
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-step screening to optimize the signal response of an auto-fluorescent protein-based biosensor†
Shunsuke Tajimaa,
Eiji Nakata a,
Reiko Sakaguchib,
Masayuki Saimuraa,
Yasuo Moric and
Takashi Morii*a
a,
Reiko Sakaguchib,
Masayuki Saimuraa,
Yasuo Moric and
Takashi Morii*a
aInstitute of Advanced Energy, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: t-morii@iae.kyoto-u.ac.jp
bSchool of Medicine, University of Occupational and Environmental Health, 1-1 Iseigaoka, Yahatanishi-ku, Kitakyushu, Fukuoka 807-8555, Japan
cDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Kyotodaigakukatsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 20th May 2022
Abstract
Auto-fluorescent protein (AFP)-based biosensors transduce the structural change in their embedded recognition modules induced by recognition/reaction events to fluorescence signal changes of AFP. The lack of detailed structural information on the recognition module often makes it difficult to optimize AFP-based biosensors. To enhance the signal response derived from detecting the putative structural change in the nitric oxide (NO)-sensing segment of transient receptor potential canonical 5 (TRPC5) fused to enhanced green fluorescent protein (EGFP), EGFP-TRPC5, a facile two-step screening strategy, in silico first and in vitro second, was applied to variants of EGFP-TRPC5 deletion-mutated within the recognition module. In in silico screening, the structural changes of the recognition modules were evaluated as root-mean-square-deviation (RMSD) values, and 10 candidates were efficiently selected from 47 derivatives. Through in vitro screening, four mutants were identified that showed a larger change in signal response than the parent EGFP-TRPC5. One mutant in particular, 551-575, showed four times larger change upon reaction with NO and H2O2. Furthermore, mutant 551-575 also showed a signal response upon reaction with H2O2 in mammalian HEK293 cells, indicating that the mutant has the potential to be applied as a biosensor for cell measurement. Therefore, this two-step screening method effectively allows the selection of AFP-based biosensors with sufficiently enhanced signal responses for application in mammalian cells.
Introduction
Fluorescent biosensors are widely applied to explore the cellular dynamics of ions and biomolecules by taking advantage of high spatio-temporal resolution in their detection.1 Genetically encoded biosensors constructed from the auto-fluorescent protein (AFP) are especially useful because they are easy to localize at a specific area or organelle in the cells and suitable for long-time imaging.2,3 In general, an AFP-based biosensor is designed by conjugating an appropriate recognition or reaction module for a given target to an AFP transduction module.4–6 Structural changes in the recognition module induced by the recognition/reaction event are transduced to a structural perturbation of AFP, which eventually results in fluorescence signal changes of AFP.7,8 Structurally well-characterized native proteins, such as those analyzed by X-ray crystallography,9–11 NMR spectroscopy,12,13 or cryo-EM,14–16 are suitable for use as recognition modules when conjugated to AFP by means of a structure-based design.5–8,17–21 However, information on the structural change in the recognition module for a target of interest is not always available. AFP-based biosensors can also be constructed by conjugating protein domains without structural information, making it difficult to optimize biosensors.22–25By appropriately introducing the recognition module near the chromophore of the AFP in a single AFP-based biosensor using structure-based design with structural information of the free and inositol-1,3,4,5-tetrakisphosphate-bound PH domain, even a subtle structural change in the recognition module is effectively transduced to a measurable fluorescence change.26 This fact prompted us to apply this design strategy in an AFP-based biosensor to detect plausible structural changes in a partial segment of the native protein as a target of interest with changes in fluorescence. The transient receptor potential canonical 5 (TRPC5) channel is a nonselective calcium-permeant cation channel, which is proposed to open its gate upon disulfide bond formation between Cys558 and S-nitrosylated Cys533 induced by a reaction with second messenger nitric oxide (NO).27,28 Disulfide bond formation is proposed to cause putative structural changes near the channel pore. A three-dimensional model deduced from the cryo-EM analysis of TRPC5 in the oxidized state did not provide detailed information on the structural change of the segment.29,30 To evaluate this putative NO-induced structural change in TRPC5, we constructed EGFP-TRPC5 by fusing a segment of the putative NO-sensing module of the TRPC5 channel containing Cys553 and Cys558 residues as a loop structure to enhanced green fluorescent protein (EGFP) by means of a structure-based design.34 EGFP-TRPC5 successfully detected the putative structural change upon disulfide bond formation induced by the reaction with NO as a change in the fluorescence intensity ratio of EGFP. The fact that NO-induced disulfide bond formation was detected by EGFP-TRPC5 implied the potential of EGFP-TRPC5 as a biosensor in mammalian cells by enhancing its ratiometric signal.
Here, we report a facile two-step strategy, in silico first and in vitro second, for enhancing the signal response of an AFP-based biosensor using a part of the protein domains without detailed information on its structural changes. The first screening based on in silico simulation effectively provides the candidates and avoid the laborious optimization process to enhance the signal responses. Our approach allows the selection of AFP-based biosensors suitable for application in mammalian cells.
Results
First screening: in silico root-mean-square-deviation (RMSD) evaluation of segment structural change induced by disulfide bond formation
In EGFP-TRPC5, a partial segment of the putative NO-sensing region ranging from Leu538 to Leu575 of TRPC5 was incorporated into EGFP to monitor its putative structural changes upon disulfide bond formation in response to NO. This segment was inserted between Asn144 and Phe145 of EGFP to form a loop-like structure with Cys553 and Cys558 located around its center (Fig. 1A and S1†).34 Based on the general action mechanism of a single AFP-based biosensor, structural changes in this segment after disulfide bond formation of two cysteines were transduced to the structural perturbation around the chromophore of EGFP to modify its emission properties. The chromophore of EGFP exists in equilibrium between protonated and deprotonated forms, showing absorption peaks at around 400 nm and 470 nm, respectively.35–37 Structural changes in this segment upon disulfide bond formation were successfully detected as changes in fluorescence emission ratio between the protonated and deprotonated states of EGFP chromophore, albeit to a small degree.34To design a fluorescent biosensor based on EGFP-TRPC5 for application in-cell measurement, the structural change associated with disulfide bond formation should be transduced more efficiently to cause structural perturbation near the EGFP chromophore. One of the strategies to efficiently transduce structural changes upon disulfide bond formation to the EGFP chromophore is the deletion of amino acid residues between the disulfide bond formation site and EGFP (Fig. 1B). The NO-sensing segment was shortened from the N- and/or C-terminal region (Fig. 1C). However, even with the simplest one-by-one deletion from the N- and/or C-terminal of NO-sensing segment, a total of 47 mutants were identified as candidate of deletion mutants. Instead of performing bacterial expression, purification, and evaluation of all 47 mutants, we first screened mutants based on in silico simulations.
 | ||
| Fig. 1 (A) (Top) A cryo-EM structure of the TRPC5 homotetramer (PDB ID: 6AEI).29 One monomer was represented in orange and Cys553 and Cys558 forming the disulfide bond were shown in the CPK representation (red). In the zoomed up view, the region locating between two helices S5 and S6 (in orange) contribute to form the pore structure. (Center) The plausible reaction scheme of TRPC5 with NO. Cys553 was S-nitrosylated with NO and nucleophilically attacked by Cys558 to form a disulfide bond.28,31,32 (Bottom) The segment of TRPC5, from Leu538 to Leu575 (purple), was embedded into EGFP (green, PDB ID: 2B3P)33 between Asn144 and Phe145 through linkers (blue). The chromophore of EGFP is represented by ball and stick model (red). (B) Schematic illustration of the parent EGFP-TRPC5 construct and its mutants. Molecular models of reduced (green) and oxidized (red) forms are shown. Images were superimposed by matching the coordinates of Cα atoms of residues from L119 to I128, which were opposite to the NO-sensing segment. The chromophore, Cys553, and Cys558 are shown as CPK representations. (C) NO-sensing segment of TRPC5 (from Leu538 to Leu575) was inserted by splitting EGFP (between 144 and 145).33 EGFP-TRPC5 mutants were designed to delete the NO-sensing segment from the N- and/or C-terminus. (D–F) (D) Plots of RMSD values of N-terminal versus C-terminal sides of the TRPC5 segment of the parent EGFP-TRPC5 and its mutants selected from the first screening (the plots of all of the mutants designed are shown in Fig. S5†). Black circle: parent EGFP-TRPC5; red circles: 10 mutants showing large RMSD values; blue circles: three mutants showing low RMSD values. (E) Amino acid sequences of the NO-sensing segment of the EGFP-TRPC5 mutants selected after the first screening and (F) their molecular models. Black box: parent EGFP-TRPC5; red box: 10 mutants showing large RMSD values; blue box: three mutants showing low RMSD values. Cys553 and Cys558 are shown in CPK representation. | ||
The structures of the 47 deletion mutants in their reduced and oxidized forms were simulated and each reduced and oxidized forms were superimposed on the EGFP domain (Fig. S2–S4†). The folded structure of EGFP was fixed during the simulation to avoid an unnecessary deformation from its proper folding. The structural change in the appended TRPC5 partial segment would be crucial to evaluate the structural perturbation of the EGFP. Therefore, the degrees of structural change in the appended TRPC5 partial segment upon disulfide bond formation were evaluated using the RMSD of the coordinates of the backbone of amino acid residues in the NO-sensing segment region between that of the reduced (Fig. 1B and S2–S4,† structure in green) and oxidized (Fig. 1B and S2–S4,† structure in red) forms. The RMSD values for the N-terminal residues of Cys553 and the C-terminal residues of C558 were calculated separately, that is, GKL-L538-N552 and D559-L575-GSG, where GKL and GSG were the linkers on each side in the case of EGFP-TRPC5 (Fig. S1 and Table S1†). RMSD values on the N-terminal side were plotted against the C-terminal side for all simulated mutants (Fig. S5†). RMSD values in the N-terminal side were distributed from 1.2 Å to 4.5 Å and those in the C-terminal side from 0.5 Å to 3.9 Å. EGFP-TRPC5 showed small RMSD values compared to those of the other deletion mutants (Fig. S5,† black filled circles). Among these, 10 mutants showing large RMSD values were chosen for subsequent screening (Fig. S5† and 1D–F, plotted in red filled circles or shown in red boxes). Three mutants with small RMSD values, 538-567, 538-565, and 538-560, were also selected for comparison (Fig. S5† and 1D–F, plotted in blue filled circles or shown in blue boxes).
Second screening: construction and expression of crude deletion mutants
Genes encoding 13 mutants of the parent EGFP-TRPC5 deleted at TRPC5 (L538-L575) were constructed (Table S2†) and expressed in Escherichia coli (E. coli). For high-throughput screening, the expressed mutant proteins were partially purified by omitting several purification steps as described previously (Fig. S6†) (see Materials and methods).34 We previously reported that most of the parent EGFP-TRPC5 formed a disulfide bond upon purification as evaluated by the conventional DTNB (5,5′-dithio-bis-(2-nitrobenzoic acid)) method38 with a purity of over 95% by SDS-PAGE analysis.34 By assuming that the parent EGFP-TRPC5 mutants also formed a disulfide bond through partial purification, changes in the fluorescence emission ratio (ΔR) upon reduction of the disulfide bond would provide opposite signal changes in the responses to NO.First, the ΔR value of the parent EGFP-TRPC5, defined as the change in the fluorescence emission ratio without or with DTT, was measured at pH 6.9 (Table 1). The partially purified parent EGFP-TRPC5 showed a −0.013 change in ΔR upon reduction of the disulfide bond, which is consistent with the value determined using highly purified parent EGFP-TRPC5 (−0.010).34 This result indicates that the parent EGFP-TRPC5 mainly formed disulfide bonds after partial purification. Next, ΔR of all the constructed mutants was determined using the same procedure (Table 1). Among the 10 mutants with greater RMSD, five mutants, 545-575, 551-575, 552-575, 553-575, and 548-565, showed larger ΔR values than that of the parent EGFP-TRPC5. However, a comparison of ΔR/Roxi, which is the ratio of ΔR to the fluorescence emission ratio (R) of the oxidized form, should be used to evaluate the enhancement of the signal response. Four mutants, 545-575 (−14%), 551-575 (−31%), 552-575 (−22%) and 553-575 (−17%) showed larger ΔR/Roxi than that of the parent EGFP-TRPC5 (−9%), whereas 548-565 (−8%) showed a similar ΔR/Roxi to that of the parent EGFP-TRPC5. The signal response was enhanced in these four mutants; in particular, mutants 551-575, 552-575, and 553-575 showed a 2 to 4 times larger signal response compared to that of the parent EGFP-TRPC5. In contrast, no mutant showed larger ΔR/Roxi values among the three mutants with the smaller RMSD (Table 1, constructs described in blue letters). Therefore, in silico screening provided candidates for enhanced signal response.
| Constructsa | R | ΔR (Rred − Roxi) | ΔR/Roxi (Rred − Roxi)/Roxi × 100 (%) | |
|---|---|---|---|---|
| w/o DTT (Roxib) | w/DTT (Rredc) | |||
| a Rows 2–11 are shown in red in Fig. 1E, and rows 12–14 are shown in blue in Fig. 1E.b Fluorescence emission ratio (R) in oxidized form.c Fluorescence emission ratio (R) in reduced form. | ||||
| EGFP-TRPC5 | 0.147 | 0.134 | −0.013 | −9% |
| 545-575 | 0.143 | 0.123 | −0.020 | −14% |
| 551-575 | 0.258 | 0.177 | −0.081 | −31% |
| 552-575 | 0.179 | 0.140 | −0.039 | −22% |
| 553-575 | 0.234 | 0.195 | −0.039 | −17% |
| 538-572 | 0.178 | 0.182 | 0.004 | 3% |
| 538-559 | 0.240 | 0.251 | 0.011 | 5% |
| 538-558 | 0.204 | 0.201 | −0.003 | −1% |
| 541-572 | 0.144 | 0.136 | −0.008 | −5% |
| 545-568 | 0.189 | 0.187 | −0.002 | −1% |
| 548-565 | 0.331 | 0.305 | −0.026 | −8% |
| 538-567 | 0.210 | 0.209 | −0.001 | 0% |
| 538-565 | 0.215 | 0.221 | 0.006 | 3% |
| 538-560 | 0.232 | 0.229 | −0.003 | −1% |
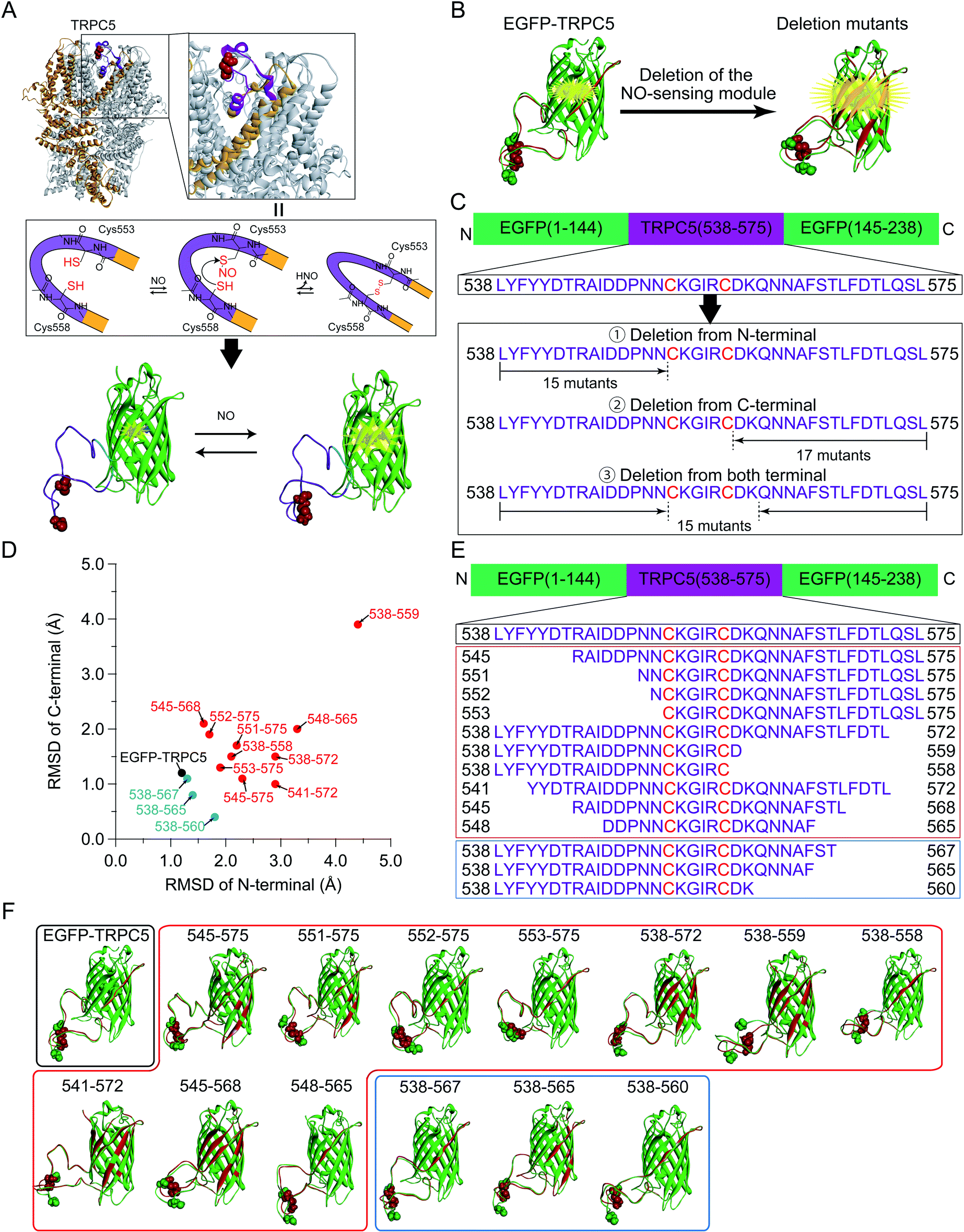
The dependence of ΔR/Roxi on the solution pH, from 5.5 to 8.0, was also measured for all the constructs because the R values between the protonated and deprotonated states of the chromophore depend on pH.7,39 Except for 545-575, 551-575, 552-575, and 553-575, the constructs showed smaller ΔR/Roxi than those of the parent EGFP-TRPC5 at all pH conditions (Fig. S7†). In contrast, 545-575, 551-575, 552-575, and 553-575, which showed larger ΔR/Roxi values at pH 6.9, also showed a larger decrease in ΔR/Roxi values than that of the parent EGFP-TRPC5 from pH 5.5 to 8.0 (Fig. 2). Based on these results, mutants 551-575, 552-575 and 553-575, showing larger ΔR/Roxi than EGFP-TRPC5 under all pH conditions tested, were further investigated as redox sensors.
 | ||
| Fig. 2 pH dependency of the R values of reduced (red) and oxidized (blue) forms. (A) Parent EGFP-TRPC5 and mutants (B) 545-575, (C) 551-575, (D) 552-575, and (E) 553-575, which show a larger ΔR/Roxi than that of the parent EGFP-TRPC5. Reduced forms were prepared by DTT treatment at pH 8.0, then diluted six times to adjust to each pH condition. pH 5.6 or 6.1: buffer containing 100 mM citrate, 500 mM NaCl, and 0.005% Tween-20 (triangles). pH 6.1, 6.5, 6.9, 7.4, or 8.0: buffer containing 100 mM phosphate, 500 mM NaCl, and 0.005% Tween-20 (circles). Oxidized forms were prepared with the same procedure without DTT. At pH 6.1, two independent data sets with different buffer compositions are shown. Data points are mean ± s.e.m. (n = 3). | ||
NO-sensing of mutants selected during the two-step screening
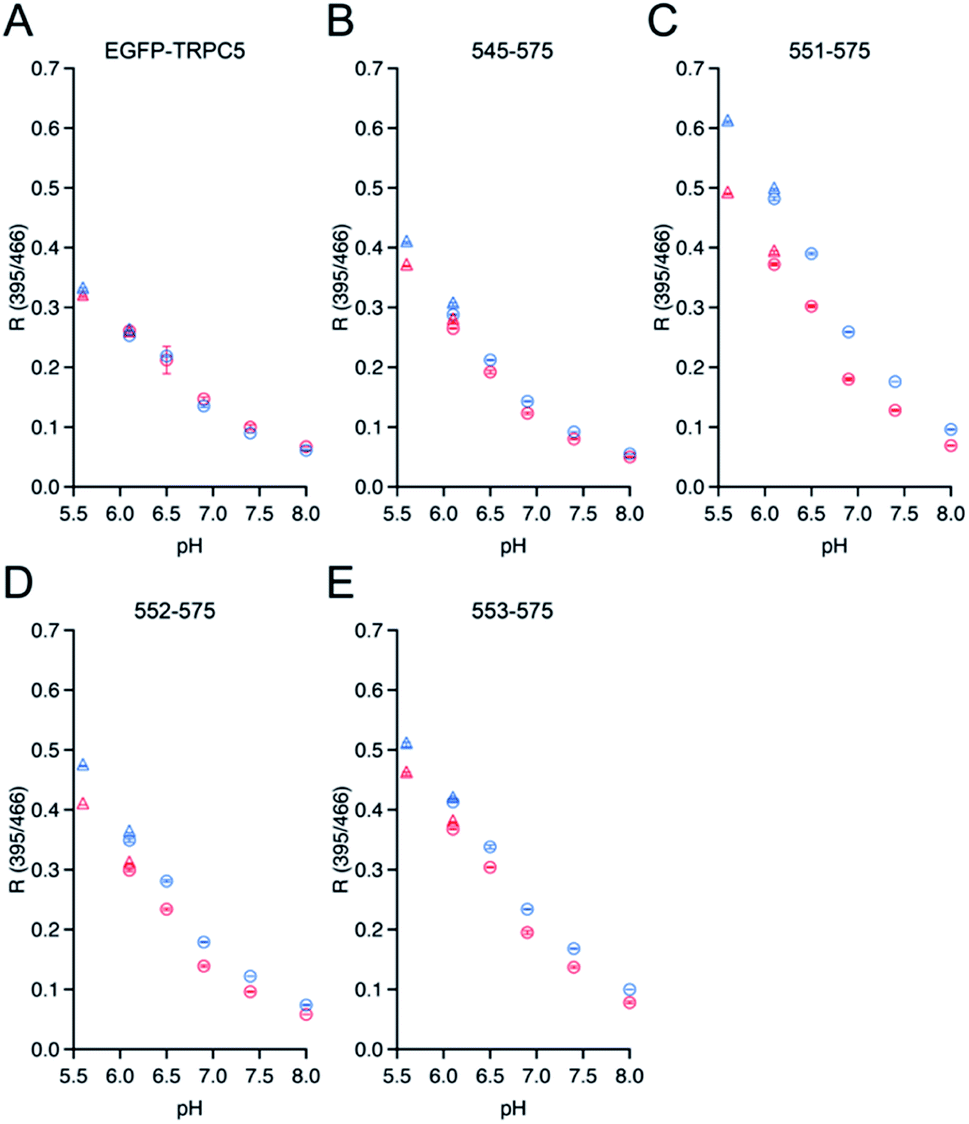
To investigate whether the three mutants 551-575, 552-575, and 553-575 could detect NO by forming disulfide bonds, the R values and the free thiol group contents of these mutants upon reaction with NO were evaluated. The three mutants were purified in the same manner as that described previously for the parent EGFP-TRPC5 (Fig. S8†).34 These mutants showed over 95% purity in SDS-PAGE analyses (Fig. S8A†). The samples were characterized by MALDI-TOF mass spectroscopy (see the experimental section). The UV-Vis absorption spectra indicated two absorption maxima at approximately 395 nm and 490 nm (Fig. S8B†). The excitation spectra monitored at 509 nm emission showed two excitation maxima at approximately 395 nm and 466 nm (Fig. S8C†). The emission spectra of the mutants excited at 395 nm and 466 nm showed an emission peak at 509 nm (Fig. S8D and E†). These optical properties of the mutants were comparable to those of the parent EGFP-TRPC5.34The free thiol group contents in purified 551-575, 552-575, and 553-575 evaluated using the DTNB method were less than 10%, 13%, and 11%, respectively. It should be noted that the two cysteine residues, Cys49 and Cys71, in the original EGFP were substituted with Ser49 and Val71,40 respectively, as described in a previous report (Fig. S9†).34 Thus, these results indicate that most Cys553 and Cys558 residues in the TRPC5 partial segment of the purified mutants formed disulfide bonds. Next, correlations between the R values and the free thiol group content were investigated. These mutants were reduced with DTT, and excess DTT was removed using size exclusion chromatography. The free thiol group contents of the mutants 551-575, 552-575, and 553-575 were then determined to be 100% in the reduced form (Table 2). The change in fluorescence emission upon disulfide bond cleavage was analyzed by comparing the fluorescence spectra of the reduced and oxidized forms (Fig. 3). When excited at 395 nm, the fluorescence emission intensity at 509 nm of the mutants 551-575 and 552-575 decreased in the reduced form compared to that in the oxidized form, and that of the mutant 553-575 did not change upon disulfide bond cleavage. The emission intensity at 509 nm when excited at 466 nm increased for the reduced form in all of the mutants. Therefore, the R values of all mutants decreased upon reduction with DTT, as in the in vitro screening (Fig. 3). The R values decreased to 31%, 20%, and 14% for mutants 551-575, 552-575, and 553-575, respectively, which were slightly lower than those observed in the in vitro screening. Among them, the mutant 551-575 showed four times larger change in the R value compared to that of the parent EGFP-TRPC5 (a decrease of 8%).
| Conditions | Constructs | ||
|---|---|---|---|
| 551-575 | 552-575 | 553-575 | |
| Without DTT | <10% | <13% | <11% |
| With 1 mM DTT | 100% | 100% | 100% |
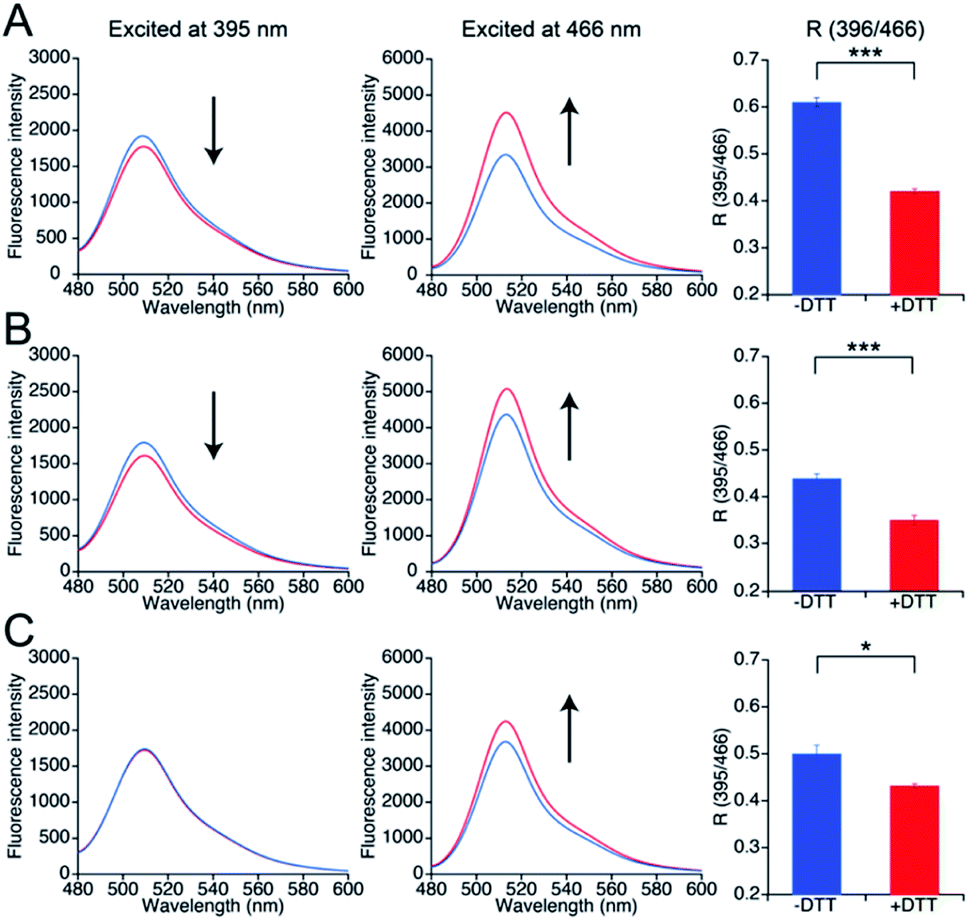 | ||
| Fig. 3 Spectral changes upon reduction of disulfide bonds in mutants (A) 551-575, (B) 552-575, and (C) 553-575 with DTT. Left: emission spectra when excited at 395 nm; center: emission spectra when excited at 466 nm; right: R values in reduced (blue) and oxidized (red) forms. R values were calculated by dividing the fluorescence intensity at 535 nm excited at 395 nm by that excited at 466 nm. *p < 0.05 and ***p < 0.001. | ||
Next, the responses of the mutants in the reduced form to NO were evaluated. Upon DTT reduction, the free thiol group content of mutants 551-575, 552-575, and 553-575 was determined to be 100%, 71%, and 74%, respectively, using DTNB quantitation (Table 3). The R values of the reduced mutants were lower than those of their respective oxidized forms (Fig. 4). The reduced mutants were treated with a NO donor, 1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC7),41 which releases two NO molecules with a half-life of 2.4 min in the reaction condition as described previously.34 In the presence of NOC7 (500 μM), all the mutants showed higher R values than the respective reduced form (Fig. 4). The free thiol group contents of these mutants were drastically decreased to 11% or less than 10% after the reaction with NO (500 μM NOC7) (Table 3). These results indicate that the mutants 551-575, 552-575, and 553-575 show higher signal responses than that of the parent EGFP-TRPC5 to NO with the reaction of their thiol groups in a ratiometric manner. The reaction pathway for disulfide bond formation upon reaction with NO was confirmed. The presence of an S-nitrosylated thiol group as an intermediate of disulfide bond formation was evaluated by the treatment with ascorbic acid that selectively reduces the S-nitrosylated thiol group in the presence of disulfide bond, as described previously.34,42 The free thiol group contents of these mutants after 4.5 h reaction in 551-575 and 2.5 h reaction in 552-575 and 553-575 with NO were similar or did not change upon treatment with ascorbic acid (5 mM) (Table 3). This result indicates that almost all cysteine residues in these mutants reacted with NO by forming a disulfide bond without preserving the S-nitrosylated form. The reaction of mutant 551-575 with NO was also analyzed at an early stage to verify the formation of S-nitrosyl cysteine. After reaction with NO for 30 min, the free thiol group content of mutant 551-575 was 43%, which was increased to 52% upon treatment with ascorbic acid (5 mM). Although the results suggest the presence of S-nitrosylated cysteine (9%), almost of the S-nitrosylated cysteine residues in 551-575 were converted to the disulfide bond form after 30 min, suggesting that disulfide bond formation was faster than the S-nitrosylation reaction. These results indicate that the increase in the signal response of these mutants was the outcome of disulfide bond formation upon reaction with NO (500 μM NOC7), with little contribution from the S-nitrosyl cysteine group to detect NO in a ratiometric manner.
| Conditions | Constructs | |||
|---|---|---|---|---|
| 551-575 | 552-575 | 553-575 | ||
| 30 min | 230 min | 150 min | 150 min | |
| Without DTT | <10% | <10% | <13% | <11% |
| With 1 mM DTT | 100% | 100% | 71 ± 2% | 74 ± 2% |
| With 500 μM NOC7 | 43 ± 3% | 11 ± 1% | <10% | <10% |
| With 5 mM ascorbic acid | 52 ± 3% | 15 ± 3% | <10% | <10% |
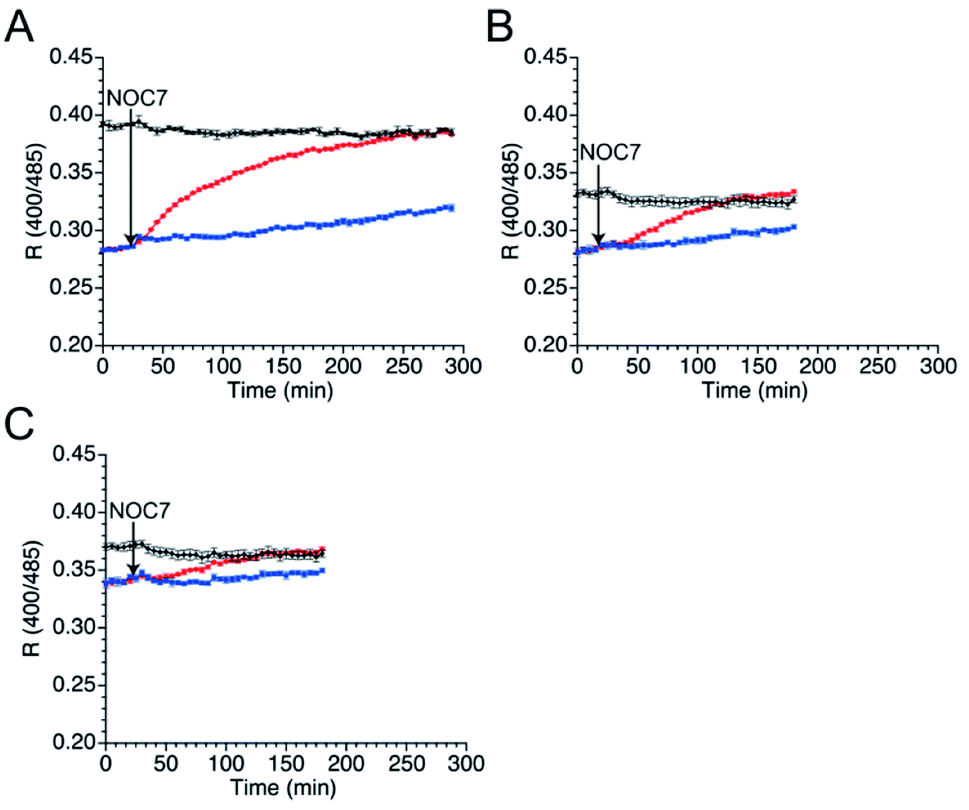 | ||
| Fig. 4 Time course changes in R values of mutants (A) 551-575, (B) 552-575, and (C) 553-575 in the presence of NO (500 μM NOC7). R values were calculated by dividing the fluorescence intensity at 535 nm excited at 400 nm by that excited at 485 nm. Reduced (blue and red) and oxidized (black) forms were treated with (red and black) or without (blue) NOC7 (500 μM) between 25 to 30 min. | ||
H2O2 detection by mutants 551-575, 552-575, and 553-575
As well as NO, H2O2 also function as second messenger upon reaction with cysteines to form sulfenic acid and subsequently disulfide bond (Fig. S10†).43,44 Wild-type TRPC5 responds more rapidly to NO over H2O2,28 but the parent EGFP-TRPC5 reacted with H2O2 as well.34 The responses of mutants 551-575, 552-575 and 553-575 to H2O2 were also evaluated. After preparation of the reduced form of these mutants in the same manner, fluorescence emission in the presence of H2O2 (500 μM) was measured (Fig. 5). The R values of all mutants in the reduced forms increased to those of the respective oxidized forms (Fig. 5), and the free thiol group content decreased to less than 11% for all mutants (Table 4). These results indicate that mutants 551-575, 552-575, and 553-575 also detected H2O2 in a ratiometric manner by forming disulfide bonds. These results are consistent with those of the parent EGFP-TRPC5.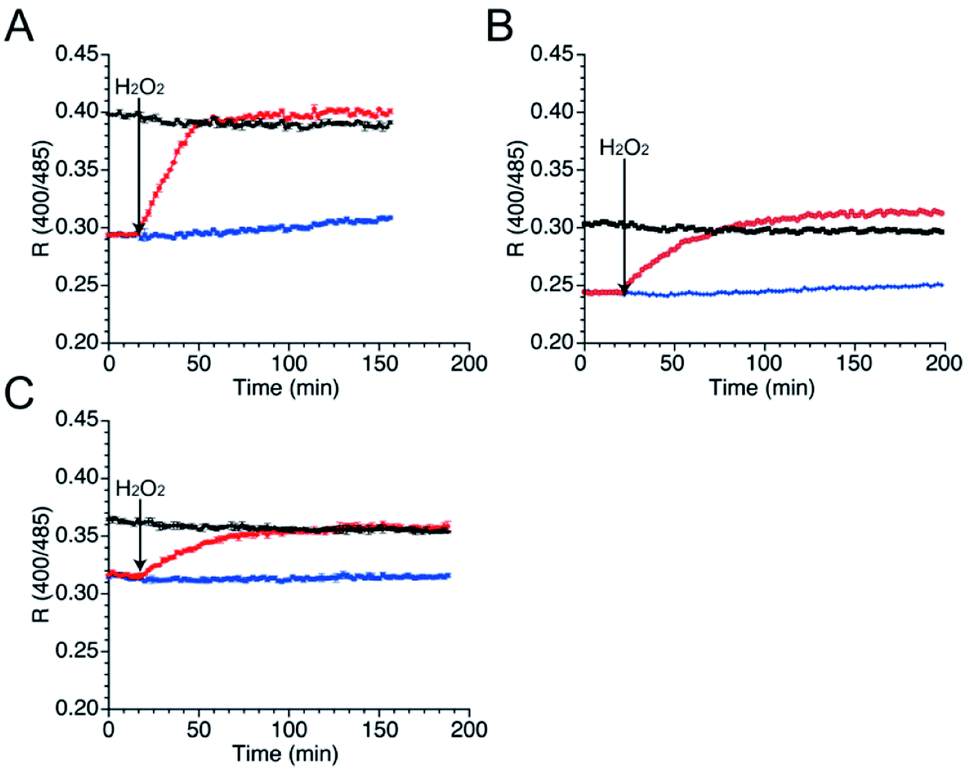 | ||
| Fig. 5 Time course changes in R values of mutants (A) 551-575, (B) 552-575, and (C) 553-575 in the presence of H2O2 (500 μM). R values were calculated by dividing the fluorescence intensity at 535 nm excited at 400 nm by that excited at 485 nm. Reduced (blue and red) and oxidized (black) forms were treated with (red and black) or without (blue) H2O2 (500 μM) between 16 to 18 min. | ||
| Conditions | Constructs | ||
|---|---|---|---|
| 551-575 | 552-575 | 553-575 | |
| Without DTT | <10% | <13% | <11% |
| With 1 mM DTT | 100% | 100% | 100% |
| With 500 μM H2O2 | <11% | <10% | <10% |
Kinetics of mutants 551-575, 552-575, and 553-575 upon reaction with NO and H2O2
To compare the kinetics and specificity of mutants 551-575, 552-575, and 553-575 and the parent EGFP-TRPC5 for reactions with NO and H2O2, the half-maximum times of the reactions were estimated. The half-maximum times were defined as the time when R values reach the half of their maximum values (Table 5). Comparison of the half-maximum times upon reaction with NO and H2O2 within the same construct shows that the response to 500 μM H2O2 was faster than that to NO (500 μM NOC7) for all constructs, indicating that they were more prone to react with H2O2 than the original TRPC5. Furthermore, the half-maximum times of mutants 551-575, 552-575, and 553-575 were three to five times longer than that of the parent EGFP-TRPC5. Based on the fact that the S-nitrosylation step was slower than the disulfide bond formation step as described in the previous section (Table 3), the S-nitrosylation step, not the disulfide bond formation step, in these mutants was slower than that in the parent EGFP-TRPC5. A shift in the pKa of cysteines to a basic pH in these mutants could decrease the S-nitrosylation rate of the thiolate group.In-cell application of mutant 551-575
The mutant 551-575 was applied in human embryonic kidney (HEK) 293 cells to investigate whether it could be utilized as a redox sensor in mammalian cells. The mutant 551-575 was subcloned into the mammalian expression vector pCI-neo and expressed in HEK293 cells. The mutant 551-575 showed sufficient fluorescence intensity at 535 nm when excited at 403 nm and 480 nm, corresponding to the protonated and deprotonated states of EGFP chromophore, respectively. In contrast, autofluorescence derived from intracellular molecules of mammalian cells, such as NADH or flavin, was not displayed in the same degree of contrast applied to monitor 551-575 (Fig. 6A).45,46 Extracellular H2O2 (500 μM) was added to the bulk solution where the cells were soaked, causing H2O2 to diffuse freely and invade the cells. Upon addition of H2O2, the cells were not damaged severely because their plasma membranes were intact with slight change in the cell size (Fig. 6B). Although the fluorescence intensity varied depending on the cells, the R values of the cells indicated the similar extent, ranging from 0.12 to 0.15 (Fig. 6B and C). Analysis of R values revealed the increase ranging from 18% in cells showing a minimum increase, to 49% in cells showing a maximum increase upon addition of H2O2, with 34% as the average (Fig. 6D and E). Considering the 31% increase in R values upon reaction with H2O2 in the mutant 551-575 under in vitro measurement, the signal responses of mutant 551-575 upon reaction with H2O2 in cells were comparable with those of the in vitro measurements, although the signal responses varied in different cells. In contrast, the original EGFP did not show changes in fluorescence emission ratios. These results indicate that mutant 551-575 detected H2O2 with an increase in R value in mammalian cells, and therefore can be used as a redox biosensor in a cellular system.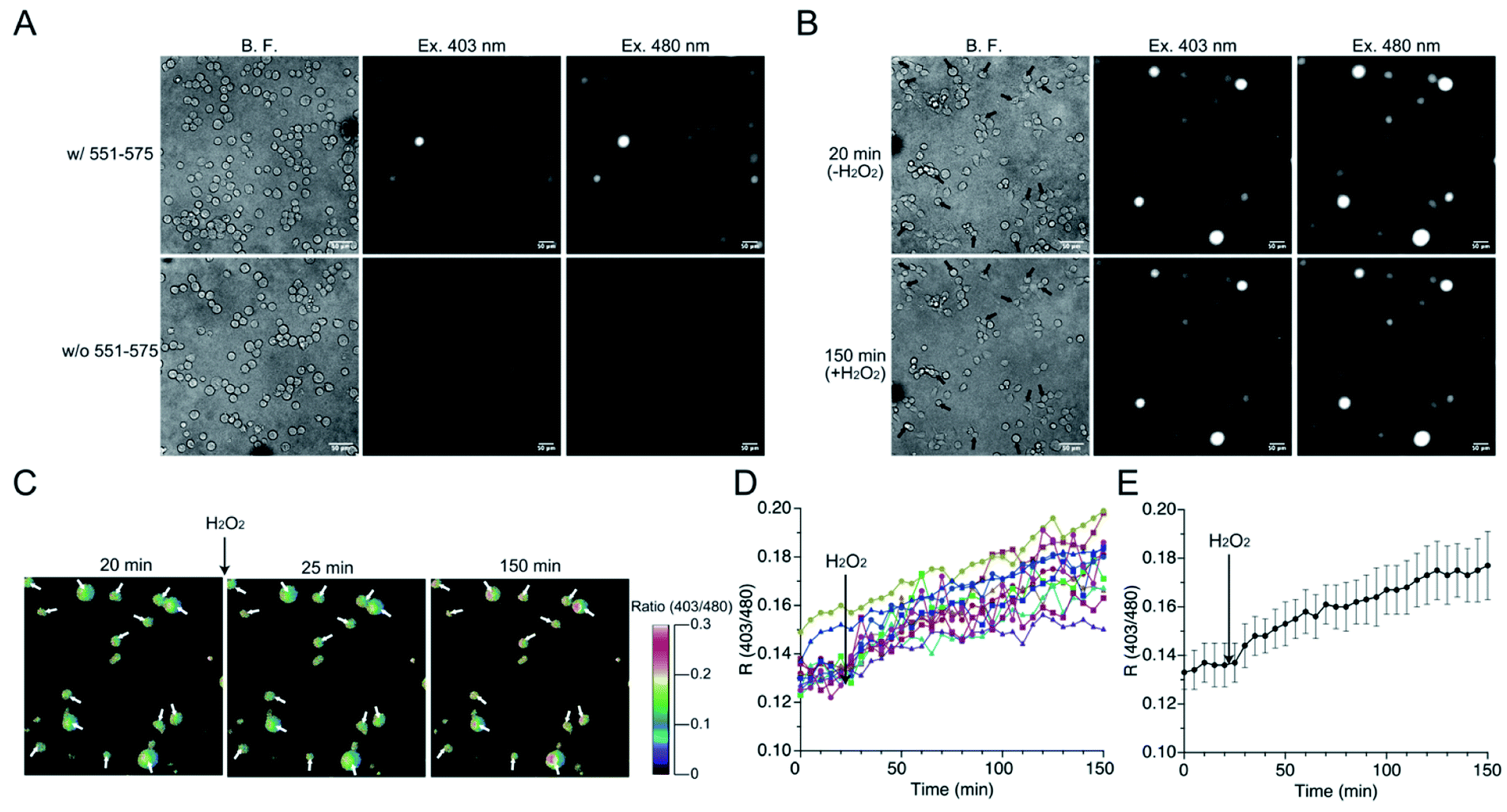 | ||
| Fig. 6 (A) Bright field (left), fluorescence images at 535 nm when excited at 403 nm (center) and 480 nm (right) of HEK293 cells with (top) or without (bottom) expressing mutant 551-575. (B) Bright field images (left) and fluorescent images of fluorescence emission at 535 nm when excited at 403 nm (center) and 480 nm (right) of mutant 551-575 expressed in HEK293 cells in response to H2O2. Cells indicated by arrows show sufficient fluorescence. Half of the pool volume of 1.5 mM H2O2 was added to a final concentration of 500 μM H2O2, causing H2O2 to diffuse freely and uniformly spread, between 20 and 25 min. (C) Time-lapse of pseudo color images of fluorescence emission ratios of mutant 551-575 expressed in HEK 293 cells in response to H2O2. Fluorescence emission ratio was calculated by dividing the fluorescence intensity when excited at 403 nm by that when excited at 480 nm from which the background intensities were subtracted. (D and E) Analysis of the fluorescence response of mutant 551-575 expressed in HEK293 cells upon addition of extracellular H2O2 of (D) individual cells and (E) average. Time-lapses of fluorescence images when excited at 403 nm and 480 nm were analyzed with ImageJ software (NIH) (n = 17 for two cultures). | ||
The responses to extracellular NO of the mutant 551-575 expressed in HEK293 cells were also evaluated (Fig. S11†) by the addition of NO (500 μM NOC7 in HBS) to the outer cellular solution. Analysis of cells revealed about 30% of cells expressing the mutant 551-575 increase in the R values ranging from 5 to 20%. The results indicated the potential reactivity of 551-575 to NO in mammalian cells. However, the changes of R values were smaller than that observed in the reaction of H2O2. This result could be caused by the slower kinetics of the mutant 551-575 to NO than that to H2O2 as observed in the in vitro reactions (Table 5).
Discussions
A structural change in the sensing module induced by a recognition and/or reaction event is transduced to fluorescence emission changes of the AFP signal transduction module in AFP-based biosensors.7,8 The lack of structural information in the recognition module often hinders structure-based design of AFP-based biosensors. Previously, the putative NO-sensing segment of TRPC5, L538-L575, was embedded into EGFP, generating EGFP-TRPC5, to monitor the putative structural change in the TRPC5 segment upon disulfide bond formation between Cys553 and Cys558 after reaction with NO. EGFP-TRPC5 showed a fluorescence intensity change with the formation of a disulfide bond in the presence of NO, but the ratiometric signal response for NO was far smaller than that applicable for a biosensor.34 In this study, a new facile screening strategy to increase the signal response of an AFP-based biosensor is proposed. We reasoned that appropriate deletion of the amino acid residues from the putative NO-sensing segment of TRPC5, L538-L575, could promote transduction of structural changes at the putative NO-sensing segment associated with disulfide bond formation to a structural perturbation of EGFP. The strategy consists of two screening steps: first in silico and then in vitro. In silico screening consisted of structural change evaluation in 47 deletion mutants of the putative NO-sensing segment upon disulfide bond formation using RMSD between the reduced and oxidized forms. Many of the deletion mutants showed larger RMSD than that of the parent EGFP-TRPC5. Among the 47 mutants, 10 mutants showing a larger RMSD were chosen for the second screening (group A). In addition, three mutants that showed RMSD similar to that of the parent EGFP-TRPC5 were chosen (538-565, 538-567, and 538-560) to validate the in silico screening (group B). For the in vitro screening, the selected mutants were expressed in E. coli to evaluate the ΔR/Roxi values upon their reduction in crude oxidized forms. None of the mutants in group B showed a larger ΔR/Roxi than that of parent EGFP-TRPC5. In contrast, the four mutants in group A showed larger ΔR/Roxi than that of the parent EGFP-TRPC5. These results indicate that in silico screening based on RMSD helped to successfully identify candidates, and this two-step screening successfully provided the mutants showing enhanced signal response without the detailed information on the structural change. Of course, it should be mentioned that the large RMSD did not always correspond to a large signal response, because the in silico simulation applied in this study was a quite simple structural optimization. Thus, the screening for the optimization of signal response was highly required. This facile two-step screening method could contribute to the efficient optimization of deletion mutation.After the screening, further evaluations of mutants 551-575, 552-575, and 553-575 with respect to the reactivity were conducted under in vitro conditions. As a result, these mutants showed larger ΔR/Roxi, as in the in vitro screening, upon reduction of the disulfide bond and formation of the disulfide bond in response to NO and H2O2. Furthermore, mutant 551-575 also changed its R value upon reaction with H2O2 in mammalian HEK293 cells, with a 34% increase as high as that under in vitro conditions (Fig. 6). Mutant 551-575 also showed the response to NO in mammalian HEK293 cells (Fig. S11†), while its reactivity was smaller than that to H2O2. These results show that mutant 551-575, which was obtained from a facile two-step screening, could be applied as a redox biosensor for in-cell measurements. Therefore, our screening strategy for successfully enhancing the signal response provided mutants with sufficiently strong signals for cell application. Considering that mutant 551-575 showed a change in R values upon reduction from pH 5.5 to 8.0 in the in vitro screening, it has the potential to be applied as a redox sensor in many pH ranges. Due to the biological importance of cellular NO dynamics,47–49 there are demands for the development of genetically encoded NO sensors. The AFP based genetically encoded NO sensor has been developed to detect endogenous NO production in living cells.50 However, the NO sensor required the addition of relatively high concentration of iron(II) that could cause toxicity to the cell. While the mutant 551-575 requiring no additives for NO detection is potentially applicable as a genetically encoded NO sensor in the cell, its lack of the specificity to NO and H2O2 has to be compensated by a simultaneous usage of specific H2O2 sensor.51
Wild-type TRPC5 expressed in HEK293 cells shows a specific reaction to NO (10 μM SNAP as an NO donor) over 100 μM H2O2.28 In our previous report, the parent EGFP-TRPC5 containing the putative NO-sensing segment did not show specificity to NO over H2O2. Mutants 551-575, 552-575, and 553-575, which contained the partial segments of TRPC5, N551-L575, N552-L575, and C553-L575 as the sensing module, respectively, also did not have the specificity to NO over H2O2. We had suggested that the selectivity of wild-type TRPC5 to NO against H2O2 should be caused by the difference in hydrophobicity or polarity of NO and H2O2.34 That is, more hydrophobic NO would be more accessible to the transmembrane domain of wild-type TRPC5 in the plasma membrane. From the cryo-EM structure of wild-type TRPC5,29 the partial segment L538-L575 is located in the extracellular region near the plasma membrane (Fig. S12,† represented in purple). In detail, the C-terminal side of this segment buried in the plasma membrane showed lower charge density and higher hydrophobicity and the N-terminal side of this segment exposed outside the plasma membrane showed higher charge density and lower hydrophobicity (Fig. S12B and C†). However, mutants remaining higher hydrophobic region still less selective toward NO against H2O2, indicating that the hydrophobic transmembrane domain with plasma membrane should be required to reinforce the accessibility of NO over H2O2.
The longer half-maximum times of three mutants 551-575, 552-575, and 553-575 compared to that of the parent EGFP-TRPC5 suggest that the pKa of the thiol group in Cys553 or Cys558 would shift to the acidic side upon deletion of the N-terminal side of the partial segment, L538-N552. The pKa of the thiol group is modulated by the interaction with the surrounded positive amino acid residues.52,53 However, because of the presence of both positively and negatively charged residues around Cys553 and Cys558, as observed in the surface charge distribution of L538-L575 (Fig. S13†), a clear insight into the effect of the amino acid residue(s) on the shift of pKa still remains to be obtained. Alternatively, the pKa shift of the cysteine thiol groups would not reflect the effect of amino acid residue(s) of TRPC5 in nature, but would take place only in constructs where the extracted NO-sensing segment of TRPC5 was embedded into EGFP and exposed to the solution.
Conclusions
Partial deletion of the recognition module of EGFP-TRPC5 enhanced the signal response by efficiently transducing the structural change in the recognition module associated with disulfide bond formation to the structural perturbation of the EGFP chromophore. A two-step screening strategy was developed to obtain deletion mutants that enhanced the signal response of EGFP-TRPC5. In the first screening, evaluation of the structural change of the recognition module based on RMSD with in silico simulations provided 10 candidates from 47 possible mutants. As a result, this screening strategy successfully provided mutants that showed two to four times larger ratiometric signal response than that of the parent EGFP-TRPC5 upon reaction with NO and H2O2. Finally, we demonstrated that mutant 551-575 could be applicable as a redox sensor for cell experiments. Mutant 551-575 would be applicable as NO biosensor in the cell without requiring any additives. While the two-step screening revealed a limitation to optimize the specificity of the sensor to NO over H2O2, the strategy successfully enhanced the signal response of parent AFP-based biosensor. In the construction of AFP-based biosensors, enhancement of the signal responses requires multi-step or large-scale screening even when structural information is available, that is, optimization of the introduction site in the structural change domain or the linker length and sequence between AFP and the structural change domain. The two-step screening strategy described herein provides a convenient and high-throughput screening method for these optimizations in the construction of AFP-based biosensors. Although NO-sensing segment exhibited specificity for NO over H2O2 in wild-type TRPC5, the deletion of the N-terminal side of Cys553, corresponding to the extracellular region of TRPC5, from the recognition module of the mutants resulted similar reactivity to NO and H2O2 as has been observed for the parent EGFP-TRPC5. Therefore, not the hydrophilicity and charge of the N-terminal side of the TRPC5 NO-sensing segment, but the hydrophobicity and lack of charge of the transmembrane domain would contribute to the specificity and accessibility to NO in the TRPC5 NO-sensing segment.Materials and methods
Materials
Restriction enzymes (EcoRI, SalI, and DpnI) were purchased from New England BioLabs. Purified oligonucleotide primers for gene construction were purchased from Thermo Fisher Scientific (Waltham, MA, USA). E. coli BL21 (DE3) competent cells were purchased from Invitrogen (Carlsbad, CA, USA). The QIAprep Spin Miniprep Kit and Mini Elute Gel Extraction Kit were purchased from Qiagen K.K. (Tokyo, Japan). His GraviTrap™, PD-10, HiTrap™ Butyl HP (5 mL), and HisTrap™ HP (5 mL) columns were purchased from GE Healthcare Japan (Tokyo, Japan). PrimeSTAR HS DNA polymerase, T4 DNA ligase, and E. coli DH5α competent cells were purchased from TaKaRa Bio (Shiga, Japan). NOC7 was purchased from Dojindo (Kumamoto, Japan). All other chemicals were purchased from Wako Chemicals (Osaka, Japan), Tokyo Chemical Industry (Tokyo, Japan), Sigma-Aldrich (Tokyo, Japan), and Nacalai Tesque (Kyoto, Japan).In silico design of deletion mutants in the first in silico screening
Discovery Studio (version 3.1, Accelrys) was used for in silico simulations. The parent EGFP-TRPC5 in the reduced form was designed by embedding the amino acid sequence of Leu538 to Leu575 into EGFP (PDB ID: 2B3P) between Asn144 and Phe145, as described in a previous report.34 All deletion mutants were simulated based on the structure of the parent EGFP-TRPC5 in the reduced form. Fixed-atom constraints (tool in Discovery Studio) were applied to the EGFP structure. Amino acids in the recognition module were deleted from the structure of the parent EGFP-TRPC5 in the reduced form. Sketches tool (tool in Discovery Studio) was applied to create new peptide bonds between the carbon atom in the N-terminal side and the nitrogen atom at the C-terminal side of the deleted amino acids. Clean geometry tool (tool in Discovery Studio) was applied to each deletion mutant 10 times to predict sterically acceptable structures just after creating the connection between the carbon and nitrogen atoms. A standard dynamic cascade tool (tool in Discovery Studio) was applied to perform molecular mechanics (MM) and molecular dynamics (MD) calculations on the structures after the application of the clean geometry tool. The structures of the mutants in oxidized form were obtained with same procedure after creating disulfide bonds between the thiols of Cys553 and Cys558.To compare the structural changes upon formation of disulfide bonds, the obtained structures of each mutant in the reduced and oxidized forms were superimposed by matching the coordinates of the Cα atoms in each residue from Leu119 to Ile128, whose residues were opposite to the conformational change domain. To estimate the structural changes upon quantitative formation of the disulfide bonds, Root Means Square Deviation (RMSD) tool (Discovery Studio) was applied to the backbone of amino acids of the N-terminal side of Cys553 and the C-terminal side of Cys558 of the loop region. RMSD values of the difference in coordinates between the reduced and oxidized forms of each region were calculated. RMSD values of the N-terminal side were plotted against RMSD values of the C-terminal side for each mutant.
Plasmid construction
Plasmids encoding mutants selected from the in silico screening, in which the NO-sensing segment of TRPC5 (L538-L575) of the parent EGFP-TRPC5 was shortened to 545-575, 551-575, 552-575, 553-575, 538-572, 538-567, 538-565, 538-560, 538-559, 538-558, 541-572, 545-568, or 548-565, were sequentially constructed. First, the plasmids encoding mutants in which one side was deleted from the parent EGFP-TRPC5 were constructed. The plasmids encoding EGFP-TRPC5 was amplified by PCR using primers 1 and 2, 3 and 4, 5 and 6, 7 and 8, 9 and 10, 11 and 12, 13 and 14, 15 and 16, 17 and 18, or 19 and 20 (Table S2†) to generate mutants, 545-575, 551-575, 552-575, 553-575, 538-572, 538-567, 538-565, 538-560, 538-559, or 538-558, respectively. The PCR product was treated with DpnI (New England Biolabs Japan) and transformed into E. coli DH5α competent cells. Vectors encoding mutants were collected, purified and sequenced. Plasmids encoding 541-572, 545-568, and 548-565 were constructed using primers 21 and 22, 23 and 24, or 25 and 26 (Table S2†) and the plasmids encoding 538-572, 545-575, or 538-565, respectively, with following the same procedure.To sub-clone mutant 551-575 to mammalian expression vector pCI-neo, the gene encoding EGFP-TRPC5 was obtained by digesting peT29a encoding the parent EGFP-TRPC5 with NdeI and XhoI and sequentially amplified by PCR using the primers listed in Table S3.† The PCR products were run on 1% agarose gels (TAE) and purified using a Mini Elute Gel Extraction Kit. The PCR products and pCI-neo were digested with EcoRI and SalI, purified in the same manner, and incubated with T4 DNA ligase. The mixture was transformed into E. coli DH5α competent cells for amplification. The NO-sensing segments of the parent EGFP-TRPC5 were shortened to 551-575 with same procedure to that of in vitro measurement.
Expression and purification of proteins
Plasmids encoding the 13 mutants were transformed into E. coli BL21 (DE3) competent cells.For partial purification for in vitro screening, the transformed cells were grown at 37 °C until the OD600 reached 0.6–0.8, and protein expression was induced with 1 mM IPTG for 24 h at 25 °C. The soluble fractions of the cell lysates containing the target proteins were loaded onto a His GraviTrap™ column with 60 mM imidazole in 50 mM phosphate and 500 mM NaCl (pH 8.0) and eluted with 100 mM imidazole, 50 mM phosphate, and 500 mM NaCl (pH 8.0). Each elution sample containing each target protein was collected and loaded onto a PD-10 column equilibrated with 100 mM phosphate and 0.005% Tween-20 (pH 8.0).
For further investigation of mutants 551-575, 552-575, or 553-575, cells were grown at 37 °C until the OD600 reached 0.5–0.7, and protein expression was induced with 1 mM IPTG for 24 h at 18 °C. The soluble fractions of cell lysates containing the target proteins were loaded onto a HisTrap™ HP column with a linear gradient of 60–225 mM imidazole in 50 mM phosphate and 500 mM NaCl (pH 8.0). Fractions containing mutants 551-575, 552-575, or 553-575 were collected and diluted to the initial condition of the next purification step (50 mM phosphate, 900 mM (NH4)2SO4 (pH 8.0)). The resultant products were loaded onto HiTrap™ Butyl HP and eluted using an (NH4)2SO4 gradient (from 900 to 0 mM). Fractions containing 551-575, 552-575, or 553-575 were collected and dialyzed against a solution containing 100 mM phosphate, 200 mM NaCl, and 50% glycerol (pH 8.0), and stored at −20 °C. Concentrations of mutants 551-575, 552-575, or 553-575 in the glycerol stocks were determined using absorbance at 440 nm derived from the isosbestic point of the EGFP chromophore (molecular coefficient of EGFP-TRPC5 at 440 nm: 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1).
000 M−1 cm−1).
Measurement of the fluorescence intensity change upon reduction in in vitro screening
After PD-10 treatment, mutants were reduced in a solution containing 100 mM phosphate, 0.005% Tween 20, and 1 mM DTT (pH 8.0) for 2 h at 25 °C. The reduced solutions were diluted six times to shift the pH to 5.6 or 6.1 with buffer containing 100 mM citrate, 500 mM NaCl, and 0.005% Tween 20 (pH 5.5 or 5.9) or to 6.1, 6.5, 6.9, 7.4, or 8 with buffer containing 100 mM phosphate, 500 mM NaCl, and 0.005% Tween 20 (pH 5.8, 6.3, 7.0, 7.3, or 8.0). Oxidized solutions were prepared using the same procedure but without DTT treatment. The emission spectra of the reduced and oxidized solutions were measured by excitation at 395 nm and 466 nm with an Infinite M200 PRO (TECAN, Zürich, Switzerland) at 20 °C. The fluorescence emission ratios were calculated by dividing the fluorescence intensity at 510 nm excited at 395 nm by that excited at 466 nm.Characterization of 551-575, 552-575, and 553-575 in in vitro
Mutants 551-575, 552-575, and 553-575 were characterized by MALDI-TOF mass spectrometry (AXIMA-LNR, SA matrix, Shimadzu, Kyoto, Japan). Mutant 551-575: m/z calcd 31602; observed 31586. Mutant 552-575: m/z calcd 31488; observed 31475. Mutant 553-575: m/z calcd 31374; observed 31356. Absorption spectra were measured using a UV-2550 UV-Vis spectrometer (Shimadzu, Kyoto, Japan). The excitation and emission spectra were measured using an F-7000 fluorescence spectrometer (Hitachi High-Tech Science, Tokyo, Japan) at 20 °C. Samples contained 4 μM 551-575, 552-575, or 553-575 in a buffer containing 100 mM phosphate buffer, 500 mM NaCl, and 0.005% Tween 20 (pH 6.8). The emission spectra were measured by excitation at 395 and 466 nm. The fluorescence emission ratios were calculated by dividing the fluorescence intensity at 509 nm excited at 395 nm by that excited at 466 nm.
Change in fluorescence intensity of mutants 551-575, 552-575, and 553-575 upon reduction for further in vitro measurement
The disulfide bonds of mutants 551-575, 552-575, or 553-575 were reduced in a solution containing 70 μM 551-575, 552-575, or 553-575, 100 mM phosphate (pH 8.0), 0.005% Tween 20, and 3.5 mM DTT for 2–3 h at 25 °C. After reduction, the solutions containing mutants 551-575, 552-575, or 553-575 were loaded onto a Micro Bio-Spin Chromatography Column P-6 (Bio-Rad) equilibrated with a buffer containing 100 mM phosphate, 500 mM NaCl, and 0.005% Tween 20 (pH 6.8). Concentrations of free thiol groups in mutants 551-575, 552-575, or 553-575 were measured using the DTNB method with Ellman's reagent (see Quantitation of the free thiol group). Concentrations of mutants 551-575, 552-575, or 553-575 were determined with absorbance at 425 nm derived from the isosbestic point of the EGFP chromophore from pH 5.5 to 7.0 (molecular coefficient of EGFP-TRPC5 at 425 nm: 21000 M−1 cm−1). The excitation and emission spectra were measured using an F-7000 fluorescence spectrometer at 20 °C. Samples contained 4 μM 551-575, 552-575, or 553-575 in a buffer containing 100 mM phosphate buffer, 500 mM NaCl, and 0.005% Tween 20 (pH 6.8). Oxidized solutions were prepared using the same procedure without DTT treatment. The emission spectra were measured by excitation at 395 and 466 nm, and the excitation spectra were measured at an emission wavelength of 509 nm.
Reaction of 551-575, 552-575, and 553-575 to NO and H2O2
The disulfide bonds of mutants 551-575, 552-575, or 553-575 were reduced in a solution containing 100 μM 551-575, 552-575, or 553-575, 100 mM phosphate, 0.005% Tween 20, and 5 mM DTT (pH 8.0) for 3 h at 25 °C. After reduction, the solutions containing mutants 551-575, 552-575, or 553-575 were loaded onto a Micro Bio-Spin Chromatography Column P-6 (Bio-Rad) equilibrated with a buffer containing 20 mM citrate, 500 mM NaCl, and 0.005% Tween 20 (pH 5.5). Concentrations of free thiol groups in mutants 551-575, 552-575, or 553-575 were measured using the DTNB method with Ellman's reagent (see next section). Concentrations of mutants 551-575, 552-575, or 553-575 were determined with absorbance at 425 nm derived from the isosbestic point of the EGFP chromophore from pH 5.5 to 7.0 (molecular coefficient of EGFP-TRPC5 at 425 nm: 21000 M−1 cm−1). Reactions of mutants 551-575, 552-575, or 553-575 with NO were monitored by measuring changes in fluorescence emission upon addition of NOC7 (dissolved in 0.01 M NaOH) to the test solution, which was diluted to 12 μM 551-575, 552-575, or 553-575 with buffer containing 100 mM phosphate, 500 mM NaCl, and 0.005% Tween 20 (pH 6.8), at 20 °C with Infinite F PLEX (TECAN, Zürich, Switzerland). Instead of addition of NOC7, an equal amount of 0.01 M NaOH was added to the reaction solutions for control (0 M NOC7). It should be noted that the pH value of the buffer did not change before and after the addition of NOC7 in 0.01 M NaOH. After measurement, the concentrations of free thiol groups of mutants 551-575, 552-575, or 553-575 in response to NO were determined using the DTNB method (see next section). In the case of H2O2, mutants 551-575, 552-575, or 553-575 were treated with H2O2.
Quantitation of the free thiol group
The concentrations of free thiol groups in mutants 551-575, 552-575, or 553-575 were measured using the DTNB method. All samples were first purified by size exclusion chromatography to remove thiol-containing reagents such as DTT. An assay solution was prepared by mixing 100 μM DTNB in a solution containing 100 mM phosphate, 500 mM NaCl, and 0.005% Tween 20 (pH 6.8) with an aliquot of mutants 551-575, 552-575, or 553-575 solutions. The absorption spectra of the assay solutions were measured using an Infinite M200 PRO spectrophotometer (Tecan, Zürich, Switzerland). The concentrations of free thiol groups in mutants 551-575, 552-575, or 553-575 were determined by measuring the absorbance at 412 nm, which was derived from 2-nitro-5-mercaptobenzoic acid. The concentrations of mutants 551-575, 552-575, or 553-575 were also determined from absorbance at 425 nm, which is the isosbestic point of the EGFP chromophore. The concentration of free thiol groups was determined using the following equation:| y = 0.004x + 0.005 |
To mutants 551-575, 552-575, or 553-575 treated with NOC7, solutions containing ascorbic acid (final concentration: 5 mM) were added to specifically reduce the S-nitrosylated thiol. After 30 min, the reaction mixture was treated as described above to determine the concentration of free thiol groups using the DTNB method.
Determination of the half-maximum times
The R values of mutants 551-575, 552-575, or 553-575 under air oxidation conditions (0 M NOC7 or H2O2) were subtracted from the R values upon reaction with NO or H2O2 (500 μM NOC7, 100 μM, 250 μM or 500 μM H2O2) to remove the air oxidation effects. The subtracted R values were normalized by dividing by ΔR upon reduction, which is called the normalized R. The minimum and maximum R values were determined as the average values of normalized R values before the addition of NOC7 or H2O2 and after saturation of the reactions, respectively. The half-maximum values were determined as the mean of the minimum and maximum R values. The average values of the half-maximum times were determined as the mean times of the measurement points before and after the normalized R reached the half-maximum R values, and intervals of measurement points were determined as error bars of the half-maximum times. In the parent EGFP-TRPC5, the normalized R values were smoothed with a linear weighted moving average of three points to remove the fluctuation of the normalized R values resulting from slight ΔR.Cell culture and transfection of vectors
HEK293 cells (ATCC) were cultured in DMEM (Gibco) supplemented with 10% FBS, 30 units per mL penicillin, and 30 μg mL−1 streptomycin at 37 °C under 5% CO2 with 100% humidity. pCI-neo encoding mutant 551-575 was transfected using Lipofectamine 2000 (Invitrogen). The medium was replaced 3 h after transfection.Measurement of fluorescence responses of redox sensors
HEK293 cells were seeded onto a poly L-lysine-coated 35 mm glass-bottom dish (IWAKI) and incubated for 1 h. Cells were washed with HBS containing 107 mM NaCl, 6 mM KCl, 11.5 mM glucose, 20 mM HEPES, 1.2 mM MgSO4, and 2 mM CaCl2, pH 7.4, and 200 μL HBS were added to the dish. Fluorescence was observed using an IX-81 fluorescence microscope (Olympus) equipped with a 20× objective lens (UPlanSApo) and Xe lamp. Fluorescence images when excited at 403 nm and 480 nm was acquired with electron multiplier CCD camera (Hamamatsu Photonics K.K.) by using a 381 nm to 415 nm and a 457 nm to 507 nm band-pass filter for excitation, respectively, and a 505 nm dichroic mirror, and 522 nm to 548 nm band-pass filter for emission. For time-lapse imaging to observe the fluorescence response to H2O2, 100 μL of 1.5 mM of H2O2 dissolved in HBS were gently added to the dish. In the case of NO, 500 μM NOC7 dissolved in HBS was applied. Time-lapse images were acquired every 5 min.Analysis of the fluorescence images and responses of redox sensors
Pseudo images of fluorescence emission ratios were modified with MetaFluor (Molecular devices). First, average background fluorescence intensities, where cell did not exist, were subtracted from fluorescence intensity of whole region. Fluorescence emission ratios were calculated by dividing the subtracted fluorescence intensity when excited at 403 nm by that when excited at 480 nm in each pixel. Pseudo color images of fluorescence emission ratios were displayed proportional to fluorescence emission ratio from 0 to 0.3 in each pixel. Fluorescence images and intensities of cells were analyzed using ImageJ software (NIH). First, cells were specified based on bright field images to determine the area for calculating the fluorescence intensity. The average fluorescence intensity within each cell was then calculated. The background signal was also estimated by specifying regions of any size without cells in each captured image. Fluorescence intensity of cells was determined by subtracting the background intensity from the raw fluorescence intensity of cells.Author contributions
S. T., Y. M., and T. M. designed and initiated the study. S. T. performed all experiments. R. S. assisted in the cell experiments. M. S. assisted with protein expression and purification experiments. S. T. analyzed the data with the help of E. N. and T. M. S. T. wrote the manuscript, and all the authors revised it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant number 17H05529 (T. M.) and by JST CREST grant number JPMJCR18H5 (T. M.), Japan.Notes and references
- B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- A. E. Palmer, Y. Qin, J. G. Park and J. E. McCombs, Trends Biotechnol., 2011, 29, 144–152 CrossRef CAS PubMed.
- L. Wenfeng, M. Deng, C. Yang, F. Liu, X. Guan, Y. Du, L. Wang and J. Chu, J. Phys. D: Appl. Phys., 2020, 53, 113001 CrossRef.
- Y. N. Tallini, M. Ohkura, B. R. Choi, G. Ji, K. Imoto, R. Doran, J. Lee, P. Plan, J. Wilson, H. B. Xin, A. Sanbe, J. Gulick, J. Mathai, J. Robbins, G. Salama, J. Nakai and M. I. Kotlikoff, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4753–4758 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- J. Berg, Y. P. Hung and G. Yellen, Nat. Methods, 2009, 6, 161–166 CrossRef CAS PubMed.
- J. Nakai, M. Ohkura and K. Imoto, Nat. Biotechnol., 2001, 19, 137–141 CrossRef CAS PubMed.
- T. Nagai, A. Sawano, E. S. Park and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3197–3202 CrossRef CAS PubMed.
- S. A. Yigong, Cell, 2014, 159, 995–1014 CrossRef PubMed.
- H. A. Hauptman, Struct. Chem., 1990, 6, 617–620 CrossRef.
- P. A. Cramer, Cell, 2014, 159, 985–994 CrossRef CAS PubMed.
- D. Marion, Mol. Cell. Proteomics, 2013, 12, 3006–3025 CrossRef CAS PubMed.
- K. Müthrich, Nat. Struct. Mol. Biol., 2001, 8, 923–925 CrossRef PubMed.
- Y. Cheng, Science, 2018, 361, 876–880 CrossRef CAS PubMed.
- X. C. Bai and G. McMullan, Trends Biochem. Sci., 2015, 40, 49–57 CrossRef CAS PubMed.
- E. Nogales and S. H. W. Scheres, Mol. Cell, 2015, 58, 677–689 CrossRef CAS PubMed.
- T. Kitaguchi, M. Oya, Y. Wada, T. Tsuboi and A. Miyawaki, Biochem. J., 2013, 450, 365–373 CrossRef CAS PubMed.
- Y. Qin, D. W. Sammond, E. Braselmann, M. C. Carpenter and A. M. Palmer, ACS Chem. Biol., 2016, 11, 2744–2751 CrossRef CAS PubMed.
- S. Matsuda, K. Harada, M. Ito, M. Takizawa, D. Wongso, T. Tsuboi and T. Kitaguchi, ACS Sens., 2017, 2, 46–51 CrossRef CAS PubMed.
- K. Tainaka, R. Sakaguchi, H. Hayashi, S. Nakano, F. F. Liew and T. Morii, Sensors, 2010, 10, 1355–1376 CrossRef CAS PubMed.
- E. Nakata, F. F. Liew, S. Nakano and T. Morii, in Biosensors-Emerging materials and Applications, ed. P. A. Serra, IntechOpen, 2011, pp. 123–140 Search PubMed.
- J. S. Marvin, B. G. Borghuis, L. Tian, J. Cichon, M. T. Harnett, J. Akerboom, A. Gordus, S. L. Renninger, T. W. Chen, C. Bargmann, M. B. Orger, E. R. Schreiter, J. B. Demb, W. B. Gan, S. A. Hires and L. L. Looger, Nat. Methods, 2013, 10, 162–170 CrossRef CAS PubMed.
- T. Patriarchi, J. R. Cho, K. Merten, M. W. Howe, A. Marley, W.-H. Xiong, R. W. Folk, G. J. Broussard, R. Liang, M. J. Jang, H. Zhong, D. Dombeck, M. von Zastrow, A. Nimmerjahn, V. Gradinaru, J. T. Williams and L. Tian, Science, 2018, 360, eaat4422 CrossRef PubMed.
- M. Jing, P. Zhang, G. Wang, J. Feng, L. Mesik, J. Zeng, H. Jiang, S. Wang, J. C. Looby, N. A. Guagliardo, L. W. Langma, J. Lu, Y. Zuo, D. A. Talmage, L. W. Role, P. Q. Barrett, L. I. Zhang, M. Luo, Y. Song, J. J. Zhu and Y. Li, Nat. Biotechnol., 2018, 36, 726–737 CrossRef CAS PubMed.
- J. S. Marvin, Y. Shimoda, V. Magloire, M. Leite, T. Kawashima, T. P. Jensen, I. Kolb, E. L. Knott, O. Novak, K. Podgorski, N. J. Leidenheimer, D. A. Rusakov, M. B. Ahrens, D. M. Kullman and L. L. Looger, Nat. Methods, 2019, 16, 763–770 CrossRef CAS PubMed.
- R. Sakaguchi, T. Endoh, S. Yamamoto, K. Tainaka, K. Sugimoto, N. Fujieda, S. Kiyonaka, Y. Mori and T. Morii, Bioorg. Med. Chem., 2009, 17, 7381–7386 CrossRef CAS PubMed.
- M. Gees, B. Colsoul and B. Nilius, Cold Spring Harbor Perspect. Biol., 2010, 2, a003962 CAS.
- T. Yoshida, R. Inoue, T. Morii, N. Takahashi, S. Yamamoto, Y. Hara, M. Tominaga, S. Shimizu, Y. Sato and Y. Mori, Nat. Chem. Biol., 2006, 2, 596–607 CrossRef CAS PubMed.
- J. Duan, J. Li, G. L. Chen, Y. Ge, J. Liu, K. Xie, X. Peng, W. Zhou, J. Zhong, Y. Zhang, J. Xu, C. Xue, B. Liang, L. Zhu, W. Liu, C. Zhang, X. L. Tian, J. Wang, D. E. Clapham, B. Zeng, Z. Li and J. Zhang, Sci. Adv., 2019, 5, eaaw7935 CrossRef CAS PubMed.
- D. J. Wright, K. J. Simmons, R. M. Johnson, D. J. Beech, S. P. Muench and R. S. Bon, Commun. Biol., 2020, 3, 704 CrossRef CAS PubMed.
- P. S. Y. Wong, J. Hyun, J. M. Fukuto, F. N. Shirota, E. G. DeMaster, D. W. Shoeman and H. T. Nagasawa, Biochemistry, 1998, 37, 5362–5371 CrossRef CAS PubMed.
- M. D. Percival, M. Ouellet, C. Campagnolo, D. Claveau and C. Li, Biochemistry, 1999, 38, 13574–13583 CrossRef CAS PubMed.
- J. D. Pédelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- S. Tajima, E. Nakata, R. Sakaguchi, M. Saimura, Y. Mori and T. Morii, Bioorg. Med. Chem., 2020, 28, 115430 CrossRef CAS PubMed.
- H. Morise, O. Shimomura, F. H. Johnson and J. Winant, Biochemistry, 1974, 13, 2656–2662 CrossRef CAS PubMed.
- K. Bejec, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2306–2311 CrossRef PubMed.
- B. P. Cormack, R. H. Valdivia and S. Falkow, Gene, 1996, 173, 33–38 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- M. A. Elsliger, R. M. Wachter, G. T. Hanson, K. Kallio and J. Remington, Biochemistry, 1999, 38, 5296–5301 CrossRef CAS PubMed.
- L. M. Costantini, M. Baloban, M. L. Markwardt, M. Rizzo, F. Guo, V. V. Verkhusha and E. L. Snapp, Nat. Commun., 2015, 6, 7670 CrossRef PubMed.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- S. R. Jaffrey, H. Erdjument-Bromage, C. D. Ferris, P. Tempst and S. H. Snyder, Nat. Cell Biol., 2001, 3, 193–197 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- S. García-Santamaria, S. Boronat and E. Hidalgo, Biochemistry, 2014, 53, 2560–2580 CrossRef PubMed.
- P. M. Schaefer, S. Kalinina, A. Rueck, C. A. F. von Arnim and B. von Einem, Cytometry, Part A, 2019, 95, 34–46 CrossRef PubMed.
- G. A. Wagnieres, W. M. Star and B. C. Wilson, Photochem. Photobiol., 1998, 68, 603–632 CrossRef CAS PubMed.
- J. Loscalzo and G. Welch, Prog. Cardiovasc. Dis., 1995, 38, 87–104 CrossRef CAS PubMed.
- S. A. Bradley and J. R. Steinert, Oxid. Med. Cell. Longevity, 2016, 2016, 5681036 Search PubMed.
- A. L. Horenberg, A. M. Houghton, S. Pandey, V. Seshadri and W. H. Guilford, Cytoskeleton, 2019, 76, 243–252 CrossRef CAS PubMed.
- E. Eroglu, B. Gottschalk, S. Charoensin, S. Blass, H. Bischof, R. Rost, C. T. Madreiter-Sokolowski, B. Pelzmann, E. Bernhart, W. Sattler, S. Hallström, T. Malinski, M. Waldeck-Weiermair, W. F. Graier and R. Malli, Nat. Commun., 2016, 7, 10623 CrossRef CAS PubMed.
- V. V. Belousov, A. F. Fradkov, K. A. Lukyanov, D. B. Staroverov, K. S. Shakhbazov, A. V. Terskikh and S. Lukyanov, Nat. Methods, 2006, 3, 281 CrossRef CAS PubMed.
- D. T. Hess, A. Matsumoto, S. O. Kim, H. E. Marshall and J. S. Stamler, Nat. Rev. Mol. Cell Biol., 2005, 6, 150–166 CrossRef CAS PubMed.
- G. Roos, N. Foloppe and J. Messens, Antioxid. Redox Signaling, 2012, 18, 94–127 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02226e |
| This journal is © The Royal Society of Chemistry 2022 |