
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent trends for chemoselectivity modulation in one-pot organic transformations
Hiren R. Chaudhary
and
Divyang M. Patel
 *
*
Department of Chemistry, Sankalchand Patel University, Visnagar 384315, Gujarat, India. E-mail: divyangpatel.dm@gmail.com; dmpatel.fsh@spu.ac.in
First published on 30th September 2024
Abstract
In organic reactions, chemoselectivity refers to the selective reactivity of one functional group in the presence of another. This can be more successful if the reagent and reaction parameters are appropriately chosen. One-pot reactions have been shown to be an effective structural variety technique for the development of novel heterocyclic or carbocyclic compounds. This review article focuses on recent efforts by researchers from around the world to synthesise novel organic molecules utilising these methodologies (2013–2024), as well as their mechanism insights. The substrate, catalyst, solvent, and temperature conditions all have a significant impact on chemoselectivity in the organic reactions described here. The manipulation of chemoselectivity in organic processes creates new potential for the production of novel heterocycles and carbocycles.
 Hiren R. Chaudhary | Hiren R. Chaudhary graduated in chemistry from Shree B.A.P.K.M. Science College, Palanpur in 2020 and received a postgraduate degree from the Department of chemistry, Shri Maneklal M. Patel Institute of Sciences & Research (Kadi Sarva Vishwavidyalaya), in 2022. He is currently pursuing PhD (Chemistry) at the Department of Chemistry, Smt. S. S. Patel Nootan Science and commerce college, affiliated with Sankalchand Patel University, Visnagar, 384315, Gujarat, India. |
 Divyang M. Patel | Dr. Divyang M. Patel graduated in chemistry from M. B. Patel Science College, Anand, in 2013 and received a postgraduate degree from the Department of Chemistry, Sardar Patel University, in 2015. In 2016, he qualified for GSET in chemical science. DMP received his PhD in organic chemistry from the Department of Chemistry, Sardar Patel University, in 2021. He is currently an Assistant Professor at the Department of Chemistry, Smt. S. S. Patel Nootan Science and Commerce College, affiliated with Sankalchand Patel University, Visnagar, Gujarat, India. His current research interests include organocatalysis, multicomponent reactions (MCRs), green chemistry, and the synthesis of medicinally privileged heterocycles and hybrid molecules. |
1. Introduction
In synthetic organic chemistry one-pot reactions are widely spread throughout medicinal,1–9 combinatorial,10–12 and heterocyclic chemistry.13–15 These reactions have been receiving considerable attention.16–18 The core advantages of these are the formation of complex molecular structures with multiple bonds forming and/or rupturing in a single-step,19,20 as well as high atom economy.21–23 Many of these are well-known name reactions used for the construction of diverse heterocyclic molecules of biological importance, including the Strecker reaction24 for the synthesis of α-aminonitriles (1850), the Hantzsch reaction25 for dihydropyridine synthesis (1882), the Biginelli reaction26 for dihydropyrimidone synthesis (1891), the Mannich reaction27 for the synthesis of β-amino-carbonyl derivatives, Passerini reaction28 for construction of α-acyloxy amides (1921), Kabachnik–Fields reaction29 for α-aminomethylphosphonates synthesis (1952), Asinger reaction30 for the preparation of 3-thiazolines and related heterocycles (1956), Ugi reaction31 for bis-amides (1959), Gewald reaction32 for 2-amino-thiophene derivatives (1966), Van Leusen reaction33 for the synthesis of nitriles (1977), and Groebke–Blackburn–Bienaymé reaction34 for the synthesis of fused imidazole (1998). One-pot reactions have been used successfully over the last few decades to produce huge quantities of drug candidates at high throughput.35,36 An estimated 5% of commercially available medications, such as Crixivan (by Ugi reaction), Telaprevir (by Passerini reaction), and Nifedipine (by Hantzsch reaction), were produced using one-pot multicomponent reaction (MCR)-based procedures.36 In these reactions, chemoselectivity is essentially important for developing new techniques for different target organic molecules with appropriate chemical modifications.37 Modulating chemoselectivity is a strategic goal in organic synthesis because it has been proven to be a powerful skeletal diversity tool in diversity-oriented synthesis (DOS) for the search for new, therapeutically active small heterocycles.38–41 Over the past few decades, a large number of researchers have reported the chemoselectively controlled one-pot reactions using various catalysts, solvents, and substrate topologies (Fig. 1). By carefully regulating the reaction conditions, these chemoselective one-pot reactions are in reality unique procedures that combine a variety of easily accessible building blocks to produce different molecular assemblies. Many heterocyclic molecules are substructures of several natural products, medicines, and synthetic organic moieties; many of them are regarded as “privileged medicinal scaffolds”. 42–44 | ||
| Fig. 1 Modulation of chemoselectivity in organic transformations. | ||
In organic chemistry the retrosynthetic procedures are typically selected when coming up with a strategy for a challenging goal to achieve maximum skeletal simplification, but particularly in cases where the proposed chemistry has little or no context.45 Such a protocol makes a revolution and reveals the main objectives of organic synthesis, as opposed to adapting a strategy to serve known reactions. The expected outcome of such efforts is the provision of the target architecture in a convergent manner, and creativity becomes a necessity for triumph.46
With the continuous progress in this field, new synergistic chemoselective one-pot methods have been frequently explored. However, a comprehensive overview of the progress and current status of these methods is lacking. The authors emphasise the efforts of the global scientific community in the development and implementation of the new chemoselective organic one-pot reactions discussed in this review. In this article, we will present those methods that were described in the literature between 2013 and 2024. The chemoselectivity of most organic reactions is primarily determined by factors such as substrates, reaction medium (solvent), catalysts or additives, and reaction temperature. To the best of our knowledge, no bibliometric study has been documented that focuses on the breakthroughs in chemoselective one-pot reactions considerably; the current review outlines the pioneering advances made in this field. To give readers a good outline, this review is organised into four groups based on the tuning of chemoselectivity in one-pot reactions. The first section focuses on substrate-modulated chemoselectivity in these reactions. The second section includes one-pot reactions with solvent-controlled chemoselectivity. The third section discusses catalyst-driven chemoselective one-pot reactions. Temperature-dependent chemoselectivity is addressed in the last section.
2. Substrate-modulated chemoselectivity in one-pot reactions
In the majority of organic transformations, chemoselectivity is modulated by varying the substrate for efficient development of complex molecular skeletons from the cost-effective and readily accessible precursors. Wang and colleagues47 developed an efficient and chemoselective reaction for the synthesis of functionalized pyrrole derivatives (1 and 2) through the reaction of alkyl and aryl isocyanides, primary and secondary amines, and 2-benzylidenemalononitrile derivatives in refluxing acetonitrile. The majority of these reactions showed successful transformations within 30 minutes with no catalyst or additive. Under well-established reaction parameters, the substrate scope with commercially available alkyl and aryl cyanides, primary and secondary amines, and 2-benzylidenemalononitrile derivatives has been successfully explored. When secondary cyclic amines such as pyrrolidine, morpholine, and 1,2,3,4-tetrahydroisoquinoline are utilised, they exclusively produce functionalized pyrrole derivatives (1) in a 23–79% yield. On the other hand, use of primary amines such as benzyl amine and phenylethylamine selectively produces poly-substituted pyrrole derivatives (2) in 55–78% yield (Scheme 1). In the proposed mechanistic route for polysubstituted pyrrole derivatives (1) or (2) initially, nucleophilic addition on 2-benzylidenemalononitrile by isocyanide forms an intermediate, which undergoes nucleophilic attack by primary amines to form functionalized pyrrole derivatives (1); the use of a secondary amine exclusively produces polysubstituted pyrrole derivatives (2).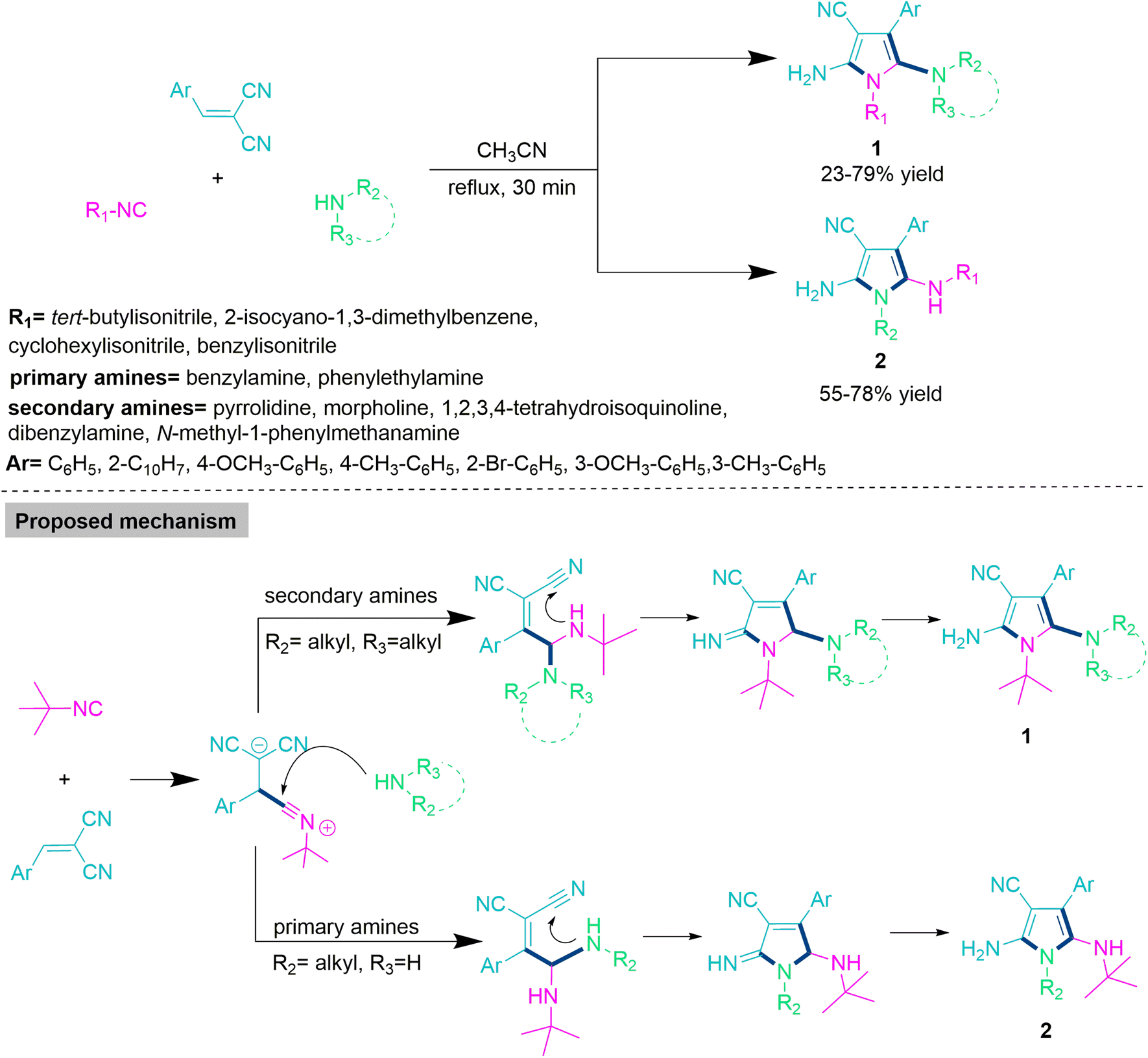 | ||
| Scheme 1 Synthesis of polysubstituted pyrrole derivatives (1) or (2). | ||
2-Aminopyrrole and 2-aminopyridine derivatives are important heterocycles because of their remarkable biological properties and their use as precursors in the development of diverse heterocycles.48–50 Thus, Yang and co-workers51 developed a chemoselective protocol for the synthesis of these heterocycles using a one-pot reaction of 3-halochromone and amidine promoted by K2CO3 in DMSO at 60 °C. In the described MCR, the formation of 2-aminopyrrole (3) or 2-aminopyridine (4) is exclusively governed by the type of 3-halochromone. Here, use of the 3-iodochromone derivative in the title MCR selectively yields 2-aminopyrroles (3), while 3-chlorochromone derivatives solely produce 2-aminopyridines (4). Yang and co-workers explored the substrate scope and limitations with the help of functionalized chromone derivatives and observed that chromone moiety-bearing electron-withdrawing groups offer good yields of target products. This is because the presence of electron-withdrawing functionality enhances the electrophilicity of the carbonyl functional group more than that of electron-releasing groups. Yang et al. reacted C, N-bis-nucleophiles (ethyl 2-amidinoacetate hydrochloride) with 3-halochromone, in which carbanion makes nucleophilic attack on electrophilic 3-chlorochromone and produce an intermediate that undergoes direct displacement reaction to form product 3. On the other hand, use of 3-iodochromone leads to carbonyl addition in the generated intermediate to produce product 4 (Scheme 2).
 | ||
| Scheme 2 Synthesis of 2-aminopyrrole (3) and 2-aminopyridine (4) derivatives. | ||
Next, Khan and colleagues52 performed MCR of primary aliphatic amines, aldehydes, and malononitrile using 4-dimethylaminopyridine (DMAP) to produce pyridines (5) and dihydropyridines (6) at ambient temperature. The reaction of primary amines like unsubstituted benzyl amine and furan-3-ylmethanamine solely produces dihydropyridines (6), while the reaction of amines such as cyclohexylamine, cyclopropylamine, butylamine, hexadecyl amine, piperidine, pyrrolidine, cyclohexylmethanamine, and methylated benzylamines exclusively forms pyridines (5). Under the optimised reaction conditions, most of the substrate with electron-releasing or -withdrawing functionalities shows successful reaction transformations to form pyridines (5) in 62–86% yield and dihyropyridine derivatives (6) in 51–65% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of pyridine (5) and dihydropyridine derivatives (6). | ||
1,2,3-Triazoles are important five-membered heterocycles, which are extensively utilised as precursors in synthetic organic chemistry.53 Many of them exhibit potential biological activities, and these heterocycles are also important constituents of pharmaceutical drugs as well.54,55 In 2015, Wang, Ji, and co-workers56 successfully performed aerobic oxidative cycloaddition between primary aromatic amine and α-chlorotosylhydrazone to synthesise 1,4-disubstituted and 1,5-disubstituted 1,2,3-triazole derivatives (7 and 8). In the literature, rare procedures have been reported that involve the formation of target products (7) and (8) under catalyst, metal, azide, and peroxide-free reaction conditions.
Under the optimal reaction conditions, reactions flow smoothly in DMF at 50 °C to form target products with acceptable yields. However, the presence of ring-deactivating functional groups lowered the yields of products (7 and 8). On the basis of control experiments, Wang et al. proposed a mechanism in which chlorotosylhydrazone effortlessly transformed into azoalkene and left aniline hydrochloride salt behind. This azoalkene undergoes 1,4-addition with amine to give an adduct, which, upon protonation by benzenaminium, forms an intermediate. This intermediate, endures subsequent cyclization, deprotonation, and oxidation, produces 1,2,3-triazoles (Scheme 4).
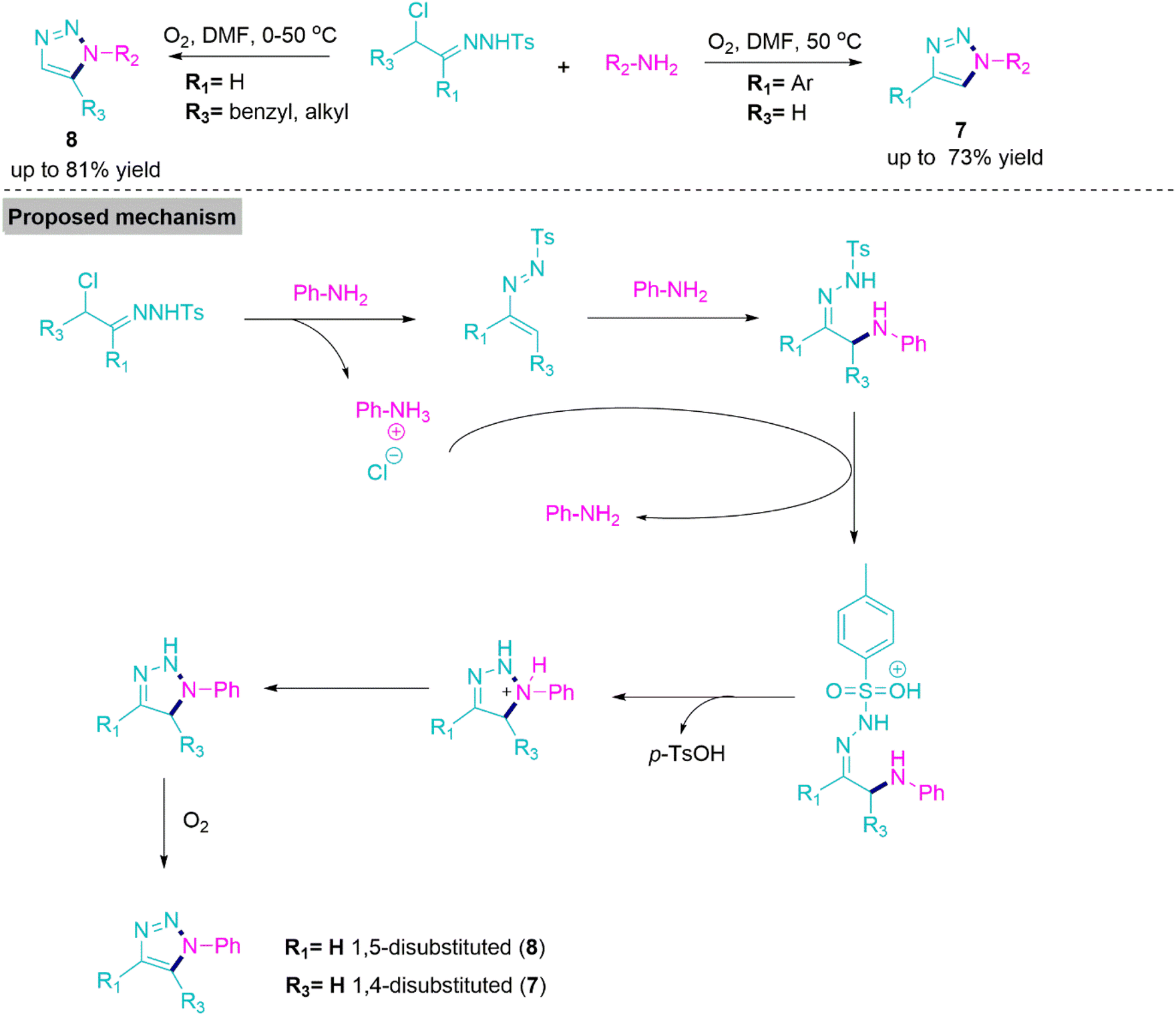 | ||
| Scheme 4 Synthesise of 1,4-disubstituted and 1,5-disubstituted 1,2,3-triazole (7) and (8) derivatives. | ||
In 2016, Li et al.57 explored a substrate-driven domino reaction for the stereoselective synthesis of chiral cyclohexane derivatives (9) and (10) using thiourea-tethered amine catalyst. The reaction of alkyl γ-nitro ketones and aryl enones using 10 mol% of 1-aryl-3-(2-(piperidin-1-yl)cyclohexyl)thiourea catalyst in ethylacetate at 50 °C temperature selectively produces enantioenriched 1,2,5,6-tetrasubstituted cyclohexane derivatives (yield up to 91%, ee up to 93%, and dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (9). While the reaction of aryl γ-nitro ketones and alkyl enones exclusively forms 1,2,4,5-tetrasubstituted cyclohexane derivatives (10) (yield up to 92%, ee up to 94%, and dr > 20:1) using a cinchona or quinine-based catalyst along with lithium salt of 1-naphthoic acid (as an additive) in THF solvent at 80 °C temperature, both reactions work efficiently with different aryl and alkyl substituents present in γ-nitro ketones and enones (Scheme 5).
:1) (9). While the reaction of aryl γ-nitro ketones and alkyl enones exclusively forms 1,2,4,5-tetrasubstituted cyclohexane derivatives (10) (yield up to 92%, ee up to 94%, and dr > 20:1) using a cinchona or quinine-based catalyst along with lithium salt of 1-naphthoic acid (as an additive) in THF solvent at 80 °C temperature, both reactions work efficiently with different aryl and alkyl substituents present in γ-nitro ketones and enones (Scheme 5).
 | ||
| Scheme 5 Synthesis of enantioenriched 1,2,5,6 and 1,2,4,5-tetrasubstituted cyclohexane derivatives (9) and (10). | ||
Amino acid derivatives are important building blocks for the preparation of N-heterocycles via multiple bonds forming reactions.58,59 Wu and colleagues60 developed 2,4,6-trisubstituted and 3,5-disubstituted pyridine derivatives (11) and (12). These modifications included a catabolism and reconstruction reaction type with the first-ever molecular iodine-mediated oxidative dissociation of inert C–N bonds. Aside from the intrinsic usefulness of this reaction as a bioinspired process, one of the key benefits of this sustainable technique is that it eliminates the usage of harmful aldehydes and rigorous reaction conditions. Furthermore, the conversion of biomass materials into chemically valuable frameworks is both environmentally and economically beneficial. 2,4,6-trisubstituted pyridines (11) was achieved in acceptable yields (up to 85%) using 1.0 equivalent of I2 in pyridine at 120 °C temperature. On the other hand, 3,5-disubstituted pyridine derivatives were successfully synthesized using 0.5 equivalent of I2, 1.0 equivalent of AcOH, in DMSO at 115 °C temperature (Scheme 6).
 | ||
| Scheme 6 Synthesis of 2,4,6-trisubstituted and 3,5-disubstituted pyridine derivatives (11) and (12). | ||
Next, Huang and co-workers61 developed a straightforward strategy for the synthesis of 5-methoxyoxazoles (13) and bicyclic imidazolines (14) through one-pot reductive addition of isocyanoacetates with extremely stable and conveniently available tert-amides or sec-amides to produce a range of 5-methoxyoxazoles (13). In contrast, the reaction of sec-lactams with isocyanoacetates offered a novel and practical method for the synthesis of a bicyclic imidazoline skeleton (14) under identical reaction parameters. The reaction shows successful transformation by stirring isocyanoacetates and tert-amides or sec-amides with 1.5 equivalent of Cp2ZrHCl, 0.2 equivalent of ZnCl2 and 2.0 equivalent of Et3N (2.0 equiv.) at room temperature for 2 h (Scheme 7). This reaction attained mild conditions, a wide range of substrates, acceptable yields, and good substrate-controlled chemoselectivity.
 | ||
| Scheme 7 Synthesis of 5-methoxyoxazole (13) and bicyclic imidazoline (14) derivatives. | ||
Isocyanides are versatile building blocks in synthetic organic chemistry owing to their distinctive structural features and reactivities.62–64 These are important substrates in well-known one-pot MCRs like Ugi and Passerini reactions. In 2017, Xu and colleagues65 developed a silver carbonate-aided domino protocol for the chemoselective annulation of o-acylaryl isocyanides and α-trifluoromethylated isocyanide derivatives. The synthesis of biologically viable trifluoromethylatedoxadiazino[3,2-a]indoles (15) in a single step from easily available starting materials is made possible by this novel reaction, which offers a quick, effective, and full atom-economic method (Scheme 8). Followed by the proton abstraction in the interim, silver carbonate reacts with a phenyl isocyanide derivative to form a silver-coordinated phenyl isocyanide derivative. Next, deprotonated trifluoromethylated isocyanide makes a nucleophilic approach to a silver-coordinated isocyanide derivative to generate an imidoyl silver adduct. This undergoes bicyclization, followed by intramolecular annulation, generating indole epoxide. Upon protonation, silver carbonate is regenerated, and subsequent epoxide ring opening takes place to produce a zwitterionic imidazole intermediate, which forms product 15.
 | ||
| Scheme 8 Synthesis of trifluoromethylatedoxadiazino[3,2-a]indoles (15). | ||
New reactivity profiles were observed in this domino transformation, including isocyanide insertion into C–O bonds, chemoselective heterodimerization of two distinct isocyanides, and indole-2,3-epoxide development and rearrangement.
One-pot transformations forming diastereoenriched products with multiple stereocenters are remarkably important. The synthesis of such products is a demanding and challenging job for the organic chemist owing to the difficult diastereocontrol of their relative configuration. In 2019, Xie and colleagues66 reported a substrate-modulated [4 + 1] cycloaddition of thioamides and sulphur ylides under a catalyst-free condition in ethanol at room temperature to produce trans-dihydrothieno[3,4-c]coumarin (16) derivatives through a Michael addition-triggered cascade process. Here, trans geometry is favoured due to steric repulsion between the benzoyl group and 2H-chromen-2-one. When the sulphur ylides were switched with acetophenones in the [4 + 1] annulation process, nucleophilic displacement was observed as the essential step, while a double π–π stacking donor–acceptor interaction stabilised the transition state that resulted in the cis-dihydrothieno[3,4-c]coumarin (16′) in acetonitrile using sodium hydroxide as catalyst (Scheme 9). In 2019, Samanta and colleagues67 developed a DBU-induced reaction of cyclic N-sulfonyl ketimines with a variety of unsaturated sulfonyl fluorides as 1,2-bielectrophiles. By a selective 3-exo-tet ring annulation using 4-methyl-N-sulfonyl ketimines as carbon donors, this novel substrate-controlled cyclization method provides a new synthetic approach for the construction of an important class of di- or trisubstituted trans-cyclopropanes (17) with an N-sulfonyl ketimine moiety in acceptable yields and outstanding diastereomeric ratio (dr up to 99:1). In contrast, a simple growing alkyl-chain from methyl to ethyl/propyl in the same system results in acceptable yields of fused dihydropyrroles (18). Samantha et al. performed a couple of control experiments and show that at first, the reaction between N-sulfonyl ketimine and, -unsaturated sulfonyl fluorides form an adduct, which upon deprotonation and subsequent 1,3-elimination reactions generates di- or trisubstituted trans-cyclopropanes (17). On the other hand, fused dihydropyrrole (18) derivatives can be achieved via a double Michael addition reaction of the adduct with unsaturated sulfonyl fluorides. This undergoes 5-exo-tet C–N bond annulation under the influence of DBU base (Scheme 10). Pyrazole-based heterocycles are recognised as significant structural motifs that are present in many pharmaceuticals, both natural and synthetic.68,69 Moreover, it has been discovered that these derivatives are frequently present in a variety of substances that have been linked to a wide range of pharmacological70,71 and agricultural properties.72,73 Moreover, they have been effectively used as ligands in coordination chemistry and as building blocks in supramolecular frameworks.
 | ||
| Scheme 9 Synthesis of thieno[3,4-c]coumarin derivatives (16). | ||
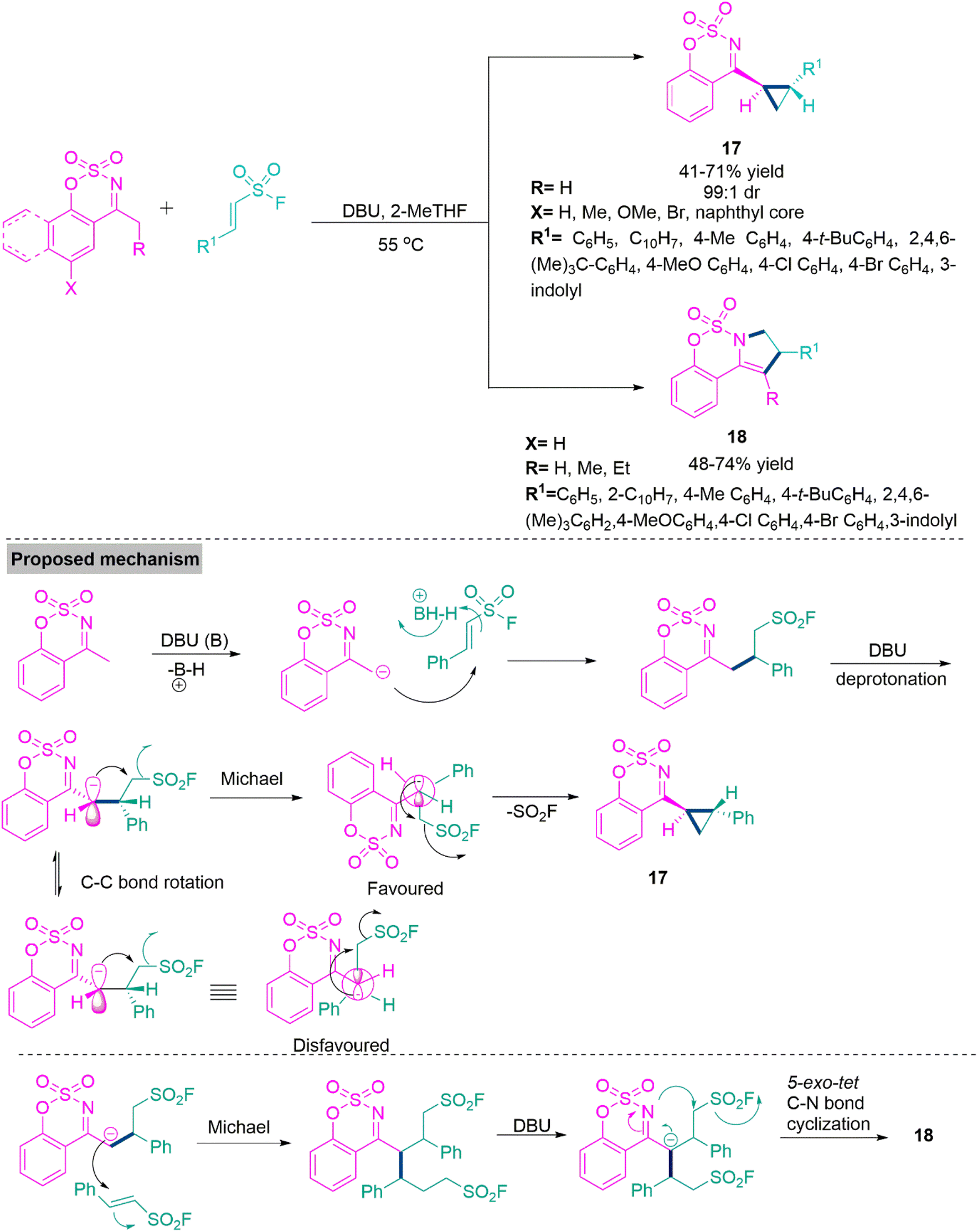 | ||
| Scheme 10 Synthesis of di- or tri-substituted trans-cyclopropanes (17) and fused dihydropyrroles (18). | ||
In 2019, He, Shang, and colleagues74 reported a straightforward chemoselective synthetic route for the synthesis of nitrogen functionalized 3-aryl-1H-pyrazole derivatives. By selecting the appropriate diazo compounds, it is possible to selectively produce both N–H insertion and Wolff rearrangement products. Under the influence of a copper catalyst, cyclic 2-diazo-1,3-diketones were used to generate N-cyclohexenone 3-aryl-1H-pyrazoles (19) while, under thermal conditions, α-carbonyl 3-aryl-1H-pyrazoles (20) could well be generated through a Wolff-rearrangement procedure with no catalyst using open chain 2-diazo-1,3-diketones. These reactions also showed strong functional group tolerance and may have been performed within acceptable reaction yields (58–93%) (Scheme 11).
 | ||
| Scheme 11 Synthesis of N-cyclohexenone (19) and α-carbonyl 3-aryl-1H-pyrazoles (20). | ||
Spiro heterocycles exhibits various pharmacological properties. Many of them known for their excellent antibacterial,75 antifungal,76 anticancer,77,78 anti-HIV79 properties, and cytotoxic agents.80 Thus, the synthesis of such heterocycles is quite demanding and challenging task for organic chemist. In 2019, Patel et al. developed an original, straightforward, and green synthetic procedure81 for the diastereoselective spiro[[1,3]dioxolo[4,5-g]quinoline-7,5′-pyrimidine] (21) derivatives through pseudo-four components and one-flask reaction of N,N-dimethylbarbituric acid, 3,4-methylenedioxyaniline, and aromatic aldehyde. The title reaction proceeds smoothly in ethanol using trimethylglycinebetaine:oxalic acid (1:1) based deep eutectic mixture (DEM) at 70 °C temperature with 1:1:1 molar proportion of N,N-dimethylbarbituric acid, 3,4-methylenedioxyaniline, and aromatic aldehyde respectively. Among all tested catalyst TMGB:oxalic acid DEM shows excellent reaction transformations and diastereoselectivities (68–94% yield of product 21, dr > 50:1). From the experimental observations it has been found that initially, aldehyde and dimethylbarbituric acid react together to form an adduct through Knoevenagel reaction. Next, this Knoevenagel adduct undergoes Michael type addition reaction with 3,4-methylenedioxyaniline and generates an adduct with free amino functionality. This adduct produces a Schiff's base like derivative using reaction with second equivalent of aldehyde and subsequent cyclization leads to generation of product 21 (Scheme 12). Next enthusiastically, Patel et al. tested the impact of substrate on multicomponent reaction of carbaldehyde, 5-aminoindazole, and barbituric acid derivative.20 Here, Patel et al. observed that MCR of barbituric acid, carbaldehyde, and 5-aminoindazole forms tetrahydro-pyrazolo[4,3-f]– pyrimido[4,5-b] quinolines (22) on the other hand tetrahydro-spiro[pyrazolo[4,3-f]quinoline]-8,5′-pyrimidine (23) (dr > 20:1) obtained by reacting N,N-barbituric acid, carbaldehyde, and 5-aminoindazole in refluxing ethanol using TMGB:oxalic acid deep eutectic mixture. Under the optimized reaction conditions both products 22 and 23 formed in 64–89% and 68–91% yields accordingly.
 | ||
| Scheme 12 Synthesis of spiro[[1,3]dioxolo[4,5-g]quinoline-7,5′-pyrimidine] (21). | ||
This methodology well tolerates electron-releasing and -withdrawing functionalities present in carbaldehydes. On the fact of experimental studies, initially, carbaldehyde reacts with barbituric acid derivative to form Knoevenagel adduct. This reacts with 5-aminoindazole and produce an amine intermediate which undergoes subsequent ring closing steps through carbon–nitrogen bond forming reaction to form product 22. In case of N,N-dimethyl barbituric acid, amine intermediate reacts with second equivalent of carbaldehyde and generates Schiff's base like benzylidene derivative. This forms spiro product 23 via intramolecular ring closing step (Scheme 13).
 | ||
| Scheme 13 Synthesis of tetrahydro-pyrazolo[4,3-f]-pyrimido[4,5-b] quinoline (22) and tetrahydro-spiro[pyrazolo[4,3-f]quinoline]-8,5′-pyrimidine (23). | ||
Next, Alizadeh and colleagues82 reported synthesis of substituted phthalimides (24) and (25) using triethyl amine promoted reaction of α,α-dicyanoalkene and 2-(4-oxo-2-thioxothiazolidin-5-ylidene)acetates. This cascade sequence involves Michael addition/intramolecular ring annulation/[1,5-H] shift/cleavage of CS2/aromatization and nucleophilic acyl substitution under ultrasound irradiation. Notably, 2-(4-oxo-2-thioxothiazolidin-5-ylidene) acetates might be produced either in situ or prior to the process. The synthesis of various derivatives under mild and environmentally friendly conditions allowed investigators to determine the substrate range of this flexible technique. In proposed mechanism, α,α-dicyanoalkene undergoes Michael addition with 2-(4-oxo-2-thioxothiazolidin-5-ylidene)acetates followed by intramolecular ring annulation produces spiro intermediate. This intermediate further follows [1,5-H]-shift, elimination of CS2 molecule and nucleophilic acyl substitution to produce substituted phthalimides (24) and (25) (Scheme 14).
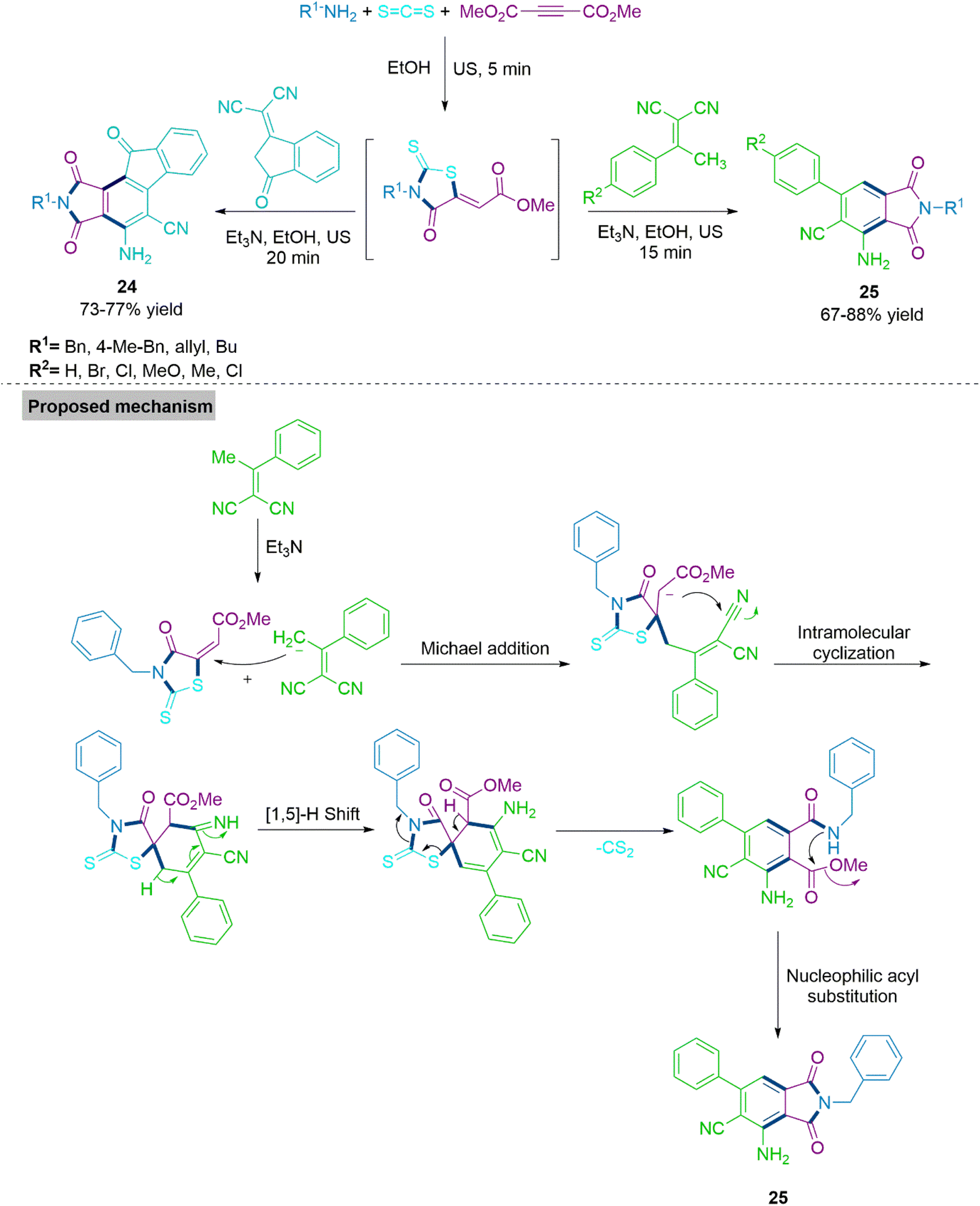 | ||
| Scheme 14 Synthesis of functionalized phthalimides derivatives (24) and (25). | ||
Thioimidates are common in biologically active compounds and crucial precursors in the synthesis of many N- and S-containing chemical compounds. Traditional techniques for producing these structural units relied on interactions between sulphur nucleophiles and substances that included C–N multiple bonds, such as imidoyl chlorides, ketenimines, or nitriles. Here, reacting carbon sources with thioamide molecules was an alternative strategy. The synthesis of aryl thioimidates via chemoselective S-arylation remains underdeveloped, despite numerous developments in S-alkylation processes to access alkyl thioimidates. The side N-arylation reaction would present a significant barrier because thioamides are bidentate-type nucleophiles that include both N–H and thio-carbonyl groups. In 2020, Tan et al.83 developed a straightforward strategy for chemoselective S-arylation reaction of thio-oxindoles with arynes to synthesize 2-(arylthio)indolenine derivative (26) under mild reaction conditions. This benign procedure has been successfully scaled to gram-scale and applied to a variety of architecturally different building blocks. The robustness screen and derivatization experiments further illustrated its capabilities in exploring chemical space for medicinal chemistry use (Scheme 15).
 | ||
| Scheme 15 Synthesis of 2-(arylthio)indolenine (26). | ||
Numerous naturally occurring substances and biologically active compounds include the pyridone core, a distinctive heterocyclic unit. Particularly, derivatives containing pyridone frequently display a wide range of biological properties, including antifungal, antibacterial, insecticidal, antiviral, anti-inflammatory, and anti-HIV effects.84–86 The pyridine skeletone has drawn a lot of attention in recent years from the scientific community, and numerous synthetic methods have been investigated to produce these compounds. In 2020, Liu, Huang and colleagues87 developed a substrate controlled functionalized pyridines (27) through three components cascade reaction of aniline, diethylacetylenedicarboxylate, and diethylethoxymethylene-malonate promoted by CsCO3 at room temperature under solvent-free conditions. This process involves Michael addition reaction/ethanol removal and intermolecular ring annulation. The chemoselectivity was modulated by substituted aniline derivative, offering functionalized 2-pyridone (27) (52–85% yield) and 4-pyridone derivatives (28) (56–72% yield) in acceptable reaction yields. From the control experimental results, the proposed mechanistic path for the synthesis of products 27 and 28 is shown in Scheme 16.
 | ||
| Scheme 16 Synthesis of 2-pyridone (27) and 4-pyridone (28) derivatives. | ||
Numerous organic substances with medicinal value contain N-arylamide and similar chemical compounds. Especially the therapeutic benefits of arylamides, including their anti-inflammatory, anti-tumour, and potassium channel activating effects, are widely recognised.88–90 As a result, organic and pharmaceutical chemists have shown a stronger interest in the synthesis of N-arylamide. The Goldberg reaction, which involves the catalyst-promoted coupling of amides and aryl halides in the presence of a base at an elevated temperature, is one of the most frequently used synthesis techniques to produce N-arylamides. In 2021, Kumar and colleagues91 explored the Cu-catalyst promoted reactions of commercially available indole-3-carbonitriles, α-cyano ketones, and diaryliodonium salts to synthesise N-arylindole-3-carboxamides (29), β-oxo amides (30) and N-arylindole-3-carbonitriles (31). Under neutral reaction conditions, a variety of N-arylindole-3-carboxamides and β-oxo amides were effectively produced with significant yields, and the addition of DIPEA base produced N-arylindole-3-carbonitriles with a completely new selectivity pattern. To check the feasibility of the reaction, Kumar et al. performed a couple of control experiments and proposed a conceivable amidation mechanism. Where the substrate reduces the Cu-catalyst and generates an active Cu(I)OTf catalyst. In the second step, oxidative addition by diaryliodonium triflate produces Cu(III) intermediate. This undergoes a ligand exchange reaction with nitrile, followed by the addition of a water molecule and reductive elimination, to furnish amide derivatives (Scheme 17).
 | ||
| Scheme 17 Synthesis of amide derivatives (29-31). | ||
Chalcogens are exceptional substances with important applications in the synthesis of organic compounds92 (as electrophiles, nucleophiles, catalyst, and ligands) life sciences,93 pharmaceutical chemistry,94 and materials science.95 As a result of its widespread use across many scientific domains, green techniques for the synthesis of chalcogens comprising heterocyclic molecules have drawn significant interest from chemists. The versatile structural scaffold pyrrolidine, which can be found in many natural products and has a wide range of pharmacological effects.
Improving the reactivity of native molecules or medicinal products may be possible by adding the chalcogen functionality to the pyrrolidine skeleton (32 and 33). Several efforts have been done up to this point to look into and create innovative methods for creating more valuable chalcogen substituted organic compounds. In 2020, Wang and colleagues96 developed an additive and oxidant free chemoselective procedure which involves radical ring annulation reaction of 1,6-enynes and chalcogens. This procedure shows reactivity of non-activated 1,6-enynes to alkenes and alkynes. Under optimized reaction conditions, Wang et al. tested a variety of substrates and observed that it flows well with internal and terminal alkyne. The successful reaction transformation involves formation of free radicals from the Se-phenyl 4-methylbenzenesulfonoselenoate. Newly generated aryl sulphonyl radical reacts with 1,6-enyne to form an intermediate which undergoes 5-exo-dig cascade annulation followed by Se-phenyl free radical capture generates product 32. On the other hand, free radical addition on alkene site by thiophenol radical produces an alkyl radical intermediate followed by 5-exo-dig cascade ring annulation and hydrogen atom transfer furnishes product 33 (Scheme 18).
 | ||
| Scheme 18 Synthesis of pyrrolidine derivatives (32) and (33). | ||
3. Solvent modulated chemoselectivity in one-pot reactions
Solvent-controlled chemoselectivity in organic reactions is a significant aspect in the formation of complex molecular structures. In these cases, changing the solvents results in the formation of two different molecular entities without affecting the other reaction parameters. In 2014, Ji and colleagues97 reported a solvent-driven synthesis of spiroindoline (34) and functionalized pyrrole derivatives (35). The reaction of gem-diactivated alkene with 2-isocyanoethylindoles in refluxing ethanol selectively forms product 34 in 35–92% yield and dr > 20:1. While the three-component reaction of 2-isocyanoethylindoles, gem-diactivated alkene, and secondary amine in refluxing acetonitrile exclusively produces product 35 in moderate yields (22–59%). In the conceived mechanism, first isocyanide makes a nucleophilic approach on gem-diactivated alkene to generate an active intermediate, which undergoes intramolecular ring annulation to generate product 34. The reaction of this active intermediate with secondary amine followed by subsequent cyclization forms product 35 (Scheme 19).
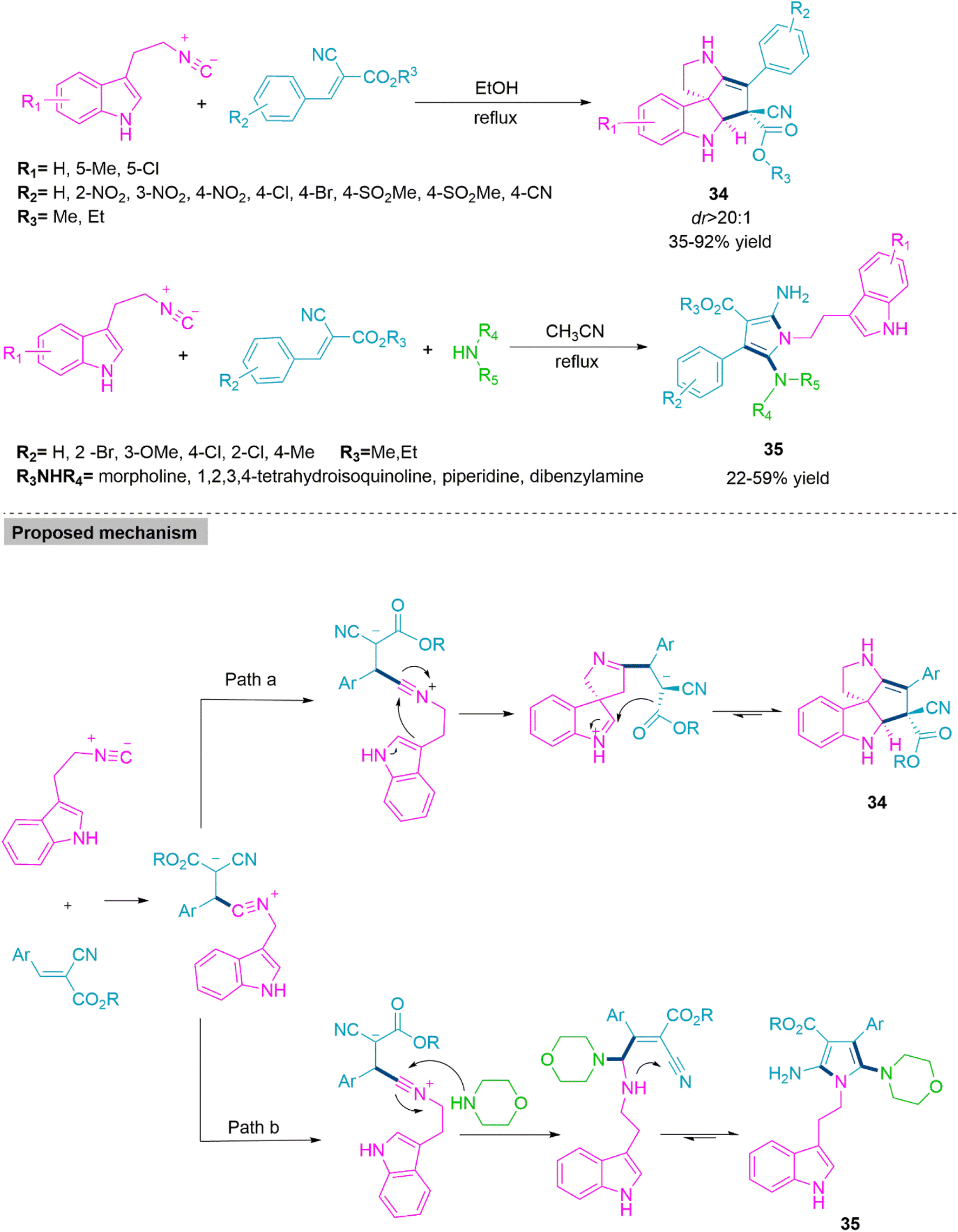 | ||
| Scheme 19 Synthesis of spiroindoline (34) and functionalized pyrrole derivatives (35). | ||
Uracil based compounds are important pharmacophore in many bioactive compounds and pharmaceuticals.98 It is not surprising that various studies have been conducted on the selective synthesis of 5- and 6-substituted uracil derivatives. Several approaches based on dearomatization, electrophilic displacement, and condensation have been developed; though, these strategies are restricted to a single kind of substitution, necessitate the preassembly of the uracil framework before the production of the desired analogues, or proceed under challenging conditions.99,100 Direct conversion of hemiaminals derived from barbituric acids to diverse products from the same synthetic precursor would be a potent stratagem for expanding the range of accessible uracil analogues. Nevertheless, nowadays such hemiaminals were unavailable due to a lack of procedures for the monoreduction of barbituric acids. Thus Szostak, Procter, and colleagues101 established a SmI2-mediated synthesis of uracil derivatives 36 and 37 in 62–85% and 76–89% yield. Here, chemoselective switching between two reaction pathways by an alcohol co-solvent effect is shown. In order to raise the redox potential of Sm(II), coordinating solvents are used in the procedure, which leads to either a chemoselective reduction using SmI2-H2O or a 1,2-migration through in situ formed N-acyliminium ions using SmI2-ethylene glycol. This study takes advantage of the benign conditions of SmI2-mediated monoreduction of barbituric acids and provides a promising procedure for the synthesis of uracil derivatives of biological interest. The proposed reaction mechanism using kinetic isotope effects is show in Scheme 20.
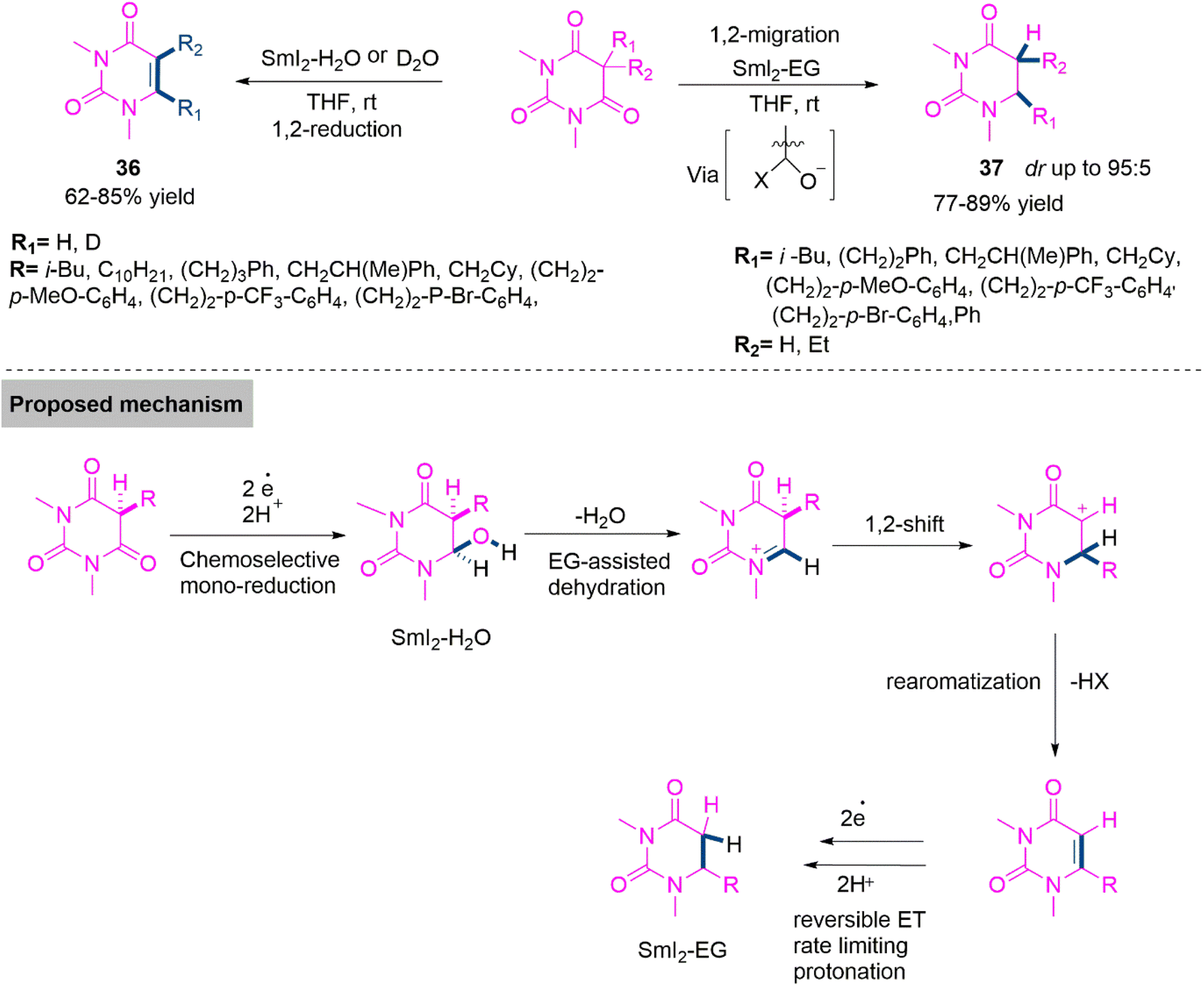 | ||
| Scheme 20 Synthesis of uracil derivatives (36) and (37). | ||
Multicomponent domino reactions (MDRs) have become widely used in recent years to synthesise a variety of complex molecules by consecutively combining three or more simple substrates.102,103 Most reported MDRs followed a linear synthetic route. The derivatives of pyrrolo[1,2-c]imidazole have numerous applications in pharmaceutical104 and agrochemical chemistry.105 Nevertheless, only a few cases of their synthesis have been described so far, which involved the formation of a pyrrolidine ring from the confined precursors imidazolidine-2,4-dione or 2-thioxoimidazolidin-4-one.106,107 Significantly substituted pyrrolothiazoles serve as the structural base for many alkaloids and have been conferred with a variety of pharmacological potencies.108 In addition, drug development109 and agrochemical applications110 benefit from the use of pyrrolo[1,2-c]thiazole derivatives. In 2015, Wu and colleagues111 reported a solvent-controlled MDR procedure involving nitriles, arylglyoxal monohydrates, and thourea. In refluxing acetic acid, Wu et al. achieved pyrrolo[1,2-c]imidazole derivative (38) on the other hand, reacting these substrates in refluxing ethanol generates pyrrolo[1,2-c]thiazoles (39). To get a clear idea about mechanistic paths, Wu et al. performed several control experiments and observed that aryl glyoxal monohydrate initially reacts with malononitrile and thiourea to form gem-diactivated olefin and 5-phenyl-2-thioxoimidazolidin-4-one. In the next step, newly generated thioxoimidazolidinone makes a nucleophilic attack on gem-diactivated olefin and follows subsequent cyclization and tautomerization to produce product 38. While, in refluxing ethanol, 2-aminothiazol-5(4H)-one is generated, which further reacts with gem-diactivated olefin and follows subsequent cyclization and tautomerization to yield product 39 (Scheme 21).
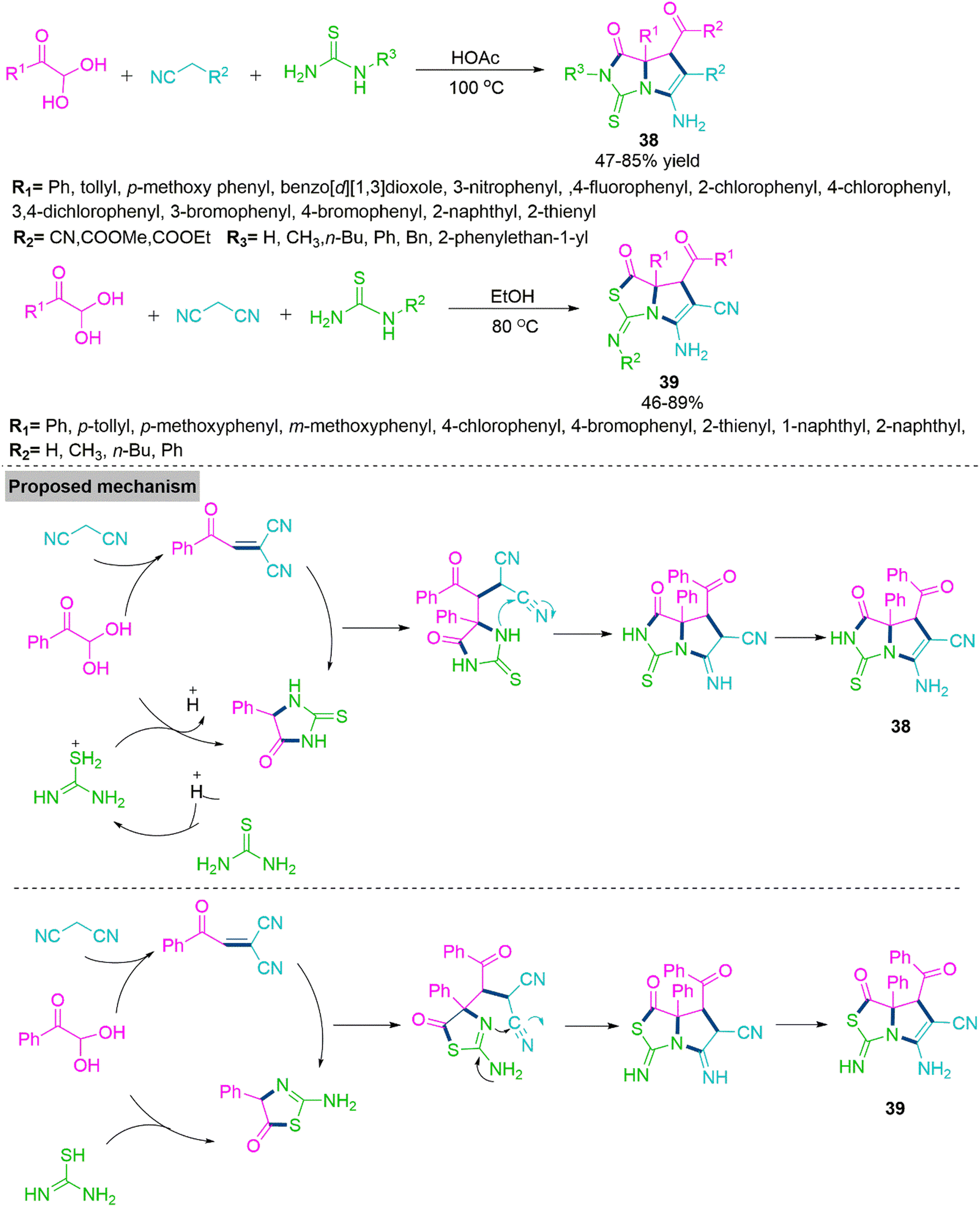 | ||
| Scheme 21 Synthesis of pyrrolo[1,2-c]imidazole (38) and pyrrolo[1,2-c]thiazoles (39). | ||
Nitrogen bearing fused heterocycles especially quinoline derivatives shows applications in medicinal chemistry,112 optoelectrical materials,113 and in synthetic organic chemistry.114 Functionalized quinoline compounds have recently drawn the interest of an increasing number of chemical researchers. In 2019, Quan and co-workers115 developed a K2CO3 promoted chemoselective C–S bond forming reaction for the synthesis of sulphur functionalized quinoline derivatives (40) (62–93% yield) in ethanol by reacting quinoline-2-thione and acrylonitrile. While, use of dioxane as a reaction media produces disulphide derivatives of quinolines (41) (63–82% yield) by chemoselective S–S bond formation. In suggested mechanism, a sulphur anion intermediate generated by proton abstraction from enolized thiol derivative makes nucleophilic addition on acrylonitrile followed by proton abstraction forms product 40. While enolized thiol derivative in presence of atmospheric oxygen forms S–S bond to give disulphide derivatives 41 (Scheme 22).
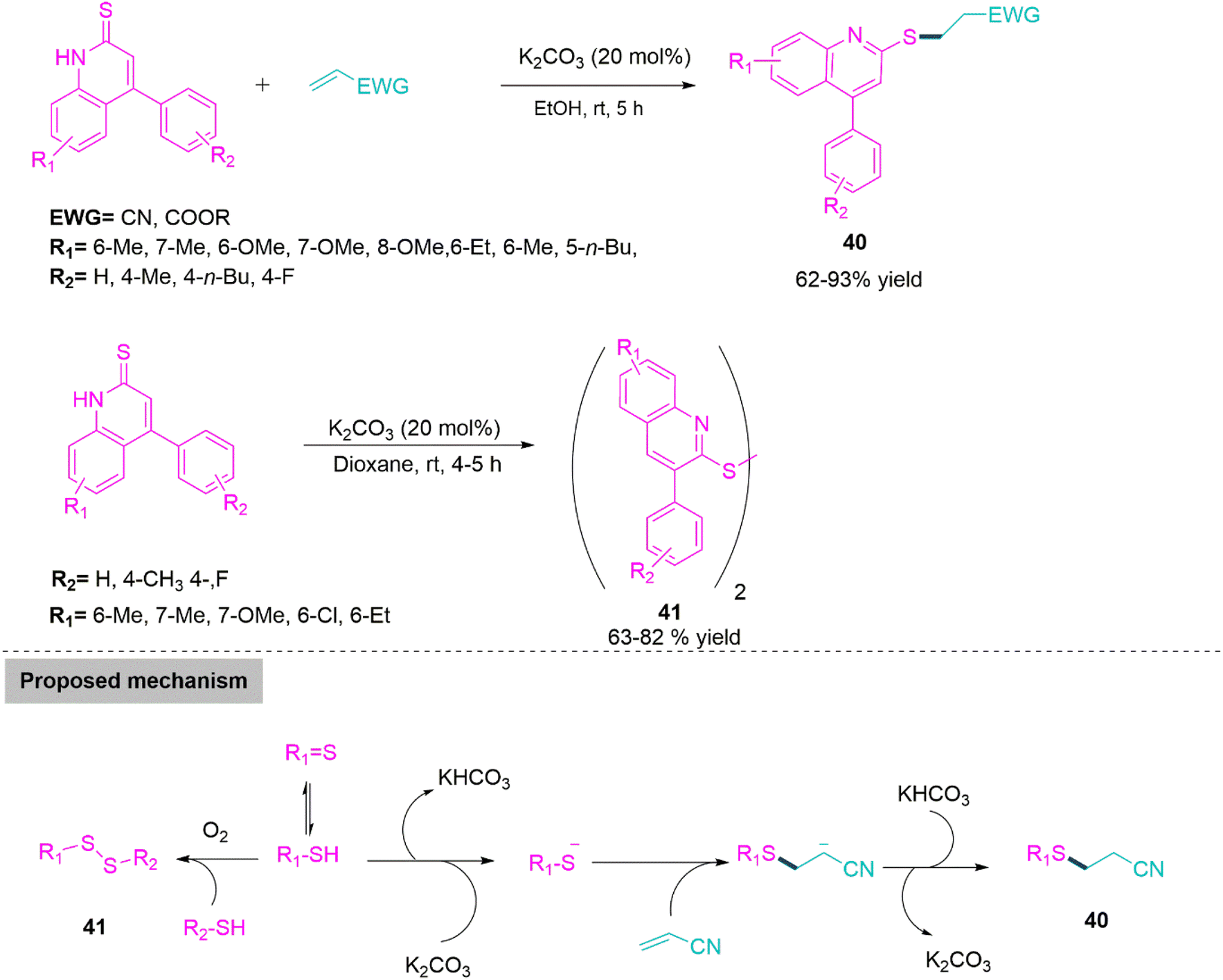 | ||
| Scheme 22 Synthesis of sulphur functionalized quinoline (40) and disulphide derivatives of quinoline (41). | ||
Barbituric acid derived spiro-heterocycles are basic constituent of many bioactive compounds, natural products. 116,117 Many of them are recognized as an inhibitor of matrix metalloproteinases (MMP),118 antitumor,119 anti-bacterial,120 inhibitors of α-glucosidase,121 dihydro-orotatedehydrogenase.122 In 2019, Yan and colleagues123 established a one-flask method involving different reaction conditions such as DBU-CH2Cl2 and K2CO3-EtOAc, a reaction of barbiturate-based olefins with acetylacetone chemoselectively produce spirodihydrofuryl barbiturates (42) (57–94% yield) and spirocyclopropylbarbiturates (43) (53–88% yield). Under optimized reaction conditions a range of barbiturate-fused spirodihydrofuran and spirocyclopropane derivatives were prepared rapidly and efficiently using this NBS-mediated and Michael addition triggered ring annulation, with acceptable yields. In proposed reaction mechanism acetylacetone makes Michael addition on benzylidene derivative of N,N-dimethylbarbituric acid derivative produces an intermediate. Next, this intermediate upon bromination by NBS generates a key reactive bromo intermediate which undergoes intramolecular ring closure step with help of DBU in CH2Cl2 to form product 42. While in the presence of K2CO3 active C–H carbon undergoes proton abstraction and generate enol form which makes intramolecular ring closure by direct displacement of leaving group (Br) generates 43 (Scheme 23).
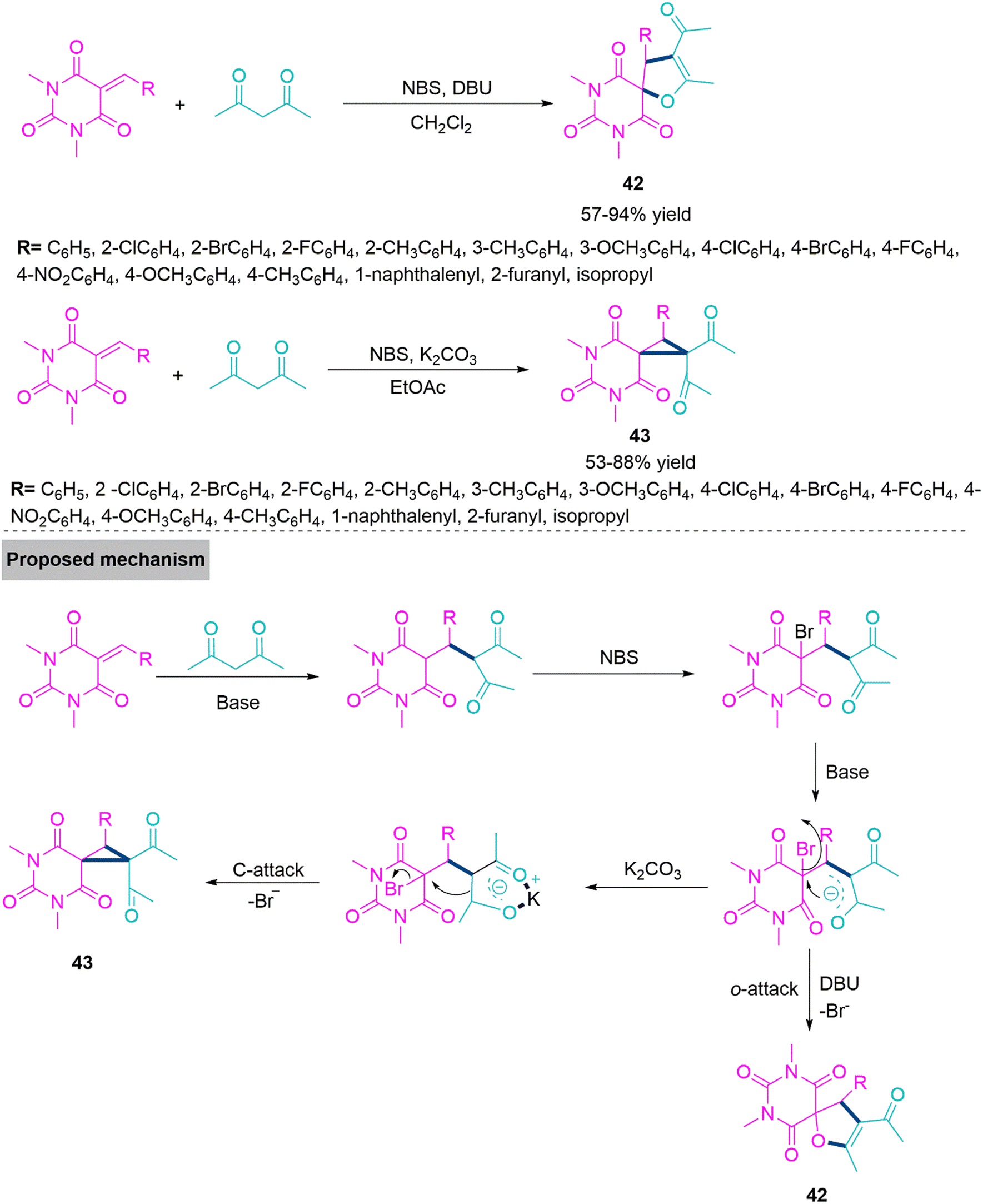 | ||
| Scheme 23 Synthesis of spirocyclopropyl barbiturates (42) and spirodihydrofuryl barbiturates (43). | ||
In organic synthesis and medicinal chemistry, convergent synthesis has been proven to be a reliable method for creating structurally different molecules from the same basic components. Understanding the reactivity of intermediates and precursors is crucial to the success of this method, which also benefits from fine-tuning the reaction conditions like solvent, catalyst, ligand, temperature, etc. 124 A variety of bioactive chemicals and related medications, including dup-697, raltitrexed, raloxifene, clopidogrel, and duloxetine contain the extremely important class of heterocyclic compounds known as thiophenes.125 Additionally, as adaptable building blocks for the development of liquid crystals, organic semiconductors, solar cells, light-emitting diodes etc. 126 These frameworks are essential in the field of materials science. Considering their significance, the discovery of new sustainable techniques for the manufacture of structurally varied thiophenes remains an essential research area in modern chemical synthesis. In 2020, Jafarpour and colleagues127 established a chemodivergent reduction versus cascade ring annulation controlled by solvent. Here, the reaction of α,β-unsaturated carbonyls with elemental sulphur using KOH in refluxing DMF produces aryl ketones (44) by the reduction of α,β-unsaturated carbonyls. On the other hand, switching DMF by DMSO produces thiophene derivatives (45). Mechanistic evaluations revealed that each procedure followed radical routes, with two hydrogen atoms originating from water throughout the reduction process (Scheme 24).
 | ||
| Scheme 24 Synthesis of aryl ketones (44) and thiophenes (45 and 45′). | ||
With a variety of biological functions, spiro-γ-lactam is a preferred scaffold that is frequently found in both natural and manufactured products. 128,129 Many pharmacologically potent alkaloids have been found to include the spiro-γ-lactam structural core. 130,131 Chemists have paid close attention to developing procedures for the synthesis of this spiro motif due to its unique three-dimensional structure and biological activity. The scaffolds that resulted increased the diversity of libraries for physiological assessment. In 2021, Li, Xu, and co-workers132 developed a sequential, solvent-driven chemoselective procedure for the synthesis of spiro-γ-lactams (46) (64–83% yield) and (47) (59–81% yield) by base promoted Michael addition reactions with weak amide nucleophiles. To generalise the reaction scope of this Ugi-cascade reaction sequence, Li et al. switch the solvent from ethanol to DMF and produce a new series of bioactive oxidised chromone derivatives. In the proposed mechanism, a unique carbon-nitrogen and carbon–carbon bond formation process on the chromone nucleus is described. Through the successive synthesis of a carbon–carbon bond and the development of a carbon–nitrogen bond between an amide and an olefin, generates the new diastereoselective spiro-γ-lactam pyrroline heterocycles (Scheme 25).
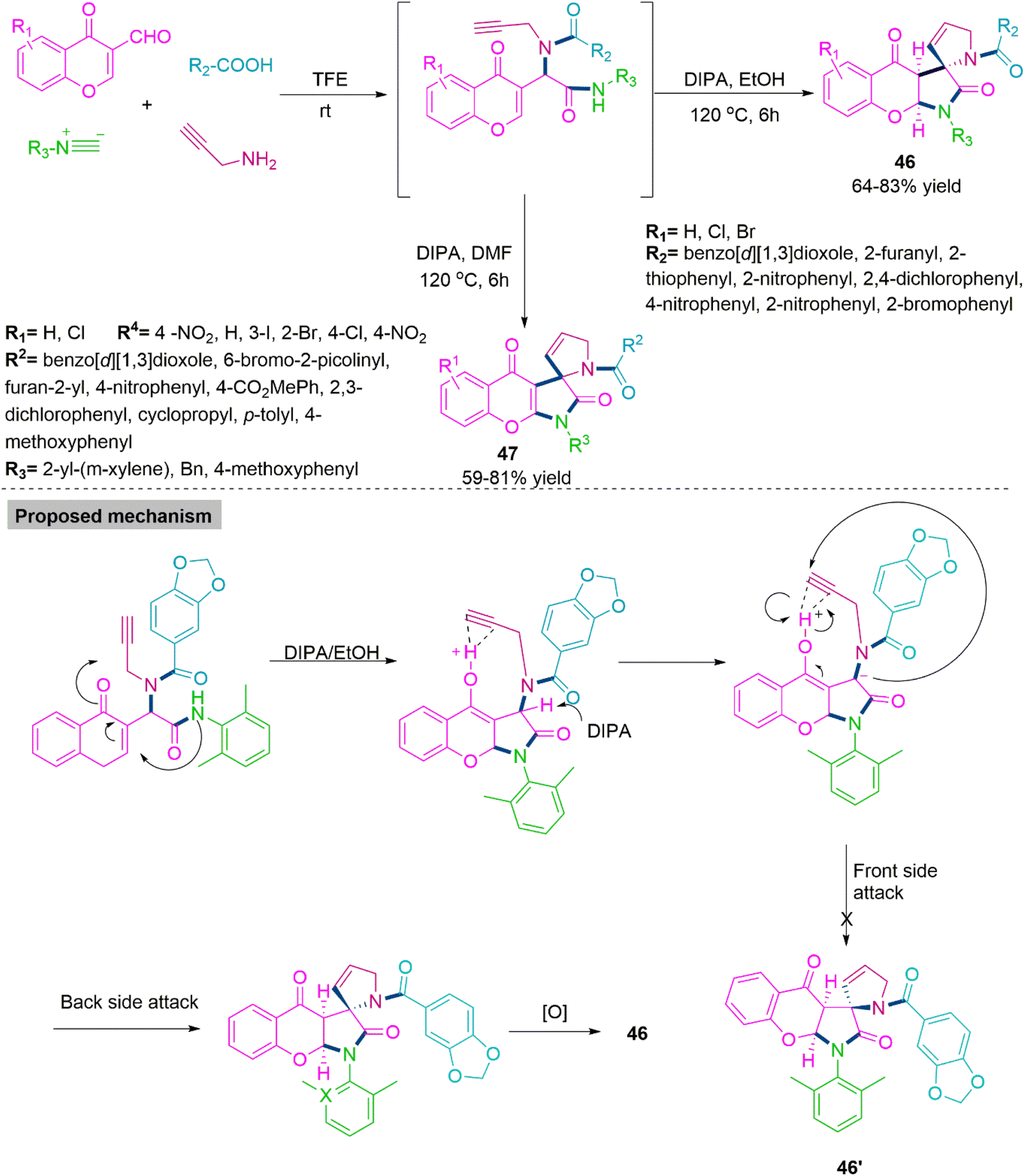 | ||
| Scheme 25 Synthesis of spiro-γ-lactams (46) and (47). | ||
One of the most diverse groups of heterocycles, which are frequently found in a variety of medicines, agrochemicals, and biologically active compounds, is sulphur-based heterocycles. 133,134 Penams with a five-membered thiazolidine ring were the first beta-lactam antibiotics licenced by the FDA.133 Anticonvulsant properties are exhibited by thiopen-2-ones. Next, diltiazem, an inhibitor of calcium channels with a seven-member ring, is prescribed to treat hypertension. In recent decades, the significance of S-heterocycles in the development of novel drug programmes has resulted in increased interest in S-heterocycle production and the study of their chemical and physiological behaviour.135 As a result, significant effort has been made in the formation of S-heterocycles via carbon–sulphur bond formation procedures or alternative reactions employing S-based synthons.136,137 Notwithstanding enormous achievements, there is still a significant need for the development of novel, efficient, and selective approaches towards S-heterocycles with distinctive structures.
Thus, in 2021, Yu and co-workers138 developed a solvent driven and additive-free synthesis of 3-difluoroalkyl-functionalized 3-hydroxylbenzothiophen-2-ones (48), 2-difluoroalkyl-substituted 2-hydroxylbenzothiophen-3-one (49) and seven membered benzo[d][1,3]-oxathiepin-4(5H)-ones (50). The reaction of gem-difluoroenoxysilane and benzo[b]thiophene-2,3-dione in methanol at 30 °C temperature forms product 48 (yield up to 79%) and 49 (yield up to 41%). Whereas, switching the solvent from methanol to water in this reaction selectively yield product 50 (yield up to 81%). Both reactions procced smoothly under the optimized reaction parameters and tolerates both electron-donating and -releasing functional groups well (Scheme 26).
 | ||
| Scheme 26 Synthesis of difluoromethylene-based S-heterocycle derivatives (48-50). | ||
Chromones are privileged class of heterocycles that present in many natural products, bioactive products, and pharmaceuticals.139 Teimouri and colleagues140 reported water oriented three-component reaction of alkyl isocanides, open chain anhydrides of carboxylic acid, and 3-formylchromones in CH2Cl2. When title MCR performed in dry CH2Cl2 it produces (acyloxymethylidene)chromonyl-furochromone derivatives (51) (60–81%yield). Remarkably when Teimouri et al. used water contained CH2Cl2 in the title MCR, it exclusively forms 2-alkylamido-3-(acyloxymethylidene)chromones (52) (63–82% yield). From the experimental results, a mechanistic path for this solvent-driven procedure in which 3-formyl chromone derivative undergoes o-acylation with anhydride and generates o-acylated 3-formyl chromone derivative. This upon Friedel–Crafts alkylation reaction with alkylaminofurochromone (generated by [4 + 1] cycloaddition between 3-formyl chromone and alkyl isocyanides) forms product 51. Whereas in moisturised CH2Cl2 reaction between o-acylated 3-formyl chromone intermediate and alkyl isocyanide selectively produce product 52 (Scheme 27).
 | ||
| Scheme 27 Synthesis of (acyloxymethylidene)chromonyl-furochromones (51) and 2-alkylamido-3-(acyloxymethylidene) chromones (52). | ||
4. Catalyst modulated chemoselectivity in one-pot reactions
Catalytic divergent multicomponent reactions describe the formation of two or more different molecular architectures from the different catalytic chemical process that involve the same starting substrates under adjustable reaction conditions. Under distinct catalytic conditions, different substrates have been identified to work with flexible reactivity, resulting in the synthesis of diverse products. In 2014, Xu and colleagues141 reported a base driven ring annulation which involves the use of 2-chloropyridine promoted reaction of ethyl malonyl chloride and N-benzylidene benzylamine to form trans-β-lactam-3-carboxylates derivatives (53) (yield up to 93%) via [2 + 2] cycloaddition. On the other hand, use of N-methylimidazole led to formation of 2,3-dihydro-1,3-oxazin-4-one derivatives (54) (yield up to 99%) through a [2 + 2 + 2] cycloaddition accomplished by two ketene molecules produced from malonyl chloride and one imine molecule. Under optimal reaction conditions both reactions flow smoothly and bear a variety of substitution present in substrate molecules. To give clear idea about this base-controlled cyclization reaction, Xu et al. proposed a mechanistic path showing the formation of ketene from the reaction of ethyl malonyl chloride with base. This in situ formed ketene undergoes [2 + 2] conrotatory ring annulation with N-benzylidenebenzylamine in presence of 2-chloropyridine base to give product 53. Where as in the presence of base like N-methylimidazole furnishes product 54 (Scheme 28).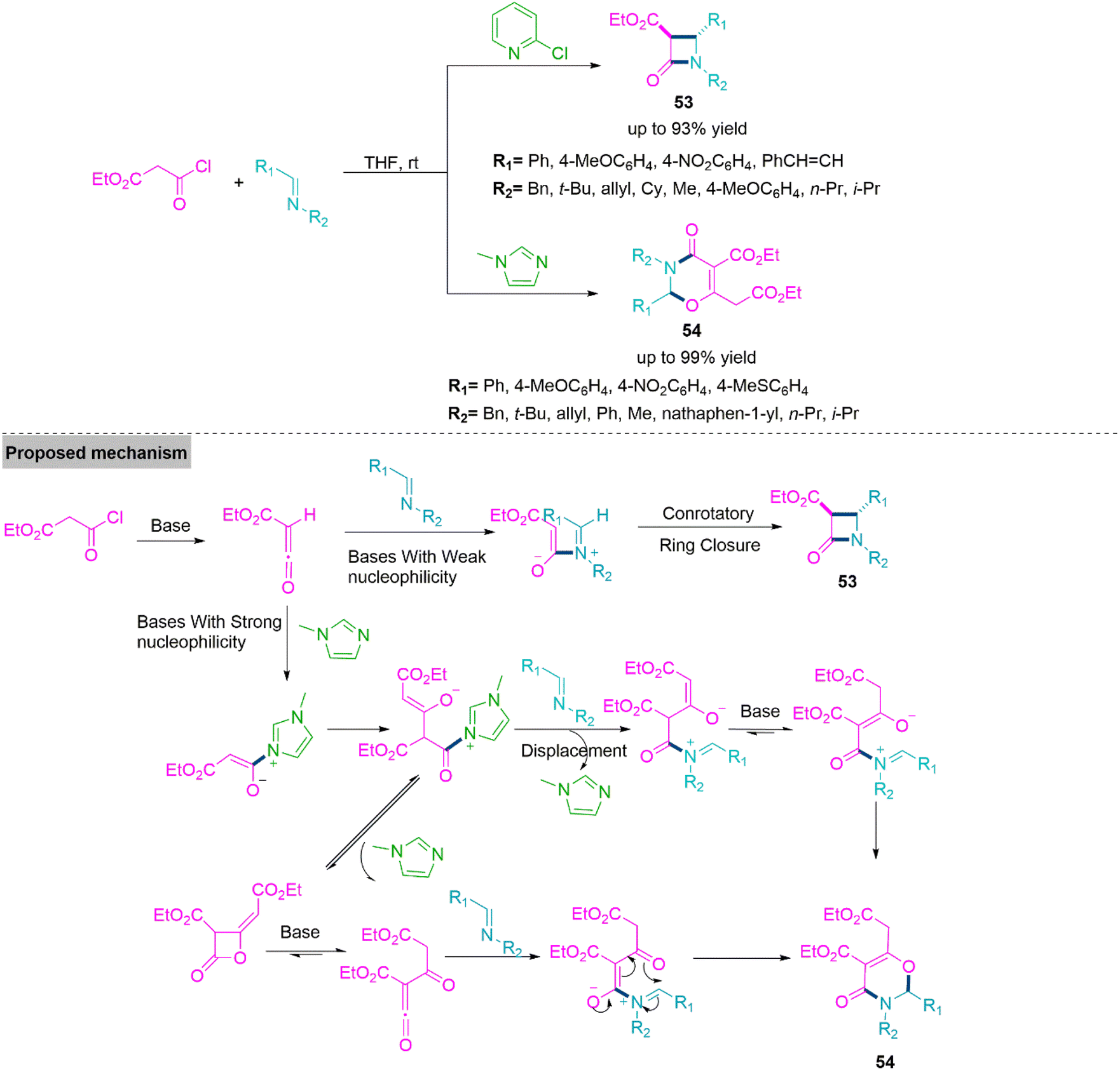 | ||
| Scheme 28 Synthesis of trans-β-lactam-3-carboxylates (53) and 2,3-dihydro-1,3-oxazin-4-one (54). | ||
Next, Zhang and colleagues142 developed a procedure that was associated with catalyst-oriented carbon–sulphur bond formation for the synthesis of C2- and C3-sulphonyl indole derivatives using indole and sodium sulphate. The C2-sulphonyl indole (55) was produced solely using 10 mol% ammonium iodide (a catalyst) and one equivalent of tert-butyl perbenzoate as an additive in refluxing acetic acid. While C3-sulphonyl indole (56) derivatives were synthesised using 20 mol% of CuCl (a catalyst), 10 mol% of KI (an additive), and 10 mol% of 1,10-phenantroline (a ligand). Both reactions work flawlessly under established reaction conditions and tolerate a broad substrate scope. Subsequently, Zhang et al. investigated the reaction mechanism on the basis of experimental evidence and found that molecular oxygen played an important role in the CuI-catalysed reaction as it oxidised the Cu(I) into Cu(II) and formed a -coordinated copper–indole complex. Next electrophilic addition eliminates the HX molecule to form a new Cu(II) complex. Then anion exchange between this complex and sodium sulfinate takes place. Finally, the reaction cycle ends with the help of molecular oxygen (Scheme 29).
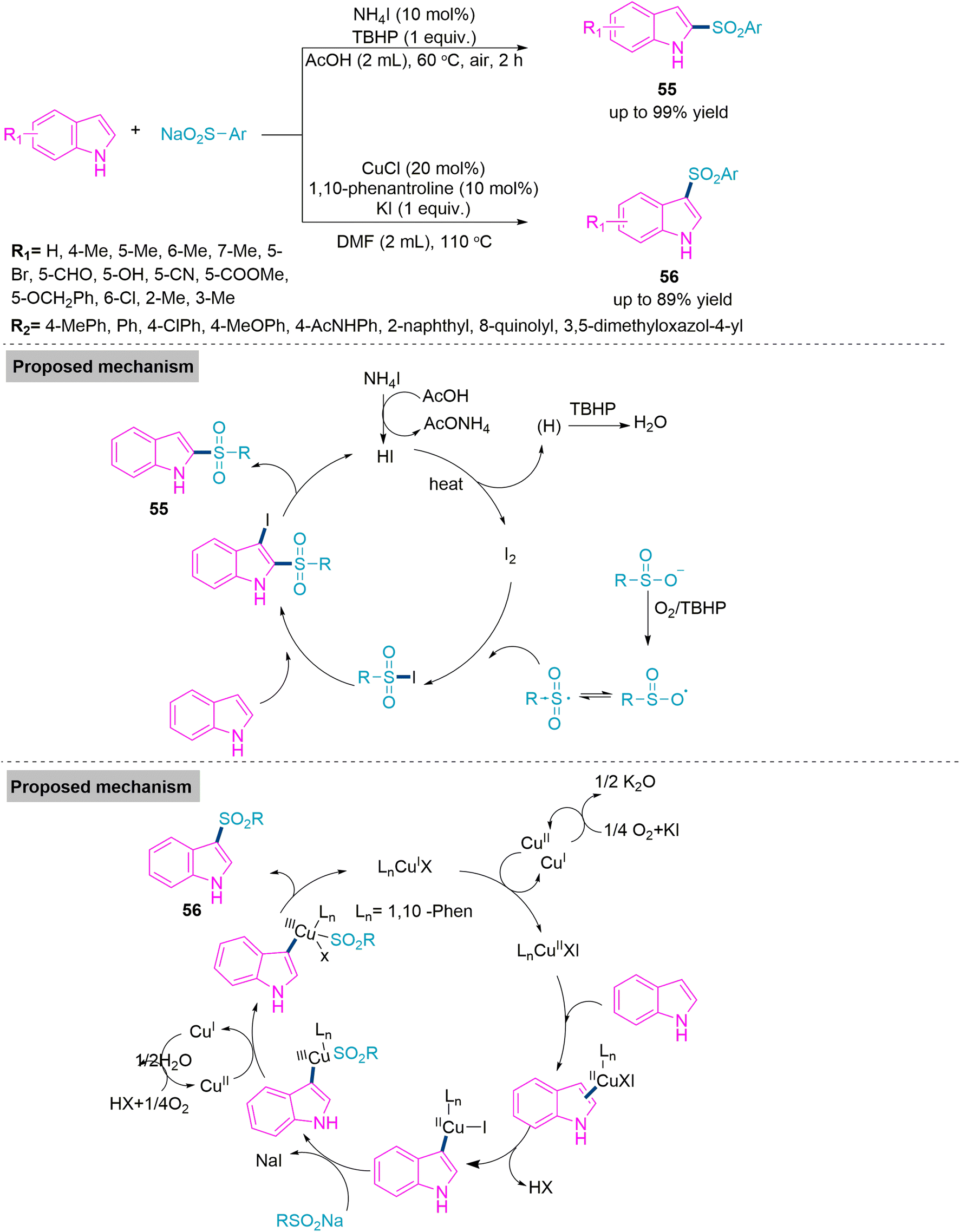 | ||
| Scheme 29 Synthesis of C2- and C3-sulphonyl indole derivatives (55-56). | ||
Nitrogen containing aromatic compounds are abundant in nature and many of them are used as significant building block in organic synthesis.143 Moreover, these derivatives are key intermediates pharmaceuticals, agrochemicals, and pigments.144 Functionalized pyrrole derivatives are also crucial structural units in naturally occurring products, bioactive compounds, and functional materials. 145,146 There has been a lot of interest in the development of viable and efficient processes for the formation of arylamines and pyrrole derivatives. In 2016, Li et al.147 developed a solvent and metal controlled chemodivergent synthesis of 1-naphthylamine (57) and indeno[1,2-c]pyrrolone (58) through cycloisomerization of (o-alkynyl)phenyl enaminones. Significantly, product 57 synthesized using AgNO3 catalyst in refluxing DMF. Whereas product 58 was achieved in high yield using CuCl and benzoyl peroxide catalysis. Under ideal reaction parameters both procedures were tested with enaminones having various substituents of different electronic properties and promising results were achieved. Conceivable mechanistic path for both products were shown in Scheme 30.
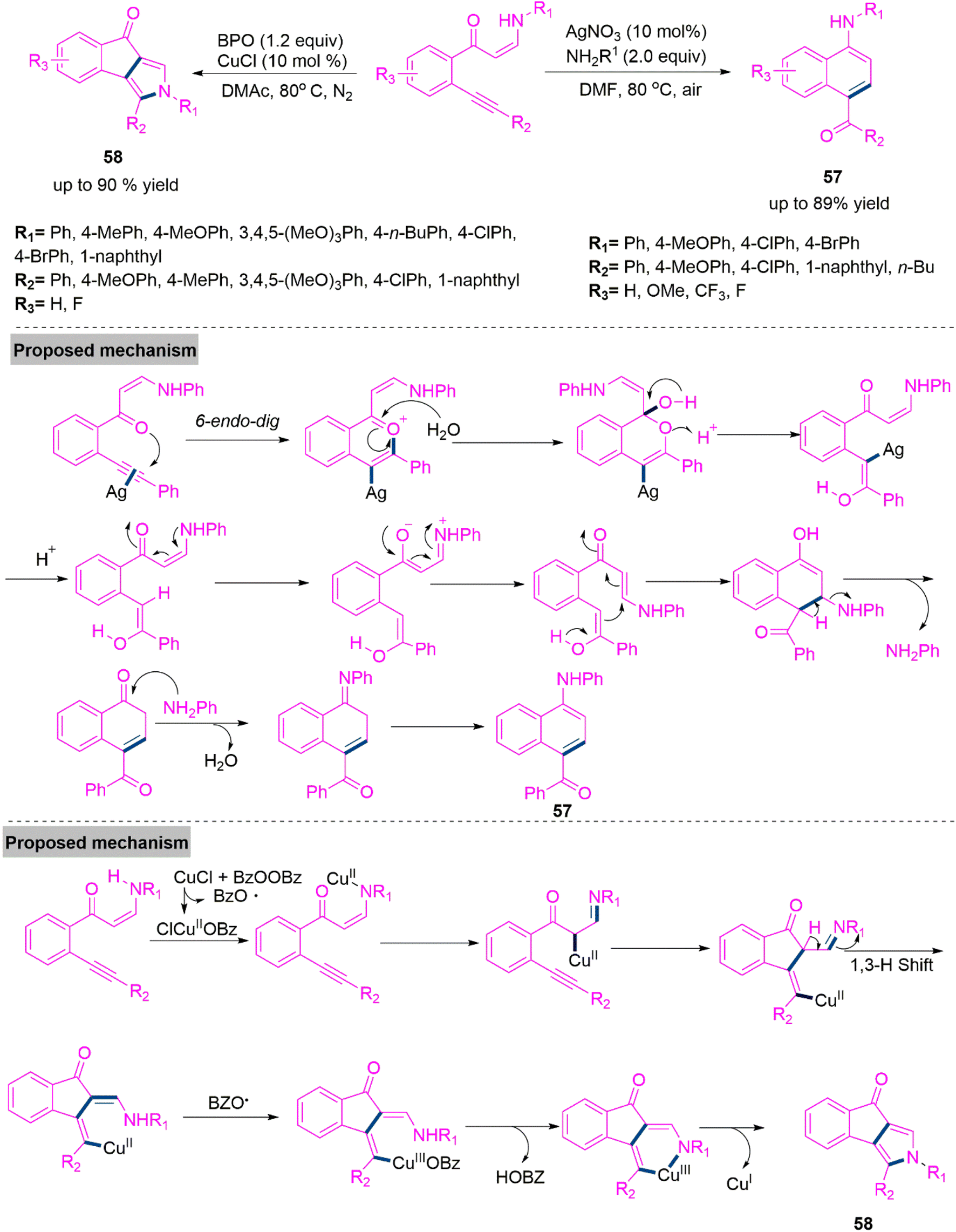 | ||
| Scheme 30 Synthesis of 1-naphthylamine (57) and indeno[1,2-c]pyrrolone (58). | ||
In the synthesis of biologically active heterocyclic molecules, the highly strained 2H-azirines are significant precursors that have found widespread use.148 Vinyl nitrenes compounds are frequently synthesised via a transition metal-initiated ring–opening reaction of 2H-azirines. Such a transition was thought to occur by the intramolecular shift of an alkenyl nitrene to an alkyne in the case of the gold-catalysed conversion of 2-propargyl-2H-azirine analogues to functionalized pyridines.149 In 2018, Chen, Wu, and colleagues150 established Pd- and Rh-based metal catalysts promoted the synthesis of α-aminooxime esters (59) and (60) from a one-pot reaction of N-(acyloxy)amides and 2H-azirines. In this reaction, regioselectivity is solely driven by metal catalysts, and two different α-aminooxime esters were produced in high yields. The room temperature reaction of 2H-azirine and N-(acyloxy)amide in dioxane using Pd-catalyst and CsOAc forms product 59 (yield up to 95%). Whereas, use of the Rh catalyst and DCE solvent led to the formation of product 60 (yield up to 99%). In the proposed catalytic cycle, first, a palladacyclic intermediate would be generated via trans-annulation and the oxidative addition of 2H-azirine to Pd(0). Next, this intermediate undergoes oxidative addition of the N–O bond, and subsequent nitrene group insertion forms a heptacyclic complex of Pd(II). In the last stage of the catalyst cycle, the base promotes acyl exchange and subsequent reductive elimination to furnish product 59. Whereas in the case of the Rh(III) catalyst, oxidative addition of the N–O bond takes place to form the Rh(V) complex. Next, this complex reacts with 2H-azirine and undergoes consecutive rearrangement to form Rh-(nitrene) amidate. This follows nitrene insertion in the Rh–O bond, 1,3-mirgartion, and reductive elimination events to generate product 60 (Scheme 31). Because of their widespread use in biomedicinally natural molecules and medicines, benzoxepines and benzofurans, which are major seven- and five-membered oxygen-containing heterocyclic compounds, serve as vital structural units. 151,152 As a result, numerous techniques for producing benzoxepines or benzofurans via intramolecular or intermolecular cyclization, respectively, from commercially available materials have been established.153–157 Although the described approaches are excellent, it is still a difficult but appealing challenge to design an effective and metal-free method for the diversity-oriented construction of both derivatives. In 2019, a base switched reaction of ynone and 2-bromophenylacetonitrile for the synthesis of 5-cyanobenzoxepines (61) and benzofuro[2,3-b]pyridines (62) reported by Wang and colleagues.158 A series of 5-cyanobenzoxepines (61) (yield up to 96%) were synthesized by Li-OtBu base promoted formal [4 + 3] cycloaddition between ynone and 2-bromophenylacetonitrile in refluxing N-methyl-2-pyrrolidone. In contrast, product benzofuro[2,3-b]pyridines (62) (yield up to 84%) were obtained through an unexpected O-rearrangement reaction using DBU in refluxing DMSO. Under optimized reaction conditions both procedures flow smoothly and generates the target products in moderate to high yield. From the foundation of labelling experiments, Wang et al. proposed a conceivable mechanism in which initially nucleophilic addition of 2-bromophenylacetonitrile on ynone in presence of Li-OtBu base generates an addition intermediate which undergo intramolecular SNAr process to form product 61.
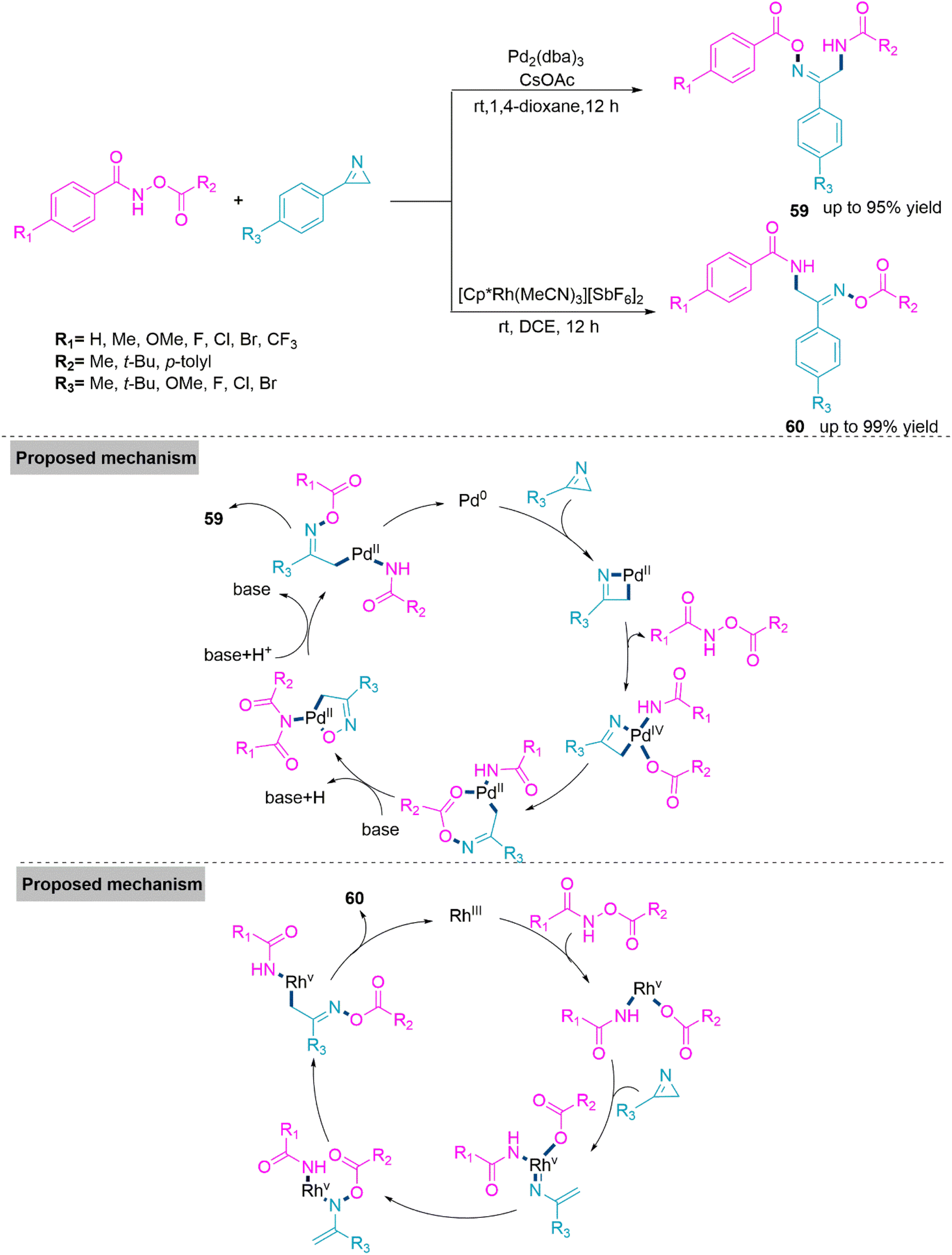 | ||
| Scheme 31 Synthesis of α-aminooxime esters (59) and (60). | ||
In DBU promoted reactions addition intermediate undergo hydrogen transfer to form ethenimines. Next, intramolecular cyclization between imine bond and carbonyl functional group takes place to form a bridged intermediate. This in presence of DBU undergoes O-rearrangement and exclusively produce product 62 (Scheme 32).
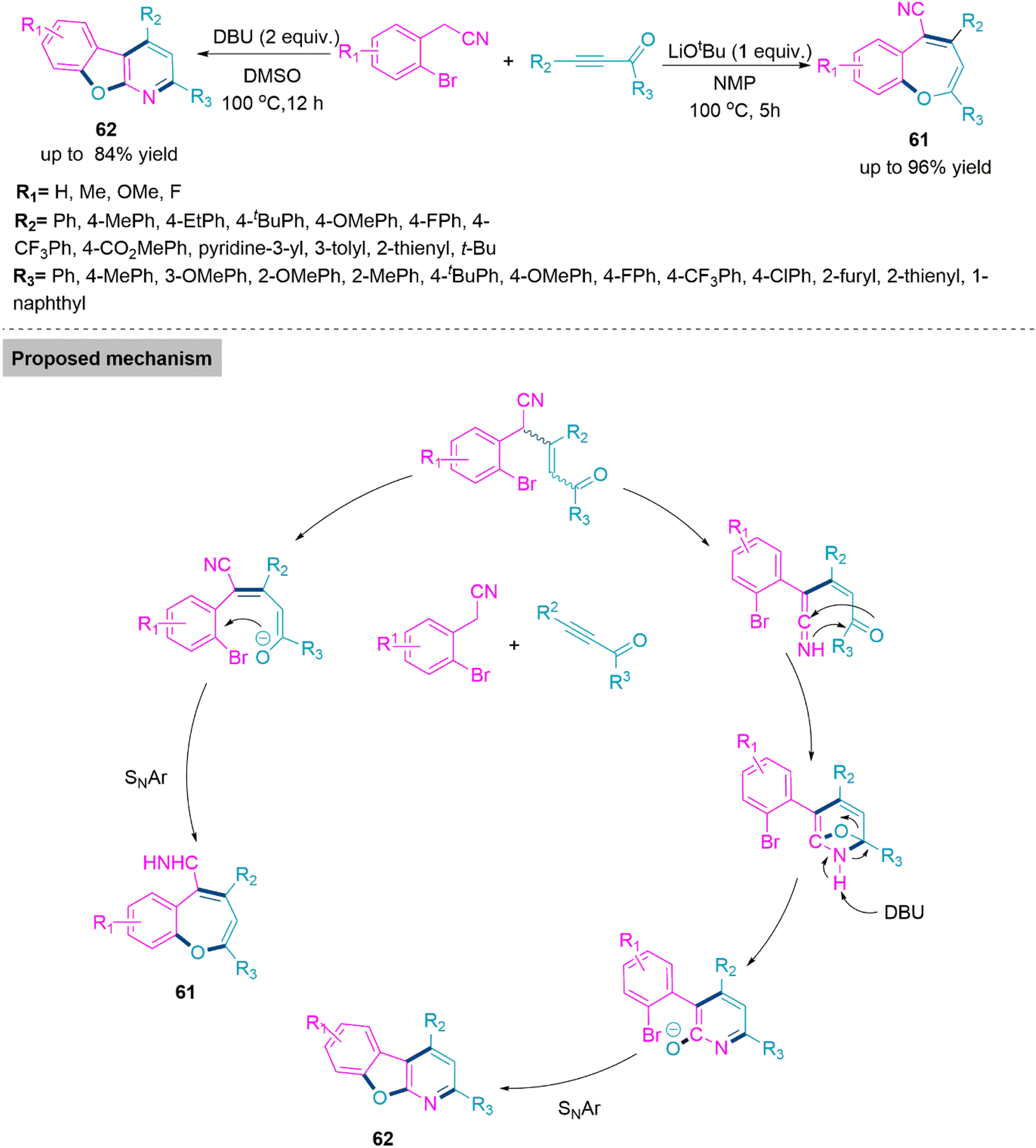 | ||
| Scheme 32 Synthesis of 5-cyanobenzoxepines (61) and benzofuro[2,3-b]pyridines (62). | ||
Next, Ji and colleagues159 developed a copper iodide-initiated ring annulation reaction of 2-isocyanoacetophenone derivatives for the synthesis of substituted 4-hydroxyquinoline derivatives (63). This transformation involves enol-tautomerism and subsequent carbon–carbon bond formation. Under ideal reaction parameters, the reaction proceeds smoothly and produces the target products with a maximum yield up to 86%. In the proposed mechanism, Cu(I) generated during the reaction course, interacts with isocyanide to form an intermediate, which undergoes keto–enol tautomerism, intramolecular ring annulation and subsequent aromatization to give product 63 with a reliving active copper complex (Scheme 33).
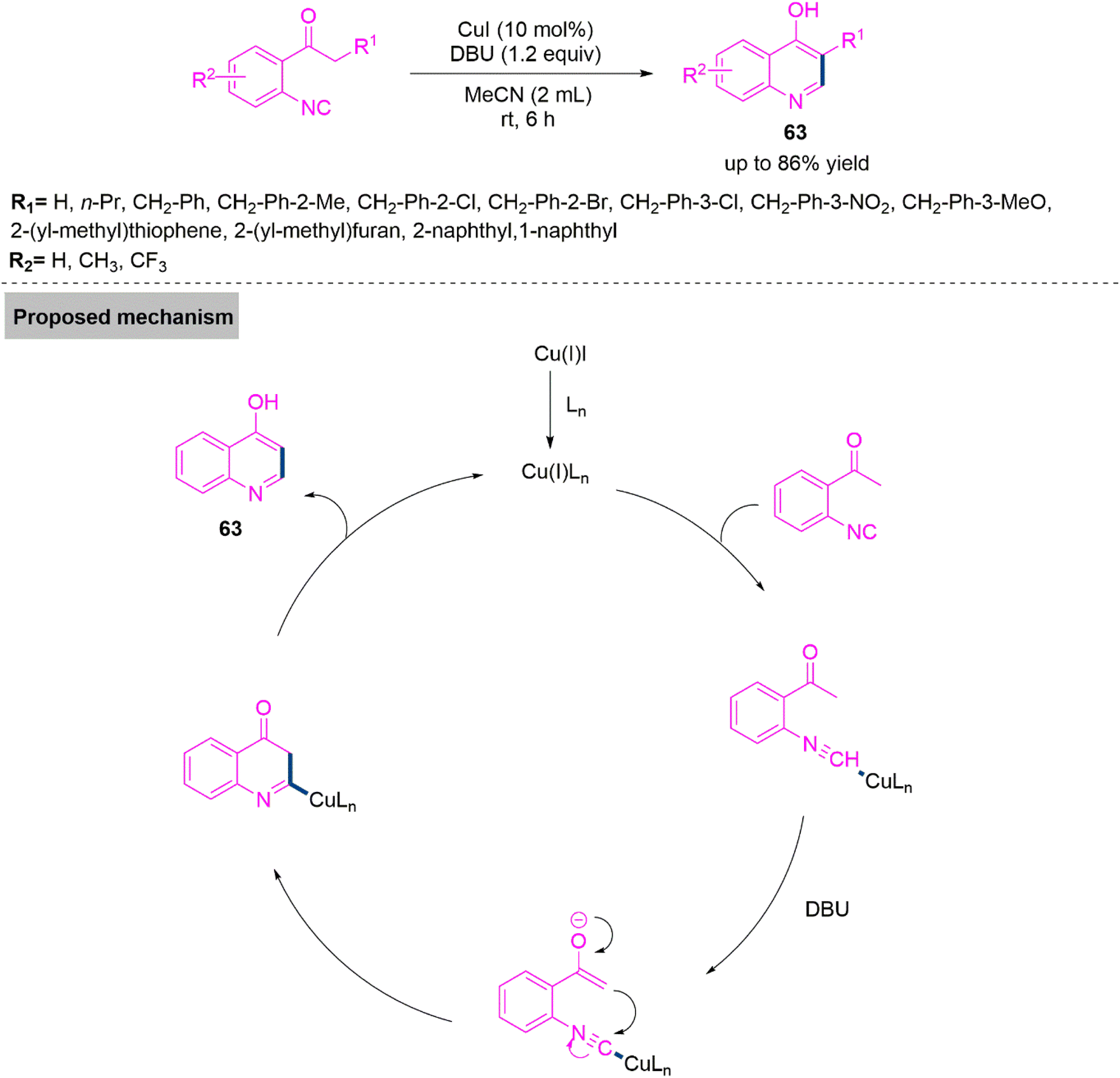 | ||
| Scheme 33 Synthesis of substituted 4-hydroxyquinoline derivatives (63). | ||
Natural products, bioactive substances, and potential drugs often include butenolide and its derivatives, especially those with hydroxyl functionality.160 Examples include the Cdc25A inhibitor dysidiolide,161 the hsPLA2 inhibitor cacospongionolide E,162 and the antifungal agent pseudomonas aureofaciens strain.163 They are important synthetic precursors that help to generate molecular complexity. Therefore, the focus of organic synthesis continues to be on developing efficient synthetic methods for the butenolide scaffolds. In 2020, Baidya and colleagues164 described a strategy that involve catalyst-driven nitroso carbonyl aldol reaction of γ-functionalized non-conjugated butenolides. A Lewis base catalyst quinidine and MnO2 oxidant facilitate the formation of γ-aminoxylation derivatives (64) through the reaction of in situ formed nitroso carbonyl and γ-substituted deconjugated butanolide. While use of Cu(OTf)2 in DCM selectively form β,γ-disubstituted butanolide derivatives (65) using nitroso carbonyl Aldol Reaction of the substrates. In proposed mechanism copper-based nitroso carbonyl complex undergo nucleophilic attack by butanolide substrate to form oxonium ion intermediate. This intermediate upon intramolecular nucleophilic addition give four-five membered fused-ring intermediate and form product 65 upon rearrangement reaction (Scheme 34).
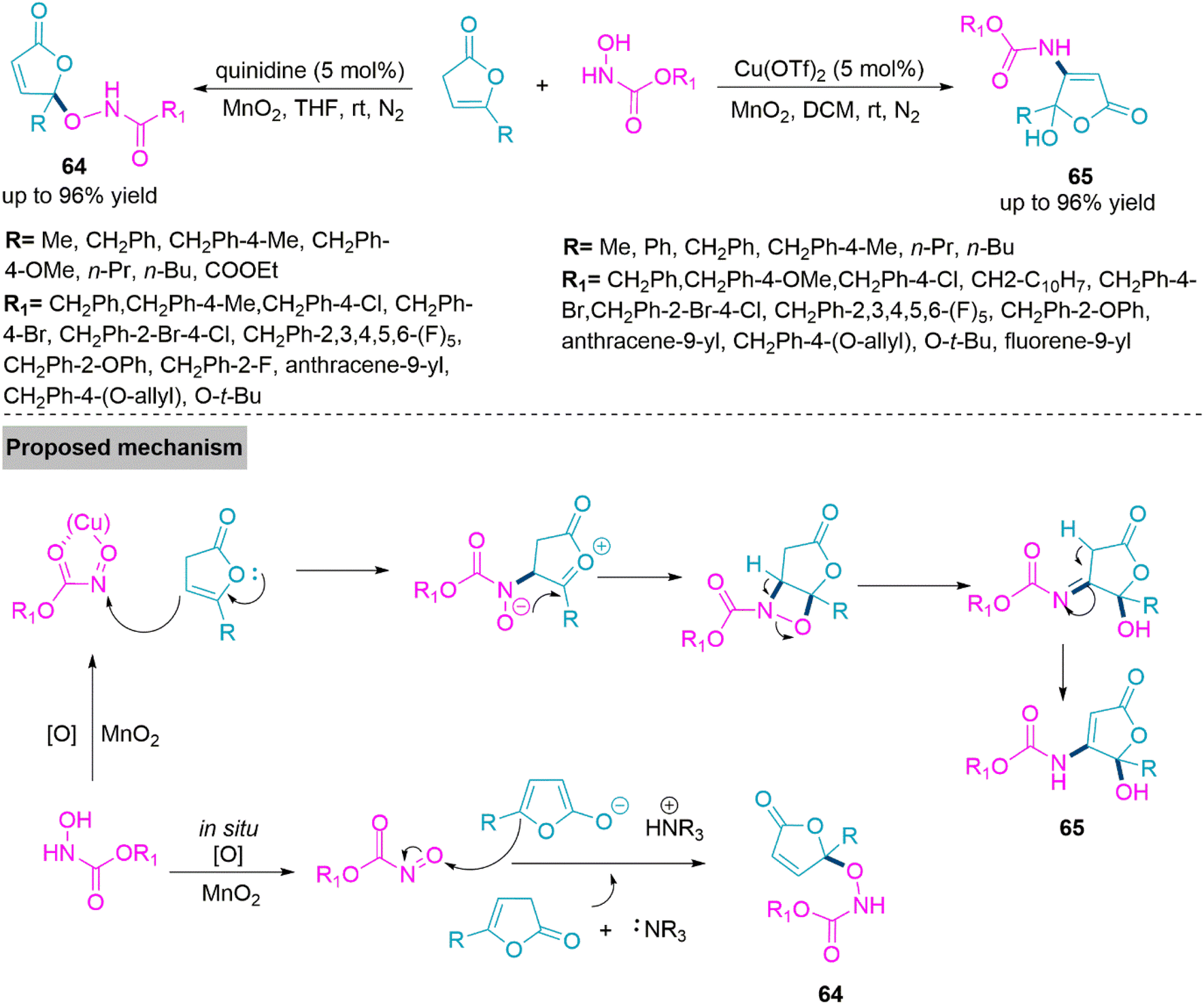 | ||
| Scheme 34 Synthesis of γ-aminoxylation derivatives (64) and β, γ-disubstituted butanolide derivatives (65). | ||
5. Temperature modulated one-pot reactions
In organic reactions, temperature has a great influence on the orientation and reactivity of the substrates. Thus, modulation of the temperature in organic reactions produces complex molecular architectures with significant biological applications.165 Significantly, tuning of regioselectivity is one of the most noteworthy facets in the development of natural products and pharmaceutically active compounds.166–168 The same starting substrates can be used in selective ring closing reactions to create compounds with different cyclic structures, such as the 5-membered phthalide and the 6-membered isocoumarin. They serve as crucial building elements for several biological, material, and naturally occurring compounds. Bioactivities of isocoumarins include anticancer,169 antifungal,170 anti-HIV,171 antimicrobial,172 and enzyme inhibition.173Lee and colleagues174 developed a temperature-modulated strategy for the synthesis of isocoumarins (66) and phthalide derivatives (67) from alkynes and 2-iodobenzoic acid. This reaction proceeds smoothly and shows successful transformation using CsCO3, in refluxing DMSO. At 100 °C, isocoumarin derivatives were formed as a 6-endo-dig product. Whereas, at 25 °C temperature, 5-exo-dig product forms. A mechanism is proposed whereby the synthesis of 2-alkynylbenzoic acid as an intermediary via Sonogashira-type coupling has been eliminated from the reaction pathway, and a variety of alkynes gave the corresponding products 66 and 67 in acceptable yields (Scheme 35).
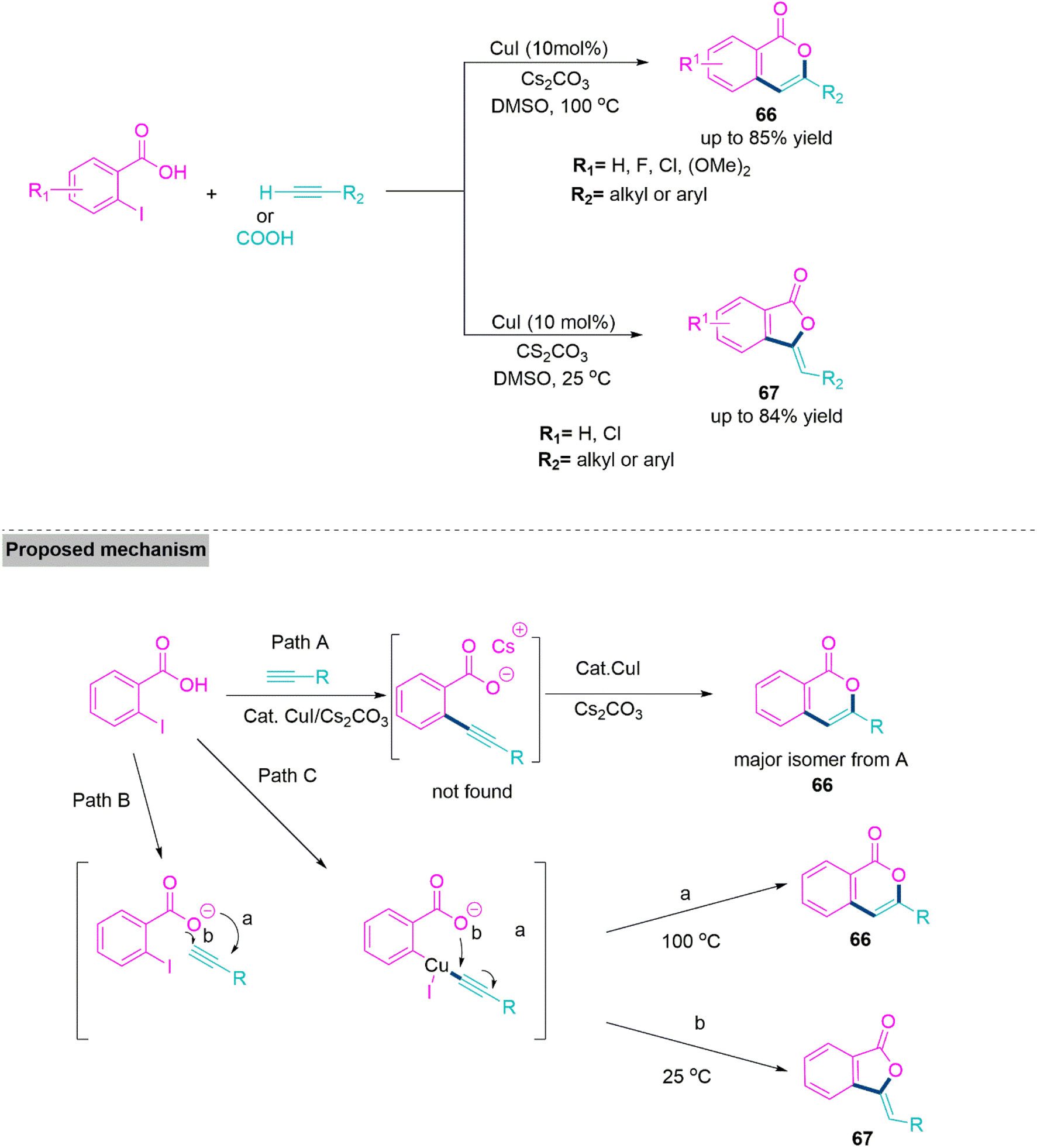 | ||
| Scheme 35 Synthesis of isocoumarins (66) and phthalide derivatives (67). | ||
Benzodiazepines are well recognized for their biological properties and commercial feat.175 A successful approach with excellent atom economy for the synthesis of functionalized benzodiazepines (68) and conjugated enaminones (69) through the reaction of alk-3-yn-1-ones and o-phenylenediamines developed by Török, Dembinski, and colleagues.176 In the absence of a catalyst, this microwave-accelerated synthesis flows smoothly in ethanol and produces product 68 in good yields (up to 92%). Whereas ambient temperature (25 °C) approach using the same substrates (stabilised with triethylamine) forms product 69 in yield up to 99%. Probably, the reaction pathway comprises a generation of allene, which corresponds to alk-3-ynone, followed by conjugate addition and isomerization to produce a conjugated enaminone (69). A parallel pathway can also be postulated by first an isomerization of alk-3-ynone to alk-2-ynone. The sequential synthesis of an imine in an intramolecular ring annulation process and isomerization of vinamidine generate product 68 due to the improved stability of the carbon-nitrogen double bond (Scheme 36).
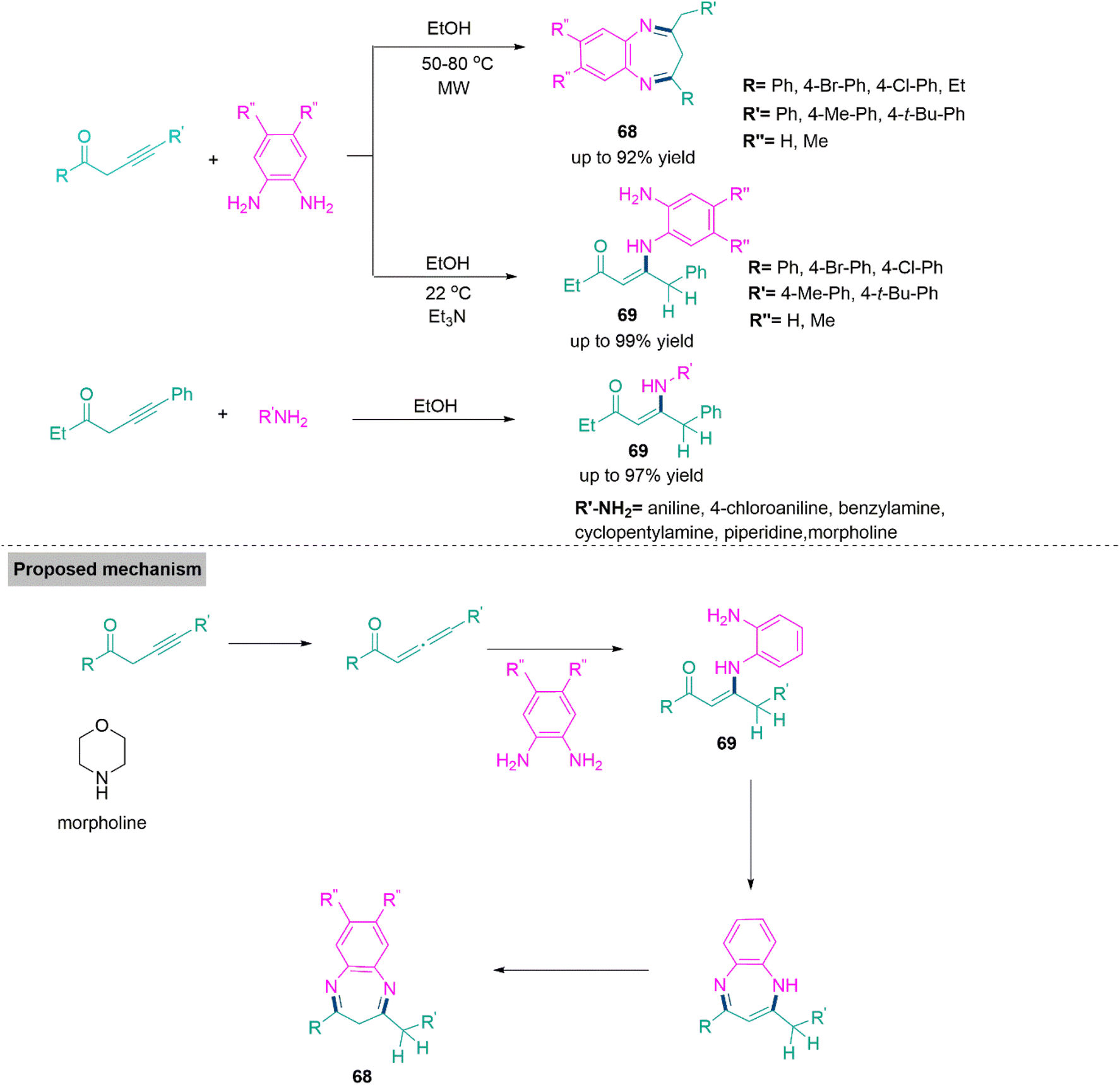 | ||
| Scheme 36 Synthesis of functionalized benzodiazepines (68) and conjugated enaminones (69). | ||
Next, Yang, Xu, et al.177 reported a synthetic methodology that involves the reaction of imines with, methanesulfonylsulfene to produce trans-β-sultams (70) and 4-aza-δ-sultams (71). Here, the annuloselectivity of methanesulfonylsulfene and imines reactions depends on temperature. The [2s + 2i] cyclization of various N-alkyl imines proceeds solely at a significantly greater temperature of 20 °C, yielding four-membered trans-β-sultams (70) in maximum 69% yields. The [2s + 2i + 2i] cyclization of N-methyl imines occurs specifically at −78 °C, yielding six-membered 4-aza-δ-sultams (71) in maximum yield up to 57%, with different configurations at the carbon-3, -5, and -6 stereocenters. The trans stereochemistry of product 70 formed via [2s + 2i] cyclization is credited to 2,3-thiazabutadiene-like zwitterionic adduct which is thermodynamically stable and endures conrotatory ring closure step. The iminium isomerization in the successive nucleophilic [4 + 2] cyclization between the identical zwitterionic adduct and an additional molecule of N-methyl imines causes multiple stereochemical outcomes of the [2s + 2i + 2i] annulations process (Scheme 37).
 | ||
| Scheme 37 Synthesis of trans-β-sultams (70) and 4-aza-δ-sultams (71). | ||
In the finding of novel bioactive compounds,43 benzo-fused nitrogen heterocycles have been proved privilege frameworks. Pyrrolobenzodiazepines (73) and pyrroloquinazoline (74) are constituents of many biologically potent compounds. Thus, Valverde and team178 explored a chemical methodology describing the synthesis of such heterocycles through efficient post-Ugi cascade cyclization process which is exclusively controlled by the temperature. The reduced form of the Ugi product (72) upon heating at 50 °C in ethanol selectively form pyrrolobenzodiazepines (73). While, pyrroloquinazolines (74) were synthesized by intramolecular ring annulation reaction of reduced Ugi product (72) in refluxing n-BuOH solvent (Scheme 38).
 | ||
| Scheme 38 Synthesis of pyrrolobenzodiazepines (73) and pyrroloquinazoline (74). | ||
Many natural compounds with remarkable biological properties contain the tetrahydrocarbazole chemical structure. 179,180 The [4 + 2] cyclic addition of 2-vinylindoles with competent dienophiles is prominent among the several synthetic methods that have been established over time to obtain tetrahydrocarbazoles.181–184 In 2018, Ghorai and colleagues185 explored the synthesis of such heterocycles through a two-fold divergent diastereoselective (dr up to >99:1) transformation of 2-vinylindoles, making it possible to quickly and directly access highly substituted tetrahydrocycloheptadiindoles (75) and tetrahydrocarbazoles (76) with several continuous stereocenters. A wide variety of functionalized 2-vinylindoles are prone to high-yielding conversions (yields up to 87%), which take place through the Lewis acid-catalysed [4 + 2] and [4 + 3] ring annulation-aromatization cascade events, respectively. These reactions involve a never-before-seen reversal of the polarities (Umpolung) of 2-vinylindoles. Two feasible molecular approaches for the synthesis of products 75 and 76 from 2-vinylindoles are suggested in Scheme 39. The 2-vinylindole precursor first undergoes activation by the Fe(III)-based Lewis acid by coordinating to the indolyl nitrogen in order to generate an intermediate product, and during this step, the polarity of the olefin component in the 2-vinyl group is switched. At the same time, another 2-vinylindole molecule uses its C3 centre to attack the olefinic component of the activated indole molecule at the 2′-position, thereby producing an active intermediate. A [4 + 2] type ring-closing is caused by the incipient negative charge accumulated at the 1′-position of the activated indole species attacking the 2′-position of the other indole fragment using an alternate Umpolung process, resulting in the intermediate, which is then rearomatized to produce the desired product 76. Alternatively, the initial steps of the synthesis of tetrahydrocycloheptadiindole are identical and result in the formation of a zwitterionic intermediate with Fe(III). The second indole fragment's 1′-centre is attacked by the developing negative charge on the indole moiety's C3, causing a ring closure and the formation of the bis-indole clubbed 7-membered ring intermediate, which then aromatizes to form the unexpected product 75.
 | ||
| Scheme 39 Synthesis of functionalized tetrahydrocyclohepta-diindoles (75) and tetrahydrocarbazoles (76). | ||
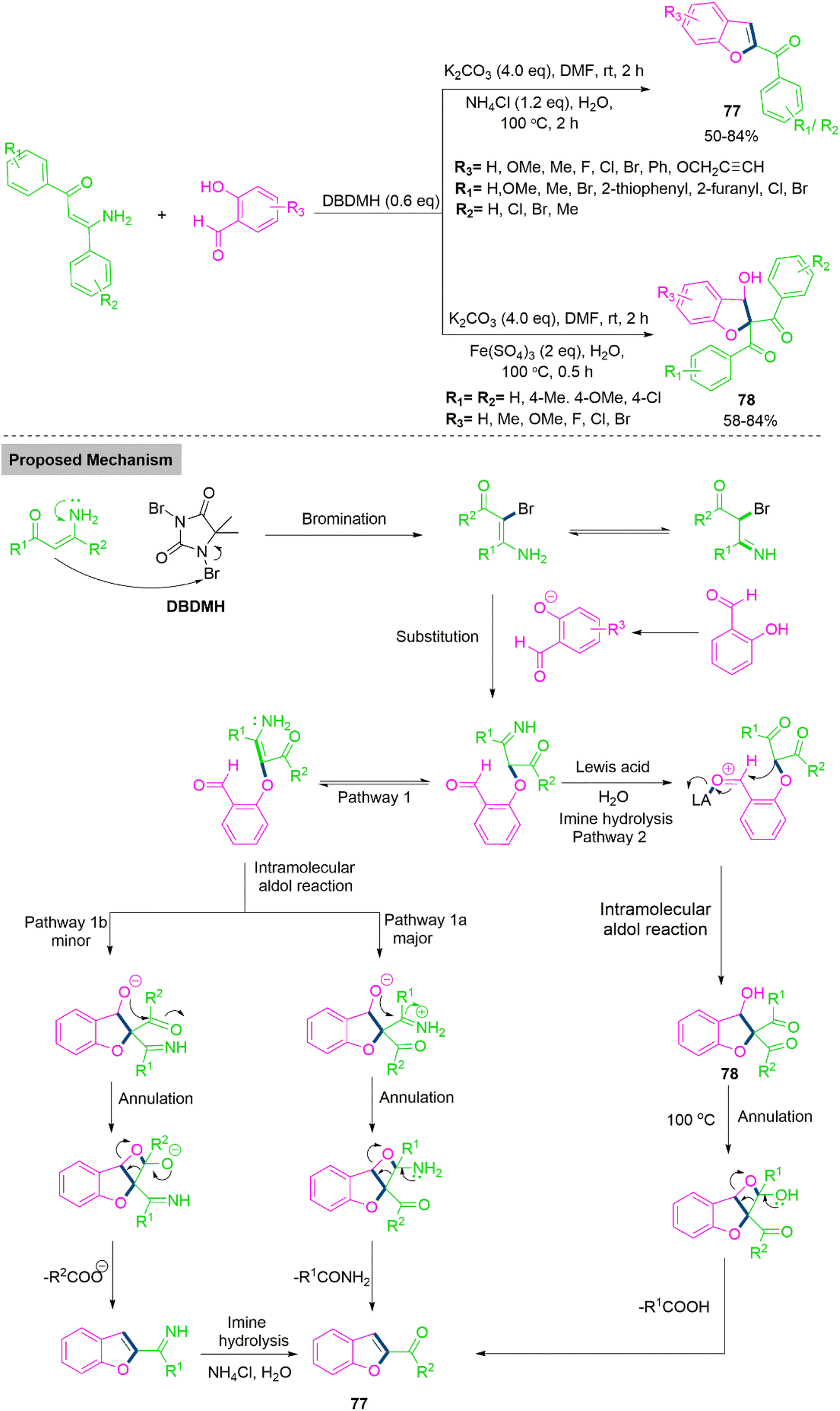
Benzofurans and 2,3-dihydrobenzofurans are important building blocks in natural products with bioactive properties and potential for pharmaceutical applications.186 Duan, Liu, and Colleagues187 developed a chemoselective method for the synthesis of benzofurans (77) and 2,3-dihydrobenzofuran (78). In this reaction system, a combination of DBDMH (1,3-Dibromo-5,5-dimethylhydantoin) and K2CO3 serves as key promotors, facilitating the controlled transformation of enaminones and salicylaldehydes into two distinct sets of valuable heterocyclic compounds 77 and 78. The system's success hinges on the precise identification and manipulation of reaction parameters. After optimizing the reaction conditions for this transformation, the scope of the reaction was explored by testing various salicylaldehyde substrates. The results showed the remarkable substrate tolerance of the transformation, demonstrating its compatibility with a wide range of salicylaldehyde derivatives. Both electron-donating and electron-withdrawing substituents on the benzene ring of salicylaldehydes were tolerated, though the nature of these substituents significantly influenced the reaction's outcome. Generally, salicylaldehydes bearing electron-donating groups on the aromatic ring performed better, yielding the desired benzofuran products (77) in moderate to good yields. In contrast, when salicylaldehydes with electron-withdrawing groups were used, the reaction yields noticeably decreased. Furthermore, the reaction showed excellent compatibility with a propargyl derivative that included a sensitive functional group, efficiently producing the valuable product in good yield. This investigation highlights the broad applicability of the reaction, particularly in the synthesis of structurally diverse benzofuran derivatives. The reaction scope was further extended to investigate the reactivity of various enaminones under the optimized conditions. Overall, in both cases the results showed that the transformation exhibited considerable flexibility with respect to the salicylaldehyde and enaminone substrates, producing the desired product 77 in yields ranging from 34% to 84%. Duan et al. introduced a slight modification to the reaction conditions by incorporating an acid, which successfully suppressed the formation of benzofuran 77 and shifted the reaction pathway toward the formation of the functionalized 2,3-dihydrobenzofuran product 78. As shown in Scheme 40, replacing NH4Cl with Fe2(SO4)3 as an additive under these conditions enabled the selective formation of 2,3-dihydrobenzofuran products in yields ranging from 62% to 84% at room temperature. The reaction demonstrated broad substrate compatibility, efficiently accommodating functional groups such as alkynyl, alkenyl, and triazole rings. These functionalized substrates underwent transformation to deliver the desired 2,3-dihydrobenzofuran 78 in moderate to good yields. Additionally, disubstituted enaminones were tolerated under these conditions, although they afforded slightly lower yields compared to their monosubstituted counterparts. This modification broadens the scope of the reaction, providing access to a new class of valuable dihydrobenzofuran derivatives with diverse functional groups. In the proposed mechanism, reaction begins with the formation of an imine intermediate, which is generated through transient halogenation coupling and substitution processes. This imine intermediate serves as a crucial branching point, where it can undergo one of two distinct pathways: either aldol condensation followed by annulation or imine hydrolysis followed by aldol condensation. The selectivity between these pathways is meticulously controlled by the choice of additives. Specifically, the use of NH4Cl or Fe2(SO4)3 plays a pivotal role in directing the reaction toward different outcomes. NH4Cl favours one pathway, while Fe2(SO4)3 encourages the other, thus determining whether the reaction leads to the formation of benzofurans (77) or dihydrobenzofurans (78). This precise manipulation of reaction parameters and intermediate selectivity highlights the importance of additives in achieving the unique outcome of this tandem transformation.
 | ||
| Scheme 40 Synthesis of benzofurans (77) and 2,3-dihydrobenzofurans (78). | ||
6. Conclusions and outlooks
Fine-tuning chemoselectivity in organic reactions is a highly effective strategy for generating complex molecular structures. Chemists may achieve significant control over reaction outcomes by modulating reaction parameters such as substrate type (including substituent alterations), solvent choice, catalyst, ligand, or additive type, and reaction temperature. This adaptability allows a single set of starting materials to produce a wide range of products, emphasizing the importance of reaction circumstances in chemoselectivity. Methods that provide such precise control over reaction pathways have made significant progress in the synthesis of new heterocyclic and carbocyclic molecules. These compounds are critical because they are found in many natural products and have numerous applications in pharmaceuticals, agrochemicals, and materials science. Thus, acquiring and optimizing chemoselectivity is more than just a theoretical exercise; it is critical for enabling practical applications in a wide range of industries.6.1 The multi-faceted nature of chemoselectivity modulation
Achieving a high level of chemoselectivity necessitates a thorough understanding of how various reaction parameters interact with one another. Synthetic organic chemists face several challenges when attempting to modulate these conditions in order to favour desired reaction pathways. The intrinsic characteristics of the starting substrates, such as their electronic and steric properties, frequently play an important role in directing the reaction. Solvents are also important because they affect reactant solubility, intermediate stability, and the entire reaction rate. Catalysts and ligands are crucial for their ability to speed up reactions and impart selectivity, often by stabilising specific transition states or intermediates. Additives can also be used strategically to modify reaction pathways, either by neutralising undesirable reactive species or stabilising preferred intermediates. Temperature is another important factor that influences the energy barriers between competing reaction paths. Higher temperatures may accelerate reactions, but they also increase the likelihood of side reactions, reducing chemoselectivity. Lower temperatures, on the other hand, may favour the formation of specific products while also resulting in longer reaction times and incomplete reactions.6.2 The importance of chemoselectivity in modern organic chemistry
The ability to control chemoselectivity has laid the way for the creation of a wide range of novel molecular structures. These advancements have made a significant impact on the synthesis of heterocycles and carbocycles, two classes of compounds commonly found in bioactive molecules. Many pharmaceuticals and natural products contain heterocycles, which are rings with one or more non-carbon atoms (such as nitrogen, oxygen, or sulphur). Meanwhile, carbocycles, which are made entirely of carbon atoms, are essential structural units in a wide range of complex organic molecules. Leading researchers from around the world have demonstrated that by carefully altering reaction conditions, it is possible to selectively form these rings, often from basic starting materials. The ability to fine-tune reactions opens up new possibilities for the precise synthesis of complex molecules.6.3 Looking ahead: chemoselectivity and molecular diversity
In the future, chemoselectivity is expected to become a guiding principle in the development of new compounds in organic synthesis. Chemists will be able to create heterocyclic and carbocyclic compounds with diverse structural features by employing their ability to control reaction outcomes through careful conditional modifications. These compounds will find use not only in the pharmaceutical and agricultural industries but also in material science, where their unique properties can be used to create innovative materials. The continuing study of chemoselective reactions will also add to our understanding of fundamental chemical reactivity. Chemists can develop new catalysts, solvents, and additives with even finer control over chemoselectivity by investigating how different factors influence reaction pathways. Such advancements will make it easier to synthesise more complex molecules with high precision, opening up new possibilities for the design of functional molecules intended for specific applications.Abbreviations
| AcOH: | Acetic acid |
| CsF: | Caesium fluoride |
| CS2: | Carbon disulphide |
| DBU: | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCE: | Dichloroethane |
| DEM: | Deep eutectic mixture |
| DIPEA: | N,N-Diisopropylethylamine |
| DMSO: | Dimethyl sulfoxide |
| DMAP: | 4-dimethylaminopyridine |
| DMF: | Dimethyl formamide |
| DOS: | Diversity-oriented synthesis |
| EG: | Ethylene glycol |
| Et3N: | Triethylamine |
| EtOAc: | Ethyl acetate |
| EtOH: | Ethanol |
| FDA: | Food and Drug Administration |
| HIV: | Human immunodeficiency virus |
| LED: | Light emitting diode |
| MCRs: | Multicomponent reactions |
| MDRs: | Multicomponent domino reactions |
| MeOH: | Methanol |
| MMP: | Matrix metalloproteinases |
| NBS: | N-Bromosuccinimide |
| SNAr: | Nucleophilic Aromatic Substitution |
| THF: | Tetrahydrofuran |
| TMGB: | Trimethylglycine betaine |
| TSOH: | Toluene sulphonic acid |
| US: | Ultrasound |
| W: | watt |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the technical support from the Smt. S. S. Patel Nootan Science and Commerce College, Sankalchand Patel University, Visnagar-384315, Gujarat, India.References
- M. Buskes, A. Coffin, D. Troast, R. Stein and M. Blanco, ACS Med. Chem. Lett., 2023, 14, 376–385 CrossRef CAS PubMed.
- D. M. Patel, M. G. Sharma, R. M. Vala, I. Lagunes, A. Puerta, J. M. Padrón, D. P. Rajani and H. M. Patel, Bioorg. Chem., 2019, 86, 137–150 CrossRef CAS PubMed.
- D. M. Patel, R. M. Vala, M. G. Sharma, D. P. Rajani and H. M. Patel, ChemistrySelect, 2019, 4, 1031–1041 CrossRef CAS.
- R. M. Vala, M. G. Sharma, D. M. Patel, A. Puerta, J. M. Padrón, V. Ramkumar, R. L. Gardas and H. M. Patel, Arch. Pharm., 2021, 354, 2000466 CrossRef CAS PubMed.
- Z. Wang and A. Domling, Multicomponent Reactions towards Heterocycles: Concepts and Applications, in Multicomponent Reactions in Medicinal Chemistry, ed. E. Van der Eycken and U. K. Sharma, John Wiley & Sons, 2022, pp. 91–137 Search PubMed.
- P. Slobbe, E. Ruijter and R. V. Orru, MedChemComm, 2012, 3, 1189–1218 RSC.
- J. Tan and A. K. Yudin, Drug Discovery Today: Technol., 2018, 29, 51–60 CrossRef.
- W. Czechtizky and P. Hamley, Small Molecule Medicinal Chemistry: Strategies and Technologies, John Wiley & Sons, Inc., 2015 Search PubMed.
- (a) H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Expert Opin. Ther. Pat., 2021, 31, 267–289 CrossRef CAS PubMed; (b) G. Graziano, A. Stefanachi, M. Contino, R. Prieto-Díaz, A. Ligresti, P. Kumar, A. Scilimati, E. Sotelo and F. Leonetti, Int. J. Mol. Sci., 2023, 24, 6581 CrossRef CAS.
- A. Jaiswal, M. Kumar and K. N. Singh, Asian J. Org. Chem., 2023, 12, e202200629 CrossRef.
- V. Tiwari, Chem. Biol. Drug Des., 2022, 99, 839–856 CrossRef CAS PubMed.
- S. Zhao, Y. Wu and L. Hu, Bioorg. Chem., 2022, 119, 105520 CrossRef CAS PubMed.
- R. P. Herrera and E. Marqués-López, Multicomponent Reactions: Concepts and Applications for Design and Synthesis, Wiley, Hoboken, 2015 Search PubMed.
- T. Nasiriani, S. Javanbakht, M. T. Nazeri, H. Farhid, V. Khodkari and A. Shaabani, Top. Curr. Chem., 2022, 380, 50 CrossRef CAS PubMed.
- Y. Zhong, Eur. J. Org Chem., 2022, 2022, e202201038 CrossRef CAS.
- (a) D. M. Patel, P. J. Patel and H. M. Patel, Eur. J. Org Chem., 2022, 2022, e202201119 CrossRef CAS; (b) S. E. John, S. Gulati and N. Shankaraiah, Org. Chem. Front., 2021, 8, 4237–4287 RSC; (c) T. Damera, R. Pagadala, S. Rana and S. B. Jonnalagadda, Catalysts, 2023, 13, 1034 CrossRef CAS.
- (a) M. T. Martins, F. R. Dias, R. S. de Moraes, M. F. da Silva, K. R. Lucio, K. D'Oliveira Góes, P. A. do Nascimento, A. S. da Silva, V. F. Ferreira and A. C. Cunha, Chem. Rec., 2022, 22, e202100251 CrossRef CAS PubMed; (b) J. C. Flores-Reyes, V. C. Cotlame-Salinas, I. A. Ibarra, E. González-Zamora and A. Islas-Jácome, RSC Adv., 2023, 13, 16091 RSC; (c) H. R. Chaudhary, P. J. Patel, V. K. Gupta and D. M. Patel, Res. Chem. Intermed., 2024, 50, 1273–1286 CrossRef CAS.
- S. E. Hooshmand, H. Yazdani and C. Hulme, Eur. J. Org Chem., 2022, 2022, e202200569 CrossRef CAS.
- S. B. Mohite, M. Bera, V. Kumar, R. Karpoormath, S. B. Baba and A. S. Kumbhar, Top. Curr. Chem., 2023, 381, 4 CrossRef CAS PubMed.
- D. M. Patel, H. J. Patel, J. M. Padrón and H. M. Patel, RSC Adv., 2020, 10, 19600–19609 RSC.
- N. Elders, D. van der Born, L. J. Hendrickx, B. J. Timmer, A. Krause, E. Janssen, F. J. de Kanter, E. Ruijter and R. V. Orru, Angew. Chem., 2009, 121, 5970–5973 CrossRef.
- B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed.
- A. Strecker, Ann. Chem. Pharm., 1850, 75, 27–45 CrossRef.
- A. Hantzsch, Chem. Ber., 1881, 14, 1637 CrossRef.
- P. Biginelli, Chem. Ber., 1891, 24, 1317 CrossRef.
- C. Mannich and V. Krösche, Arch. Pharm., 1912, 250, 647–667 CrossRef CAS.
- M. Passerini, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- E. K. Fields, J. Am. Chem. Soc., 1952, 74, 1528–1531 CrossRef CAS.
- F. Asinger, Angew. Chem., 1956, 68, 413 CrossRef CAS.
- I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 373–388 Search PubMed.
- K. Gewald, E. Schinke and H. Böttcher, Chem. Ber., 1966, 99, 94–100 CrossRef CAS.
- D. Van Leusen and A. M. Van Leusen, Org. React., 2003, 57, 419 Search PubMed.
- H. Bienaym'e and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef.
- R. Abonia, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Eur. J. Org Chem., 2010, 2010, 6454–6463 CrossRef.
- A. Boltjes and A. Dömling, Eur. J. Org Chem., 2019, 2019, 7007–7049 CrossRef CAS PubMed.
- J. Zhu and H. Bienayme, Multicomponent Reactions, John Wiley & Sons, 2005 Search PubMed.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- S. Pelliccia, J. Amato, D. Capasso, S. Di Gaetano, A. Massarotti, M. Piccolo, C. Irace, G. C. Tron, B. Pagano, A. Randazzo and E. Novellino, Eur. J. Med. Chem., 2019, 63, 2035–2050 CrossRef PubMed.
- M. Grazia, L. Martina, M. Giannessi and M. Radi, Eur. J. Org Chem., 2023, 26, e202201288 CrossRef.
- A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112(6), 3083–3135 CrossRef CAS PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- L. Costantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef CAS PubMed.
- E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, 1989 Search PubMed.
- C. E. J. Corey, B. Czakó and L. Kürti, Molecules and Medicine, Wiley, 2008 Search PubMed.
- H. Song, Y. Liu and Q. Wang, Org. Lett., 2013, 15, 3274 CrossRef CAS PubMed.
- Y. Wang, C. Cherian, S. Orr, S. Mitchell-Ryan, Z. Hou, S. Raghavan, L. H. Matherly and A. Gangjee, J. Med. Chem., 2013, 56, 8684–8695 CrossRef CAS PubMed.
- P. Norman, Expert Opin. Ther. Pat., 2014, 24, 121–125 CrossRef CAS PubMed.
- V. La Pietra, G. La Regina, A. Coluccia, V. Famiglini, S. Pelliccia, B. Plotkin, H. Eldar-Finkelman, A. Brancale, C. Ballatore, A. Crowe and K. R. Brunden, J. Med. Chem., 2013, 56, 10066–10078 CrossRef CAS PubMed.
- X. Qi, H. Xiang, Q. He and C. Yang, Org. Lett., 2014, 16, 4186–4189 CrossRef CAS PubMed.
- S. Sarkar, D. K. Das and A. T. Khan, RSC Adv., 2014, 4, 53752–53760 RSC.
- H. Wamhoff, in Comprehensive Heterocyclic Chemistry 1, ed. A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1984, vol. 5, p. 669 Search PubMed.
- P. W. Baures, Org. Lett., 1999, 1, 249–252 CrossRef CAS PubMed.
- R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, D. E. Clercq, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef CAS PubMed.
- H. W. Bai, Z. J. Cai, S. Y. Wang and S. J. Ji, Org. Lett., 2015, 17, 2898–2901 CrossRef CAS PubMed.
- L. Yu, Z. Yang, J. Peng and P. Li, Eur. J. Org Chem., 2016, 2016, 535–540 CrossRef CAS.
- H. P. Bi, L. Zhao, Y. M. Liang and C. J. Li, Angew. Chem., Int. Ed., 2009, 48, 792–795 CrossRef CAS PubMed.
- Z. Zuo and D. W. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- J. C. Xiang, M. Wang, Y. Cheng and A. X. Wu, Org. Lett., 2016, 18, 24–27 CrossRef CAS PubMed.
- J. F. Zheng, X. N. Hu, Z. Xu, D. C. Cai, T. L. Shen and P. Q. Huang, J. Org. Chem., 2017, 82, 9693–9703 CrossRef CAS PubMed.
- M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295–1357 RSC.
- T. Nasiriani, S. Javanbakht, M. T. Nazeri, H. Farhid, V. Khodkari and A. Shaabani, Top. Curr. Chem., 2022, 380, 50 CrossRef CAS PubMed.
- V. Nenajdenko, Isocyanide Chemistry: Applications in Synthesis and Material Science, John Wiley & Sons, 2012 Search PubMed.
- Y. Gao, Z. Hu, J. Dong, J. Liu and X. Xu, Org. Lett., 2017, 19, 5292–5295 CrossRef CAS PubMed.
- L. S. Yu, C. Y. Meng, J. Wang, Z. J. Gao and J. W. Xie, Adv. Synth. Catal., 2019, 361, 526–534 CrossRef CAS.
- S. K. Arupula, S. K. Gudimella, S. Guin, S. M. Mobin and S. Samanta, Org. Biomol. Chem., 2019, 17, 3451–3461 RSC.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed.
- S. Fustero, M. Sanchez-Rosello, P. Barrio and A. Simon-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- A. A. Bekhit, A. Hymete, A. E.-D. A. Bekhit, A. Damtew and H. Y. Aboul-Enein, Mini-Rev. Med. Chem., 2010, 10, 1014–1033 CrossRef CAS PubMed.
- S. Bräse, in Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation, ed. S. Bräse, RSC Cambridge, Cambridge, 2016, p. 478 Search PubMed.
- B. Yu, S. Zhou, L. Cao, Z. Hao, D. Yang, X. Guo, N. Zhang, V. A. Bakulev and Z. Fan, J. Agric. Food Chem., 2020, 68, 7093–7102 CrossRef CAS PubMed.
- Y. Zhao, N. Yang, Y. Deng, K. Tao, H. Jin and T. Hou, J. Agric. Food Chem., 2020, 68, 11068–11076 CrossRef CAS PubMed.
- Y. Zuo, X. He, Y. Ning, Q. Tang, M. Xie, W. Hu and Y. Shang, Org. Biomol. Chem., 2019, 17, 9766–9771 RSC.
- M. De Candia, C. Altamura, N. Denora, S. Cellamare, M. Nuzzolese, D. De Vito, L. G. Voskressensky, A. V. Varlamov and C. D. Altomare, Chem. Heterocycl. Compd., 2017, 53, 357–363 CrossRef CAS.
- A. Barakat, S. M. Soliman, A. M. Al-Majid, M. Ali, M. S. Islam, Y. A. Elshaier and H. A. Ghabbour, J. Mol. Struct., 2018, 1152, 101–114 CrossRef CAS.
- B. Yu, Z. Yu, P. P. Qi, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 95, 35–40 CrossRef CAS PubMed.
- K. S. Mani, W. Kaminsky and S. P. Rajendran, New J. Chem., 2018, 42, 301–310 RSC.
- P. Saraswat, G. Jeyabalan, M. Z. Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS.
- X. Cheng, P. Gao, L. Sun, Y. Tian, P. Zhan and X. Liu, Expert Opin. Ther. Pat., 2017, 27, 1277–1286 CrossRef CAS PubMed.
- D. M. Patel and H. M. Patel, ACS Sustainable Chem. Eng., 2019, 7, 18667–18676 CrossRef CAS.
- A. Alizadeh, B. Farajpour, T. O. Knedel and C. Janiak, J. Org. Chem., 2021, 86, 574–580 CrossRef CAS PubMed.
- A. Saputra, R. Fan, T. Yao, J. Chen and J. Tan, Adv. Synth. Catal., 2020, 362, 2683–2688 CrossRef CAS.
- M. M. Amer, M. A. Aziz, W. S. Shehab, M. H. Abdellattif and S. M. Mouneir, J. Saudi Chem. Soc., 2021, 25, 101259 CrossRef CAS.
- S. Lin, C. Liu, X. Zhao, X. Han, X. Li, Y. Ye and Z. Li, Front. Chem., 2022, 10, 869860 CrossRef CAS PubMed.
- Y. Zhang and A. Pike, Bioorg. Med. Chem. Lett., 2021, 38, 127849 CrossRef CAS PubMed.
- S. Liu, J. Li, J. Lin, F. Liu, T. Liu and C. Huang, Org. Biomol. Chem., 2020, 18, 1130–1134 RSC.
- Y. Arakida, K. Ohga, Y. Okada, H. Morio, K. Suwa, M. Yokota and T. Yamada, Eur. J. Pharmacol., 2000, 403, 169–179 CrossRef CAS PubMed.
- J. V. Allen, C. Bardelle, K. Blades, D. Buttar, L. Chapman, N. Colclough, A. G. Dossetter, A. P. Garner, A. Girdwood, C. Lambert and A. G. Leach, Bioorg. Med. Chem. Lett., 2011, 21, 5224–5229 CrossRef CAS PubMed.
- V. Calderone, A. Coi, F. L. Fiamingo, I. Giorgi, M. Leonardi, O. Livi, A. Martelli and E. Martinotti, Eur. J. Med. Chem., 2006, 41, 1421–1429 CrossRef CAS PubMed.
- M. K. Mehra, M. Malik, B. Kumar and D. Kumar, Org. Biomol. Chem., 2021, 19, 1109–1114 RSC.
- W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- C. Cheignon, E. Cordeau, N. Prache, S. Cantel, J. Martinez, G. Subra, C. Arnaudguilhem, B. Bouyssiere and C. Enjalbal, J. Med. Chem., 2018, 61, 10173–10184 CrossRef CAS PubMed.
- G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182–9188 RSC.
- M. R. Mutra, V. S. Kudale, J. Li, W. H. Tsai and J. Wang, Green Chem., 2020, 22, 2288–2300 RSC.
- X. Wang, S. Y. Wang and S. J. Ji, J. Org. Chem., 2014, 79, 8577–8583 CrossRef CAS PubMed.
- L. Brunton, B. Chabner, B. Knollman, and L. Goodman, Gilman's the Pharmacological Basis of Therapeutics, McGraw-Hill, New York, 2010 Search PubMed.
- F. Kopp and P. Knochel, Org. Lett., 2007, 9, 1639–1641 CrossRef CAS PubMed.
- Y. Y. Yu and G. I. Georg, Chem. Commun., 2013, 49, 3694–3696 RSC.
- M. Szostak, M. Spain, B. Sautier and D. J. Procter, Org. Lett., 2014, 16, 5694–5697 CrossRef CAS PubMed.
- B. T. Barry and G. H. Dennis, Chem. Rev., 2009, 109, 4439–4486 CrossRef PubMed.
- M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- M. Valhondo, I. Marco, M. Martín-Fontecha, H. Vázquez-Villa, J. A. Ramos, R. Berkels, T. Lauterbach, B. Benhamú and M. L. Lopez-Rodriguez, J. Med. Chem., 2013, 56, 7851–7861 CrossRef CAS PubMed.
- C. Cseke, B. C. Gerwick, G. D. Crouse, M. G. Murdoch, S. B. Green and D. R. Heim, Pestic. Biochem. Physiol., 1996, 55, 210–217 CrossRef CAS.
- B. Rajarathinam and G. Vasuki, Org. Lett., 2012, 14, 5204–5206 CrossRef CAS PubMed.
- H. A. Daboun, S. E. Abdou, M. M. Hussein and M. H. Elnagdi, Synth., 1982, 1982, 502–504 CrossRef.
- M. I. Soares, A. F. Brito, M. Laranjo, A. M. Abrantes, M. F. Botelho, J. A. Paixão, A. M. Beja, M. R. Silva and T. M. e Melo, Eur. J. Med. Chem., 2010, 45, 4676–4681 CrossRef CAS PubMed.
- K. Santos, M. Laranjo, A. M. Abrantes, A. F. Brito, C. Gonçalves, A. B. Ribeiro, M. F. Botelho, M. I. Soares, A. S. Oliveira and T. M. e Melo, Eur. J. Med. Chem., 2014, 79, 273–281 CrossRef CAS PubMed.
- W. Franke, F. Blume, F. Arndt and R. Rees, EU Pat., EP 298405 A2 19890111, Eur. Pat. Appl, 1989.
- Q. Cai, F. C. Jia, D. K. Li, C. Xu, K. R. Ding and A. X. Wu, Tetrahedron, 2015, 71, 6104–6111 CrossRef CAS.
- D. A. Ibrahim, D. A. Abou El Ella, A. M. El-Motwally and R. M. Aly, Eur. J. Med. Chem., 2015, 102, 115–131 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- P. Dziedzic, J. A. Cisneros, M. J. Robertson, A. A. Hare, N. E. Danford, R. H. Baxter and W. L. Jorgensen, J. Am. Chem. Soc., 2015, 137, 2996–3003 CrossRef CAS PubMed.
- X. Zhang, T. L. Wang, X. J. Liu, X. C. Wang and Z. J. Quan, Org. Biomol. Chem., 2019, 17, 2379–2383 RSC.
- A. Burger, Burger's Medicinal Chemistry, Drug Discovery and Development, Wiley, Hoboken, 2010 Search PubMed.
- L. Brunton, Gilman's the Pharmacological Basis of Therapeutics, McGraw-Hill, New York, 2006 Search PubMed.
- S. H. Kim, A. T. Pudzianowski, K. J. Leavitt, J. Barbosa, P. A. McDonnell, W. J. Metzler, B. M. Rankin, R. Liu, W. Vaccaro and W. Pitts, Bioorg. Med. Chem. Lett., 2005, 15, 1101 CrossRef CAS PubMed.
- J. J. W. Duan, L. Chen, Z. Lu, B. Jiang, N. Asakawa, J. E. Sheppeck II, R.-Q. Liu, M. B. Covington, W. Pitts, S.-H. Kim and C. P. Decicco, Bioorg. Med. Chem. Lett., 2007, 17, 266 CrossRef CAS PubMed.
- J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, J. Am. Chem. Soc., 2009, 131, 3991–3997 CrossRef CAS PubMed.
- A. Barakat, M. S. Islam, A. M. Al-Majid, H. A. Ghabbour, H. K. Fun, K. Javed, R. Imad, S. Yousuf, M. I. Choudhary and A. Wadood, Bioorg. Med. Chem., 2015, 23, 6740–6748 CrossRef CAS PubMed.
- W. Fraser, C. J. Suckling and H. C. Wood, J. Chem. Soc., Perkin Trans., 1990, 1, 3137–3144 RSC.
- X. Yan, P. Shao, X. Song, C. Zhang, C. Lu, S. Liu and Y. Li, Org. Biomol. Chem., 2019, 17, 2684–2690 RSC.
- C. Nájera, I. P. Beletskaya and M. Yus, Chem. Soc. Rev., 2019, 48, 4515 RSC.
- R. Mishra, K. K. Jha, S. Kumar and I. Tomer, Pharm. Chem., 2011, 3, 38–54 CAS.
- G. Rassu, F. Zanardi, L. Battistini and G. Casiraghi, Chem. Soc. Rev., 2000, 29, 109–118 RSC.
- F. Jafarpour, S. Rajai-Daryasarei and M. H. Gohari, Org. Chem. Front., 2020, 7, 3374–3381 RSC.
- C. Marti and E. M. Carreira, Eur. J. Org Chem., 2003, 2003, 2209–2219 CrossRef.
- J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- N. Ye, H. Chen, E. A. Wold, P. Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS PubMed.
- B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- J. Lei, L. J. He, D. Y. Tang, J. Wen, W. Yan, H. Y. Li, Z. Z. Chen and Z. G. Xu, Adv. Synth. Catal., 2021, 363, 2996–3000 CrossRef CAS.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- M. J. Minvielle, C. A. Bunders and C. Melander, MedChemComm, 2013, 4, 916–919 RSC.
- K. A. Scott and J. T. Njardarson, Curr. Top. Med. Chem., 2018, 376, 5 CrossRef PubMed.
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed.
- N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed.
- J. X. He, Z. H. Zhang, B. S. Mu, X. Y. Cui, J. Zhou and J. S. Yu, J. Org. Chem., 2021, 86, 9206–9217 CrossRef CAS PubMed.
- A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960–4992 CrossRef CAS PubMed.
- M. B. Teimouri, E. Batebi, S. Mohammadnia and H. R. Khavasi, Tetrahedron, 2021, 96, 132374 CrossRef CAS.
- Z. Yang, S. Li, Z. Zhang and J. Xu, Org. Biomol. Chem., 2014, 12, 9822–9830 RSC.
- Y. Yang, W. Li, C. Xia, B. Ying, C. Shen and P. Zhang, ChemCatChem, 2016, 8, 304–307 CrossRef CAS.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, 2004 Search PubMed.
- P. A. Gale, Acc. Chem. Res., 2006, 39, 465–475 CrossRef CAS PubMed.
- H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264–287 CrossRef CAS PubMed.
- F. Zhang, Z. Qin, L. Kong, Y. Zhao, Y. Liu and Y. Li, Org. Lett., 2016, 18, 5150–5153 CrossRef CAS PubMed.
- C. Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed.
- A. Prechter, G. Henrion, P. Faudot dit Bel and F. Gagosz, Angew. Chem., Int. Ed., 2014, 53, 4959–4963 CrossRef CAS PubMed.
- F. Wu, W. Chen, M. Liu and H. Wu, Eur. J. Org Chem., 2018, 2018, 5553–5557 CrossRef CAS.
- P. Spiteller, Chem.–Eur. J., 2008, 14, 9100–9110 CrossRef CAS PubMed.
- H. Khanam, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed.
- M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932–6933 CrossRef CAS PubMed.
- A. Varela-Fernández, C. García-Yebra, J. A. Varela, M. A. Esteruelas and C. Saá, Angew. Chem., Int. Ed., 2010, 49, 4278–4281 CrossRef PubMed.
- A. Seoane, N. Casanova, N. Quinones, J. L. Mascarenas and M. Gulias, J. Am. Chem. Soc., 2014, 136, 834–837 CrossRef CAS PubMed.
- J. H. Lee, M. Kim and I. Kim, J. Org. Chem., 2014, 79, 6153–6163 CrossRef CAS PubMed.
- B. K. Villuri, S. S. Ichake, S. R. Reddy, V. Kavala, V. Bandi, C. W. Kuo and C. F. Yao, J. Org. Chem., 2018, 83, 10241–10247 CrossRef CAS PubMed.
- L. L. Chen, J. W. Zhang, P. Chen, S. Zhang, W. W. Yang and J. Y. Fu, et al., Org. Lett., 2019, 21, 5457–5461 CrossRef CAS PubMed.
- Q. Yuan, W. Rao, S. Y. Wang and S. J. Ji, J. Org. Chem., 2020, 85, 1279–1284 CrossRef CAS PubMed.
- T. C. Silva, D. M. Pereira, P. Valentão and P. B. Andrade, “Phospholipase A2 Inhibitors of Marine Origin”, in Natural Products Targeting Clinically Relevant Enzymes, Wiley, 2017, pp. 69−92 Search PubMed.
- R. Shimazawa, T. Suzuki, K. Dodo and R. Shirai, Bioorg. Med. Chem. Lett., 2004, 14, 3291–3294 CrossRef CAS PubMed.
- S. De Rosa, A. Crispino, A. De Giulio, C. Iodice, P. Amodeo and T. Tancredi, J. Nat. Prod., 1999, 62, 1316–1318 CrossRef PubMed.
- G. Pascale, F. Sauriol, N. Benhamou, R. R. Bélanger and T. C. Paulitz, J. Antibiot., 1997, 50, 742–749 CrossRef PubMed.
- S. Mallik, V. Bhajammanavar and M. Baidya, Org. Lett., 2020, 22, 1437–1441 CrossRef CAS PubMed.
- A. Adhikari, S. Bhakta and T. Ghosh, Tetrahedron, 2022, 126, 133085 CrossRef CAS.
- S. Iimura, L. E. Overman, R. Paulini and A. Zakarian, J. Am. Chem. Soc., 2006, 128, 13095–13101 CrossRef CAS PubMed.
- A. R. Van Dyke and T. F. Jamison, Angew. Chem., 2009, 121, 4494–4496 CrossRef.
- I. N. Lykakis, C. Efe, C. Gryparis and M. Stratakis, Eur. J. Org Chem., 2011, 2011, 2334–2338 CrossRef.
- M. E. Riveiro, A. Moglioni, R. Vazquez, N. Gomez, G. Facorro, L. Piehl, E. R. de Celis, C. Shayo and C. Davio, Bioorg. Med. Chem., 2008, 16, 2665–2675 CrossRef CAS PubMed.
- K. Nozawa, M. Yamada, Y. Tsuda, K. Kawai and S. Nakajima, Chem. Pharm. Bull., 1981, 29, 2689–2691 CrossRef CAS PubMed.
- Y. Shikishima, Y. Takaishi, G. Honda, M. Ito, Y. Takeda, O. K. Kodzhimatov, O. Ashurmetov and K. H. Lee, Chem. Pharm. Bull., 2001, 49, 877–880 CrossRef CAS PubMed.
- M. T. Hussain, N. H. Rama and A. Malik, Indian J. Chem., Sect. B, 2001, 40, 372–376 Search PubMed.
- J. J. Heynekamp, L. A. Hunsaker, T. A. Vander Jagt, R. E. Royer, L. M. Deck and D. L. Vander Jagt, Bioorg. Med. Chem., 2008, 16, 5285–5294 CrossRef CAS PubMed.
- M. R. Kumar, F. M. Irudayanathan, J. H. Moon, S. Lee and Ad. Synt, Catal., 2013, 355, 3221–3230 CAS.
- D. L. Comins, S. O'Connor and R. S. Al-Awar, “Chemistry of Pyridines at Ring Positions”, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, UK, 2008, vol. 7, pp. 41−100 Search PubMed.
- A. Solan, B. Nişanci, M. Belcher, J. Young, C. Schäfer, K. A. Wheeler, B. Török and R. Dembinski, Green Chem., 2014, 16, 1120–1124 RSC.
- Q. Wu, Z. Yang and J. Xu, Org. Biomol. Chem., 2016, 14, 7258–7267 RSC.
- P. Pertejo, P. Peña-Calleja, I. Carreira-Barral, R. Quesada, N. A. Cordero, F. J. Rodríguez and M. García-Valverde, Org. Biomol. Chem., 2017, 15, 7549–7557 RSC.
- V. Capra, M. Mauri, F. Guzzi, M. Busnelli, M. R. Accomazzo, P. Gaussem, S. P. Nisar, S. J. Mundell, M. Parenti and G. E. Rovati, Biochem. Pharmacol., 2017, 124, 43–56 CrossRef CAS PubMed.
- J. Y. Su, Y. Zhu, L. M. Zeng and X. H. Xu, J. Nat. Prod., 1997, 60, 1043–1044 CrossRef CAS.
- V. Pirovano, E. Arpini, M. Dell'Acqua, R. Vicente, G. Abbiati and E. Rossi, Adv. Synth. Catal., 2016, 358, 403–409 CrossRef CAS.
- J. W. Ren, Z. F. Zhou, J. A. Xiao, X. Q. Chen and H. Yang, Eur. J. Org Chem., 2016, 2016, 1264–1268 CrossRef CAS.
- S. Zhao and R. B. Andrade, J. Org. Chem., 2017, 82, 521–531 CrossRef CAS PubMed.
- Q. J. Liu, W. G. Yan, L. Wang, X. P. Zhang and Y. Tang, Org. Lett., 2015, 17, 4014–4017 CrossRef CAS PubMed.
- I. A. Wani, et al., Org. Biomol. Chem., 2018, 16, 2910–2922 RSC.
- (a) Z. Xu, S. Zhao, Z. Lv, L. Feng, Y. Wang, F. Zhang, L. Bai and J. Deng, Eur. J. Med. Chem., 2019, 162, 266–276 CrossRef CAS PubMed; (b) H. K. Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef PubMed.
- X. Duan, H. Li, J. Wang, K. Liu, M. Shi, W. Lian, R. Chen and P. Liu, Chin. J. Chem., 2024, 42, 1727–1733 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |