
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper and electrocatalytic synergy for the construction of fused quinazolinones with 2-aminobenzaldehydes and cyclic amines†
Yujie Shia,
Ganpeng Lia,
Ruirui Wang*b,
Xiao-jing Zhao *a and
Yonghui He*a
*a and
Yonghui He*a
aKey Laboratory of Chemistry in Ethnic Medicinal Resources, State Ethnic Affairs Commission & Ministry of Education, Key Laboratory of Natural Products Synthetic Biology of Ethnic Medicinal Endophytes, State Ethnic Affairs Commission, School of Ethnic Medicine, Yunnan Minzu University, Kunming, 650500, China. E-mail: Zhaoxj@ymu.edu.cn; heyonghui@ymu.edu.cn
bCollege of Chinese Materia Medica, Yunnan University of Chinese Medicine, Kunming 650000, P. R. China. E-mail: wangrryucm@126.com
First published on 11th October 2024
Abstract
A new copper and electrocatalytic synergy strategy for efficiently constructing fused quinazolinones has been developed. In the presence of cupric acetate and oxygen, aryl ketones and 1,2,3,4-tetrahydroisoquinoline can smoothly participate in this transformation, thus providing a variety of substituted quinazolones in an undivided cell. The reaction shows good functional group tolerance and provides universal quinazolinones at a good yield under mild conditions.
Nitrogen heterocyclic compounds have played pivotal roles in malaria chemotherapy progressing from quinine to chloroquine, mefloquine, and amodiaquine in the last century.1 Studies have shown that quinazolinone alkaloids and their analogs have good antimalarial activity.2 At the same time, quinazolinones also demonstrate a series of pharmacological and biological activities among various nitrogen-containing heterocyclic compounds.3 These include anti-cancer,4 anti-inflammatory,5 anticonvulsant,6 and anti-allergy effects.7 Especially, due to their wide pharmacological activities, fused quinazolinones have been regarded as potential drug molecules against various types of diseases (Fig. 1).
 | ||
| Fig. 1 Representative drugs and bioactive quinazolinones. | ||
In light of this utility, many methods for synthesizing quinazolinones have been developed over the last decade. Among them, traditional synthetic methods for fused quinazolinones involve the oxidative decarboxylation of isatin and tetrahydroisoquinoline catalyzed by Cu2+ or peroxide (Scheme 1a).8 In addition, 2-aminoarylmenthanol and isoquinoline can be converted to quinazolinone in the presence of MOF-derived cobalt nanoparticles (Scheme 1b).9 However, these methods require large amounts of oxidants and additives, high reaction temperatures, and pre-functionalized complex substrates, which limits their usefulness. Interestingly, Xie and their coworkers reported a visible-light-induced cyclization of 2-aminobenzaldehydes with tetrahydroisoquinoline for the synthesis of fused quinazolinones (Scheme 1c).10 Although this reaction provided a green, simple, and oxidant-free strategy, further research for overcoming the limited substrate scope and ambiguous transformation process is still needed.
 | ||
| Scheme 1 Strategies for the synthesis of quinazolinones. | ||
Organic mechatronic synthesis has attracted much attention in recent years due to its environmental friendliness, sustainability and mild reaction conditions.11,12 Organic conversion triggered by electricity is becoming a uniquely powerful tool for building new chemical bonds in organic synthesis.13 Recently, our research group have developed some organic electrochemical strategies for the efficient construction of N-containing heterocyclic compounds.14 Based on our precious works, in this work, we developed a new copper and electrocatalytic synergy strategy for the efficient construction of fused quinazolinones. The reaction shows good functional group tolerance and provides universal quinazolinones at a good yield at mild conditions (Scheme 1d).
We began our study by choosing 2-aminobenzaldehyde 1a and tetrahydroisoquinoline 3a as template substrates to screen the optimal reaction conditions. The reaction was carried out in an undivided three-necked flask with an aluminum rod cathode and a carbon rod anode, nBu4NBF4 as the supporting electrolyte, and a mixture of Cu(AcO)2 with these compounds in 10 mL MeOH solution at room temperature (Table 1). To our delight, the reaction performed very well under the original electrized conditions, leading to obtaining the product 4a with a 94% yield in 13 hours (entry 1). A shorter or longer reaction time was insufficient to effectively form the product (entry 2 and 3). Changing the electrolyte from nBu4NBF4 to nBu4LiClO4 or nBu4NPF6 resulted in a reduced product yield (entries 4 and 5). Both increasing or decreasing the current afforded the final product in lower yield (entries 6 and 7). As for the reaction solvent, DMSO, DMA, DMF and CH3CN exhibited lower product yields when compared with CH3OH (entries 8–11). Further increase the polar of solvent by adding 0.5 mL H2O, the reaction will be suppressed (entry 12). When using KI or NaI instead of NH4I, the reaction proceeded with poor reactivity (entries 13 and 14). Using N2 or air instead of O2, the reactivity will be affected (entry 15 and 16). This result indicated that O2 are significant for this transformation. Using the Pt plate as electrode led to poor reaction efficiency (entry 17 and 18). When there is no Cu(CH3COO)2, the yield drops to 57% (entry 19). Finally, the electrolyte or electricity are necessary for this transformation (entry 21).
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Reaction conditions: aluminum rod cathode, carbon rod anode, constant current = 5 mA, 1a (0.2 mmol), 3a (0.3 mmol), nBu4NBF4 (0.5 mmol), NH4I (0.5 mmol), Cu(AcO)2 (1% mmol), MeOH (10 mL), O2, 30 °C, undivided cell.b Isolated yield. | ||
| 1 | None | 94% |
| 2 | 10 h instead of 13 h | 60% |
| 3 | 18 h instead of 13 h | 90% |
| 4 | nBu4LiClO4 instead of nBu4NBF4 | N.R. |
| 5 | nBu4NPF6 instead of nBu4NBF4 | 63% |
| 6 | 3 mA instead of 5 mA | 79% |
| 7 | 9 mA instead of 5 mA | 85% |
| 8 | DMSO as solvent | N.R. |
| 9 | DMA as solvent | 31% |
| 10 | DMF as solvent | 22% |
| 11 | CH3CN as solvent | 45% |
| 12 | 9.5 mL CH3OH + 0.5 mL H2O as solvent | N.R. |
| 13 | KI instead of NH4I | 32% |
| 14 | NaI instead of NH4I | 41% |
| 15 | N2 instead of O2 | N.R. |
| 16 | Air instead of O2 | 22% |
| 17 | C(+)|Pt(−) instead of C(+)|Al(−) | 30% |
| 18 | Pt(+)|Pt(−) instead of C(+)|Al(−) | 26% |
| 19 | Without Cu(AcO)2 | 57% |
| 20 | Without electrolytes or electricity | N.R. |
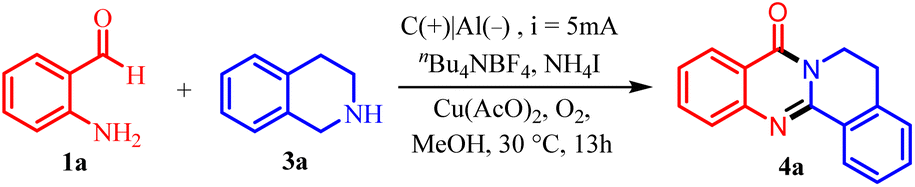
Under optimal conditions, the application range of substrates for electrolytic synthesis of quinazolinones was studied. Firstly, the conversion of various substituted 2-aminobenzenaldehyde was investigated. Of particular note, functional groups such as Me, MeO, F, Cl and Br were well tolerated in the reaction (Table 2, 4b–m). When the functional group is located in C-5 position, the yield of the electron-donating group is higher than that of the electron-withdrawing group. When the functional group is located at the C-4 position, the yield of the reaction is higher than that at the C-3 and C-5 positions. When substituent group is located in C-3 position, the corresponding electron-withdrawing group substitution products 4j, 4k and 4l are obtained with yields of 67%, 81% and 77%, respectively (Table 2, 4j–l). The electron donor substitution product 4m with 91% yield was also obtained. The yield of di-substituted quinazolinone 4n was 63% (Table 2, 4n). It is a pity that 6-aminobenzo[d][1,3]dioxole-5-carbaldehyde couldn't participate in this transformation (Table 2, 4n′).
| a Reaction conditions: aluminum rod cathode, carbon rod anode, constant current = 5 mA, 1 (0.2 mmol), 3a (0.3 mmol), nBu4NBF4 (0.5 mmol), NH4I (0.5 mmol), Cu(AcO)2 (1% mmol), MeOH (10 mL), O2, 30 °C, undivided cell. |
|---|
 |
Then, we continued to investigate the substrate range of tetrahydroisoquinoline (Table 3). When C-7 is linked to Br substituent, the reactivity is very well, and gave the yield 86% (Table 3, 4t). While other tetrahydroisoquinolines with electron-attracting or electron-donating groups reduced the reactivity (Table 3, 4o–s, 4u). The yield of di-substituted quinazolinone 4v was 43% (Table 3, 4v). It is noteworthy that other cyclic amine substrates tetrahydropyrrole and hexamimide were also compatible with the reaction (Table 3, 4w and 4x). Interestingly, the natural active alkaloid red pine was obtained in this reaction (Table 3, 4y).
| a Reaction conditions: aluminum rod cathode, carbon rod anode, constant current = 5 mA, 1a (0.2 mmol), 3 (0.3 mmol), nBu4NBF4 (0.5 mmol), NH4I (0.5 mmol), Cu(AcO)2 (1% mmol), MeOH (10 mL), O2, 30 °C, undivided cell. |
|---|
 |
Surprisingly, when 2-aminophenone 2a was used instead of the substrate 2-aminobenzaldehyde 1a in this reaction, the corresponding fused quinazolinones have been obtained (Table 4). With –F, –Cl and –Br substituents, it effectively provides the required quinazolines under given conditions, with moderate to good yield (Table 4, 4b, 4c, 4f, 4h and 4k).
| a Reaction conditions: aluminum rod cathode, carbon rod anode, constant current = 5 mA, 2 (0.2 mmol), 3a (0.3 mmol), nBu4NBF4 (0.5 mmol), NH4I (0.5 mmol), Cu(AcO)2 (1% mmol), MeOH (10 mL), O2, 30 °C, undivided cell. |
|---|
 |
To further emphasize the practical application of the above method, we first carried out gram preparation and obtained the corresponding product 4a with a 45% (0.92 g) yield (Scheme 2a). The phenylation of product 4c could be smoothly carried out in the meantime, resulting in the corresponding product 6a (Scheme 2b).
 | ||
| Scheme 2 Synthetic applications. | ||
Due to their good biological activity of quinazolinones, we have performed the antibacterial activity research of the synthetic products in this work. Among the synthetic quinazolinones compounds, 19 compounds show good effect of anti-resistant strains of Candida albicans, and had the synergistic antibacterial effect when combined with fluconazole (Table S1†). This method provides a new tool for the development of antifungal drugs and the overcoming of drug resistance.
To further understand the reaction mechanism, we conducted several control reactions (Scheme 3). Firstly, the free radical inhibitor 2,2,6,6-tetramethylpiperidine (TEMPO) and dibutylhydroxytoluene (BHT) can suppressed this reaction in part. This suggests that the reaction could be carried out via a radical pathway (Scheme 3a and b). Secondly, when the compound 5 was used as an intermediate, the product 4a was obtained in good yield, which further indicated that compound 5 was the required intermediate (Scheme 3c). Thirdly, when O2 is replaced by air, the yield is reduced to 22%; and using N2 atmosphere, no desired product can be obtained (Scheme 3d). The results revealed that O2 is very essential for this transformation. Finally, cyclic voltammetry experiment result suggested that the oxidation of I− initiates this reaction (Scheme S3†).
 | ||
| Scheme 3 The control experiments. | ||
Based on the above observations and the precious literature,8b,10 a plausible reaction mechanism was proposed (Scheme 4). Initially, the iodine anion is oxidized to an iodine radical, which reacts with 3a to form the radical intermediate A. A undergoes anodization and deprotonation to give the imide intermediate 5. Subsequently, a formal [4 + 2] cycloaddition of 5 and 2-aminophenylacetaldehyde would produce intermediate D, which was further hydrolyzed to give intermediate E which undergoes multiple oxidizations to furnish the product 4a. Finally, H+ is reduced to hydrogen gas on the cathode. Cu could be stripped from the cathode to regenerate the catalyst.15
 | ||
| Scheme 4 Plausible mechanism for the reaction. | ||
Additionally, 2-aminophenone would be firstly oxidized to give 2-aminobenzaldehyde 1a catalyzed by the Cu and molecular oxygen in this reaction.16 Subsequently, 2-aminobenzaldehyde reacts with intermediate 5, thus giving the final product 4a.
In summary, we have successfully developed an efficient copper and electrocatalytic synergy strategy for the synthesis of quinazolinones via the intermolecular oxidation cyclization reaction. It is worth noting that the reaction is characterized by broad substrate scope and mild reaction conditions. At the same time, the synthesized products have good antifungal activity.
Data availability
All relevant data are within the manuscript and its additional files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Natural Science Foundation of Yunnan (2016FB149, 202101AT070079), “Xingdian” Talent Support Program of Yunnan Province, The Key Research and Development Program of Yunnan Province (202103AC100005), and Yunnan Provincial Science and Technology Department-Applied Basic Research Joint Special Funds of Yunnan University of Chinese Medicine (202101AZ070001-001).References
- E. Fernández-Álvaro, W. D. Hong, G. L. Nixon, P. M. O'Neill and F. Calderón, J. Med. Chem., 2016, 59, 5587–5603 CrossRef PubMed.
- (a) S. Hirai, H. Kikuchi, H.-S. Kim, K. Begum, Y. Wataya, H. Tasaka, Y. Miyazawa, K. Yamamoto and Y. Oshima, J. Med. Chem., 2003, 46, 4351–4359 CrossRef PubMed; (b) M. M. Heravi, M. Zakeri and N. Mohammadi, Chin. J. Chem., 2011, 29, 1163–1166 CrossRef.
- (a) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650–668 RSC.
- Z.-W. Mei, L. Wang, W.-J. Lu, C.-Q. Pang, T. Maeda, W. Peng, M. Kaiser, I. El Sayed and T. Inokuchi, J. Med. Chem., 2013, 56, 1431–1442 CrossRef PubMed.
- M. E. Azab, Z. Naturforsch. B, 2016, 71, 803–810 CrossRef CAS.
- Z. Wang, Y. He, F. Wang, Y. Wang, H. Luo, J. Wu and J. Yang, RSC Adv., 2024, 14, 23693–23698 RSC.
- (a) K. J. Britto, M. Meenakshi and K. Srinivasan, RSC Adv., 2024, 14, 22076–22085 RSC; (b) I. Khan, A. Ibrar, W. Ahmed and A. Saeed, Eur. J. Med. Chem., 2015, 90, 124–169 CrossRef CAS PubMed.
- (a) F.-C. Jia, T.-Z. Chen and X.-Q. Hu, Org. Chem. Front., 2020, 7, 1635–1639 RSC; (b) D. Wang, F. Xiao, F. Zhang, H. Huang and G.-J. Deng, Chin. J. Chem., 2021, 39, 87–92 CrossRef CAS.
- F. Xie, Q.-H. Chen, R. Xie, H.-F. Jiang and M. Zhang, ACS Catal., 2018, 8, 5869–5874 CrossRef CAS.
- X. Chen, L. Jin, Y. Wang, H. Yang, Z. Le and Z. Xie, Org. Biomol. Chem., 2023, 21, 3863–3870 RSC.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (b) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS; (c) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS; (d) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS; (e) S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS; (f) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed; (g) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (h) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (i) Z.-W. Hou, H.-C. Xu and L. Wang, Curr. Opin. Electrochem., 2024, 44, 101447 CrossRef CAS.
- Y. Li, L. Wen and W. Guo, Chem. Soc. Rev., 2023, 52, 1168–1188 RSC.
- (a) F. Ke, Y. Xu, S. Zhu, X. Lin, C. Lin, S. Zhou and H. Su, Green Chem., 2019, 21, 4329–4333 RSC; (b) Z.-L. Wu, J.-Y. Chen, X.-Z. Tian, W.-T. Ouyang, Z.-T. Zhang and W.-M. He, Chin. Chem. Lett., 2022, 33, 1501–1504 CrossRef; (c) Y.-H. Lu, S.-Y. Mu, H.-X. Li, J. Jiang, C. Wu, M.-H. Zhou, W.-T. Ouyang and W.-M. He, Green Chem., 2023, 25, 5539–5542 RSC; (d) H.-T. Tang, Y.-Z. Pan and Y.-M. Pan, Green Chem., 2023, 25, 8313–8327 RSC; (e) T.-K. Xiong, Q. Xia, X.-Q. Zhou, S.-H. Li, F.-H. Cui, H.-T. Tang, Y.-M. Pan and Y. Liang, Adv. Synth. Catal., 2023, 365, 2183–2187 CrossRef; (f) X.-Q. Zhou, H.-T. Tang, F.-H. Cui, Y. Liang, S.-H. Li and Y.-M. Pan, Green Chem., 2023, 25, 5024–5029 RSC.
- (a) Y. Cao, Y. Yuan, Y. Lin, X. Jiang, Y. Weng, T. Wang, F. Bu, L. Zeng and A. Lei, Green Chem., 2020, 22, 1548–1552 RSC; (b) L. Gu, G. Li, L. Zhang, Y. He, Y. Zhou, M. Li and Z. Zhao, Chin. J. Org. Chem., 2021, 41, 2476 CrossRef; (c) Y. Wang, X.-J. Zhao, X. Wu, L. Zhang, G. Li and Y. He, Chemelectrochem, 2022, 9, e202200378 CrossRef; (d) D. Zeng, L. Zhang, W. Wang, G. Li, X.-J. Zhao and Y. He, Eur. J. Org Chem., 2022, 2022, e202200679 CrossRef; (e) M. Wang, Y. Gao, X.-J. Zhao, L. Gao and Y. He, Chem. Commun., 2024, 60, 2677–2680 RSC; (f) Z.-M. Zong, L. Zhang, G.-P. Li, W. Wang, X.-J. Zhao and Y. He, Org. Lett., 2024, 26, 1271–1276 CrossRef.
- B. R. Walker, S. Manabe, A. T. Brusoe and C. S. Sevov, J. Am. Chem. Soc., 2021, 143, 6257–6265 CrossRef.
- L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303–11307 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06539e |
| This journal is © The Royal Society of Chemistry 2024 |