol mainly studied the α-cyano-TMS capped alkynyl aryl alkyl ketones substrates, paid little attention to α-cyano-aryl-capped alkynyl aryl alkyl ketones.
 | ||
| Scheme 16 Synthesis of ADHNs via intramolecular radical cascade of aryl alkyl ketones. | ||
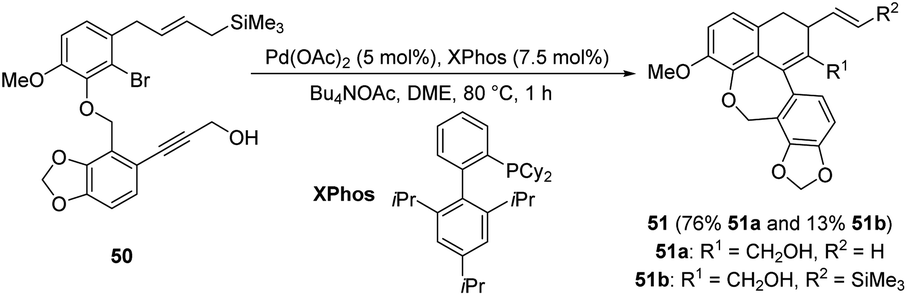 | ||
| Scheme 17 Synthesis of ADHNs via domino carbopalladation/Heck reaction of allylsilanes. | ||
Given the potential biological activity of the natural product linoxepin and the related synthetic challenges, this field has garnered significant attention from chemists. In the same year, Lautens et al. reported a palladium-catalyzed and base-facilitated intramolecular Mizoroki–Heck reaction of the dihydronaphthalene 52, affording the target (+)-linoxepin 53 in 76% yield (Scheme 18).30 This is the first asymmetric synthesis of (+)-linoxepin in eight steps using the authors' modified version of the Catellani reaction. The entire process is characterized by an overall yield of 30%, protecting-group-free and good enantioselectivity.
 | ||
| Scheme 18 Synthesis of ADHNs via intramolecular Mizoroki–Heck reaction of dihydronaphthalene. | ||
Recently, in 2022, Peng and co-workers took the same final intramolecular Heck reaction as reported by Lautens's group (Scheme 18) to prepare linoxepin.31 Differed from Lautens's work, Peng's group adopted a new nickel-catalyzed reductive cyclization as a key step in building a tricyclic core embedded in linoxepin (not shown here). This strategy facilitated the production of several linoxepin analogues.
In 2017, Hajra and co-workers proposed an intramolecular Heck coupling reaction of 54 to afford ADHNs 55 and dihydronaphthalene 56 (Scheme 19).32 Among the screened Pd catalysts, Pd2dba3 was found to be the most effective one. The ligand had strong effect on the yields of the desired products, the use of suitable ligand and base produced 55 as the major product. The suppression of Pd catalyst loading gave better yields by inhibiting the side products 56 formation. Compound 55 was a crucial synthetic intermediate. Subsequently, the authors' team furnished the total synthesis of (−)-podophyllotoxin, (−)-picropodophyllin, (+)-isopicropodophyllin, and (+)-isopicropodophyllone from the molecule 55.
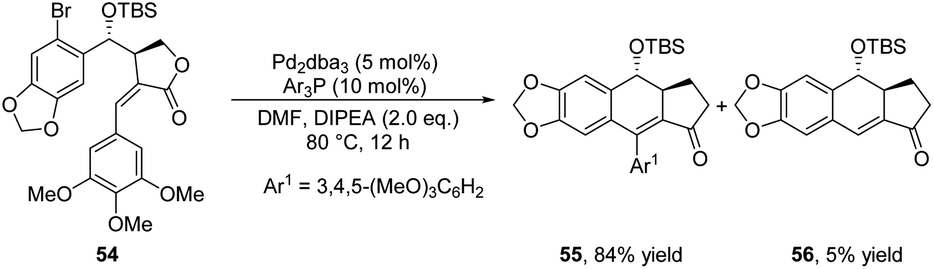 | ||
| Scheme 19 Synthesis of ADHNs via intramolecular Heck reaction. | ||
In 2018, Pinney and collaborators introduced two alternatives for the synthesis of ADHNs, one of them was the coupling reaction of 3,4-dihydronaphthalen-1(2H)-one 57 and 5-bromo-1,2,3-trimethoxybenzene, providing the ADHN product 58 with 55% yield (Scheme 20).33 This reaction necessitates the utilization of n-butyllithium (n-BuLi), a chemical whose handling demands rigorous safety precautions.
 | ||
| Scheme 20 Synthesis of ADHNs via coupling reaction of ketone and bromo-benzene. | ||
 | ||
| Scheme 21 Synthesis of ADHNs via AcOH mediated elimination of azabenzonorbornadienes with N-sulfonyl ketimines. | ||
 | ||
| Scheme 22 Synthesis of ADHNs via elimination reaction of alcohol. | ||
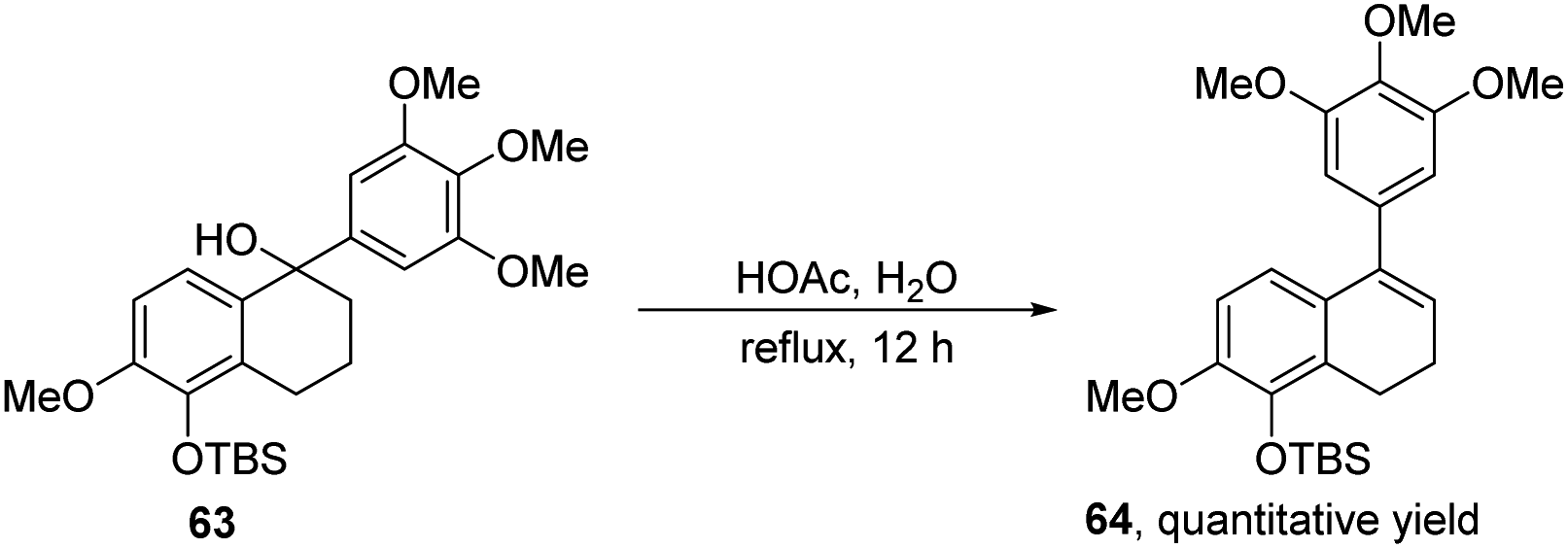
Recently, in 2024, Peng, Xiao and co-workers reported the synthesis of dextrorotatory and levorotatory linoxepin (66 and 66′) through the elimination of the phenyl sulfoxides from compounds 65 and 65′, with total yields of 50% and 55%, respectively (Scheme 23).35 In this protocol, the authors employed a previously reported nickel-catalyzed intramolecular reductive cyclization cascade reaction of sophisticated β-bromo acetals with allyl tethers as the key step. In contrast to the previously reported methods for linoxepin synthesis, this work initially utilized nickel catalysis to construct the tricyclic core, which is subsequently elaborated into linoxepin via an intramolecular elimination reaction. Conversely, the prior method first synthesized the three-membered ring via nickel catalysis, and ultimately synthesized linoxepin through an intramolecular Heck reaction.
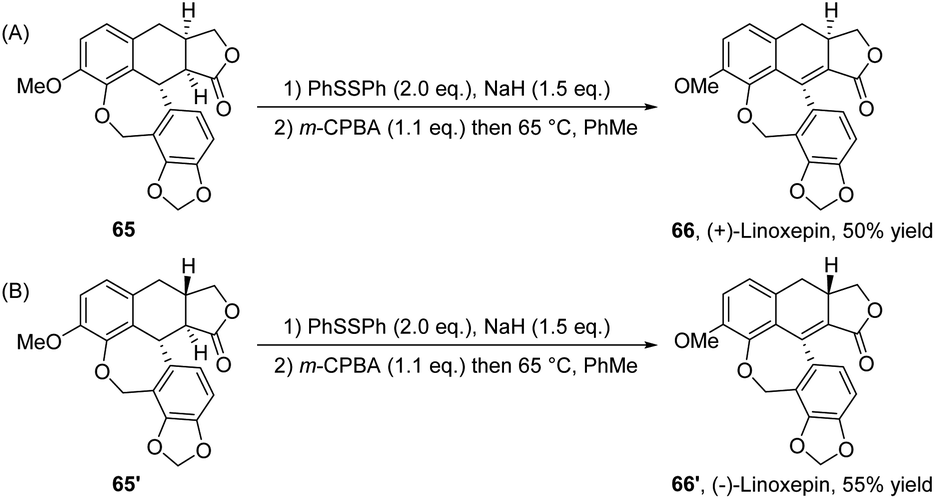 | ||
| Scheme 23 Synthesis of ADHNs via intramolecular elimination of phenyl sulfoxides. | ||
2.2 Intermolecular reactions
 | ||
| Scheme 24 Synthesis of ADHNs via oxidative coupling of caffeic acid phenethyl ester. | ||
In 2013, Alami, Provot and co-workers presented the coupling reaction of N-tosylhydrazone 73 with aryl iodides under palladium catalysis, providing the key intermediates 76a, b (Scheme 25).38 Then, a series of coupling reactions were performed, such as the Sonogashira–Linstrumelle reaction of 74a with propargylic and homopropargylic alcohols, the Heck coupling reaction of 74a with methyl acrylate, and other reactions involving 74a, to obtain ADHN analogues with different substitutions (not shown here). These diverse ADHN analogues provide a material basis for studying their structure–activity relationships.
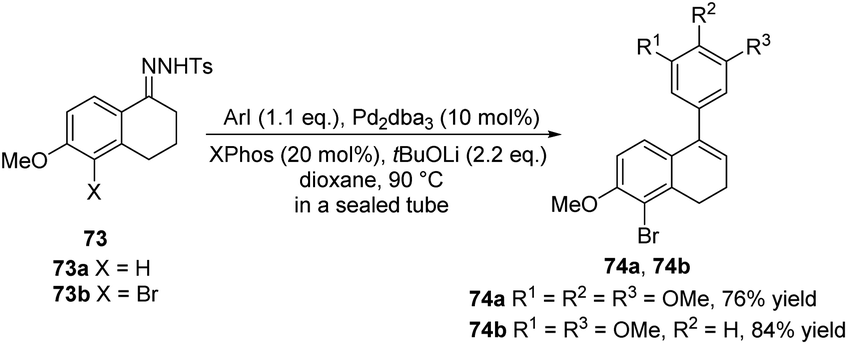 | ||
| Scheme 25 Synthesis of ADHNs via coupling reaction of N-tosylhydrazone with aryl iodides. | ||
In 2015, the group of Li discovered a FeCl3·6H2O catalyzed oxidative coupling reaction of 75 and 79 (Scheme 26).39 In contrast to the oxidation dimerization of 75 and 79 facilitated by conventional oxidants such as HPR-H2O2 or Cu(NO3)2·3H2O, this approach boasts superior yields and selectivity, thereby broadening its applicability in the synthesis of natural products. The reaction mechanism can be best explained by the presence of the unstable intermediate 76, which was easily generated at the start and underwent further 6π electrocyclization to form dihydronaphthalene structures under the acidic oxidative condition. The author also described the removal of tert-butyl protecting groups from the two coupling intermediates 77 or 78 and other related transformations. Further treatment of compound 80 with excess LiAlH4 in THF for global debromination ultimately accomplished the concise synthesis of racemic natural liganamide.
 | ||
| Scheme 26 Synthesis of ADHNs via regioselective oxidative coupling reaction. | ||
A combination Lewis acid system composed of two or more Lewis acids occasionally exhibits enhanced catalytic activity in organic transformations that cannot be achieved by any single component alone. In 2018, Loh and co-workers disclosed the construction of ADHNs 83 via a cascade cyclization reaction of diarylalkynes 81 with acrylates 82 under the catalysis of a combined Lewis acid derived from In(III) salt and TMSBr (Scheme 27).40 Both indium(III) and TMSBr are indispensable for the efficient progress of the reaction. This reaction sequence completed the formation of multiple new C–C bonds in minimal steps, aligns well with the principles of green chemistry.
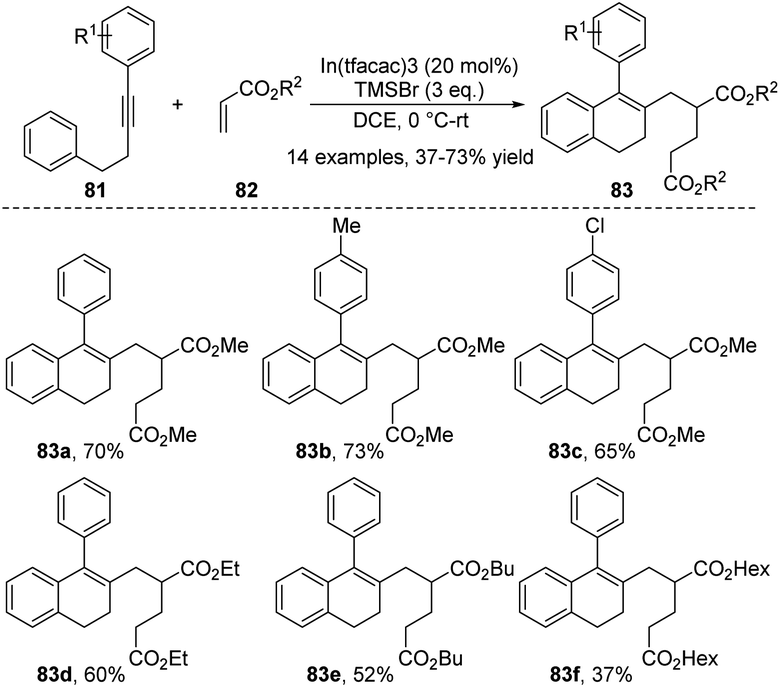 | ||
| Scheme 27 Synthesis of ADHNs via Lewis acid catalyzed cascade cyclization reaction. | ||
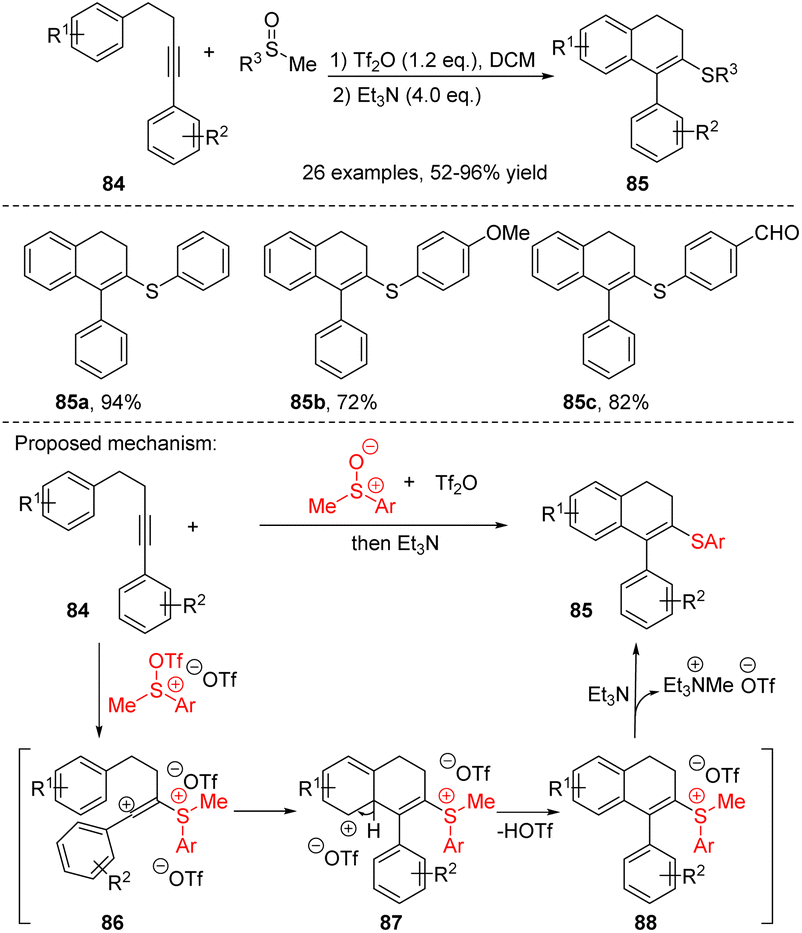
During the synthesis of 3,4-dihydronaphthalene derivatives from 4-aryl alkynes, diverse electrophilic reagents can induce the cyclization process, thereby allowing the incorporation of various functional groups into the products. In 2019, Li, Xu, Du et al. reported a sulfur-mediated electrophilic cyclization reaction of aryl-tethered internal alkynes 84, leading to the synthesis of 3-sulfenyl-1,2-dihydronaphthalenes 85 (Scheme 28).41 In this process, the sulfoxide activated by anhydride serves as a sulfur electrophile to trigger the intramolecular electrophilic cyclization of the reactive aryl-tethered alkynes. This reaction exhibits broad functional group tolerance and excellent yields, enabling the synthesis of sulfenyl-substituted phenanthrenes, dihydroquinolines, 2H-chromenes, and coumarins with yields ranging from high to moderate. The proposed mechanism is illustrated in Scheme 32, the strongly electrophilic reagent sulfoxide activated by anhydride reacted with a carbon–carbon triple bond to form a vinyl carbocation intermediate 86. The intermediate 86 can be captured by the tethered aryl ring, followed by deprotonation to generate the intermediate 87 and sulfonium salt 88. Subsequently, demethylation was achieved using Et3N (SN2 process) to obtain the desired product.
 | ||
| Scheme 28 Synthesis of ADHNs via sulfur-mediated electrophilic cyclization reaction of aryl-tethered internal alkynes. | ||
In 2020, Beeler and co-workers presented a study on one-pot synthesis of ADHNs 91 via a concerted [3 + 2] dipolar cycloaddition reaction between epoxide 89 and dimethyl fumarate 90 (Scheme 29).42 In this transformation, dimethyl fumarate served as a dipolarophile. It is also noteworthy that temperature control is crucial for minimizing the undesired side products and obtaining high selectivity. In some cases, the lower yield can be rationally explained by the dearylation decomposition pathway of the resulting dihydronaphthalene into naphthalene. The obtained product 91 can be mapped as a naturally occurring lignan, which provides a basis for further study of the biological activities of ADHN derivatives.
 | ||
| Scheme 29 Synthesis of ADHNs via a concerted [3 + 2] dipolar cycloaddition of epoxide and dimethyl fumarate. | ||
In 2021, Gao and co-workers revealed a palladium-catalyzed Suzuki–Miyaura coupling of 92 with aromatic boronic acid 93 resulting in 94 (Scheme 30A). Subsequently, a Pd0 catalyzed Suzuki–Miyaura coupling of 95 with aryl boronic acid pinacol ester 96 or 98 afforded (−)-7,8-dihydroisojusticidin B (97) and the desired coupled product 99 in 70% and 60% yield, respectively (Scheme 30B and C).43 The potentiality of this strategy has been well demonstrated by the preparation of ADHN-type lignans, such as linoxepin and 7,8-dihydroisojusticidin B, through the cross-coupling reactions. The ADHNs generated by this process can be further converted into aryltetralin cyclic ether lignans aglacins A, B and E via asymmetric photoenolization/Diels–Alder (APEDA) reactions.
 | ||
| Scheme 30 Synthesis of ADHNs via coupling reaction of N-tosylhydrazone with aryl iodides. | ||
In 2002, Lautens and co-workers reported the first rhodium(I)-catalyzed asymmetric addition of organoboronic acids or boronic ester 101 to oxabicyclic alkenes 100 (Scheme 31).45 The reaction features very mild reaction conditions, generating multiple stereocenters with high yields and excellent diastereoselectivity and enantioselectivity. In this scheme, arylboronic species served as nucleophillic partners. The optimization process replied that a minimal amount of water was required for the catalytic turnover of the reaction. The substrate scope study indicated that substituents on the phenyl ring ortho to boron were intolerant under the optimized reaction conditions. Unfortunately, only two examples of ADHN derivatives were shown in this protocol, and the reaction with 100b led to a complex mixture of products.
 | ||
| Scheme 31 Synthesis of ADHNs via rhodium(I)-catalysed asymmetric addition of organoboronic ester with oxabicyclic alkenes. | ||
Subsequently, in the same year, using the similar reagents, Murakami and co-workers achieved a rhodium(I)-catalysed addition reaction of arylboronic acids 103 with oxabenzonorbornadienes 104 to afford cis-2-aryl-1,2-dihydro-1-naphthol stereoselectively, a series of products were obtained in good yields without concomitant deboronation of the boronic acid (Scheme 32).46 In this reaction, the rhodium complex was prepared in situ by refluxing [Rh(cod)Cl]2 and P(OEt)3 (Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P = 1:2) in MeOH under a nitrogen atmosphere. Interestingly, the hydrolysis deboronation of phenylboronic acid was minimized under the current conditions, as only 1.1 equivalents of boronic acid 103a were sufficient to obtain the product 105a in 86% yield. In previously reported rhodium-catalyzed addition reactions of this type, competitive hydrolysis degradation reactions demanded the use of excess arylboronic acids, ranging from 1.4 to 10 equivalents. The authors proposed a plausible mechanism, which initially involves a metal transformation of rhodium(I) with phenylboronic acid 103a to generate phenylrhodium(I) 106. This process might be promoted by a base. Subsequently, the addition of the phenyl–rhodium linkage across a carbon–carbon double bond of 104a from the exo-side to give 107. β-Oxygen elimination then proceeds to open the furyl ring. The resulting rhodium(I) alkoxide 108 underwent protonolysis with methanol or phenylboronic acid 103a to produce the alcohol 105a and regenerate rhodium(I) for a new catalytic cycle.
:P = 1:2) in MeOH under a nitrogen atmosphere. Interestingly, the hydrolysis deboronation of phenylboronic acid was minimized under the current conditions, as only 1.1 equivalents of boronic acid 103a were sufficient to obtain the product 105a in 86% yield. In previously reported rhodium-catalyzed addition reactions of this type, competitive hydrolysis degradation reactions demanded the use of excess arylboronic acids, ranging from 1.4 to 10 equivalents. The authors proposed a plausible mechanism, which initially involves a metal transformation of rhodium(I) with phenylboronic acid 103a to generate phenylrhodium(I) 106. This process might be promoted by a base. Subsequently, the addition of the phenyl–rhodium linkage across a carbon–carbon double bond of 104a from the exo-side to give 107. β-Oxygen elimination then proceeds to open the furyl ring. The resulting rhodium(I) alkoxide 108 underwent protonolysis with methanol or phenylboronic acid 103a to produce the alcohol 105a and regenerate rhodium(I) for a new catalytic cycle.
 | ||
| Scheme 32 Synthesis of ADHNs via rhodium(I)-catalysed addition reaction of arylboronic acids with oxabenzonorbornadienes. | ||
Due to the challenges encountered in Lautens' previously reported reaction (Scheme 26), particularly pertaining to the addition of heteroaryl boronic acids, which frequently leads to the formation of unreacted addition products and oligomers. Lautens and his colleagues have continued their efforts to further enhance the yield of these reactions. They broadened their studies by investigating the boronic acid addition reaction with different metals. In 2003, the authors' group reported a palladium(II) catalyzed ring-opening addition of arylboronic acids 109 to heterobicyclic alkenes 110 (Scheme 33).47 Under these optimized condition, a wide variety of arylboronic acids were tolerated, leading to the target products with excellent yields (71–98%). The reaction optimization process revealed that metal catalysts such as nickel and platinum were unreactive, inorganic bases and water were necessary for the good reactivity, and methanol had a promoting effect on the reaction. Among the ligands screened, DPPP (1,3-bisdiphenylphosphinopropane) showed the best effect. The authors' group proposed a plausible mechanism, firstly, in methanol solvent, the reactive Pd(ligand)(OMe)2 112 was generated. Then, the aryl boron underwent a transmetalation process to give intermediate 113. After the dissociation of the X ligand, the heterobicyclic alkene substrate (Y = oxygen or protected nitrogen) complexes to form the cationic complex 114. Finally, β-heteroatom elimination after carbopalladation results in the ring-opening product.
 | ||
| Scheme 33 Synthesis of ADHNs via palladium(II) catalyzed ring-opening addition of arylboronic acids with heterobicyclic alkenes. | ||
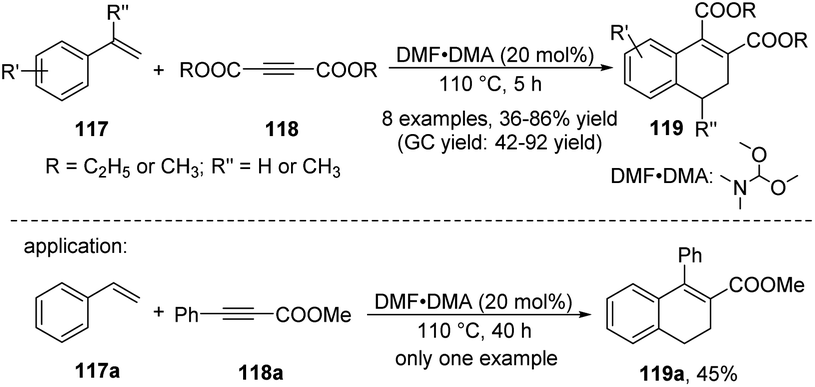
In 2007, Hua et al. demonstrated the DMF·DMA (N,N-dimethylformamide dimethyl acetal) catalyzed cycloaddition reaction of vinylarenes 117 with electron-deficient alkynes 118 for the highly efficient synthesis of 1,2-disubstituted-3,4-dihydronaphthalenes 119 (Scheme 34).48 The main advantages of this protocol are the availability of substrates, the low cost and stability of the DMF·DMA, and the ability to conduct the catalytic reaction under air. The screening of reaction conditions showed that the catalysts used and the ratio of 117 to 118 had significant effects on the product yield. Notably, the reaction exhibited complete regioselectivity, with only 119a being detected by GC and GC-MS.
 | ||
| Scheme 34 Synthesis of ADHNs via DMF·DMA-catalysed cyclic addition of vinylarenes with acetylenedicarboxylate. | ||
In 2019, Fan, Chen et al. reported the formation of 2-aryl dihydronaphthalene derivatives 122 through a rhodium(III)-catalyzed ring-opening addition reaction between azabenzonorbornadienes 120 and cyclic N-sulfonyl ketimines 121 (Scheme 35).49 The reaction exhibited good efficiency and excellent regioselectivity under the redox-neutral conditions. It has proved that the use of the additive is essential, solvents other than DCE, including tetrahydrofuran, acetonitrile, 1,4-dioxane, and toluene, are detrimental to the conversion process. The assessment of substrate scope showed that under optimized conditions, various electron-donating substituents such as methyl, ethyl, tertiary butyl, methoxyl, or phenoxyl on the phenyl ring of the N-sulfonyl ketimines were well tolerated, yielding the products in good to excellent yields. Nevertheless, the conversion of oxabenzonorbornadienes proved unsuccessful, resulting in the formation of naphthalen-1-ol as the major byproduct.
 | ||
| Scheme 35 Synthesis of ADHNs via rhodium(III)-catalyzed ring-opening addition reaction of azabenzonorbornadienes with N-sulfonyl ketimines. | ||
3. Applications of ADHNs
3.1 Synthetic applications
The ADHN, arylnaphthalene and aryltetrahedronaphthalene skeletons are widely existed in natural products and bioactive compounds. Among which the ADHN skeletons are especially useful because it can be transferred into naturally occurring arylnaphthalene and aryltetrahedronaphthalene molecules via a one-step reaction. Therefore, the synthesis and transformation of ADHNs for the establishment of arylnaphthalene and aryltetrahedronaphthalene have attracted much attention among organic chemists, and many elegant synthetic methods for constructing ADHN-type natural products have been reported.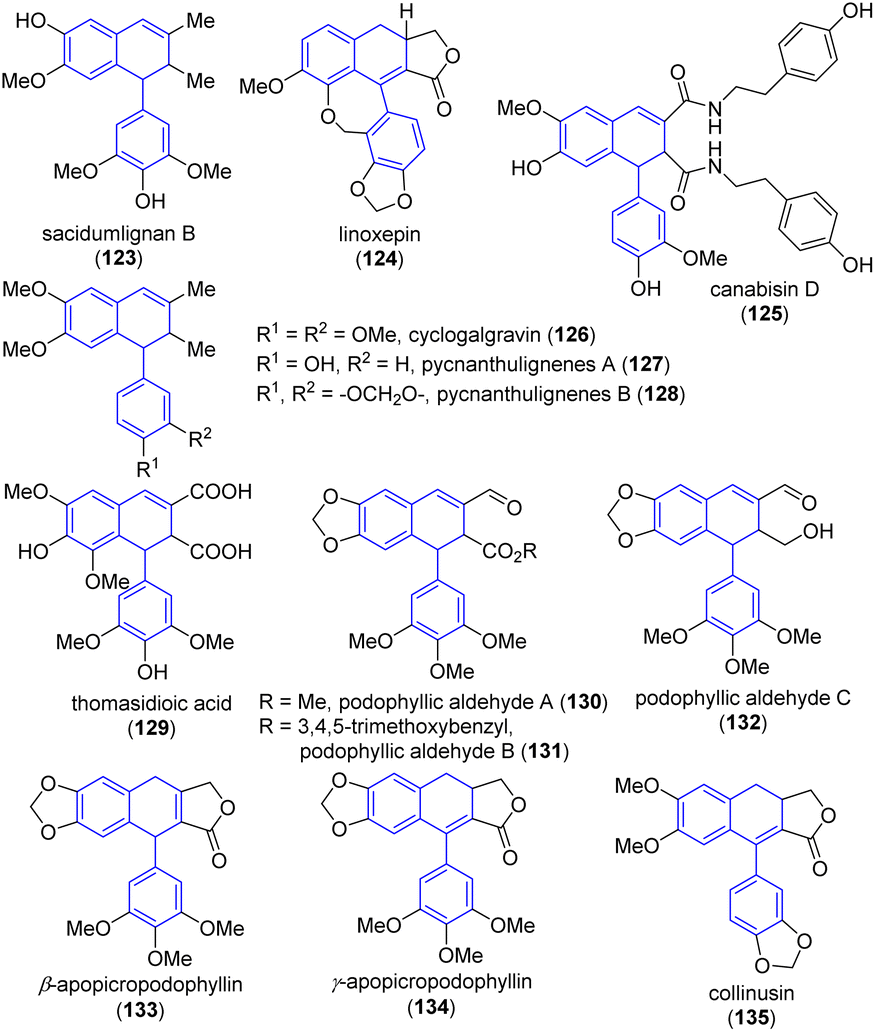 | ||
| Fig. 2 Natural products or bioactive compounds containing ADHN skeletons. | ||
Sacidumlignan B (123) (Fig. 2) is a natural product belong to typical 2,7′-cyclolignans, which have already attracted broad attention from the synthetic community. In 2005, Yue and co-workers isolated sacidumlignan B from the EtOH extract of the plant Sarcostemma acidum (Roxb.) in Hainan Island of China and confirmed its relative configuration.50 In 2012, Ramana and co-workers first reported the asymmetric total synthesis of the naturally occurring sacidumlignan B and determined its absolute configuration.51 In 2020, the Peng group reported a base-promoted addition of N,N-dimethylacetamide (DMA) to 1,1-diarylethylenes, and successfully applied this method to the total synthesis of (−)-sacidumlignan B with an overall yield of 3%.52 Although this protocol avoids the use of transition metal catalysts, providing simple operation and broad substrate scope, the route comprises 16 steps. Therefore, they continued their research to explore more efficient strategies. Encouragingly, in 2022, the same group first reported the racemic synthesis of this molecule, featuring seven steps (46% overall yield) from a known compound (38% overall yield, nine steps from commercially available syringol/syringaldehyde).53
Linoxepin (124) is a natural ADHN lignan, which was first isolated from the dichloromethane extract of aerial parts of Linum perenne L. (Linaceae) and first named by Schmidt's group in 2007. Its absolute configuration was determined using CD spectroscopy combined with DFT calculations.54 In 2013, Tietze et al. reported the first total synthesis of (+)- and (−)-linoxepin in only ten steps without the use of any protecting groups, achieving an overall yield of 30%.29 In this transformation, the Sonogashira reaction and the domino carbopalladation/Heck reaction of allylsilane played significant roles in delivering the target compound. Later, in the same year, Lautens and co-workers reported for the first time an eight-step asymmetric synthesis method of (+)-linoxepin, which was achieved through the modified version of Catellani reaction for the synthesis of this natural product with high enantioselectivity and without protective groups.30 Shortly thereafter, in 2014, the Tietze group further reported the enantioselective total synthesis of the natural lignan (+)-linoxepin, starting from commercially available raw materials through a total of 11 steps with an overall yield of 27%.55 The key steps of which include the domino carbopalladation/Mizoroki–Heck reaction for the formation of the pentacyclic system, asymmetric hydroboration, and oxidative lactonization. In 2015, Nagasawa, Yamanaka and co-workers reported an organocatalytic oxidative kinetic resolution approach for the enantioselective synthesis of (+)-linoxepin from tetralone-derived β-ketoesters in only six steps.56 In 2021, Gao et al. reported an asymmetric photoenolization/Diels–Alder (PEDA) reaction between electron-rich 2-methylbenzaldehydes and unsaturated γ-lactones, which allowed the construction of (+)-linoxepin in six steps.43 Recently, in 2024, Peng, Xiao and co-workers reported a nickel-catalyzed reductive tandem cyclization of the elaborated β-bromo acetal with a dibenzoxepin scaffold to realize enantioselective total synthesis of linoxepin.35
Canabisin D (125) is a naturally occurring liganamide which can be found in fruits or seeds of several plants. In 2015, Li et al. reported a regioselective oxidative coupling reaction for the synthesis of natural (±)-canabisin D, in which, tert-butyl or bromine atom was introduced into the C-5 of N-trans-feruloyltramine precursor to hamper the 8–5 coupling product.39 This biomimetic synthetic strategy can be extensively investigated in the synthesis of a variety of naturally occurring liganamides for the further study of their structure–activity relationships.
Cyclogalgravin (126) is a natural product which was extracted from Araucaria angustifolia knots.57 The air-dried and ground roots of P. angolensis were extracted with a mixture of CH2Cl2–CH3OH (1:1) at room temperature to provide pycnanthulignenes A (127) and B (128).58 In 2011, the Barker and Rye group reported the first asymmetric synthesis of three types of natural ADHNs such as cyclogalgravin (125), pycnanthulignene A (127) and pycnanthulignene B (128).59 A common chiral precursor, aza-Claisen-derived amide, was the starting material for each of these structurally ADHNs. In more detail, the synthesis of pycnanthulignenes A and B began from aldehyde, then it led to the corresponding alcohol, which was then be treated with mesyl chloride to give the ADHN intermediate, the further deprotection of the methoxymethyl (MOM) ether or treatment of alcohol with mesyl chloride gave the desired pycnanthulignene A and pycnanthulignene B in 86% and 95% yield, respectively. In 2020, Beeler and co-workers reported an in situ Finkelstein reaction with NaI and single electron transfer (SET) reduction with Zn of mesylated alcohol to offer pycnanthulignene B in 73% yield.42
In 2002, Niwa and co-workers reported the synthesis of thomasidioic acid 129 from dehydrosinapinic acid dilactone via an α,β-unsaturated γ-lactone-type dimer in phosphate buffer (pH 7.4).60 In this work, sinapinic acid was used as the starting material. Differed from the previous reported synthetic methods (under acid conditions),5b this approach finished the target conversion under neutral conditions. Later, in 2008, using sinapinic acid as the same starting material, Orlandi and co-workers achieved enantioselective biomimetic synthesis of thomasidioic acid through asymmetric biomimetic oxidation of the intermediate phenol, yielding a racemic mixture of trans-thomasidioic acid dimethylesters with a 70% yield.61
In 2014, Nishii and co-workers first reported the total synthesis of three (+)-podophyllic aldehydes 130–132, with corresponding yields and enantiomeric excess (ee) of 85%, 72% and 97%, 95% ee, 95% ee, and 95% ee, respectively.62 The overall yields of (+)-podophyllic aldehydes A, B, and C were 30% (16 steps), 26% (16 steps), and 43% (8 steps), respectively. The key steps of which were the organocatalyzed highly enantioselective cyclopropanation and Lewis acid-mediated chiral transfer ring expansion of the cyclopropane. Notably, the transformation was accomplished with remarkable enantiomeric control.
In 2018, Peng et al. reported the regiodivergent oxidation of (+)-deoxypicropodophyllin in the synthesis of bioactive ADHNs (+)-β- and (+)-γ-apopicropodophyllins (133–134).63 The in situ syn-elimination of the phenylselenoxide generated by the oxidation of α-phenylselenide provided (+)-β-apopicropodophyllins 133 in 88% yield. In contrast, the subjection of β-phenylselenide to m-chloroperoxybenzoic acid (m-CPBA) led to (+)-γ-apopicropodophyllins 134 in 88% yield.
The same year, Giri and co-workers reported a Ni-catalyzed tandem cyclization/cross-coupling regioselective dicarbon functionalization of unactivated olefins, and applied this method to the concise synthesis of (±)-collinusin 135 on a gram-scale.64 In this protocol, the authors utilized the highly functionalized (2-aryoylaryl)zinc iodide as the coupling partner, the carbonylbutyrolactone core was synthesized in one pot and two step procedure, affording a yield of 70% (2.5 mmol scale, 0.672 g). Subsequently, the carbonylbutyrolactone was treated with lithium diisopropylamide (LDA) and SOCl2 to furnish (±)-collinusin with a 65% yield. Noteworthy, the concise synthesis of (±)-collinusin via this method is feasible due to the ready access to arylzinc reagents containing carbonyl groups.
 | ||
| Scheme 36 Synthesis of aryl-naphthalenes via DDQ-mediated oxidation of ADHNs. | ||
 | ||
| Scheme 37 Synthesis of arylnaphthalene lactones, taiwanin C and justicidin E via DDQ-mediated oxidation of ADHNs. | ||
In 2015, Shia, Lin and co-workers reported a SmI2 mediated decyanation reaction of α-cyano lactone 142, followed by DDQ oxidation, to furnish the arylnaphthalene lactones 143 (Scheme 38A).66 Then, a similar approach was represented by this group, and it was used to construct the natural arylnaphthalene lignans, retrojusticidin B 145, justicidin E 148, and helioxanthin 149 (Scheme 38B and D).
 | ||
| Scheme 38 Synthesis of arylnaphthalene lactones, retrojusticidin B, justicidin E, and helioxanthin via SmI2-mediated reaction of ADHNs. | ||
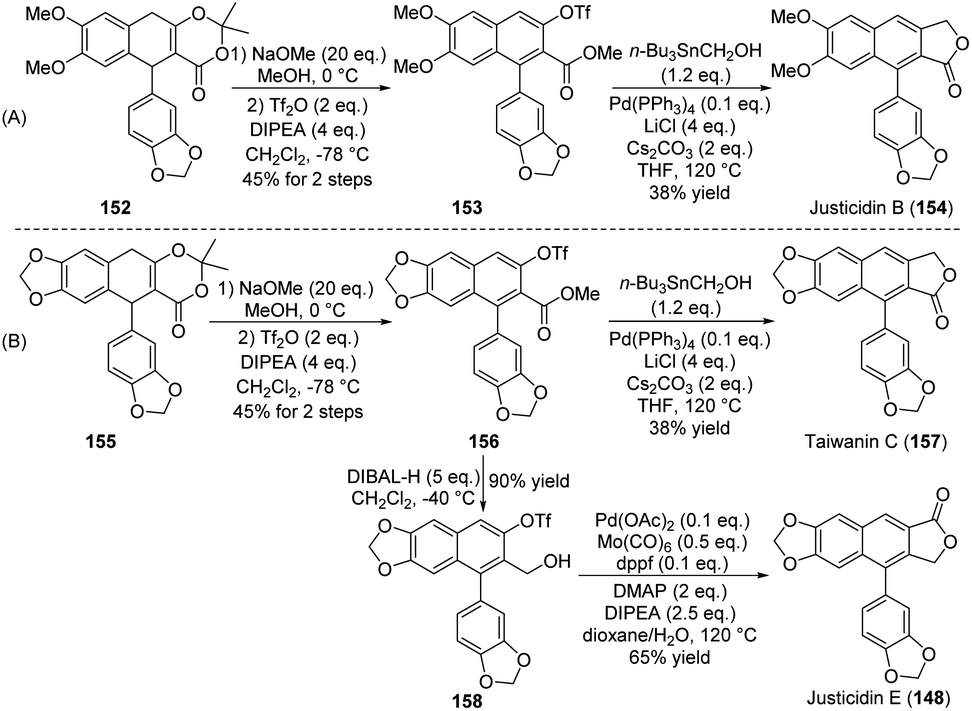
In 2022, Zhang, Chen and co-workers reported a versatile and flexible approach toward the synthesis of natural arylnaphthalene lactone lignans, including justicidin B 154, taiwanin C 157, and justicidin E 148, from ADHNs precursors (Scheme 39A and B).20 In this channel, an aryl–alkyl Suzuki cross-coupling was introduced for the formation of the dioxinone unit, a cation-induced cyclization occurred to construct the aryl dihydronaphthalene, and base-mediated oxidative aromatization to furnish the arylnaphthalene core. For example, the treatment of 152 or 155 with sodium methoxide in MeOH under air followed by the addition of Tf2O and diisopropylethylamine (DIPEA) in CH2Cl2 produced the first common intermediate 153 or 156, both with yields of 45%. In this work, an oxidative (by air) aromatization occurred under strong basic conditions. Then, in the presence of Pd(PPh3)4, Cs2CO3, and LiCl, a Pd-catalyzed Stille cross-coupling reaction was performed between triflates 153 or 156 and tributylstannyl methanol. This was followed by spontaneous lactonization reaction, providing the naturally occurring justicidin B (154) or taiwanin C (157). Subsequently, reduction of 156 using diisobutylaluminum hydride (DIBAL-H) provided the alcohol 158 in 90% yield. The natural justicidin E (148) was furnished in 38% isolated yield via an improved Pd-catalyzed carbonylative lactonization of triflate 158 with Mo(CO)6.
 | ||
| Scheme 39 Synthesis of arylnaphthalene lactones, justicidin B, taiwanin C and justicidin E. | ||
 | ||
| Scheme 40 Synthesis of aryltetrahedronaphthalen, cagayanin and galbulin via reduction reaction of ADHNs. | ||
In 2004, the same group used the same starting material, 2,3-dibenzylidenesuccinate, and showed an asymmetric photocyclization to produce a chiral ADHN. Subsequent hydrolysis and re-esterification provided the precursor 165, the reduction reaction of the double bond of 165 offered ester 166, then providing (+)-lyoniresinol dimethyl ether 167 (Scheme 41) as a major product, along with its 2,3-cis diastereomer as a minor product.17
 | ||
| Scheme 41 Synthesis of aryltetrahedronaphthalene, lyoniresinol dimethyl ether via reduction reaction of ADHNs. | ||
Similarly, in 2013, Peng and co-workers reported the synthesis of (±)-galbulin (164) via hydrogen and Pd/C catalyzed reduction of cyclogalgravin (168) (Scheme 42).67 Notably, the cyclogalgravin emerged as a pivotal precursor facilitating the collective synthesis of stereodivergent 2,7′-cyclolignans, featuring either anti–anti or (anti–)syn–anti substituent arrangements within the tetrahydronaphthalene scaffold.
 | ||
| Scheme 42 Synthesis of aryltetrahedronaphthalen, galbulin via reduction reaction of cyclogalgravin. | ||
In 2020, Studer and co-workers also presented the synthesis of natural product (+)-Galbulin (164). The remarkable transformation of the ADHN precursor was outlined in Scheme 43,68 in which the incorporation of pinacol into 169 yielded the corresponding boronic ester 170. Notably, the extreme instability of 170 necessitates its immediate progression through the subsequent reaction sequence without any further purification. Unfortunately, due to potential steric hindrance, 170 failed to transform into the boronic ester 171. Then, transesterification with pinacol yielded the pinacol boronic ester 171. Ultimately, the final protodeboronation of 171 led to (±)-galbulin (164). Despite the formidable challenge posed by the final step, they successfully achieved this through the oxidation under photoredox conditions and the subsequent capture of the generated primary alkyl radical with a thiophenol.
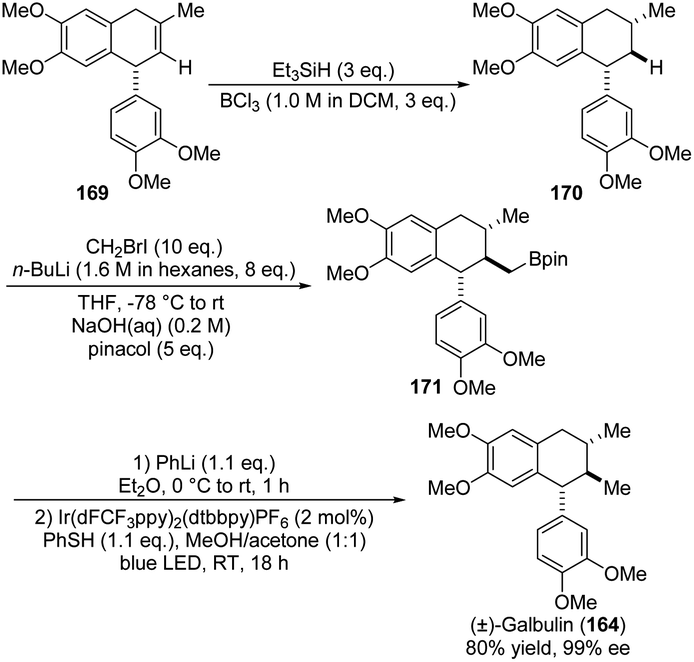 | ||
| Scheme 43 Synthesis of aryltetrahedronaphthalen, galbulin via oxidation under photoredox conditions. | ||
In 2008, Lautens and co-workers completed the first enantioselective total synthesis of aryltetrahedronaphthalen-type natural products, (+)-homochelidonine 173, (+)-chelamidine 174, (+)-chelidonine 176, (+)-chelamine 177 and (+)-norchelidonine 178 from ADHNs precursors 172 and 175 by several steps (Scheme 44A and B, the details were not shown here).69 The synthetic method reported here provided possibility to prepare structural analogues of the hexahydrobenzo[c]phenanthridine alkaloids with potentially improved pharmacological properties.
 | ||
| Scheme 44 Enantioselective total synthesis of aryltetrahedronaphthalen, (+)-homochelidonine, (+)-chelamidine, (+)-chelidonine, (+)-chelamine and (+)-norchelidonine from ADHNs. | ||
Podophyllotoxin and its natural analogues, isolated from Podophyllum species, have been extensively studied over the past two centuries due to their significant biological activities and structural diversity. However, the catalytic and enantioselective concise total synthesis of 180 remains an unmet challenge. In 2017, Hajra and co-workers reported the first catalytic enantioselective synthesis of (−)-podophyllotoxin 180, (−)-picropodophyllin 181, (+)-isopicropodophyllin 182′, (+)-isopicropodophyllone 182 (Scheme 45) from a common ADHN intermediate.32
 | ||
| Scheme 45 Synthesis of aryltetrahedronaphthalen, (−)-podophyllotoxin, (−)-picropodophyllin, (+)-isopicropodophyllin, (+)-isopicropodophyllone from ADHN. | ||
Previously, (+)-epigalcatin has not yet been synthesized or found in natural sources. In 2018, Czarnocki and Lisiecki presented the first total synthesis of (+)-epigalcatin 184 using ADHN precursor 183 as the raw material, achieving a three step synthesis yield of 40% (Scheme 46).70 With L-prolinol as the chiral source, the target product 184 was synthesized in 11 steps with an overall yield of 5%. The product 184 can be obtained in a highly stereoselective manner from piperonal, 3,4-dimethylbenzaldehyde, and diethyl succinate. In this scheme, a key intermediate cyclolignan was formed through the continuous flow photocyclization of a chiral atropisomeric 1,2-bisbenzylidenesuccinate amide ester. This strategy may allow access to cyclolignan analogues that are inaccessible from natural plant sources.
 | ||
| Scheme 46 The first total synthesis of (+)-epigalcatin from ADHN. | ||
In 2021, Gao et al. reported the first asymmetric total synthesis and absolute configuration revision of naturally occurring aryltetralin cyclic ether lignans aglacins A, B, and E from ADHN precursor 185 based on the asymmetric photoenolization/Diels–Alder (PEDA) reaction between electron-rich 2-methylbenzaldehydes and unsaturated γ-lactones (Scheme 47).43 The Lewis acid [Ti(OiPr)4] played a pivotal role in the PEDA reaction by activating the inert dienophiles and controlling the diastereoselectivity. Upon obtaining aglacin E (186), aglacins A (187) and B (188) can be derived through the manipulation of the C7 hydroxyl group of aglacin E (186).
 | ||
| Scheme 47 The first asymmetric total synthesis of aglacins A, B, and E from the ADHN precursor. | ||
3.2 Bioactivity applications
The structural diversity of ADHN-type lignans determines their extensive range of biological activities. Currently, pharmaceutical chemists have reported the antitumor, antimicrobial, antiviral and neuroprotective activities of ADHNs derivatives. The overview of their biological activities is shown in Fig. 3. | ||
| Fig. 3 The summary of biological activities of ADHNs derivatives (the images are sourced from the website: https://cn.bing.com). | ||
 | ||
| Fig. 4 Representative antitumor agents containing ADHNs. | ||
Cyclolignans podophyllotoxin and its' derivatives etoposide and teniposide are widely used anticancer drugs, exhibiting good clinical effects in treating various neoplasms, such as testicular and small-cell lung cancer, lymphoma, leukaemia, and Kaposi's sarcoma.75 Nevertheless, limitations such as myelosuppression, the emergence of drug resistance, and cytotoxicity towards normal cells still persist. Therefore, it is necessary to develop new cyclolignans.
Previously, (−)-podophyllic aldehyde A (189), a form of cyclolignans, was tested by Gordaliza's group at the National Cancer Institute (USA) against a panel of 60 diverse cancer types. Notably, it emerged as one of the most potent compounds, displaying highly selective cytotoxicity, particularly towards colon carcinoma lines.76 Subsequently, to further substantiate the impact of the aldehyde group on antineoplastic selectivity and establish the structure–activity relationships of these compounds, the same research team prepared and tested other aldehydes derived from podophyllotoxin. Gordaliza and co-workers discovered that (−)-podophyllic aldehydes B (190)77 and C (191)78 exhibit significant antineoplastic cytotoxicity and apoptosis-inducing activities (Fig. 4).62
Vascular disrupting agents (VDAs) represent one class of vascular targeting agents (VTAs) that specifically target tumor vasculature. The anti-tumor mechanism of VDAs involves inducing microtubule depolymerization and cytoskeletal rearrangement, ultimately resulting in irreversible and selective damage to the microvasculature within the tumor microenvironment.79 In 2004, Pinney and colleagues reported that dihydronaphthalene Oxi6196 (192) possesses potent cytotoxicity and can be used as an inhibitor of tubulin assembly and a VDA (Fig. 4).80
Microtubules are found in the cytoskeleton of almost all eukaryotic cell types and are directly involved in a variety of cellular functions such as cell movement, transport of organelles inside the cell, maintenance of cell shape as well as mitosis and cell replication.81 Therefore, perturbations in tubulin assembly/disassembly have emerged as a popular target for novel chemotherapeutic drugs. In 2012, Alami, Provot and co-workers designed and synthesized a series of new dihydronaphthalene derivatives. Biological evaluation of these compounds revealed that compounds 192–194 had high cell growth inhibition (at the nanomolar level) against four tumour cell lines.38 The effect of the lead compound 192 on cancer cells was associated with cell cycle arrest in the G2/M phase.
Subsequently, in 2018, Pinney and co-workers found that compound 192 is a potent tubulin polymerization inhibitor (IC50 = 1.0 μM) with low nM cytotoxicity against human cancer cell lines.33 To further improve the water-solubility for in vivo evaluation, the phosphate salt prodrug compound 195 was synthesized and subjected to preliminary in vitro and in vivo (mouse and rat) evaluation. The results showed that the compound 195 has inhibition of tubulin polymerization (cell-free assay) and cytotoxicity in vitro against NCI-H460 (non-small cell lung carcinoma), DU-145 (prostate carcinoma), and SKOV-3 (ovarian adenocarcinoma) human cancer cell lines, and disruption of tumor-associated vasculature, thereby functioning as VDAs in vivo.
Alvarez, Salas-Vidal and co-workers demonstrated that compounds 196–198 exhibit varying degrees of cytotoxic activity against human nasopharyngeal (KB), colon (HF-6), breast (MCF-7) and prostate (PC-3) cancer cell lines, with IC50 values ranging from 1.49 to 1.0 × 105 μM (Fig. 4).82 The in vivo zebrafish embryos experiments indicated that these compounds may exert an antimitotic effect by disturbing tubulin. The study also showed that compounds 196–198 promote mitotic arrest and induce morphological changes in a similar manner to that of the antimitotic drugs nocodazole and podophyllotoxin.
Four previously undescribed ADHN-type lignans, echiumins A–D (199–202) (Fig. 4), were isolated and identified from the butanol fraction of Echium angustifolium Mill, displaying strong to weak antitumor activity against HepG2 and MCF7 cancer cell lines.83 Among them, echiumins A and D have the strongest anticancer activities.
In 2010, Nkengfack and colleagues reported two novel cyclolignene derivatives, named pycnanthulignene A (203) and pycnanthulignene B (204), which were extracted and isolated from the air-dried and ground roots of Pycnanthus angolensis (Scheme 48).58 Compound 203 was evaluated in vitro for antimicrobial activity against a panel of drug-resistant bacteria and pathogenic fungi strains, including one Gram-positive bacteria, methicillin-resistant Staphylococcus aureus (MRSA, LMP805), and six Gram-negative bacteria including β-lactamase positive (L+) Escherichia coli (L+ EC, LMP701), (L+) Shigella dysenteriae (L+ SD, LMP606), ampicillin-resistant Klebsiella pneumoniae (ARKP, LMP803), carbenicillin-resistant Pseudomonas aeruginosa (CRPA, LMP804), chloramphenicol resistant Salmonella typhi (CRST, LMP706), and chloramphenicol resistant Citrobacter freundii (CRCF, LMP802). The two pathogenic fungi used were Candida albicans (C. albicans, LMP709U) and Microsporum audouinii (M. audouinii, LMP725D). Compound 203 had significant activity against all of the tested organisms, methicillin-resistant S. aureus being the most sensitive pathogen. The minimal inhibitory concentration (MIC) values for 203 varied from 28.7 μM (against S. aureus) to 230.9 μM (vs. K. pneumoniae and P. aeruginosa). In addition, the compound 203 exhibited a minimum microbicidal concentration (MMC) value against the listed bacteria and fungi that was four times lower than its corresponding MIC value. In 2016, a comprehensive review conducted by Spiteller and co-workers also concluded that the compound 203 possessed moderate antimicrobial activity against a panel of drug-resistant pathogens, particularly with an MIC of 29 μM against Staphylococcus aureus.3c
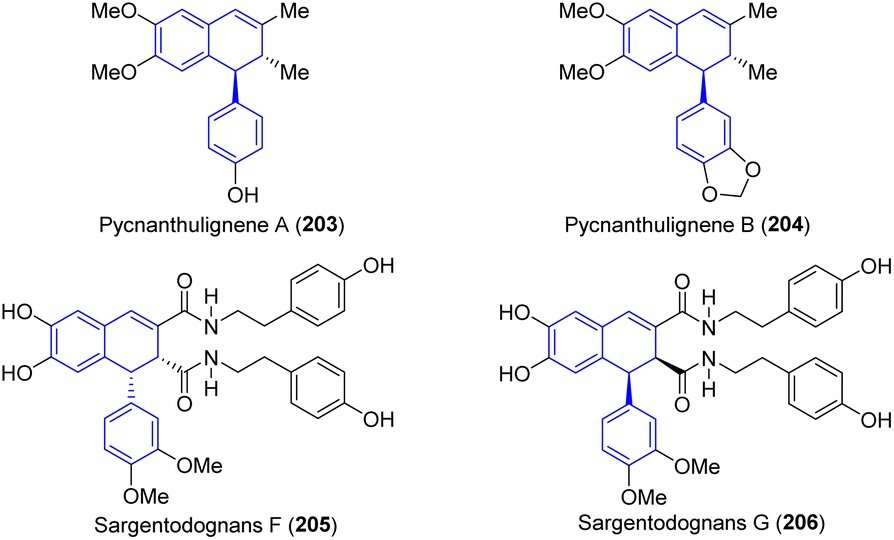 | ||
| Scheme 48 Representative antimicrobial agents containing ADHNs. | ||
In 2015, two novel phenolic glycosides, Sargentodognans F–G (205–206, Scheme 48), were extracted and isolated from a 60% ethanol extract of Sargentodoxa cuneata., and their structures and absolute configurations were determined by Tang's group.85 Antimicrobial tests were performed against S. aureus ATCC 29213, S. aureus ATCC 25923, A. baumanii ATCC 19606, and C. albicans ATCC 10231, respectively. Among them, compounds 205–206 showed antibacterial activity against S. aureus ATCC 29213.
A review on the antiviral activities of lignans87 prompted the Charlton group to explore the antiviral activities of several 1-arylnapthalenes and 1-aryl-1,2-dihydronaphthalenes lignans and their analogues. In 2000, the compounds 207–209 were tested by Charlton's group using a standard plaque reduction assay to evaluate their ability to inhibit human cytomegalovirus (Scheme 49).4a The cytotoxicity of the compounds were also measured using a tetrazolium salt MTT. The experimental results showed that the EC50s of compounds 207–209 were in low micromolar range (EC50/TC50: 207 25/>37 μM; 208 1.4/>25 μM; 209 22/>33 μM).
 | ||
| Scheme 49 Representative antiviral agents containing ADHNs. | ||
 | ||
| Scheme 50 Representative neuroprotective agents containing ADHNs. | ||
4. Conclusions
In summary, we have presented an overview of the research progress on ADHNs derivatives over the past two decades. Generally, the types of intramolecular and intermolecular synthesis methods for ADHNs derivatives, their reaction characteristics, applications in the total synthesis of natural products and bioactive molecules, and their biological applications are systematically introduced in this review.Notably, the intramolecular cyclization reaction of styrene-ynes has gained substantial attention and made much progress in recent years owing to its atom-economy, mild reaction conditions, extensive functional group tolerance and good yields. However, some substrates for the intramolecular cyclization reactions require multiple preparation steps, which affects the convenience of the synthesis process and reduces the overall yields of the synthetic routes. Despite some alternative intermolecular strategies have been established, most of them tend to provide access to the desired products, with few comprehensive methods to achieve diversified constructions. Moreover, the chemoselectivity in obtaining ADHNs skeletons exclusively remains elusive and challenging to pinpoint, leading to increased difficulty in purification and decreased yields.
Although some biological activities such as antitumor, antimicrobial, antiviral and neuroprotective activities have been well documented by researchers, offering a reference for future related biological study and drug exploitation, the scarcity of diverse ADHNs derivatives hinders pharmaceutical chemists from exploring their structure–activity relationships and impedes the development of their biological applications and potential therapeutic values. In the next stage, more attention should be paid to the exploration of new, atom- and step-economical, chemically selective, and environmentally benign synthetic methodologies to increase the diversity of ADHNs derivatives, laying a foundation for further exploration of the organic synthesis and synthetic and biological applications of ADHNs in the future.
Data availability
All data included in this study are available upon request by contact with the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 82003593)References
- J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zou, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC.
- (a) C.-Y. Li, T. J. Chow and T.-S. Wu, J. Nat. Prod., 2005, 68, 1622–1624 CrossRef PubMed; (b) V.-T. Vu, X.-L. Chen, L.-Y. Kong and J.-G. Luo, Org. Lett., 2020, 22, 1380–1384 CrossRef PubMed; (c) T. J. Schmidt, S. Hemmati, M. Klaes, B. Konuklugil, A. Mohagheghzadeh, I. Ionkova, E. Fuss and A. Wilhelm Alfermann, Phytochemistry, 2010, 71, 1714–1728 CrossRef PubMed; (d) T. J. Schmidt, M. Klaes and J. Sendker, Phytochemistry, 2012, 82, 89–99 CrossRef PubMed.
- (a) K. Kashima, K. Sano, Y. S. Yun, H. Ina and H. Inoue, Chem. Pharm. Bull., 2010, 58, 191–194 CrossRef PubMed; (b) R. G. Reynolds, H. Q. A. Nguyen, J. C. T. Reddel and R. J. Thomson, Nat. Prod. Rep., 2022, 39, 670–702 RSC; (c) R. B. Teponno, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2016, 33, 1044–1092 RSC.
- (a) C. Cow, C. Leung and J. L. Charlton, Can. J. Chem., 2000, 78, 553–561 CrossRef CAS; (b) L. S. Kocsis and K. M. Brummond, Org. Lett., 2014, 16, 4158–4161 CrossRef CAS; (c) A. F. A. Wallis, Aust. J. Chem., 1973, 26, 585–594 CrossRef CAS.
- (a) R. Ahmed, F. G. Schreiber, R. Stevenson, J. R. Williams and H. M. Yeo, Tetrahedron, 1976, 32, 1339–1344 CrossRef CAS; (b) R. Ahmed, M. Lehrer and R. Stevenson, Tetrahedron, 1973, 29, 3753–3759 CrossRef CAS.
- (a) A. F. A. Wallis, Aust. J. Chem., 1973, 26, 1571–1576 CrossRef CAS; (b) L. Lajide, P. Escoubas and J. Mizutani, Phytochemistry, 1995, 40, 1105–1112 CrossRef CAS.
- (a) L. H. Klemm and K. W. Gopinath, Tetrahedron Lett., 1963, 4, 1243–1245 CrossRef; (b) L. H. Klemm, D. H. Lee, K. W. Gopinath and C. E. Klopfenstein, J. Org. Chem., 1966, 31, 2376–2380 CrossRef CAS; (c) L. H. Klemm, P. S. Santhanam and J. Heterocyclic, Chem, 1972, 9, 423–426 CAS; (d) L. H. Klemm, T. M. McGuire and K. W. Gopinath, J. Org. Chem., 1976, 41, 2571–2579 CrossRef CAS.
- (a) K. V. Sarkanen and A. F. A. Wallis, J. Chem. Soc., Perkin Trans., 1973, 1, 1869–1878 RSC; (b) R. Stevenson and J. R. Williams, Tetrahedron, 1977, 33, 285–288 CrossRef CAS.
- Q. Wang, Y. Yang, Y. Li, W. Yu and Z. J. Hou, Tetrahedron, 2006, 62, 6107–6112 CrossRef CAS.
- (a) R. Jasti and S. D. Rychnovsky, J. Am. Chem. Soc., 2006, 128, 13640–13648 CrossRef CAS PubMed; (b) T. J. Steiman, A. E. Kalb, J. C. Coombs, J. K. Kirkland, H. Torres, D. H. Ess and C. Uyeda, ACS Catal., 2021, 11, 14408–14416 CrossRef CAS; (c) J. D. Brandt and K. D. Moeller, Org. Lett., 2005, 7, 3553–3556 CrossRef CAS PubMed.
- (a) G. Tiwari, A. Khanna, V. K. Mishra and R. Sagar, RSC Adv., 2023, 13, 32858–32892 RSC; (b) R. Javahershenas, A. Makarem and K. D. Klika, RSC Adv., 2024, 14, 5547–5565 RSC; (c) W. Zhang, C. H.-T. Chen, Y. Lu and T. Nagashima, Org. Lett., 2004, 6, 1473–1476 CrossRef CAS PubMed.
- E. Ruijter, J. Garcia-Hartjes, F. Hoffmann, L. van Wandelen, F. de Kanter, E. Janssen and R. Orru, Synlett, 2010, 2010, 2485–2489 CrossRef.
- T. Ozawa, T. Kurahashi and S. Matsubara, Org. Lett., 2011, 13, 5390–5393 CrossRef PubMed.
- X. Chen, C. Zhong, X. Duan, Z. Guan, L. Gu, Z. Luo, Y. Chen and Y. Zhang, J. Org. Chem., 2022, 87, 6601–6611 CrossRef PubMed.
- (a) X. Chen, Y. Lu, Z. Guan, L. Gu, C. Chen, H. Zhu, Z. Luo and Y. Zhang, Org. Lett., 2021, 23, 3173–3178 CrossRef PubMed; (b) X. Chen, Z. Luo, Y. Chen and Y. Zhang, Org. Lett., 2022, 24, 9200–9204 CrossRef PubMed; (c) X. Chen, C. Zhong, Y. Lu, M. Yao, Z. Guan, C. Chen, H. Zhu, Z. Luo and Y. Zhang, Chem. Commun., 2021, 57, 5155–5158 RSC.
- P. K. Datta, C. Yau, T. S. Hooper, B. L. Yvon and J. L. Charlton, J. Org. Chem., 2001, 66, 8606–8611 CrossRef.
- T. Assoumatine, P. K. Datta, T. S. Hooper, B. L. Yvon and J. L. Charlton, J. Org. Chem., 2004, 69, 4140–4144 CrossRef PubMed.
- E. Yoshida, K. Nishida, K. Toriyabe, R. Taguchi, J. Motoyoshiya and Y. Nishii, Chem. Lett., 2010, 39, 194–195 CrossRef.
- W. Li, H. Liu, J. Xu, P. Zang, Q. Liu and W. Li, Eur. J. Org Chem., 2014, 2014, 3475–3482 CrossRef.
- K. Wei, Y. Sun, Y. Xu, W. Hu, Y. Ma, Y. Lu, W. Chen and H. Zhang, Front. Chem., 2022, 10, 1103554 CrossRef.
- (a) P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef PubMed; (b) A. Kumar, Chem. Rev., 2001, 101, 1–20 CrossRef PubMed; (c) K.-i. Takao, R. Munakata and K.-i. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef.
- J. S. Tang, Y. X. Xie, Z. Q. Wang and J. H. Li, Chin. J. Org. Chem., 2011, 31, 653–658 Search PubMed.
- (a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef PubMed; (b) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef; (c) A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef PubMed; (d) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef.
- Y. C. Wong, T. T. Kao, Y. C. Yeh, B. S. Hsieh and K. S. Shia, Adv. Synth. Catal., 2013, 355, 1323–1337 CrossRef.
- Y. C. Wong, T. T. Kao, J. K. Huang, Y. W. Jhang, M. C. Chou and K. S. Shia, Adv. Synth. Catal., 2014, 356, 3025–3038 CrossRef.
- H. J. Mun, E. Y. Seong, K. H. Ahn and E. J. Kang, J. Org. Chem., 2018, 83, 1196–1203 CrossRef PubMed.
- Y.-C. Wong, C.-T. Tseng, T.-T. Kao, Y.-C. Yeh and K.-S. Shia, Org. Lett., 2012, 14, 6024–6027 CrossRef PubMed.
- V. W. Bhoyare, E. D. Sosa Carrizo, C. C. Chintawar, V. Gandon and N. T. Patil, J. Am. Chem. Soc., 2023, 145, 8810–8816 CrossRef.
- L. F. Tietze, S. C. Duefert, J. Clerc, M. Bischoff, C. Maaß and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 3191–3194 CrossRef CAS PubMed.
- H. Weinstabl, M. Suhartono, Z. Qureshi and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 5305–5308 CrossRef PubMed.
- J.-S. Cao, J. Zeng, J. Xiao, X.-H. Wang, Y.-W. Wang and Y. Peng, Chem. Commun., 2022, 58, 7273–7276 RSC.
- S. Hajra, S. Garai and S. Hazra, Org. Lett., 2017, 19, 6530–6533 CrossRef.
- C. J. Maguire, Z. Chen, V. P. Mocharla, M. Sriram, T. E. Strecker, E. Hamel, H. Zhou, R. Lopez, Y. Wang, R. P. Mason, D. J. Chaplin, M. L. Trawick and K. G. Pinney, MedChemComm, 2018, 9, 1649–1662 RSC.
- L. Devkota, C.-M. Lin, T. E. Strecker, Y. Wang, J. K. Tidmore, Z. Chen, R. Guddneppanavar, C. J. Jelinek, R. Lopez, L. Liu, E. Hamel, R. P. Mason, D. J. Chaplin, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem., 2016, 24, 938–956 CrossRef PubMed.
- Z.-H. Liu, J. Xiao, Q.-Q. Zhai, X. Tang, L.-J. Xu, Z.-Y. Zhuang, Y.-W. Wang and Y. Peng, Chem. Commun., 2024, 60, 694–697 RSC.
- (a) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (b) M. Kashihara and Y. Nakao, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS; (c) A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856–13863 CrossRef.
- C. Daquino, A. Rescifina, C. Spatafora and C. Tringali, Eur. J. Org Chem., 2009, 2009, 6289–6300 CrossRef.
- E. Rasolofonjatovo, O. Provot, A. Hamze, J. Rodrigo, J. Bignon, J. Wdzieczak-Bakala, D. Desravines, J. Dubois, J.-D. Brion and M. Alami, Eur. J. Med. Chem., 2012, 52, 22–32 CrossRef PubMed.
- W. Li, Q. Liu, H. Liu, P. Chen, X. Yang and Y. Liu, Chin. J. Chem., 2015, 33, 717–722 CrossRef.
- Q.-C. Zhang, W.-W. Zhang, L. Shen, Z.-L. Shen and T.-P. Loh, Molecules, 2018, 23, 979 CrossRef.
- Z. Zhang, P. He, H. Du, J. Xu and P. Li, J. Org. Chem., 2019, 84, 4517–4524 CrossRef PubMed.
- E. Alfonzo, A. M. Millimaci and A. B. Beeler, Org. Lett., 2020, 22, 6489–6493 CrossRef CAS PubMed.
- M. Xu, M. Hou, H. He and S. Gao, Angew. Chem., Int. Ed., 2021, 60, 16655–16660 CrossRef.
- (a) Y. Yamamoto, Y. Kubota, Y. Honda, H. Fukui, N. Asao and H. Nemoto, J. Am. Chem. Soc., 1994, 116, 3161–3162 CrossRef; (b) H. Nakamura, M. Sekido, M. Ito and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 6838–6839 CrossRef; (c) S. Sharma, Y. Oh, N. K. Mishra, U. De, H. Jo, R. Sachan, H. S. Kim, Y. H. Jung and I. S. Kim, J. Org. Chem., 2016, 82, 3359–3367 CrossRef PubMed; (d) A. S. Baghel, A. Aghi and A. Kumar, J. Org. Chem., 2021, 86, 9744–9754 CrossRef PubMed.
- M. Lautens, C. Dockendorff, K. Fagnou and A. Malicki, Org. Lett., 2002, 4, 1311–1314 CrossRef PubMed.
- M. Murakami and H. Igawa, Chem. Commun., 2002, 390–391 RSC.
- M. Lautens and C. Dockendorff, Org. Lett., 2003, 5, 3695–3698 CrossRef CAS PubMed.
- J. L. Jiang, J. Ju and R. Hua, Org. Biomol. Chem., 2007, 5, 1854–1857 RSC.
- X. Zhang, Y. Gao, J. Chen, R. Fan, G. Shi, Z. He and B. Fan, Adv. Synth. Catal., 2019, 361, 4495–4499 CrossRef.
- L.-S. Gan, S.-P. Yang, C.-Q. Fan and J.-M. Yue, J. Nat. Prod., 2005, 68, 221–225 CrossRef PubMed.
- J. K. Rout and C. V. Ramana, J. Org. Chem., 2012, 77, 1566–1571 CrossRef PubMed.
- Z.-B. Luo, Y.-W. Wang and Y. Peng, Org. Biomol. Chem., 2020, 18, 2054–2057 RSC.
- Z. Zhuang, Z. Luo, S. Yao, Y. Wang and Y. Peng, Molecules, 2022, 27, 5775 CrossRef PubMed.
- T. Schmidt, S. Vößing, M. Klaes and S. Grimme, Planta Med., 2007, 73, 1574–1580 CrossRef PubMed.
- L. F. Tietze, J. Clerc, S. Biller, S. C. Duefert and M. Bischoff, Chem.–Eur. J., 2014, 20, 17119–17124 CrossRef CAS PubMed.
- M. Odagi, K. Furukori, Y. Yamamoto, M. Sato, K. Iida, M. Yamanaka and K. Nagasawa, J. Am. Chem. Soc., 2015, 137, 1909–1915 CrossRef CAS PubMed.
- S. F. Fonseca, L. T. Nielsen and E. A. Rúveda, Phytochemistry, 1979, 18, 1703–1708 CrossRef CAS.
- E. C. N. Nono, P. Mkounga, V. Kuete, K. Marat, P. G. Hultin and A. E. Nkengfack, J. Nat. Prod., 2010, 73, 213–216 CrossRef.
- C. E. Rye and D. Barker, J. Org. Chem., 2011, 76, 6636–6648 CrossRef.
- T. Niwa, U. Doi and T. Osawa, Bioorg. Med. Chem. Lett., 2002, 12, 963–965 CrossRef PubMed.
- L. Zoia, M. Bruschi, M. Orlandi, E.-L. Tolppa and B. Rindone, Molecules, 2008, 13, 129–148 CrossRef PubMed.
- J. Ito, D. Sakuma and Y. Nishii, Chem. Lett., 2015, 44, 297–299 CrossRef.
- J. Xiao, G. Nan, Y.-W. Wang and Y. Peng, Molecules, 2018, 23, 3037 CrossRef.
- S. Kc, P. Basnet, S. Thapa, B. Shrestha and R. Giri, J. Org. Chem., 2018, 83, 2920–2936 CrossRef CAS.
- J.-E. Park, J. Lee, S.-Y. Seo and D. Shin, Tetrahedron Lett., 2014, 55, 818–820 CrossRef.
- T. T. Kao, C. C. Lin and K. S. Shia, J. Org. Chem., 2015, 80, 6708–6714 CrossRef PubMed.
- Y. Peng, Z.-B. Luo, J.-J. Zhang, L. Luo and Y.-W. Wang, Org. Biomol. Chem., 2013, 11, 7574–7586 RSC.
- F. Clausen and A. Studer, Org. Lett., 2020, 22, 6780–6783 CrossRef.
- M. J. Fleming, H. A. McManus, A. Rudolph, W. H. Chan, J. Ruiz, C. Dockendorff and M. Lautens, Chem.–Eur. J., 2008, 14, 2112–2124 CrossRef PubMed.
- K. Lisiecki and Z. Czarnocki, Org. Lett., 2018, 20, 605–607 CrossRef PubMed.
- (a) O. Vincze, F. Colchero, J.-F. Lemaître, D. A. Conde, S. Pavard, M. Bieuville, A. O. Urrutia, B. Ujvari, A. M. Boddy, C. C. Maley, F. Thomas and M. Giraudeau, Nature, 2021, 601, 263–267 CrossRef; (b) D. Wen, K. Li, R. Deng, J. Feng and H. Zhang, J. Am. Chem. Soc., 2023, 145, 3952–3960 CrossRef; (c) X. Chen, Y. Chen, C. Wang, Y. Jiang, X. Chu, F. Wu, Y. Wu, X. Cai, Y. Cao, Y. Liu and W. Bu, Angew. Chem., Int. Ed., 2021, 60, 21905–21910 CrossRef.
- R. Cai, H. Xiang, D. Yang, K.-T. Lin, Y. Wu, R. Zhou, Z. Gu, L. Yan, Y. Zhao and W. Tan, J. Am. Chem. Soc., 2021, 143, 16113–16127 CrossRef.
- J. Xu, W. Zhu, X. Yao, S. Mai, C. Li, M. Zhang, D. Shu and W. Yang, ACS Appl. Nano Mater., 2023, 6, 12029–12039 CrossRef.
- Y.-A. Lee, S. S. Lee, K. M. Kim, C. O. Lee and Y. S. Sohn, J. Med. Chem., 2000, 43, 1409–1412 CrossRef PubMed.
- M. A. Castro, J. M. Miguel del Corral, M. Gordaliza, P. A. García, M. A. Gómez-Zurita, M. D. García-Grávalos, J. de la Iglesia-Vicente, C. Gajate, F. An, F. Mollinedo and A. San Feliciano, J. Med. Chem., 2004, 47, 1214–1222 CrossRef PubMed.
- (a) A. San Feliciano, M. Gordaliza, J. M. M. Del Corral, M. A. Castro, M. D. Garcia-Gravalos and P. Ruiz-Lazaro, Planta Med., 1993, 59, 246–249 CrossRef PubMed; (b) M. Gordaliza, M. A. Castro, M. D. García-Grávalos, P. Ruiz, J. M. M. Del Corral and A. S. Feliciano, Arch. Pharm., 1994, 327, 175–179 CrossRef.
- M. Gordaliza, M. A. Castro, J. M. Miguel del Corral, M. L. López-Vázquez, P. A. García, M. D. García-Grávalos and A. San Feliciano, Eur. J. Med. Chem., 2000, 35, 691–698 CrossRef PubMed.
- M. Á. Castro, J. M. Miguel del Corral, P. A. García, M. V. Rojo, J. de la Iglesia-Vicente, F. Mollinedo, C. Cuevas and A. San Feliciano, J. Med. Chem., 2010, 53, 983–993 CrossRef PubMed.
- M. Sriram, J. J. Hall, N. C. Grohmann, T. E. Strecker, T. Wootton, A. Franken, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem., 2008, 16, 8161–8171 CrossRef PubMed.
- K. G. Pinney, V. P. Mocharla, Z. Chen, C. M. Garner, A. Ghatak, M. Hadimani, J. Kessler, J. M. Dorsey, K. Edvardsen, D. J. Chaplin, J. Prezioso, U. R. Ghatak, US Pat., Appl. Publ. 20040043969 A1, 2004 Search PubMed.
- (a) K. H. Downing and E. Nogales, Curr. Opin. Struct. Biol., 1998, 8, 785–791 CrossRef; (b) P. K. Sorger, M. Dobles, R. Tournebize and A. A. Hyman, Curr. Opin. Cell Biol., 1997, 9, 807–814 CrossRef.
- M. A. Mojica, A. León, A. M. Rojas-Sepúlveda, S. Marquina, M. A. Mendieta-Serrano, E. Salas-Vidal, M. L. Villarreal and L. Alvarez, RSC Adv., 2016, 6, 4950–4959 RSC.
- A. R. El-Rokh, A. Negm, M. El-Shamy, M. El-Gindy and M. Abdel-Mogib, Phytochemistry, 2018, 149, 155–160 CrossRef PubMed.
- (a) S. A. Polash, T. Khare, V. Kumar and R. Shukla, ACS Appl. Bio Mater., 2021, 4, 8060–8079 CrossRef; (b) P. Linciano, V. Cavalloro, E. Martino, J. Kirchmair, R. Listro, D. Rossi and S. Collina, J. Med. Chem., 2020, 63, 15243–15257 CrossRef PubMed.
- X. Zeng, H. Wang, Z. Gong, J. Huang, W. Pei, X. Wang, J. Zhang and X. Tang, Fitoterapia, 2015, 101, 153–161 CrossRef PubMed.
- (a) D. A. DeGoey, D. J. Grampovnik, H.-J. Chen, W. J. Flosi, L. L. Klein, T. Dekhtyar, V. Stoll, M. Mamo, A. Molla and D. J. Kempf, J. Med. Chem., 2011, 54, 7094–7104 CrossRef; (b) M. Kumar, K. Kuroda, K. Dhangar, P. Mazumder, C. Sonne, J. Rinklebe and M. Kitajima, Environ. Sci. Technol., 2020, 54, 8503–8505 CrossRef PubMed.
- J. L. Charlton, J. Nat. Prod., 1998, 61, 1447–1451 CrossRef PubMed.
- S. H. Mi, Y. Chang, X. Zhang, J. Y. Hou, J. Q. Niu, J. L. Hao, G. D. Yao, B. Lin, X. X. Huang, M. Bai and S. J. Song, Chem. Biodivers., 2023, 20, e202300941 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |