
Synthesis of a cyclic poly(methyl acrylate) via topological transformation of a [1]rotaxane†
Kazuko
Nakazono
 *,
Takahiro
Ogawa
and
Toshikazu
Takata
*
*,
Takahiro
Ogawa
and
Toshikazu
Takata
*
School of Materials and Chemical Technology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: takata.t.ab@m.titech.ac.jp; Fax: +81 45-924-5358
First published on 16th October 2019
Abstract
Even today, obtaining a cyclic polymer in pure form remains difficult. This paper discloses a synthetic protocol for a vinyl polymer-type cyclic polymer in pure form. A linear macromolecular [1]rotaxane containing a poly(methyl acrylate) (PMA, Mn,NMR 3200) axle and a crown ether wheel was synthesized via the terminal modification of PMA with a semi[2]rotaxane unit and subsequent linking of the wheel and axle components. The positional change of the wheel from the initial to opposite end on the PMA axle comprised the preparation of pure cyclic PMA via an efficient linear-to-cyclic polymer topological transformation.
Introduction
Cyclic polymer chemistry is a rapidly growing discipline with many recent advances in the areas of synthesis and purification techniques.1–6 Cyclic polymers are interesting because their specific topology does not contain ends. However, there are still many limitations in the selective synthesis and isolation of pure cyclic polymers. There are two typical synthetic strategies for cyclic polymer synthesis: (i) intramolecular cyclization of linear telechelic polymers7–10 or (ii) ring-expansion polymerization.11–18 Cyclization of a linear polymer is the more commonly used technique and it is applicable to many classes of polymers. However, in several cases, isolation of the desired cyclic polymer in pure form becomes difficult because residual linear polymer or intermolecular reaction products are present, even if the reaction is performed under high-dilution conditions. In the ring-expansion method, strictly controlling the molecular weight and polydispersity through polymerization from small cyclic catalysts is challenging. Outside of these methods, we recently established an efficient synthetic method for cyclic polymers by exploiting the mobility of the rotaxane mechanical bond. In particular, the initial small cyclic structure, which contains [1]rotaxane moiety19–21 (Fig. 1) or a self-assembly constructed from [c2]daisy chains,22 is installed at the polymer end. Thereafter, the small cycle is expanded by active transport of the mechanical junction on the polymer chain toward the opposite end of the polymer. Finally, the resultant polymer is confirmed to be cyclic. This method can access gram-scale synthesis in high yield without unexpected reaction products or remaining linear polymer, independent of the concentration of the reactants.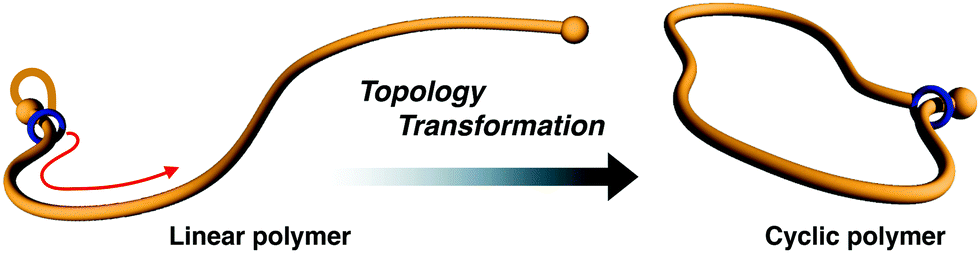 | ||
| Fig. 1 Schematic illustration for polymer topological transformation from linear-to-cyclic utilizing the rotaxane protocol, which is facilitated by the positional change of the wheel from the initial to opposite end along the polymer chain. | ||
In our rotaxane protocol, using dibenzo-24-crown-8-ether (DB24C8) as the typical wheel component, the efficient cyclization of poly(tetrahydrofuran)19 and aliphatic polyesters20–22 was achieved. Because the protocol strongly depends on whether the polymer is capable of passing through the wheel cavity, penetrating polymers must be smaller than the cavity. To expand the utility of the rotaxane protocol, we aimed to apply it for the cyclization of a vinyl polymer because there is no vinyl polymer capable of being penetrated into the crown ether cavity so far. Computational study using the CPK model, which is reliable to investigate the relative size of rotaxane components, indicated that PMA, a small vinyl polymer, has complementary size to the cavity of DB24C8. Thus, it is experimentally intriguing that the DB24C8 cavity actually accepts PMA as an axle component. Another factor for consideration is how to introduce the rotaxane unit to the polymer end. Use of a polymer prepared by living polymerization using a rotaxane initiator followed by the termination via bulky stopper would provide a useful method because gram-scale synthesis of cyclic polymer has been actually realized.20–22 Conversely, simple polymer end modification19 of a living polymer with rotaxane unit becomes a straightforward and universal method for the cyclization of various polymers by the rotaxane protocol. As for vinyl polymers, atom transfer radical polymerization (ATRP) enables the control and modification of living polymers from a diverse range of monomers.23 Furthermore, the bromoalkyl group of the propagation end of living ATRP polymers can be easily substituted with an azide group. Therefore, this strategy for creating a macromolecular building block is often utilized for constructing various polymer topologies, including cyclic polymers.24–26 To extend the synthetic scope of the rotaxane protocol, we now describe the synthesis of cyclic PMA by the cyclization of a linear PMA having a [1]rotaxane terminal unit.
Results and discussion
Based on our previous study, we designed the rotaxane building block as shown in Scheme 1.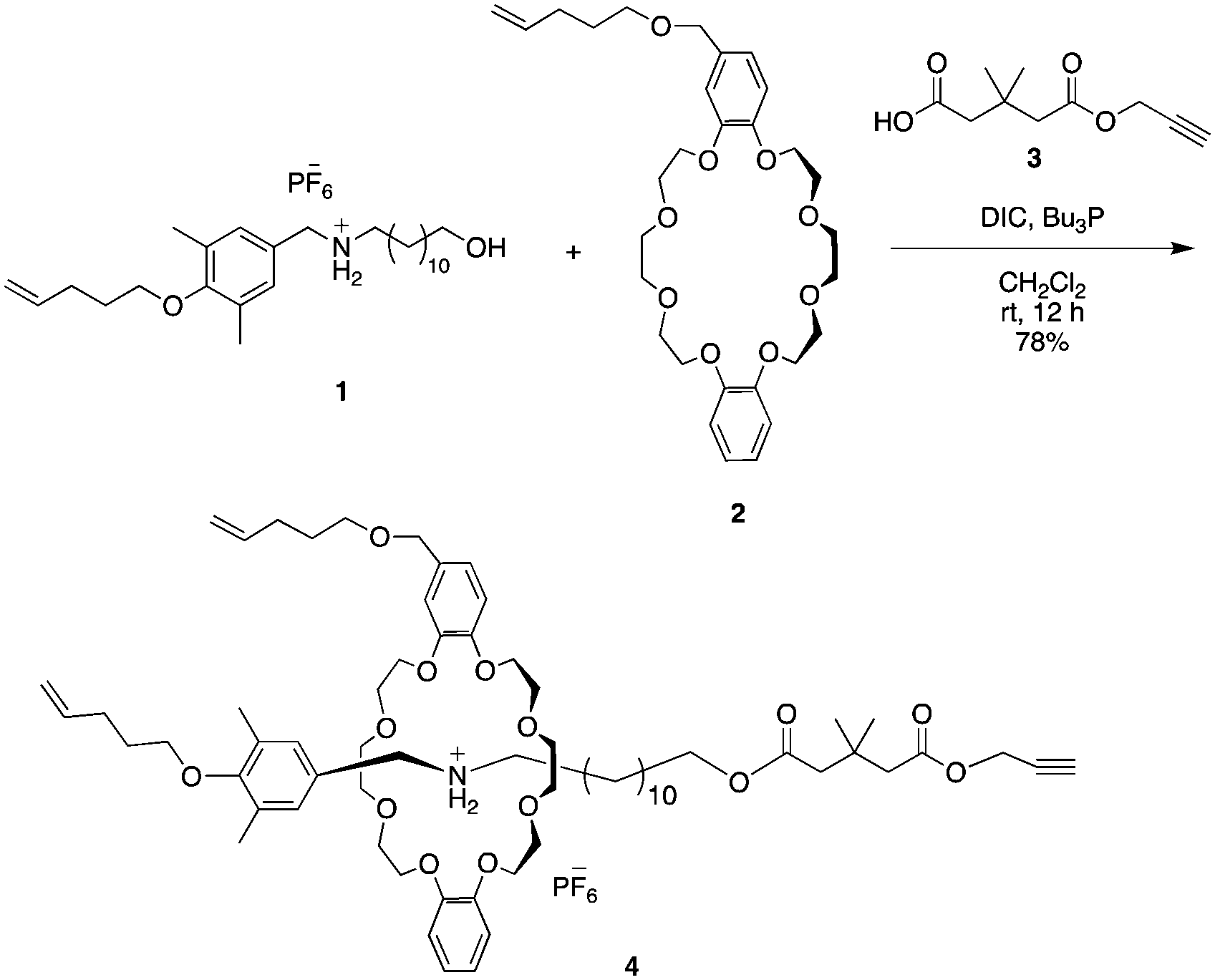 | ||
| Scheme 1 Synthesis of the temporarily stabilized rotaxane terminal unit 4: ammonium salt 1 and crown ether 2in situ generate a semi[2]rotaxane intermediate, the neopentyl group kinetically stabilizes rotaxane 4. | ||
sec-Ammonium salt 1 and a DB24C8 containing a terminal olefin 2 were complexed in situ to generate an intermediary semi[2]rotaxane in dichloromethane. Thereafter, a terminal alkyne-containing stopper was condensed to generate rotaxane 4 in 78% yield.27,28 Both the hydrogen bonding at the ammonium moiety and the neopentyl group act to thermodynamically and kinetically stabilize rotaxane 4. Therefore, once the hydrogen bond between ammonium and crown ether is destabilized by an anion exchange reaction, the rotaxane immediately dissocuates.28–30 Moreover, similar to our previous report,4 the [1]rotaxane structure was constructed by a metathesis reaction after the macromolecular [2]rotaxane structure was completed utilizing the terminal olefins on both the wheel and axle end. Such a strategy was developed to increase the selectivity of intramolecular metathesis because the proximity effect is realized at the polymer terminus and helps to inhibit intermolecular reactions.
As the second building block, PMA was prepared by polymerization from α-bromoester-derivative initiators under typical ATRP reaction conditions using a CuBr–pentamethyldiethylenetriamine (PMDETA) catalyst system, followed by substitution of the bromoalkyl group with sodium azide (Scheme 2).
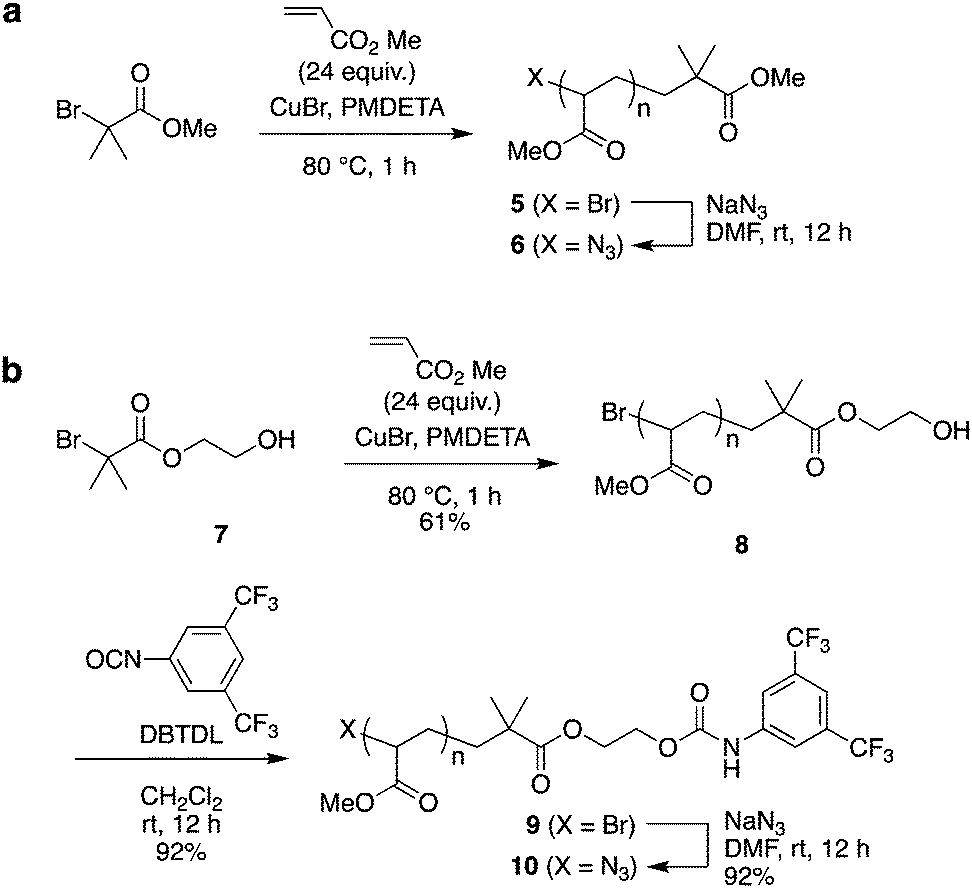 | ||
| Scheme 2 Synthesis of poly(methyl acrylate) (PMA) by ATRP: the azide-terminated PMA 6 (without a bulky stopper) and PMA 10 (bearing a bulky stopper). | ||
As the model for a dethreading study of PMA through the wheel, azide-methylester terminated PMA 6 was synthesized from methyl 2-bromoisobutyrate. The other PMA compound 10 was constructed with a stopper group starting from initiator 3. After polymerization of MA from initiator 7, the terminal hydroxyl group was reacted with 3,5-di(trifluoromethyl)phenyl isocyanate to install a bulky stopper with a carbamate group. This bulky terminal group will act as a second station for the crown ether when the ammonium moiety is removed later.
The MALDI-TOF mass spectra of polymers 1 and 5 supported the presence of the appropriate end group-bearing PMAs, as expected (Fig. S3 and S4 in ESI†). From SEC analysis, the molecular weights of PMA 5 and PMA 8 were estimated to be Mn 6100 (Mw/Mn 1.05) and Mn 5200 (Mw/Mn 1.07), respectively (Fig. S5 in ESI†), while the molecular weights calculated from their NMR spectra were Mn,NMR 3600 and Mn,NMR 3200 for 5 and 8, respectively. After the terminal substitution reaction of these polymers with sodium azide, each product was obtained in good yield and identified structurally by IR analysis (Fig. S6 and S7 in ESI†).
With the PMA and rotaxane units in hand, macromolecular [2]rotaxane was synthesized by a CuAAC reaction. We first synthesized macromolecular [2]rotaxane 11 as the model compound to study whether the wheel component could pass through the macromolecular axle (Scheme 3).
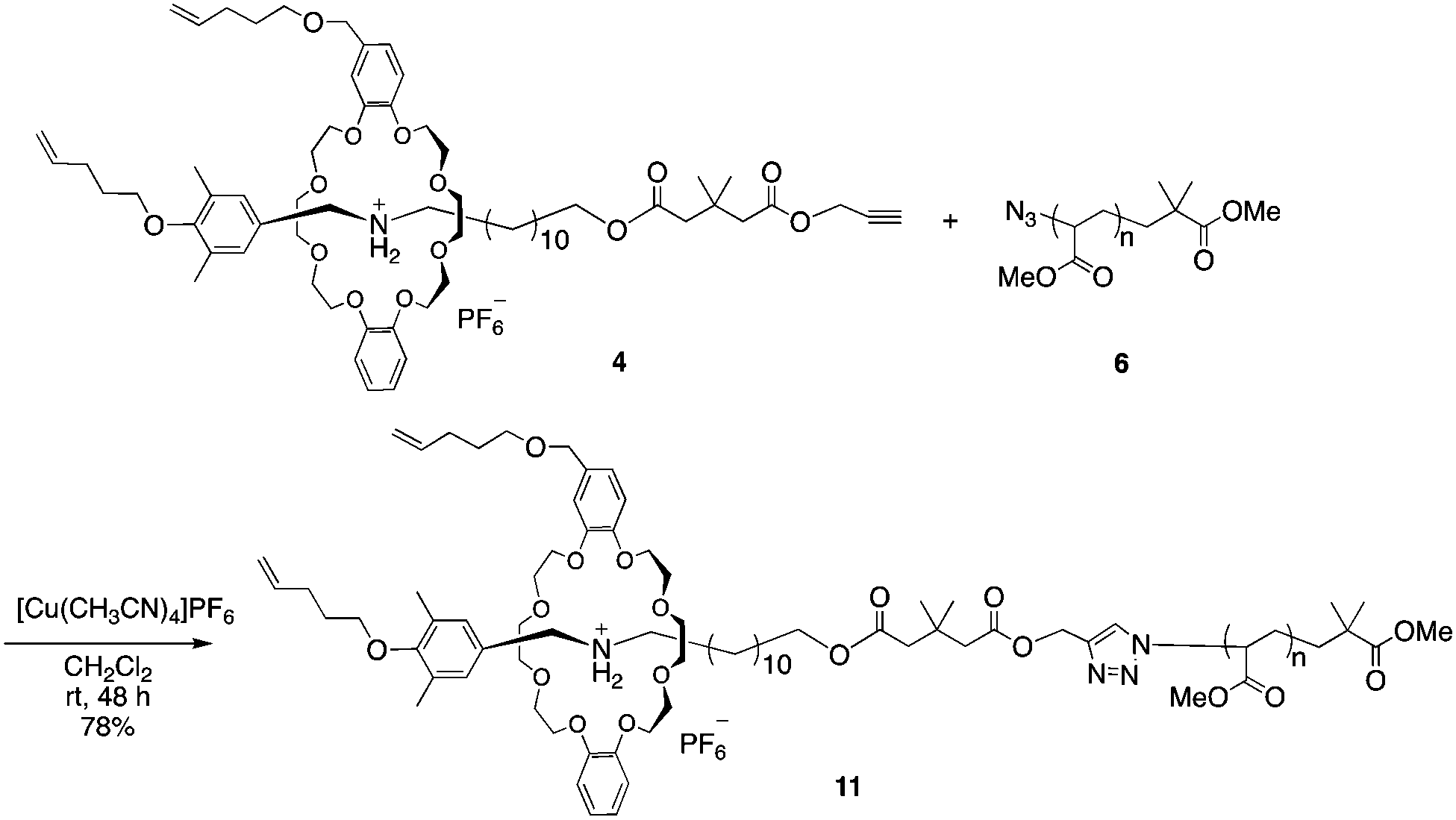 | ||
| Scheme 3 Synthesis of macromolecular semi[2]rotaxane 11 with PMA 6 as the polymer segment. | ||
To prevent the dissociation of semirotaxane 4, [Cu(CH3CN)4]PF6 was employed as the catalyst, when rotaxane 4 was ligated to PMA 6. After the removal of the catalyst and the residual rotaxane unit by preparative GPC, the desired macromolecular semi[2]rotaxane 11 was obtained in 78% yield. In the MALDI-TOF mass spectrum, the peaks of the product coincided well with the theoretical value for macromolecular [2]rotaxane 11; additional peaks for byproducts were not detected (Fig. 2a).
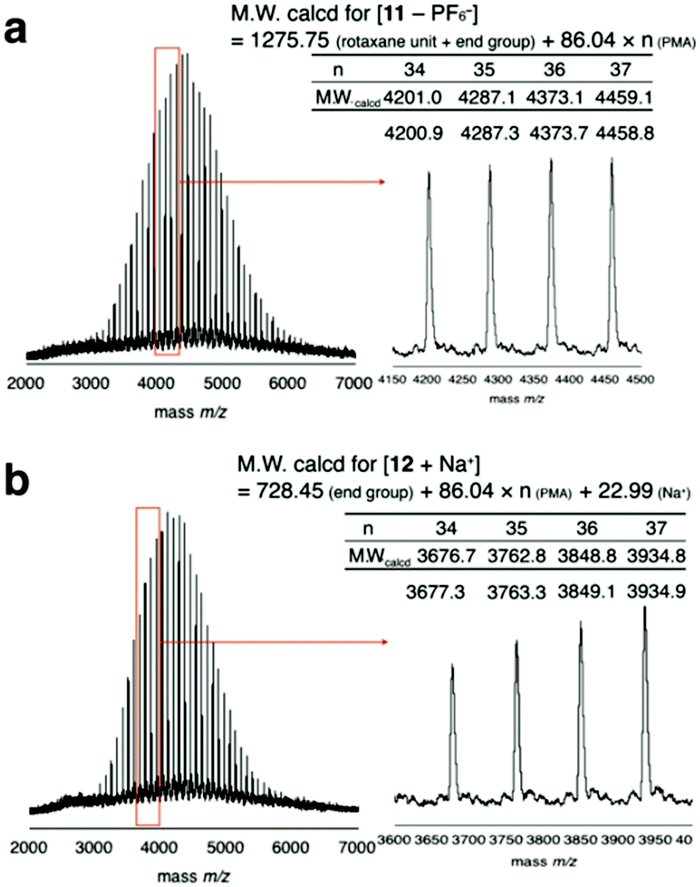 | ||
| Fig. 2 MALDI-TOF mass spectra of macromolecular semi[2]rotaxane 11 (a) and the wheel-free PMA 12 residue after the anion exchange reaction (b). | ||
Despite the reaction between a huge rotaxane unit and the polymer end, the expected product was obtained with high selectivity and high efficiency using the CuAAC reaction.
Next, the macromolecular semi[2]rotaxane 11 was treated with tetrabutylammonium chloride in THF at room temperature to exchange the counter anion (Scheme 4).
 | ||
| Scheme 4 Anion exchange reaction of macromolecular semi[2]rotaxane 11 with Bu4NCl in THF. | ||
In previous study, PF6− was essential for stabilizing the hydrogen bonding of the ammonium moiety with the wheel, however replacing PF6− with a harder anion, such as chloride ion, the equilibrium is biased to ion pairing of ammonium and the hard anion instead of hydrogen bonding with the wheel.29–31 Consequently, the wheel can overcome the complementary sized bulky group to slip out of the axle polymer.32–34 In the MALDI-TOF mass spectrum after the anion exchange reaction, a series of peaks attributable to the wheel-free PMA 12 was observed, while wheel-containing compound 11 was not (Fig. 2b). Thus, a PMA chain can move through DB24C8 as expected from our preliminary calculations.
Based on this result, the 3,5-bis(trifluoromethyl)phenyl carbamate stopper-bearing macromolecular [2]rotaxane 13 was synthesized in 80% yield using PMA 10 in the same manner as semi[2]rotaxane 11 (Scheme 5).
 | ||
| Scheme 5 Synthesis of macromolecular [1]rotaxane 14 and 15, and macromolecular [2]rotaxane 16. | ||
By MALDI-TOF mass spectrum, the polymer was identified to be macromolecular [2]rotaxane 13. Furthermore, the IR absorption peaks attributed to the azide group and terminal alkyne completely disappeared in the product (Fig. S8b in ESI†). The macromolecular [2]rotaxane 13 was then subjected to metathesis conditions (Grubbs 2nd generation catalyst) to afford macromolecular [1]rotaxane 14 in 85% yield. As intended, the obtained MALDI-TOF mass spectrum strongly supported the selective generation of macromolecular [1]rotaxane 14 as the single product, and no intermolecularly-reacted product was detected.
Finally, to expand the cyclic moiety to construct cyclic PMA, 14 was subjected to N-acetylation condition to remove the strong attractive interaction between the ammonium moiety and wheel. N-Acetylation of 14 was performed in THF with an excess of acetic anhydride and triethylamine under gentle heat to promote mobility of the polymer chain through the wheel component.28 After purification of the polymer by preparative GPC, the desired polymer product was obtained in 89% yield. N-Acetylation was confirmed by its MALDI-TOF mass spectrum. The spectrum showed a single series of peaks attributable to the theoretical values of N-acetylated macromolecular [1]rotaxane 15 (Fig. 3a).
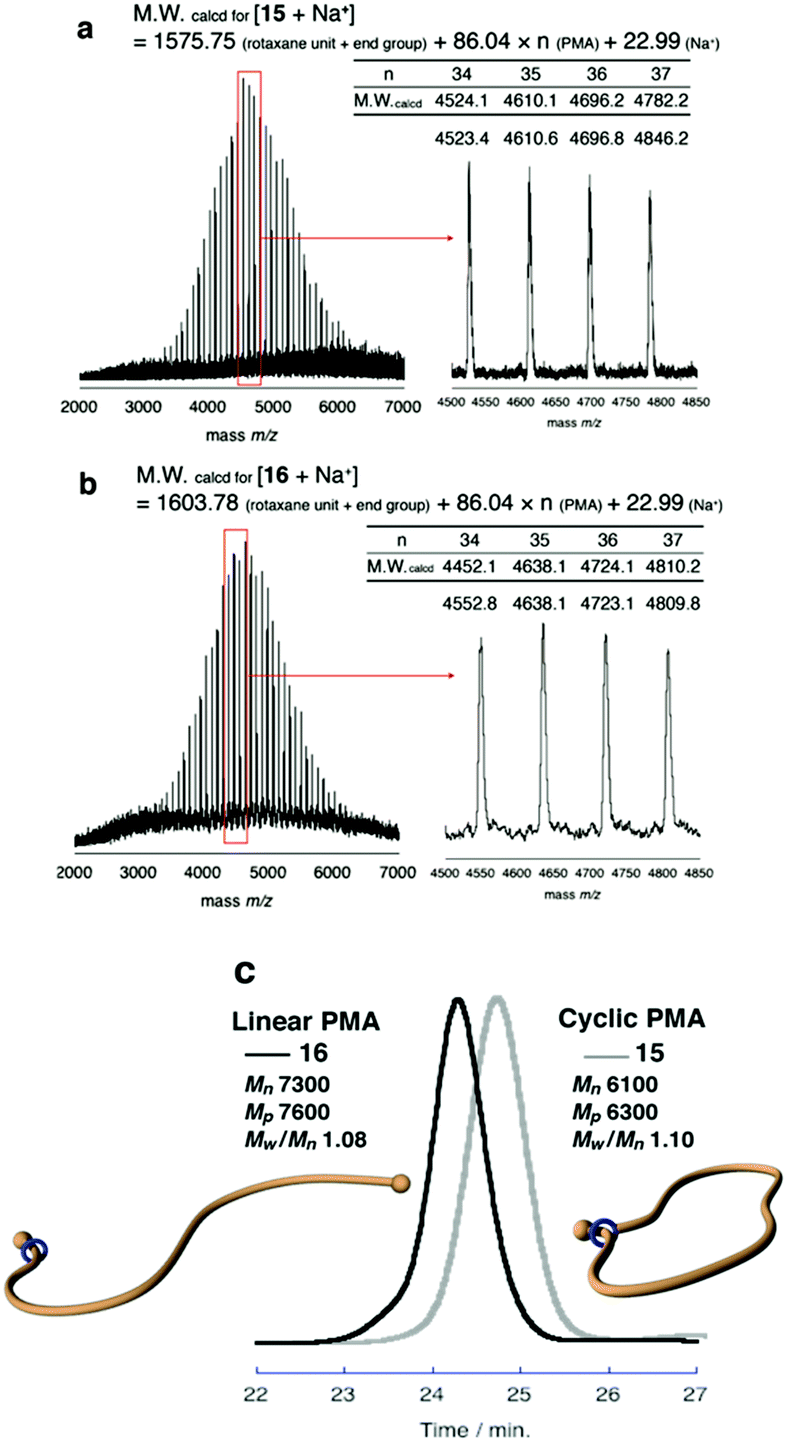 | ||
| Fig. 3 MALDI-TOF mass spectra of macromolecular [1]rotaxane 15 (a) and macromolecular [2]rotaxane 16 (b). SEC traces (c) of 15 (Light gray line) and 16 (Black line) (eluent: CHCl3, 0.85 mL min−1, PSt standards). | ||
To the best of our knowledge, ammonium salt-containing polymers are more easily detected in MALDI-TOF mass than the N-acetylated neutral polymers; thus, this spectrum strongly indicates that pure 15, entirely free from contamination with 14, was obtained.
Furthermore, SEC analysis was carried to evaluate the molecular weight of 15. Fig. 3c shows that SEC trace of the obtained 15 was unimodal peak without a shoulder to indicate that the desired reaction was successful without any decomposition and side reactions. The estimated number-average molecular weight Mn was 6100, and peak molecular weight Mp was 6300. Polydispersity was also estimated as Mw/Mn = 1.10. To confirm the cyclic topology of polymer 15, G-value (Mp,cyclic/Mp,linear) was calculated based on the Mp values.35–38 In Tillman's report on the synthesis of cyclic PMA by the combination of ATRP and a subsequent radical coupling reaction between the propagation ends,38 the G-value of cyclic PMA derived from a linear polymer having almost the same Mn as the PMA 8 utilized by us was reported as 0.80. To compare the SEC retention time of macromolecular [1]rotaxane 15 with its linear analog containing a similar composition and polarity, macromolecular [2]rotaxane 13 was N-acetylated to obtain the macromolecular [2]rotaxane 16 having a structure different from that of 15 by an ethylene unit (Scheme 5). As is obvious from Fig. 3c, the retention time of 15 (Mn 6100, Mp 6300, Mw/Mn 1.10) was longer than that of 16 (Mn 7300, Mp 7600, Mw/Mn 1.08), and the ratio of Mp,15/Mp,16 (G-value) was approximately 0.83. Therefore, we concluded that macromolecular [1]rotaxane 15 possessed the desired cyclic topology in which the wheel is localized at the carbamate moiety. By simple calculation from the degree of polymerization of PMA used, the wheel moved ca. 80 carbon atoms (approximately 120 Å) during this conversion process. The achievement of such a long-range positional change is an interesting result from the viewpoint of rotaxane chemistry.
Conclusions
In conclusion, we conducted a linear-to-cyclic polymer topological transformation utilizing the rotaxane's mobility and demonstrated a selective and high-yielding synthetic protocol for cyclic PMA. The CuAAC reaction was very effective for modifying the linear ATRP-derived polymer end with a huge rotaxane unit having various functional groups. Moreover, the dethreading study using an anion stimulus in a temporary stabilized macromolecular [2]rotaxane provided strong evidence that PMA can pass through DB24C8. Comparison of SEC traces between the linear analog and the linear-to-cyclic transformed polymer supported the successful generation of the cyclic PMA using the rotaxane protocol. The present topological transformation protocol of polymer should have highly potential applicability, although the 24-membered crown ether can only accept smallest vinyl polymer PMA in this case. Namely, larger membered crown ethers such as dibenzo-30-crown-10, bis(m-phenylene)-32-crown-10 and bis(p-phenylene)-34-crown-10 including cryptands with cation-accepting ability can be readily available and should make the topological transformation possible for a variety of guest polymers with larger repeating units. Although the subject of a reversible system remains, this cyclic polymer synthesis should prove to be an efficient protocol for other cyclic polymer syntheses and open the door for further development of applications in the field of supramolecular materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Funding Program for KAKENHI from MEXT (19K05417) and the CREST program from JST Grant Number JPMJCR1522, Japan.Notes and references
- S. M. Grayson and J. M. J. Frechet, Chem. Rev., 2001, 101, 3819 CrossRef CAS PubMed.
- T. MacLeish, Nat. Mater., 2008, 7, 933 CrossRef PubMed.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202 RSC.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4723 CrossRef CAS.
- E. M. Benetti, M. Divandari, S. N. Ramakrishna, G. Morgese, W. Yan and L. Trachsel, Chem. – Eur. J., 2017, 23, 12433 CrossRef CAS PubMed.
- T. Yamamoto and Y. Tezuka, Soft Matter, 2015, 11, 7458 RSC.
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406 CrossRef CAS.
- S. Honda, T. Yamamoto and Y. Tezuka, Nat. Commun., 2013, 4, 1574 CrossRef PubMed.
- M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2011, 133, 1630 CrossRef CAS PubMed.
- T. Yamamoto, S. Yagyu and Y. Tezuka, J. Am. Chem. Soc., 2016, 138, 3904 CrossRef CAS PubMed.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041 CrossRef CAS PubMed.
- F. M. Haque, A. Alegria, S. M. Grayson and F. Arroso-Bujans, Macromolecules, 2017, 50, 1870 CrossRef CAS.
- A. Bunha, P.-F. Cao, J. D. Mangadlao and R. C. Advincula, React. Funct. Polym., 2014, 80, 33 CrossRef CAS.
- H. Kudo, M. Sato, R. Wakai, T. Iwamoto and T. Nishikubo, Macromolecules, 2008, 41, 521 CrossRef CAS.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791 CrossRef CAS PubMed.
- D. A. Culki, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627 CrossRef PubMed.
- J. P. Edwards, W. J. Wolf and R. H. Grubbs, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 228 CrossRef CAS.
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892 CrossRef CAS.
- T. Ogawa, N. Usuki, K. Nakazono, Y. Koyama and T. Takata, Chem. Commun., 2015, 51, 5606 RSC.
- T. Ogawa, K. Nakazono, D. Aoki, S. Uchida and T. Takata, ACS Macro Lett., 2015, 4, 343 CrossRef CAS.
- S. Valentina, T. Ogawa, K. Nakazono, D. Aoki and T. Takata, Chem. – Eur. J., 2016, 22, 8759 CrossRef CAS PubMed.
- D. Aoki, G. Aibara, S. Uchida and T. Takata, J. Am. Chem. Soc., 2017, 139, 6791 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- S. Hayashi, K. Adachi and Y. Tezuka, Chem. Lett., 2007, 36, 982 CrossRef CAS.
- A. F. Voter and E. S. Tillman, Macromolecules, 2010, 43, 10304 CrossRef CAS.
- G. Y. Shi, L. P. Yang and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6496 CrossRef CAS.
- Y. Makita, N. Kihara and T. Takata, Chem. Lett., 2007, 36, 102 CrossRef CAS.
- J. Nishiyama, Y. Makita, N. Kihara and T. Takata, Chem. Lett., 2015, 44, 1428 CrossRef CAS.
- M. Montalti and L. Prodi, Chem. Commun., 1998, 1461 RSC.
- J. W. Jones and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 7001 CrossRef CAS PubMed.
- H. W. Gibson, J. W. Jones, L. N. Zakharov, A. L. Rheingold and C. Slebodnick, Chem. – Eur. J., 2011, 17, 3192 CrossRef CAS PubMed.
- K. Iijima, Y. Kohsaka, Y. Koyama, K. Nakazono, S. Uchida, S. Asai and T. Takata, Polym. J., 2014, 46, 67 CrossRef CAS.
- Y. Kohsaka, K. Nakazono, Y. Koyama, S. Asai and T. Takata, Angew. Chem., Int. Ed., 2011, 50, 4872 CrossRef CAS PubMed.
- K. Nakazono and T. Takata, Chem. – Eur. J., 2010, 16, 13783 CrossRef CAS PubMed.
- S. Hayashi, K. Adachi and Y. Tezuka, Chem. Lett., 2007, 36, 982 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238 CrossRef CAS PubMed.
- H. Li, A. Debuigne, R. Jerome and P. Lecomte, Angew. Chem., Int. Ed., 2006, 45, 2264 CrossRef CAS PubMed.
- S. C. Blackburn, K. D. Myers and E. S. Tillman, Polymer, 2015, 68, 284 CrossRef CAS : Cyclic polymer obtained from the linear PMA Mn 5000 (PDI 1.06) was Mn 5300 (PDI 1.47), the G-value calculated by these Mp values was 0.80.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for preparation and characterization of synthesized chemicals. See DOI: 10.1039/c9qm00563c |
| This journal is © the Partner Organisations 2019 |