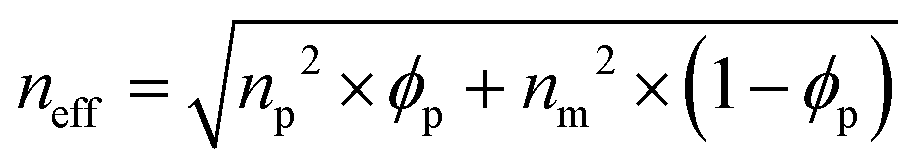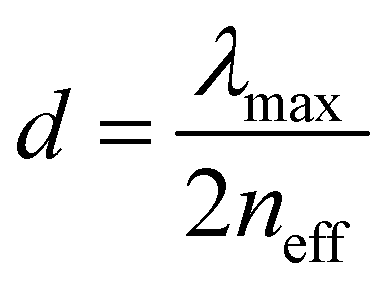DOI:
10.1039/C9QM00454H
(Research Article)
Mater. Chem. Front., 2019,
3, 2707-2715
Self-healing and recyclable photonic elastomers based on a water soluble supramolecular polymer†
Received
11th July 2019
, Accepted 8th October 2019
First published on 9th October 2019
Abstract
Inspired by the color shifting capability of chameleon skin, photonic elastomers (PEs) that can change their color with mechanical deformation have been attracting growing attention because of their potential applications in sensing and optical materials. However, it remains challenging to develop facile and environment-friendly methods for the preparation of PEs with desirable optical and mechanical properties for practical applications. Here, water-soluble supramolecular polymer-based PEs with angle-independent structural color, self-healing, and recyclable properties are reported. The PEs are prepared by incorporating isotropically arranged SiO2 nanoparticles (SiO2 NPs) into a water-soluble ureidopyrimidinone (UPy) cross-linked poly(ethylene glycol) (PEG) polymeric matrix. The stable angle-independent structural color and mechanical properties could be tuned by incorporating SiO2 NPs with different sizes and contents. The resulting flexible PEs demonstrate a fast response to mechanical strain and can be used as a visual strain sensor for monitoring finger bending motions. More importantly, the resultant elastomers exhibit improved self-healing properties due to the introduced dynamic quadruple hydrogen bonding arising from the UPy–UPy dimers and could be readily recycled via a redissolving–casting or hot-press remolding process with negligible change in the structural color and mechanical properties. This study provides an eco-friendly and sustainable strategy for the preparation of PEs with improved self-healing and recycling capabilities, potentially useful in visual sensors.
Introduction
Structural color, derived from the interactions between the incident light and periodic macro-nanostructure, is one of the prominent characteristics of photonic crystals (PCs).1–15 According to Bragg's law, the periodical structural parameters of PCs determine the wavelength of reflection. Thus, when responsive materials are introduced into PCs, visual sensors based on the color shifting of PCs can be realized by actively tuning the period parameters under external stimuli.16 Among these responsive PCs, photonic elastomers (PEs), inspired by the color shifting capability of chameleon skin, have attracted growing interest in recent years.17–21 PEs can be readily obtained by integrating colloidal crystal assemblies into an elastic polymer network and exhibit strain-induced color shifting properties similar to those of chameleon skin. Ito et al. fabricated a strain-responsive PE by fixing colloidal crystals of poly(ethyl acrylate-co-methyl methacrylate) into a poly(ethyl acrylate) elastomer.17 Lee and co-workers obtained PEs by embedding a long-range ordered array of silica nanoparticles (NPs) in a matrix of a polymerized poly(ethylene glycol) phenyl ether acrylate elastomer.19 Although these PEs exhibited good strain-induced color shifting properties, they suffer from poor mechanical durability22,23 and angle-dependent structural color24 due to their long-range periodic macro-nanostructure, which might limit their practical applications.
Recently, it has been shown that some amorphous structures with short-range order can create angle-independent structural color.22,25 By incorporating such amorphous structures into dynamic supramolecular polymeric matrices of polydimethylsiloxane, our group has successfully fabricated PEs with angle-independent structural color and self-healing capability, which are capable of changing color in response to a strain-based stimulus.26,27 The introduction of the supramolecular matrix endows the PEs with self-healing capabilities, potentially extending their service duration. However, the preparation of such polysiloxane-based supramolecular PEs involves the use of volatile organic compounds, which might obstruct their practical applications.28,29 Therefore, to extend the service lifespan, strengthen the safety profile, and reduce the contamination of the environment, it is greatly desired to further develop environment-friendly PEs with improved self-healing and recyclability.
Nowadays, many water-soluble polymers have been developed to replace organic solvent soluble polymers and can be applied in many fields such as paints, coatings, and so on.30,31 Among these water-soluble polymers that are commonly used, polyethylene glycol (PEG) exhibits many advantages, such as high water-solubility, high flexibility, nontoxicity, and transparency.32–34 By introducing ureidopyrimidinone (UPy) units into PEG chains, a cross-linked elastic PEG network by reversible quadruple hydrogen bonding can be readily obtained, which possesses water-soluble, self-healing, and recyclable merits.35–39 Herein, by exploiting the UPy cross-linked PEG (PEG–UPy) polymer as the matrix and by the incorporation of a short-range ordered arrangement of SiO2 NPs into such a matrix, PEs with angle-independent structural color, water-solubility, and improved self-healing and recyclability are prepared. Furthermore, the mechanical and optical properties could be tuned by incorporating SiO2 NPs with different sizes and contents. The resultant elastic composites show chameleon-skin-like strain-induced color shifting capability. More importantly, the PEs exhibit improved self-healing capability for superficial scratches and cuts at room temperature and are readily recycled with water by redissolving–casting and hot-press molding. The PEs developed in the current study with improved self-healing and recyclability exhibit good prospects in the area of visual strain sensors and meet the environment-friendly and resource-saving requirements.
Experimental section
Materials
Tetraethyl orthosilicate (TEOS, purity: 98%), ammonia aqueous solution (28–30%), nitric acid (65–68%) and N,N-dimethylformamide (DMF, purity: 99.5%) were purchased from China National Medicines Corporation Ltd. Ethanol (purity: 98%), dimethyl sulfoxide (DMSO), and trimethylamine (Et3N) were purchased from Beijing Chemical Works. 6-Methylisocytosine (purity: 98%), and 2-isocyanatoethyl methacrylate (purity: 98%) were purchased from Aladdin. Carbon black nanoparticles (CB NPs) (30 nm, VX72R) were purchased from Cabot Corporation. Poly(ethylene glycol) methyl ether methacrylate (PEGMA, Mn = 500 g mol−1) was purchased from Aldrich and purified through an alumina column prior to polymerization. 2,2-Azobis(isobutyronitrile) (AIBN, Aladdin) was recrystallized from ethanol before use. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was synthesized according to the reported RAFT polymerization strategy.40
Synthesis of (2-(3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido)ethyl methacrylate)
(2-(3-(6-Methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido)ethyl methacrylate) (UPyMA) was synthesized according to a literature method.41 Briefly, 6-methylisocytosine (2.0 g, 16 mmol) was first dissolved in 50 mL DMSO at 150 °C. Then, 2-isocyanatoethyl methacrylate (2.64 g, 17 mmol) was added to the above mixture, and the mixture was quickly cooled using an ice bath. After 2 h, fine white solid products were obtained and then washed with cold acetone three times. The white product was dried under a vacuum at 30 °C for 4 h to afford the target product.
Synthesis of the PEG–UPy polymer
The copolymerization of PEGMA and UPyMA was performed through the reversible addition–fragmentation chain transfer (RAFT) method.41 In a typical polymerization process, PEGMA (3.0 g, 6 mmol), UPyMA (336.0 mg, 1.2 mmol), CPADB (16.8 mg, 0.06 mmol), AIBN (3.3 mg, 0.02 mmol), and 20 mL DMF were added to a Schlenk flask equipped with a magnetic stirring bar. Oxygen was removed from the reaction mixture through three freeze–pump–thaw cycles, and then the flask was backfilled with argon gas. The reaction was carried out at 75 °C for 24 h and terminated by exposure to the air. Subsequently, the reaction mixture was purified by dialysis against deionized water for 3 days to remove the unreacted monomer. The purified polymer was obtained by freeze-drying the sample for 24 h. The molecular weight (Mn) of the UPy cross-linked PEG polymer was determined using an Agilent 1100 gel permeation chromatograph (GPC).
Synthesis of monodispersed SiO2 nanoparticles (NPs)
Monodispersed SiO2 NPs were synthesized by a previously reported method.27 Taking SiO2 NPs with a size of 204 nm as an example, ethanol (680 mL), ammonia aqueous solution (32 mL), and H2O (64 mL) were first mixed under stirring at 30 °C for 30 min, followed by the dropwise addition of TEOS (24 mL). The resulting mixture was stirred for 2 h, and the products were separated by centrifugation (TG16WS, XIANGZHI) and washed with a copious amount of water and ethanol. Similarly, SiO2 NPs with different sizes were also prepared by tuning the volumes of the ammonia aqueous solution and water.
Surface modification of CB NPs
The CB NPs were treated with nitric acid to improve their hydrophilicity.42 Briefly, CB NPs (5 g) were dispersed in 200 mL of nitric acid solution under continuous stirring at 100 °C for 2 h. The treated CB NPs were separated by centrifugation and washed with plenty of water, followed by drying in a vacuum oven at 60 °C for 48 h.
Preparation of the PEs
To prepare the PEs, SiO2 NPs (0.5 g) and CB NPs (0.005 g) were first dispersed in 5 mL of deionized water, followed by the addition of a desired amount of PEG–UPy. The mixture was sonicated for 30 min and then was cast into a Teflon mold. After the preliminary evaporation of water at 60 °C for 12 h, the sample was further dried under a vacuum at 60 °C for 48 h to obtain SiO2/PEG–UPy PEs. PEs with 40 wt%, 50 wt%, and 60 wt% SiO2 NP content were prepared and denoted as SiO2/PEG–Upy40, SiO2/PEG–Upy50, and SiO2/PEG–UPy60, respectively.
Characterization
To observe the arrangement of SiO2 NPs in the polymer matrix, the microstructure of the SiO2/PEG–UPy PE was characterized by an FEI Magellan 400 scanning electron microscope (SEM) at an acceleration voltage of 10 kV. Thermogravimetric analysis (TGA) was performed on a TGA 4000 (PerkinElmer) under an N2 flow at a heating and cooling rate of 10 °C min−1 in the range of 30–600 °C. Reflectance spectra measurements were performed on a USB4000 fiber optical spectrometer (Ocean Optics). Digital photographs were captured using a Canon PowerShot SX6 camera. Rheological tests were conducted on an MCR-302 rheometer (Anton Paar) with parallel plate geometry (25 mm diameter rotating top plate) at 30 °C. Tensile tests were carried out on a MODEL electronic universal test machine with the extension speed at 20 mm min−1. For testing the self-healing performance, the changes of surface scratches were monitored by a Dino-lite AM4113ZTL(R4) optical microscope. Small-angle X-ray scattering (SAXS) measurements were conducted on a Xeuss 2.0 system (Xenocs France). The refractive index of the PEG–UPy polymer at different humidities was measured using a SE-VE spectral ellipsometer (EOPTICS). The transmittance spectrum was obtained with a Shimadzu UV-2450 spectrophotometer.
Results and discussion
Preparation and characterization of SiO2/PEG–UPy PEs
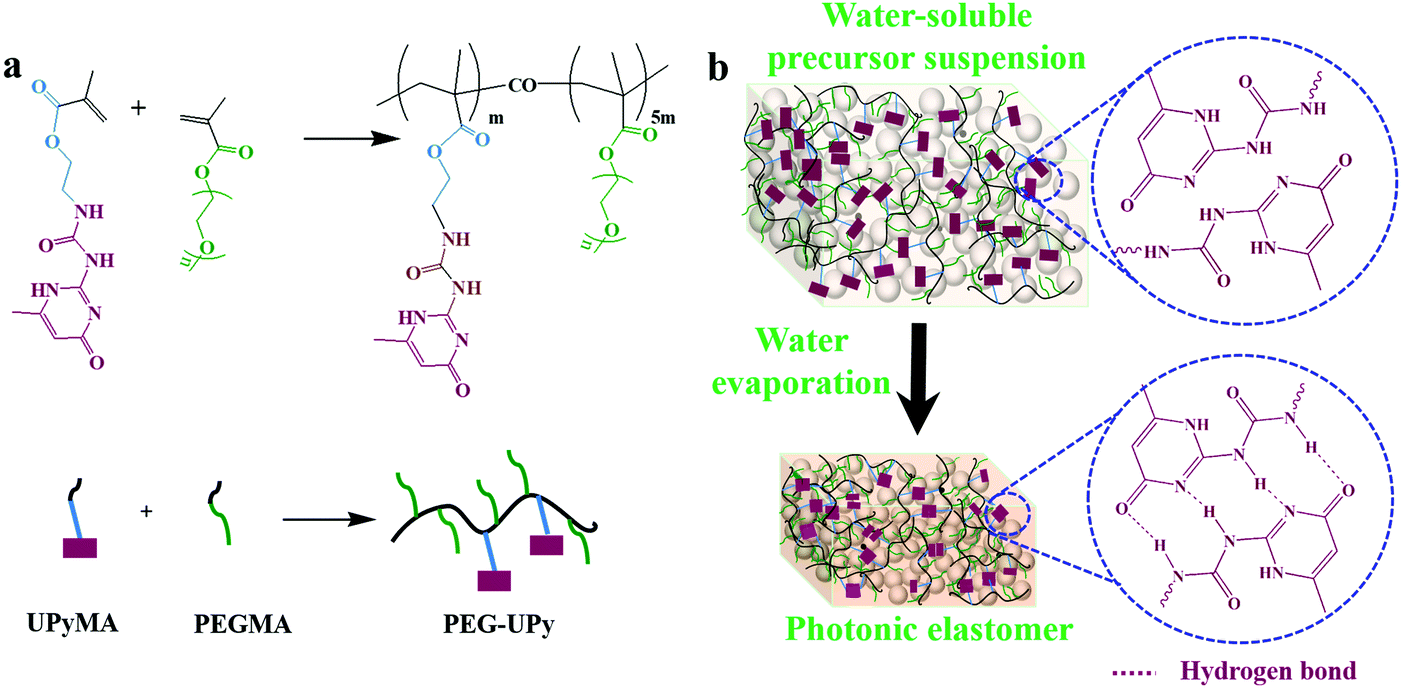
The PEG–UPy polymer with a molecular weight of 11.1 kDa was synthesized via RAFT polymerization (Fig. 1a), and it has demonstrated many advantages, such as high flexibility, water-solubility, and transparency in the visible region (Fig. S1, ESI†). Furthermore, the UPy units can form quadruple hydrogen bonding, which endows the polymer with room temperature self-healing capability.36 Such merits suggest that it can be used as an ideal polymeric matrix for preparing PEs with self-healing and recycling capabilities. In this study, an optimized ratio of PEGMA to UPyMA of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was chosen for the preparation of the PEG–UPy matrix with suitable self-healing, flexibility (glass transition temperature ∼62.7 °C) and mechanical strength.41 The procedure for fabricating PEs with the PEG–UPy polymer is schematically illustrated in Fig. 1b. Briefly, SiO2 NPs, CB NPs, and the PEG–UPy polymer were firstly dispersed in water to obtain the precursor suspension. Then, the precursor suspension was cast into a Teflon mold, followed by water evaporation to obtain the SiO2/PEG–UPy PEs. SiO2/PEG–UPy PEs with 40 wt%, 50 wt%, and 60 wt% SiO2 NP content were prepared and denoted as SiO2/PEG–UPy40, SiO2/PEG–UPy50, and SiO2/PEG–UPy60, respectively. The added CB NPs could absorb multiple and incoherent light scattering which arises inside the colloidal amorphous array and thereby improve the saturation of structural colors.43
:1 was chosen for the preparation of the PEG–UPy matrix with suitable self-healing, flexibility (glass transition temperature ∼62.7 °C) and mechanical strength.41 The procedure for fabricating PEs with the PEG–UPy polymer is schematically illustrated in Fig. 1b. Briefly, SiO2 NPs, CB NPs, and the PEG–UPy polymer were firstly dispersed in water to obtain the precursor suspension. Then, the precursor suspension was cast into a Teflon mold, followed by water evaporation to obtain the SiO2/PEG–UPy PEs. SiO2/PEG–UPy PEs with 40 wt%, 50 wt%, and 60 wt% SiO2 NP content were prepared and denoted as SiO2/PEG–UPy40, SiO2/PEG–UPy50, and SiO2/PEG–UPy60, respectively. The added CB NPs could absorb multiple and incoherent light scattering which arises inside the colloidal amorphous array and thereby improve the saturation of structural colors.43
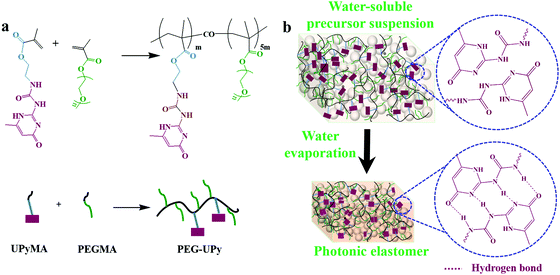 |
| | Fig. 1 Synthesis of the PEG–UPy polymer (a) and the preparation of the SiO2/PEG–UPy PEs by a solvent evaporation method (b). | |
Optical and mechanical properties of SiO2/PEG–UPy PEs
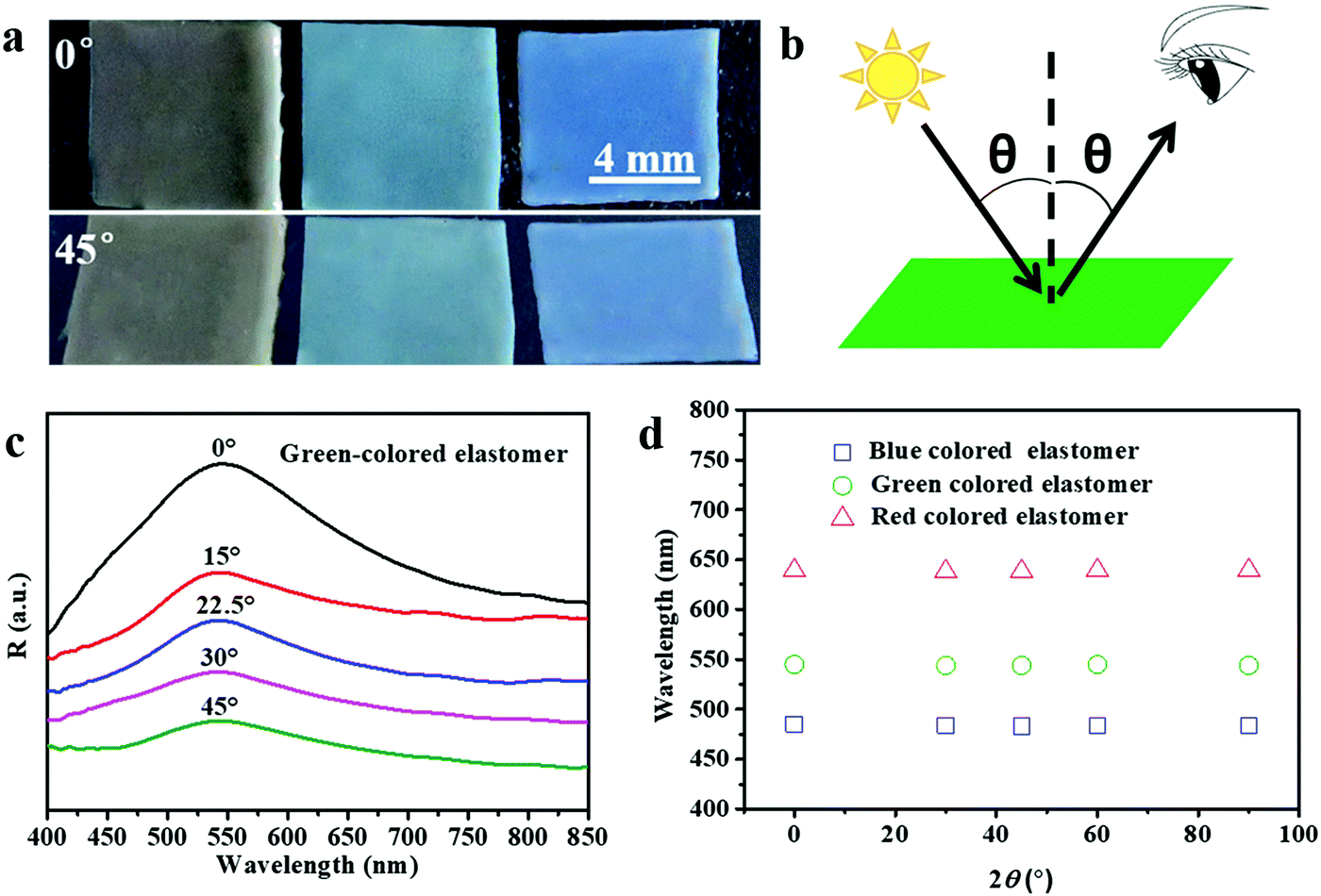
The structural colors of the PEs can be tailored by varying the size and content of the SiO2 NPs. Fig. 2a shows PEs with three structural colors that were obtained by using a SiO2 NP mass loading content of 50% and SiO2 NP sizes of 220, 204, and 180 nm, respectively. As expected, the obtained PEs show angle-independent structural color. When changing the viewing angle from 0° to 45°, the PEs show negligible color change under diffuse light illumination. To further verify the structural color with angle-independent characteristics, the reflectance spectra of the green-colored SiO2/PEG–UPy50 PE were measured at varied detection angles from 0° to 45° and the defined detection angle θ is illustrated (Fig. 2b). As shown in Fig. 2c, the positions of the reflection peak of the PEs showed negligible change with varied detection angle from 0° to 45°. Furthermore, we summarize the reflection peak positions of the as-obtained PEs with red, green, and blue colors under varied detection angles in Fig. 2d. Clearly, almost constant peak positions of 639 nm, 543 nm, and 484 nm are observed for the PEs, further confirming that the structural colors of the obtained PEs are angle-independent. Such results coincide well with the amorphous structure of colloidal particles in the polymer matrix in the previous report.27
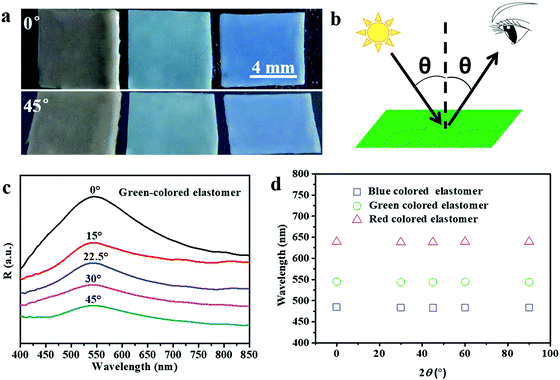 |
| | Fig. 2 (a) Photographs of SiO2/PEG–UPy50 PEs under viewing angles of 0° and 45°. SiO2 NP sizes from left to right: 220 nm, 204 nm, and 180 nm. (b) Illustration of the reflectance spectrum measurement with varied detection angles θ. The detection angle θ is the angle between the detection direction and the normal surface of the substrate. (c) Reflectance spectra of the green-colored elastomer at different detection angles, from 0° to 45°. (d) The relationship between the peak wavelength in the reflectance spectra of the three different colored elastomers and the detection angle. | |
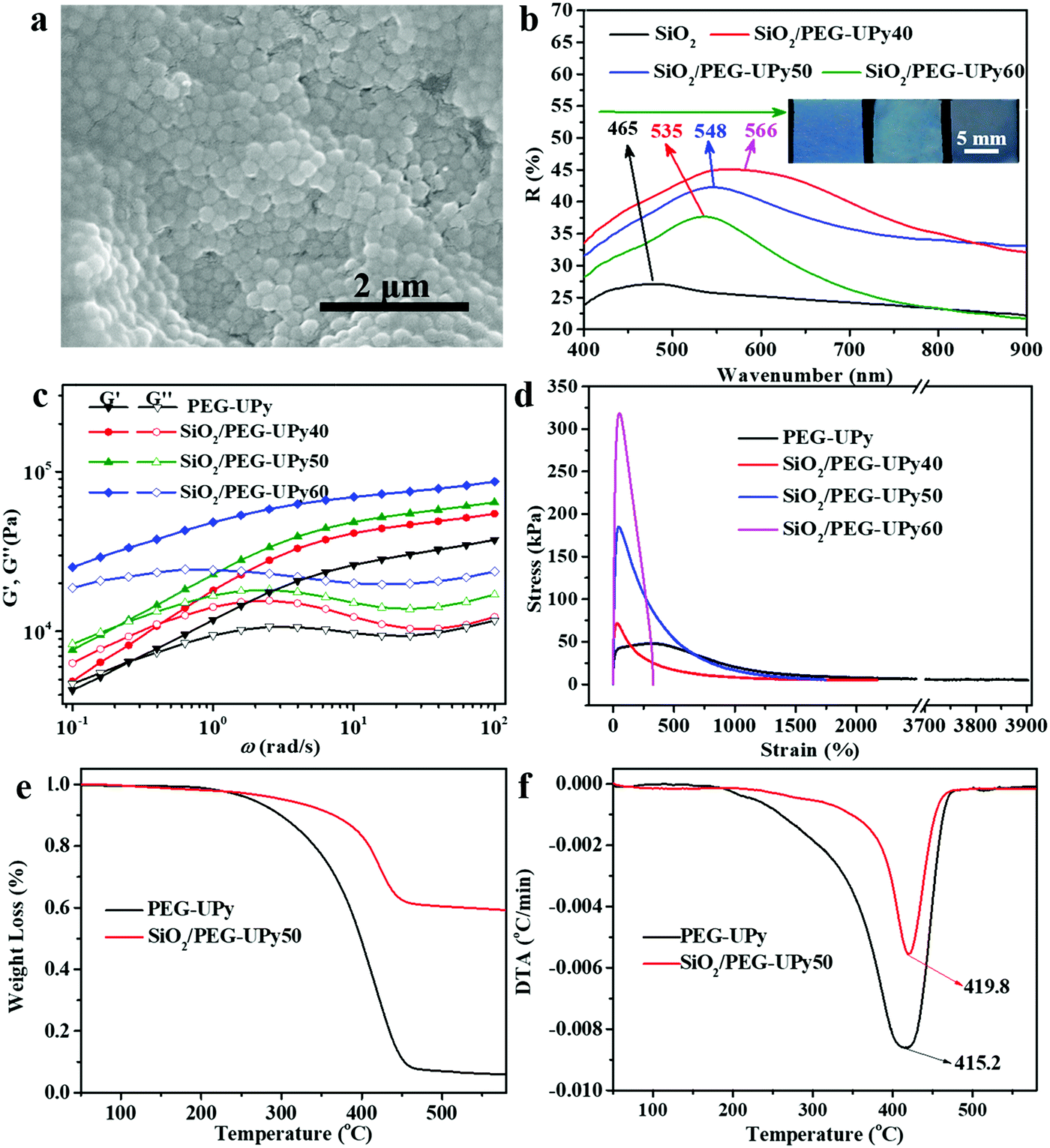
The microstructure of the resultant SiO2/PEG–UPy PE was investigated by SEM measurement (Fig. 3a). Clearly, SiO2 NPs arranged into amorphous structures when being introduced into the polymer matrix. The small-angle X-ray scattering curve can further confirm that the SiO2 NPs arranged in amorphous structures in the polymer matrix (Fig. S2, ESI†), which is critically important for producing the angle-independent structural color.26 In fact, the introduced PEG–UPy polymer not only changes the packing periodicity of the SiO2 NPs (Fig. S3, ESI†) but also increases the SiO2 NP interparticle distance.
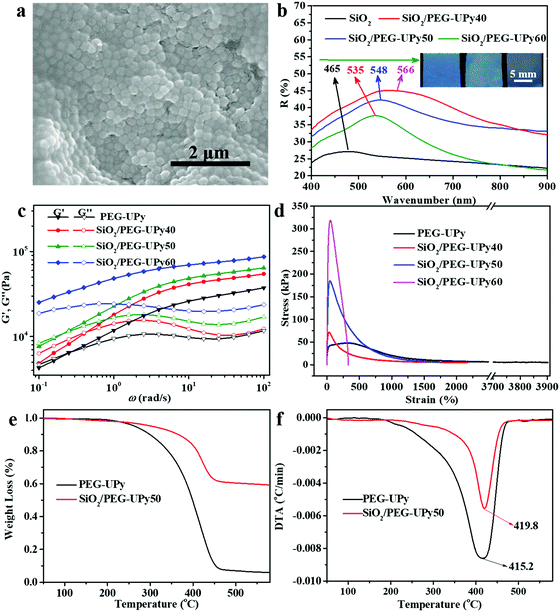 |
| | Fig. 3 (a) SEM image of the SiO2/PEG–UPy PE with a SiO2 NP content of 66.67%. (b) Reflectance spectra of SiO2, and SiO2/PEG–UPy40, SiO2/PEG–UPy50, and SiO2/PEG–UPy60 PEs (a SiO2 NP diameter of 180 nm). The inset in (b) shows the corresponding photographs of the PEs. Rheological measurements (c) and stress–strain tests (d) of the PEG–UPy polymer, and SiO2/PEG–UPy40, SiO2/PEG–UPy50, and SiO2/PEG–UPy60 PEs. TGA (e) and the corresponding DTG curves (f) of PEG–UPy and the SiO2/PEG–UPy50 PE. | |
Therefore, the structural colors of the SiO2/PEG–UPy PEs could be easily adjusted by changing the SiO2 NP content. As shown in Fig. 3b, the reflection peak position of the SiO2 NP film with a closely packed manner is located at 465 nm. After being introduced into the PEG–UPy polymer matrix, the reflection peaks shift to longer wavelengths, from 465 to 535, 548 and 566 nm for 100%, 60%, 50%, and 40% SiO2 NP content because of the increase of the center-to-center distance between two neighboring SiO2 NPs in the PEs. In addition, we found that the full width at half maximum (FWHM) of the reflection peaks of the SiO2/PEG–UPy PEs became broader with the increase of the PEG–UPy content, which can be attributed to the higher scattering intensity of amorphous structures (Fig. S4, ESI†).44 The average distance between these neighboring particles with different addition of the PEG–UPy polymer can be quantitatively calculated by eqn (1) and (2),19,26
| | 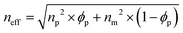 | (1) |
| | 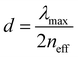 | (2) |
where
neff is the effective refractive index of the elastomer containing different contents of the PEG–UPy polymer and SiO
2 NPs.
np and
nm are the refractive indices of the SiO
2 NPs (1.420) and PEG–UPy polymer (1.457, measured by a SE-VE spectral ellipsometer), respectively.
ϕp is the volume fraction of SiO
2 NPs in the PEs.
λmax is the measured position of the reflection peak.
d is the center-to-center distance of the neighboring particles. According to the equations, the calculated
d increased from 180 nm to 186 nm, 190 nm, and 196 nm, when the corresponding SiO
2 NP content in the PEs is 100 wt%, 60 wt%, 50 wt%, and 40 wt%, respectively. These results can further confirm the increased distance of SiO
2 NPs with the PEG–UPy polymer content.
Besides their optical properties, the mechanical properties of the SiO2/PEG–UPy PEs could also be tuned by the amount of incorporated SiO2 NPs. We show that the introduction of the rigid SiO2 NPs could improve the mechanical performance of the polymeric matrix. The mechanical properties of the PEG–UPy polymer and SiO2/PEG–UPy PEs were investigated by rheological measurements and stress–strain tests (Fig. 3c and d). The storage moduli (G′) of the PEG–UPy polymer and SiO2/PEG–UPy PEs dominate the loss moduli (G′′) at >0.5 rad s−1, indicating that all these samples exhibited elastic solid behavior due to the cross-linked hydrogen bonding network. G′ and G′′ of the PEs at low angular frequency gradually enhanced with the increase of the SiO2 NP content. Stress–strain measurements could further confirm the reinforcement by incorporating rigid SiO2 NPs. The tensile strength and elongation at break are 47.3 kPa and 3900% for the PEG–UPy polymer, respectively, while the introduction of SiO2 NPs increased the tensile strength of the PEG–UPy polymer up to 70.3 kPa, 182.6 kPa, and 317.5 kPa, for SiO2/PEG–UPy40, SiO2/PEG–UPy50, and SiO2/PEG–UPy60. Meanwhile, the elongation at break of the SiO2/PEG–UPy PEs decreases rapidly with the content of SiO2 NPs, due to the confinement effect of the SiO2 NPs on the mobility of polymer chains.45 In addition, the thermal stability of the PE is also improved with the addition of SiO2 NPs. From the curves of TGA, the weight residue of the SiO2/PEG–UPy50 PE is ∼59.2%, which is attributed to the existence of the inorganic SiO2 NPs (Fig. 3e and f). From the curves of DTG shown in Fig. 2f, it can be found that the weight loss peak attributed to decomposition occurs at 415.2 °C for the PEG–UPy polymer, while that for the SiO2/PEG–UPy50 PE shifts to a high temperature of 419.8 °C. The enhanced thermal stability of SiO2/PEG–UPy can be ascribed to the formation of hydrogen bonding between the hydroxyl groups in the PEG chains and silicon hydroxyl groups on the surface of the SiO2 NPs.45 The improved mechanical and thermal properties of the PEs ensure the mechanical stability and flexibility of the PEs for practical applications.
Self-healing properties of the SiO2/PEG–UPy PEs
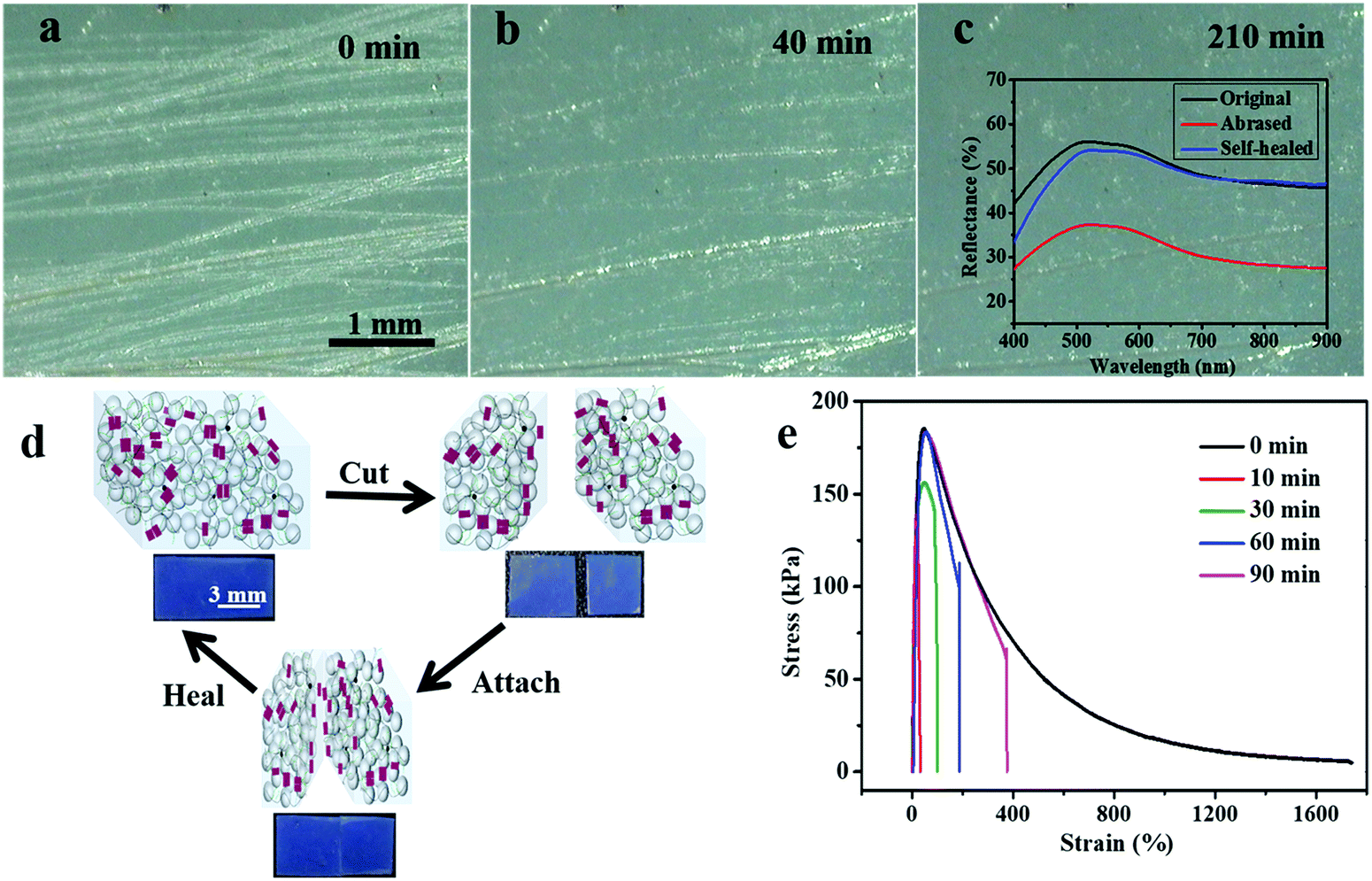
Accidental scratches or cuts would strongly interfere with the optical performance of the PE materials, therefore it is highly important for the materials to rapidly self-heal their damage without the requirement of any external stimulus.46–48 Since the inherent hydrogen bonds arising from the UPy–UPy dimers in the PEG–UPy polymer are easily broken and reconfigured, the pristine PEG–UPy film showed self-healing properties (Fig. S5, ESI†), and with the introduction of such a polymeric matrix, the SiO2/PEG–UPy PEs are endowed with the self-healing capability of healing cuts or scratches at room temperature. We simulate scratches by purposely abrasing the green-colored SiO2/PEG–UPy50 PE with 150-grade sandpaper to evaluate the self-healing properties over time under a relative humidity of 56%. As shown in Fig. 4a, after being abrased by the sandpaper, the surface of the elastomer became rough and scratches with a width of 1–100 μm were generated on the surface. Correspondingly, the reflection peak intensity of the PE decreased because of the light scattering from the surface microstructures (inset in Fig. 4c). After being stored at ambient conditions for different times, it can be observed that some minor scratches disappeared and major scratches narrowed (Fig. 4b and Fig. S6, ESI†). After ∼210 min, the scratches on the abrased SiO2/PEG–UPy50 PE almost totally disappeared and the corresponding reflection peak intensity nearly recovered to the initial position, suggesting effective self-healing behavior (Fig. 4c). Additionally, the self-healing process of the SiO2/PEG–UPy50 PE for cuts was quantitatively evaluated by the stress–strain test. We first cut the SiO2/PEG–UPy50 PE into two pieces with a blade and put them in contact with each other for different periods, and the corresponding schematic illustration is shown in the Fig. 4d inset. When the two cut surfaces physically contacted, the broken UPy–UPy dimers reconfigure rapidly and reconnect the separated elastomers by forming quadruple hydrogen bonding. The mechanical strength of the cut SiO2/PEG–UPy50 PE after 10 min healing reached 133 kPa and further reached 155 kPa after 30 min of healing. After healing for 60 min, the mechanical strength could return to the initial value of the PE. With further extending the healing duration for 90 min, the mechanical strength remains, while the elongation at break can be further improved. Compared with the PEs based on relatively strong coordination bonding in our previous work, the improved self-healing performance of the current PEs can be attributed to the weak hydrogen bonding, which can effectively facilitate molecular chain mobility.49–52
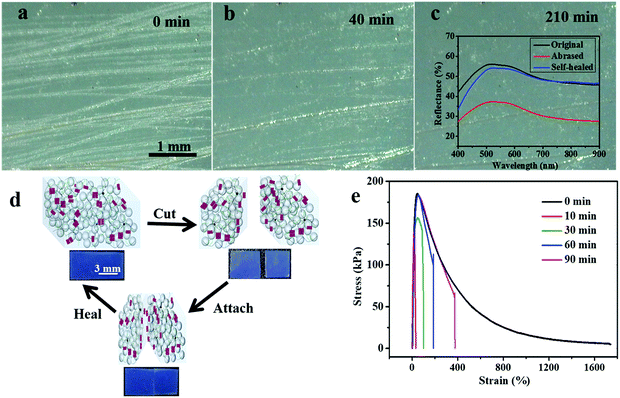 |
| | Fig. 4 Self-healing capability of the PEs. Optical microscopy images of the as-abrased green-colored elastomer (a, 0 min) and time-course snapshots of the abrased colored elastomer at 40 min (b) and 210 min (c). Inset in (c): reflectance spectra of the original, abrased, and self-healed PEs. The scale bar in (a) applies to (b) and (c). (d) The schematic illustration of the self-healing mechanism. Inset in (d): the corresponding photographs of the self-healed blue-colored elastomer. The stress–strain curves of the original and the self-healed blue-colored SiO2/PEG–UPy50 samples after different healing durations (10 min, 30 min, 60 min, and 90 min) at room temperature. The self-healing of PEs was performed under a relative humidity of ∼56%. | |
Recycling properties of SiO2/PEG–UPy PEs
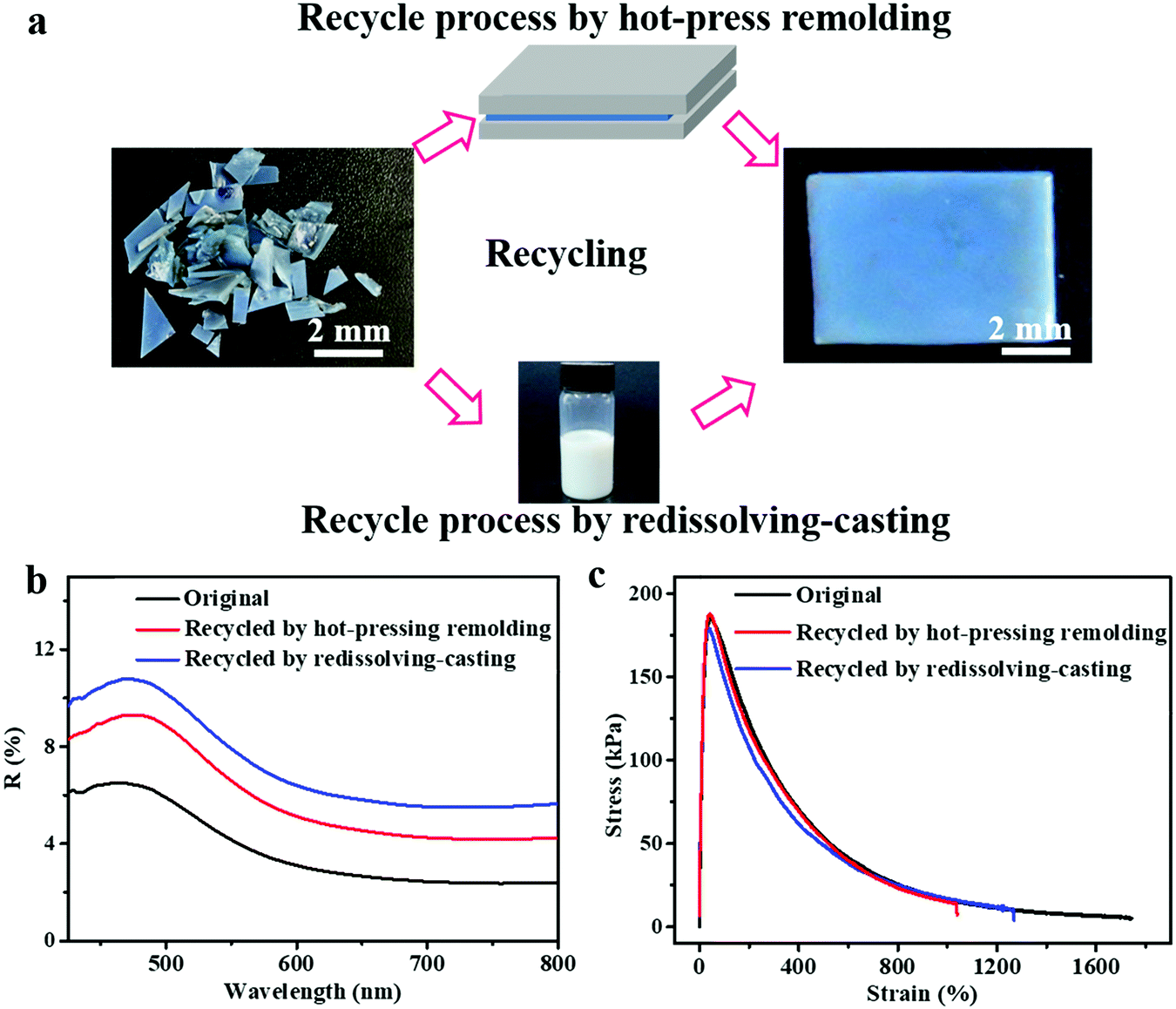
In addition to the self-healing capability of the PEs for scratches or cuts, they can be recycled and reshaped completely by a conventional polymer processing strategy, which is another environment-friendly feature of the PEs developed in this study. Because of the water-solubility and reversible crosslinking of the introduced PEG–UPy polymer, the SiO2/PEG–UPy50 PEs could be easily recycled by a redissolving–casting or hot-press remolding process. Fig. 5a, top panel, presents the hot-press remolding process for recycling the SiO2/PEG–UPy50 PE. A piece of the blue-colored SiO2/PEG–UPy50 PE was first cut into many small pieces and filled into a Teflon mold, and a pressure of ∼500 Pa was applied using a Teflon plate for 20 min at 60 °C, resulting in the recovery of the damaged PE. Additionally, it also can be easily recycled by redissolving it in water, followed by recasting into a Teflon mold. As shown in Fig. 5a, the original and recycled SiO2/PEG–UPy50 PE almost have the same appearance. Furthermore, reflectance spectrum measurements and stress–strain tests of the elastomers were further performed, revealing that not only the structural color but also the mechanical properties can be recovered to the original state. These results demonstrated that the SiO2/PEG–UPy PEs have high-efficiency and good prospects in the recovery of secondary resources, and are beneficial to meet the environment-friendly and resource-saving requirements. Notably, although the water-soluble PEG–UPy polymer is used for the preparation of the SiO2/PEG–UPy PE, the structural color of the PEs is stable in a high humidity environment, which is confirmed by the invariable reflectance peak position under different humidity (25%, 56%, and 96%) conditions (Fig. S7, ESI†). Such a stable structural color of the PEs under high humidity can be attributed to the equilibrium between the decreased effective refractive index of the PE and the increased NP distance due to the water absorption.53,54
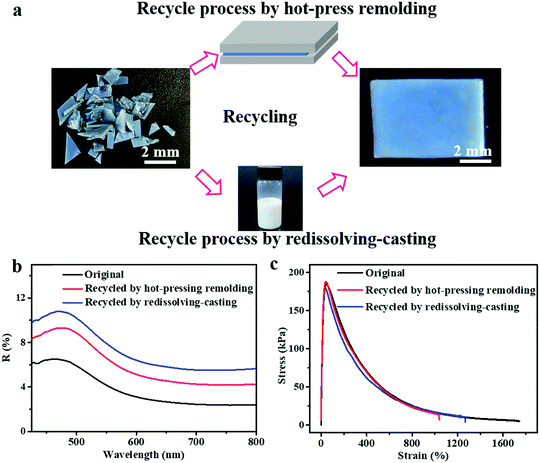 |
| | Fig. 5 (a) Recycling properties of the PEs through the hot-press molding and the redissolving–casting processes. Reflectance spectra (b) and stress–strain curves (c) of the original SiO2/PEG–UPy50 PE and the corresponding recycled samples by redissolving–casting and hot-press remolding. | |
Strain-induced color shifting properties of SiO2/PEG–UPy PEs
Similar to chameleon skin, the SiO2/PEG–UPy PEs developed in the current study exhibited strain-induced color shifting capability and therefore can be used as a visual strain sensor. As proof of concept, we show that the SiO2/PEG–UPy PEs can be used as a visual motion sensor. To this end, a piece of a red-colored SiO2/PEG–UPy50 PE with a thickness of ∼0.7 mm was attached to the joint of a finger, and the color changes through the different degrees of finger bending are monitored and shown in Fig. 6a. As the bending angle of the elastomer changed from 0° to 15°, 45° and 90°, it is observed that the color of the PE shifts from red to green. Accordingly, the corresponding reflectance peak shifts from 654 nm to 583 nm as shown in Fig. 6b due to the decrease of the lattice constant from the bend-induced deformation. When strain recovery occurs, the color of the elastomer almost returns to its original state. Notably, the SiO2/PEG–UPy50 PE which is cut into two pieces and healed for 90 min shows similar performance to that of the original one due to its excellent self-healing ability. Moreover, the reversible color change further illustrates their sensitivity and reliability in the visual strain sensing field.
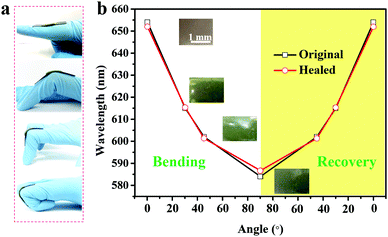 |
| | Fig. 6 Strain-induced color shifting properties of the red-colored SiO2/PEG–UPy50 PE for different bending angles. (a) Illustration of the bending angle of the elastomer and the corresponding reflectance peak position (b) of the original and self-healed SiO2/PEG–UPy50 PE. Inset in (b): photographs of the original SiO2/PEG–UPy50 PE for the different bending angles. | |
Conclusions
In summary, we have developed a facile yet environment-friendly strategy to prepare PEs by introducing isotropically arranged SiO2 NPs into a water-soluble PEG–UPy polymer matrix. The obtained SiO2/PEG–UPy PEs show angle-independent structural colors, and their optical and mechanical properties could be adjusted by incorporating different sizes and contents of SiO2 NPs. Due to the solvent-free state and the introduced PEG–UPy polymer, the SiO2/PEG–UPy PEs exhibit stable structural color even in a high humidity environment. Notably, the reversible multiple hydrogen bonds in the PEG–UPy elastic matrix endow the SiO2/PEG–UPy PEs with improved self-healing and excellent recycling performance. It is envisioned that with the stable and angle-independent structural color as well as the self-healing and the easy recycling properties, the SiO2/PEG–UPy PEs exhibit excellent prospects in strain sensor and wearable device development.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National National Science Foundation of China (51525302 and 51973075), Key R&D Program of Ministry of Science and Technology (2018YFA0209200), Open Research Fund of State Key Lab of Polym Phys & Chem, CIAC, CAS (2017-27), and Program for HUST Academic Frontier Youth Team (2015-01). We also thank the HUST Analytical and Testing Center for their help with the facilities.
Notes and references
- X. Q. Wang, C. F. Wang, Z. F. Zhou and S. Chen, Adv. Opt. Mater., 2014, 2, 652–662 CrossRef CAS.
- C. I. Aguirre, E. Reguera and A. Stein, Adv. Funct. Mater., 2010, 20, 2565–2578 CrossRef CAS.
- F. Wang, X. Zhang, Y. Lin, L. Wang and J. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 5009–5016 CrossRef CAS PubMed.
- C. Nakamura, K. Manabe, M. Tenjimbayashi, Y. Tokura, K. H. Kyung and S. Shiratori, ACS Appl. Mater. Interfaces, 2018, 10, 22731–22738 CrossRef CAS PubMed.
- J. Hou, M. Li and Y. Song, Angew. Chem., Int. Ed., 2017, 56, 2–12 CrossRef.
- P. F. Tan, C. F. Wang, X. Q. Wang, W. Q. Ji, M. Chen and S. Chen, Macromol. Mater. Eng., 2016, 301, 1363–1373 CrossRef CAS.
- D. Ge, E. Lee, L. Yang, Y. Cho, M. Li, D. S. Gianola and S. Yang, Adv. Mater., 2015, 27, 2489–2495 CrossRef CAS.
- S. Z. Yu, W. B. Niu, S. L. Wu, W. Ma and S. F. Zhang, J. Mater. Chem. C, 2018, 6, 12814–12821 RSC.
- J. D. Lin, H. Y. Lin, G. J. Wei, Y. C. Chuang, L. J. Chen, T. S. Mob and C. R. Lee, J. Mater. Chem. C, 2019, 7, 4740–4747 RSC.
- S.-H. Kim, V. Hwang, S. G. Lee, J.-W. Ha, V. N. Manoharan and G.-R. Yi, Small, 2019, 15, 1900931 CrossRef.
- S. Magkiriadou, J.-G. Park, Y.-S. Kim and V. N. Manoharan, Opt. Mater. Express, 2012, 2, 1343–1352 CrossRef CAS.
- J.-G. Park, S.-H. Kim, S. Magkiriadou, T. M. Choi, Y.-S. Kim and V. N. Manoharan, Angew. Chem., Int. Ed., 2014, 53, 2899–2903 CrossRef CAS PubMed.
- S. Magkiriadou, J.-G. Park, Y.-S. Kim and V. N. Manoharan, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 062302 CrossRef PubMed.
- D.-W. Jung, K. J. Park, S. Lee, J. Kim, G. Lee and G. R. Yi, Korean J. Chem. Eng., 2018, 35, 2138–2144 CrossRef CAS.
- S.-H. Kim, S. Magkiriadou, D. K. Rhee, D. S. Lee, P. J. Yoo, V. N. Manoharan and G.-R. Yi, ACS Appl. Mater. Interfaces, 2017, 9, 24155–24160 CrossRef CAS PubMed.
-
X. Jia, H. Tan and J. Zhu, Switzerla, Springer International Publishing, 2017, pp. 151–172 Search PubMed.
- T. Ito, C. Katsura, H. Sugimoto, E. Nakanishi and K. Inomata, Langmuir, 2013, 29, 13951–13957 CrossRef CAS PubMed.
- R. Zheng, Y. Wang, C. Jia, Z. Wan, J. Luo, H. A. Malik, X. Weng, J. Xie and L. Deng, ACS Appl. Mater. Interfaces, 2018, 10, 35533–35538 CrossRef CAS PubMed.
- G. H. Lee, T. M. Choi, B. Kim, S. H. Han, J. M. Lee and S. H. Kim, ACS Nano, 2017, 11, 11350–11357 CrossRef CAS PubMed.
- H. Fudouzi and T. Sawada, Langmuir, 2006, 22, 1365–1368 CrossRef CAS PubMed.
- T. Ding, G. Cao, C. G. Schafer, Q. Zhao, M. Gallei, S. K. Smoukov and J. J. Baumberg, ACS Appl. Mater. Interfaces, 2015, 7, 13497–13502 CrossRef CAS PubMed.
- J. Zhou, P. Han, M. Liu, H. Zhou, Y. Zhang, J. Jiang, P. Liu, Y. Wei, Y. Song and X. Yao, Angew. Chem., Int. Ed., 2017, 56, 10462–10466 CrossRef CAS PubMed.
- R. Hong, Y. Shi, X. Q. Wang, L. Peng, X. Wu, H. Cheng and S. Chen, RSC Adv., 2017, 7, 33258–33262 RSC.
- F. Fu, Z. Chen, Z. Zhao, H. Wang, L. Shang, Z. Gu and Y. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5900–5905 CrossRef CAS PubMed.
- J. D. Forster, H. Noh, S. F. Liew, V. Saranathan, C. F. Schreck, L. Yang, J. G. Park, R. O. Prum, S. G. Mochrie, C. S. O'Hern, H. Cao and E. R. Dufresne, Adv. Mater., 2010, 22, 2939–2944 CrossRef CAS PubMed.
- H. Tan, Q. Lyu, Z. Xie, M. Li, K. Wang, K. Wang, B. Xiong, L. Zhang and J. Zhu, Adv. Mater., 2019, 31, 1805496 CrossRef PubMed.
- M. M. Li, Q. Q. Lyu, J. T. Zhu and L. B. Zhang, Acta Polym. Sin., 2019, 50, 271–280 Search PubMed.
- Y. Fang, X. Du, Y. Jiang, Z. Du, P. Pan, X. Cheng and H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 14490–14500 CrossRef CAS.
- J. Zhang, Y. Niu, C. Huang, Z. Chen, C. Chen, K. Yang and Y. Wang, Polym. Chem., 2012, 3, 1390–1393 RSC.
- F. Meng, M. M. Umair, K. Iqbal, X. Jin, S. Zhang and B. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 13022–13028 CrossRef CAS PubMed.
- S. Y. Tawfik, M. W. Sabaa and R. T. Botros, Pigm. Resin Technol., 2017, 46, 408–422 CrossRef CAS.
- M. Bejaoui, E. Pantazi, M. Calvo, E. Folch-Puy, A. Serafin, G. Pasut, A. Panisello and R. Adam, Oxid. Med. Cell. Longevity, 2016, 9096549 Search PubMed.
- D. Chen, M. Wu, B. Li, K. Ren, Z. Cheng, J. Ji, Y. Li and J. Sun, Adv. Mater., 2015, 27, 5882–5888 CrossRef CAS PubMed.
- B. Q. Y. Chan, S. J. W. Heng, S. S. Liow, K. Zhang and X. J. Loh, Mater. Chem. Front., 2017, 1, 767–779 RSC.
- J. Hou, M. Liu, H. Zhang, Y. Song, X. Jiang, A. Yu, L. Jiang and B. Su, J. Mater. Chem. A, 2017, 5, 13138–13144 RSC.
- M. Liu, P. Liu, G. Lu, Z. Xu and X. Yao, Angew. Chem., Int. Ed., 2018, 57, 11242–11246 CrossRef CAS PubMed.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- G. Zhang, Y. Yang, Y. Chen, J. Huang, T. Zhang, H. Zeng, C. Wang, G. Liu and Y. Deng, Small, 2018, 1801189 CrossRef PubMed.
- Y. Chen and Z. Guan, Chem. Commun., 2014, 50, 10868–10870 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 669–692 CrossRef.
- B. Zhou, D. He, J. Hu, Y. Ye, H. Peng, X. Zhou, X. Xie and Z. Xue, J. Mater. Chem. A, 2018, 6, 11725–11733 RSC.
- X. Y. Yang, Z. L. Wang and M. C. Pan, Adv. Mater. Res., 2011, 189-193, 3836–3839 CAS.
- M. Iwata, M. Teshima, T. Seki, S. Yoshioka and Y. Takeoka, Adv. Mater., 2017, 29, 1605050 CrossRef PubMed.
- B. Yi and H. Shen, J. Mater. Chem. C, 2017, 5, 8194–8200 RSC.
- J. Li, L. He, T. Liu, X. Cao and H. Zhu, Sol. Energy Mater. Sol. Cells, 2013, 118, 48–53 CrossRef CAS.
- A. Pettignano, M. Häring, L. Bernardi, N. Tanchoux, F. Quignard and D. D. Díaz, Mater. Chem. Front., 2017, 1, 73–79 RSC.
- H. Wang, X. Ji, Z. Li, C. N. Zhu, X. Yang, T. Li, Z. L. Wu and F. Huang, Mater. Chem. Front., 2017, 1, 167–171 RSC.
- J. Cui, Z. Ma, L. Pan, C. An, J. Liu, Y. Zhou and Y. Li, Mater. Chem. Front., 2019, 3, 464–471 RSC.
- X. Wu, J. Wang, J. Huang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 7387–7396 CrossRef CAS PubMed.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J. C. Lai, L. Liu, C. H. Li, X. Yan, C. Liu, J. B. H. Tok, X. Jia and Z. Bao, Adv. Mater., 2018, 30, 1801435 CrossRef PubMed.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed.
- Y. Li, S. Chen, X. Li, M. Wu and J. Sun, ACS Nano, 2015, 9, 1005–1065 CrossRef PubMed.
- D. Shi, X. Zhang, Z. Yang, S. Liu and M. Chen, RSC Adv., 2016, 6, 85885–85890 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00454h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Lianbin
Zhang
*,
Lianbin
Zhang