
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular adducts based on weak interactions between the trimeric Lewis acid complex (perfluoro-ortho-phenylene)mercury and polypnictogen complexes†‡
Martin
Fleischmann
a,
James S.
Jones
b,
Gábor
Balázs
a,
François P.
Gabbaï
 *b and
Manfred
Scheer
*a
*b and
Manfred
Scheer
*a
aInstitut für Anorganische Chemie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: manfred.scheer@chemie.uni-regensburg.de; Fax: (+49) 941-943-4439
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA
First published on 20th July 2016
Abstract
Reactions of the trinuclear Lewis acid perfluoro-ortho-phenylene)mercury [(o-HgC6F4)3] (1) with the polypnictogen complex [CpMo(CO)2(η3-P3)] (2) containing a cyclo-P3 ligand and the series of E2 complexes [{CpMo(CO)2}2(μ,η2:η2-E2)] (E = P(3a), As(3b), Sb(3c), Bi(3d)) are reported. In all cases, the reaction products show very weak interactions between the En ligand complexes and the Lewis acid 1, as evidenced by their highly dynamic behaviour in solution and the formation of adducts in the solid state showing Hg⋯E contacts below the respective sum of the van der Waals radii. The complexes 2 (P3), 3a (P2) and 3b (As2) show interactions of only one pnictogen atom with all three Hg atoms of 1. The complex 3c (Sb2) forms two adducts with 1 showing either a side-on coordination of the Sb2 dumbbell towards Hg or an end-on coordination of both Sb atoms towards two independent molecules of 1. The Bi2 complex 3d shows an almost parallel alignment of the Bi2 dumbbell situated above the center of the planar Lewis acid 1. The arrangements of the E2 complex series towards 1 are rationalized with the help of electrostatic potential maps obtained by DFT calculations. Finally the structural characterizations of a new modification of the free Sb2 complex 3c, the Bi2 complex 3d, the starting material of its preparation [Bi{CpMo(CO)3}3] (4) and an unprecedented ‘Cr4As5’ cluster 5 are presented.
Introduction
During the last few decades supramolecular chemistry based on weakly interacting molecules instead of strong covalent bonds has gained more and more attention. The investigation of planar electron-deficient molecules has developed into a rich area of research with applications in anion recognition,1,2 molecular machines,3 or light emitting materials.4 In this context, pyridinium cations,3 or electron deficient aromatic compounds,1,2 are prototypical representatives. We became particularly interested in the chemistry of the trinuclear organometallic compound (perfluoro-ortho-phenylene)-mercury (1).5 Due to its planar geometry this molecule contains three sterically accessible mercury centers whose naturally low Lewis acidity is significantly increased by the fluorinated molecular backbone.6,7 This unusual Lewis acid builds up alternating binary stacks with aromatic hydrocarbons8–11 and forms double-sandwich complexes featuring Hg⋯Cp interactions when reacted with the metallocenes Cp2Fe and Cp2Ni (Scheme 1a).12 Additionally 1 forms weak Lewis acid/base adducts with a variety of O, N and S donors which simultaneously interact with all three Hg atoms (Scheme 1b).6,7,13 During the past few years we have extended this chemistry to polyphosphorus complexes, observing only weak P⋯Hg interactions instead of potential Hg⋯Cp interactions in the reactions of 1 and the triple-decker complex [(CpMo)2(μ,η6:η6-P6)] (Scheme 1c).14 Recently we reported the differences in coordination behavior of the aromatic cyclo-P5 and cyclo-As5 ligands from the ferrocene analogues [Cp*Fe(η5-E5)] (E = P, As)15,16 towards the Lewis acid 1.17 While the P5 ring forms a weak Lewis acid/base adduct (Scheme 1d) with one P donor atom, the As5 ring shows a rather cofacial arrangement towards the molecular plane of 1 with three As atoms interacting simultaneously with the Hg centers (Scheme 1e).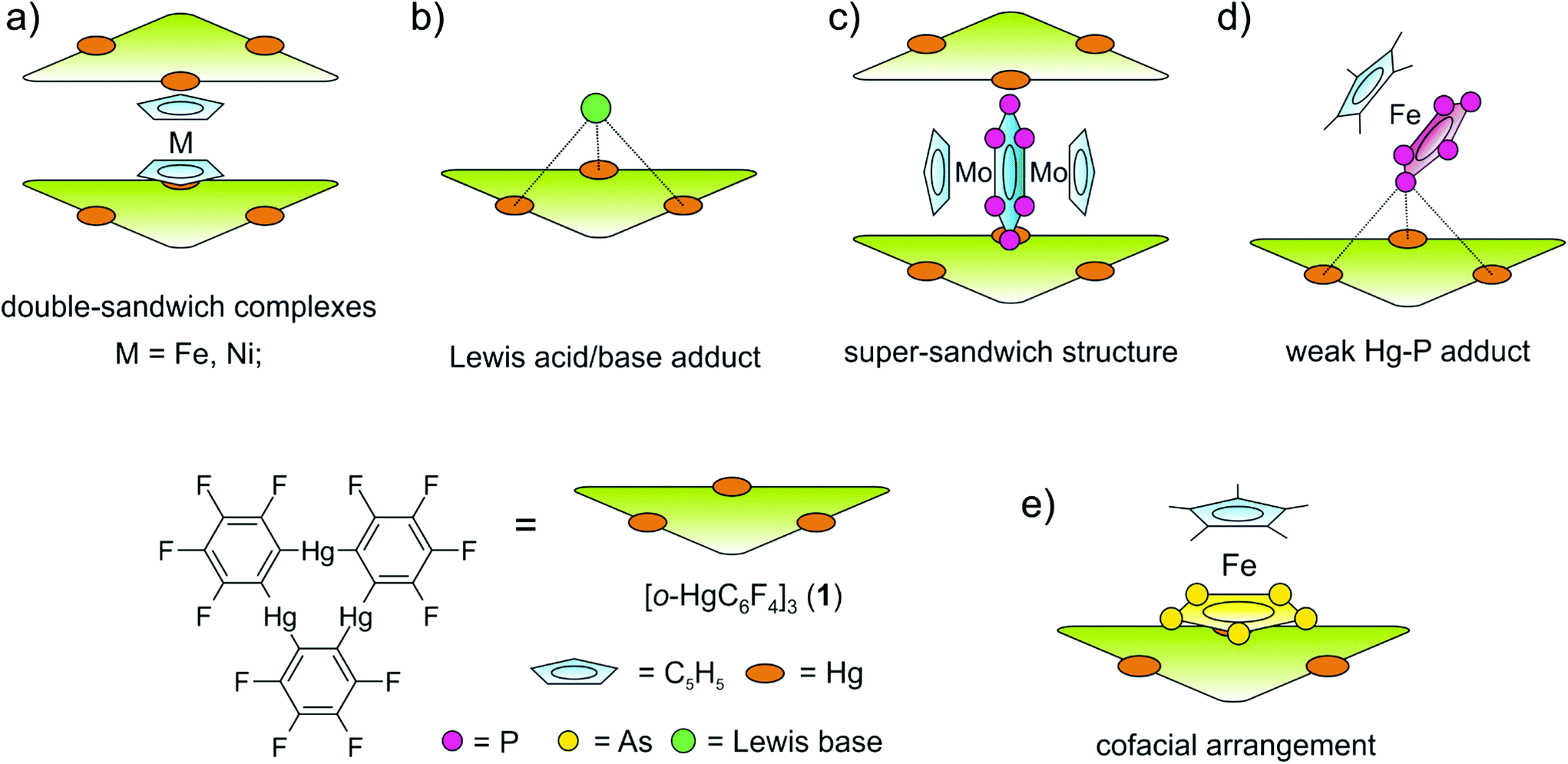 | ||
| Scheme 1 (a) Representation of the double-sandwich complexes formed from 1 and the metallocenes Cp2Fe and Cp2Ni. (b) Illustration of Lewis acid/base adducts of 1 with the Lewis bases simultaneously interacting with all three Hg centers. (c) Super-sandwich structures obtained from 1 and [(CpMo)(μ,η6:η6-P6)]. (d) Coordination of [Cp*Fe(η5-P5)] to the three Hg atoms of 1via one P atom. (e) [Cp*Fe(η5-As5)] interacting with 1via three As atoms showing an almost cofacial arrangement. | ||
Since the nature of the polypnictogen complexes has been shown to have a dramatic effect on its reactivity towards 1, we questioned how the trinuclear Lewis acid might interact with a terminal cyclo-E3 ligand and furthermore how its coordination behavior may vary when reacting 1 with complexes of the heavier pnictogen atoms Sb and Bi. To address these questions we first investigated the reactivity of 1 towards the P3 complex [CpMo(CO)2(η3-P3)] (2) and subsequently towards the E2 complex series [{CpMo(CO)2}2(μ,η2:η2-E2)] E = P(3a), As(3b), Sb(3c), Bi(3d) (Scheme 2). For the first time the Bi2 complex 3d and the starting material of its synthesis [{CpMo(CO)3}3Bi] (4), as well as a new modification of the Sb2 complex 3c could be structurally characterized. Additionally, an unprecedented Cr–As cluster [(CpCr)4(μ,η3:η3-As2)(μ,η3:η2:η3-As3)] (5), obtained from a related reaction was also characterized by single crystal X-ray diffraction.
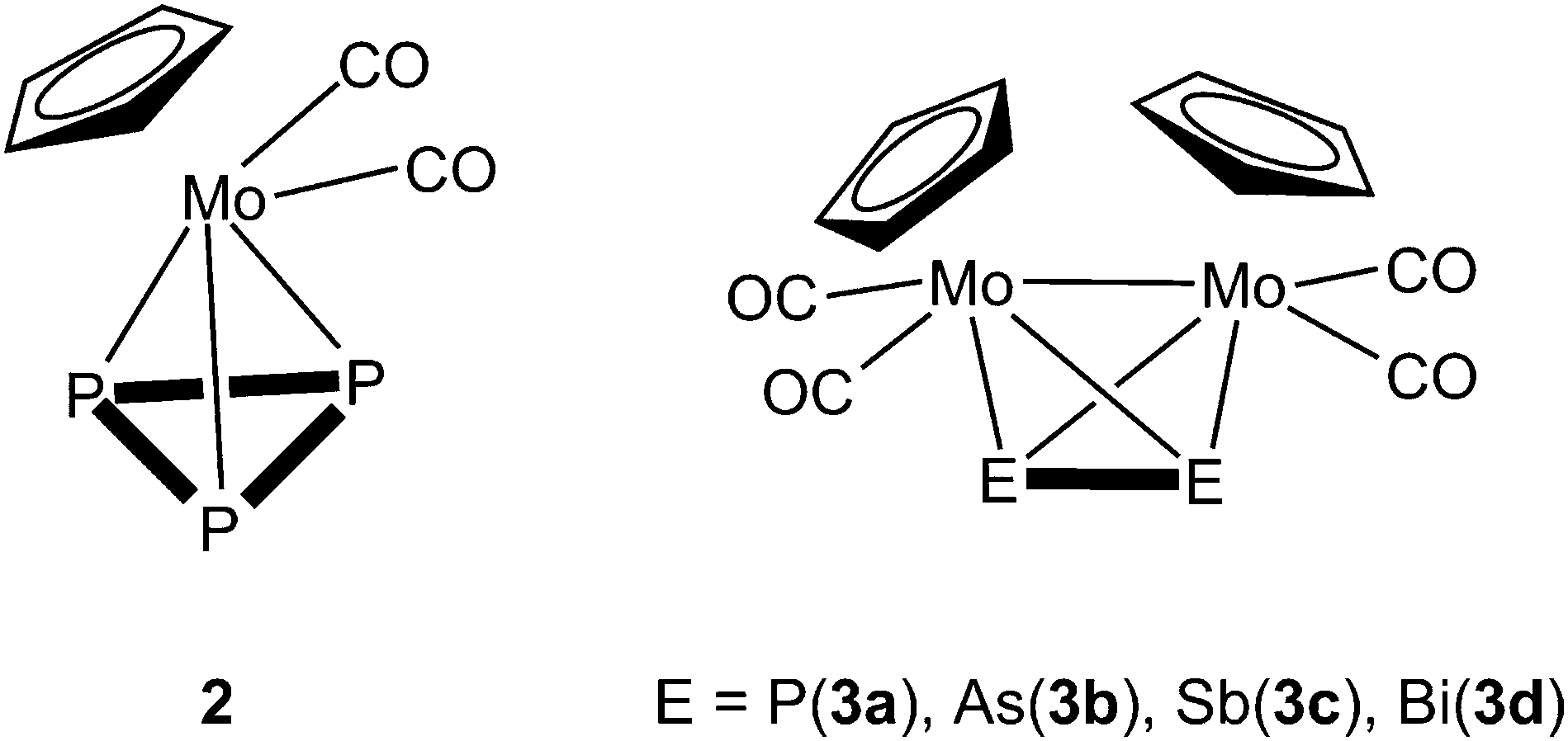 | ||
| Scheme 2 Representation of the tetrahedrane complexes [CpMo(CO)2(η3-P3)] (2) and [{CpMo(CO)2}2(μ,η2:η2-E2)] (E = P(3a), As(3b), Sb(3c), Bi(3d)). | ||
Results and discussion
Syntheses and spectroscopic characterization of the products
The adducts between the pnictogen complexes and 1 are easily prepared by dissolving 1 and an equimolar amount of the cyclo-P3 complex 2 or the E2 complexes 3a–d in 10 mL of CH2Cl2 until a clear solution is formed. After stirring at room temperature, this solution is filtered and the solvent is removed until the filtrate solution is saturated. Storing the resultant solutions at +4 °C results in product crystallization in a matter of hours to days. Following this approach one can obtain pure samples of equimolar adducts of 1 and the cyclo-P3 complex 2 [(1)·(2)] (80% isolated yield), the P2 complex 3a [(1)·(3a)] (75%) and the As2 complex 3b [(1)·(3b)] (74%), respectively. In the case of the Sb2 complex 3c two adducts with molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 [(1)·(3c)] and 2:1 [(1)2·(3c)] are detected from the same reaction. In the case of the Bi2 complex 3d we were able to determine the coexistence of the adduct [(1)·(3d)2], pure 1 and pure 3d next to each other in crystalline form from the same reaction. This observation suggests very weak interactions between 1 and 3d and prevents a successful separation of these compounds. Moreover, the Sb2 complex 3c was isolated in 56% yield after refluxing (Me3Si)2CHSbH2 and CpMo(CO)3Me in xylene for 3 h. The reaction of the triple-decker complex [(CpCr)2(μ,η5:η5-As5)] with 1 enabled the structural characterization of an unprecedented mixed Cr–As cluster 5 (for details see below). Since the described adducts are based on weak Hg⋯E (E = P, As, Sb, Bi) interactions we assume only very weak association in solution. In accordance with this, in the mass spectra we never observed any peaks assignable to any adduct but only the starting materials of the described products. The 1H and the 19F NMR spectra in CD2Cl2 solution are unchanged compared to the starting compounds. While the 31P{1H} NMR spectrum of [(1)·(2)] in CD2Cl2 solution shows a sharp singlet at −349.5 ppm which is also unchanged to the starting compound 2 we can observe an upfield shift of about 11 ppm for the singlet of the P2 ligand 3a in the case of the adduct [(1)·(3a)].
:1 [(1)·(3c)] and 2:1 [(1)2·(3c)] are detected from the same reaction. In the case of the Bi2 complex 3d we were able to determine the coexistence of the adduct [(1)·(3d)2], pure 1 and pure 3d next to each other in crystalline form from the same reaction. This observation suggests very weak interactions between 1 and 3d and prevents a successful separation of these compounds. Moreover, the Sb2 complex 3c was isolated in 56% yield after refluxing (Me3Si)2CHSbH2 and CpMo(CO)3Me in xylene for 3 h. The reaction of the triple-decker complex [(CpCr)2(μ,η5:η5-As5)] with 1 enabled the structural characterization of an unprecedented mixed Cr–As cluster 5 (for details see below). Since the described adducts are based on weak Hg⋯E (E = P, As, Sb, Bi) interactions we assume only very weak association in solution. In accordance with this, in the mass spectra we never observed any peaks assignable to any adduct but only the starting materials of the described products. The 1H and the 19F NMR spectra in CD2Cl2 solution are unchanged compared to the starting compounds. While the 31P{1H} NMR spectrum of [(1)·(2)] in CD2Cl2 solution shows a sharp singlet at −349.5 ppm which is also unchanged to the starting compound 2 we can observe an upfield shift of about 11 ppm for the singlet of the P2 ligand 3a in the case of the adduct [(1)·(3a)].
Structural characterization of the products in the solid state
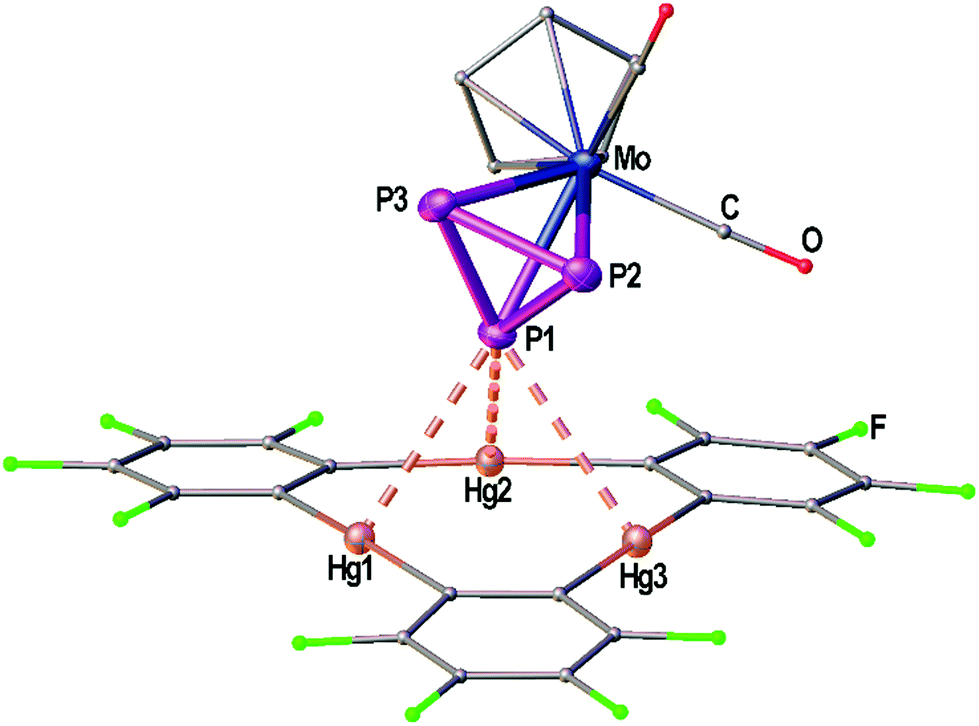 | ||
| Fig. 1 Solid state structure of compound the adduct [(1)·(2)]. Selected bond lengths [Å]: Hg1–P1 3.21821(9), Hg2–P1 3.19349(7), Hg3–P1 3.25262(6), P1–P2 2.140(2), P1–P3 2.145(2), P2–P3 2.147(3); interplanar angle P3 plane – Hg3 plane = 65.43(7)°. Ellipsoids are drawn at 50% probability. H atoms are omitted and C, F and O atoms are represented as small spheres for clarity. | ||
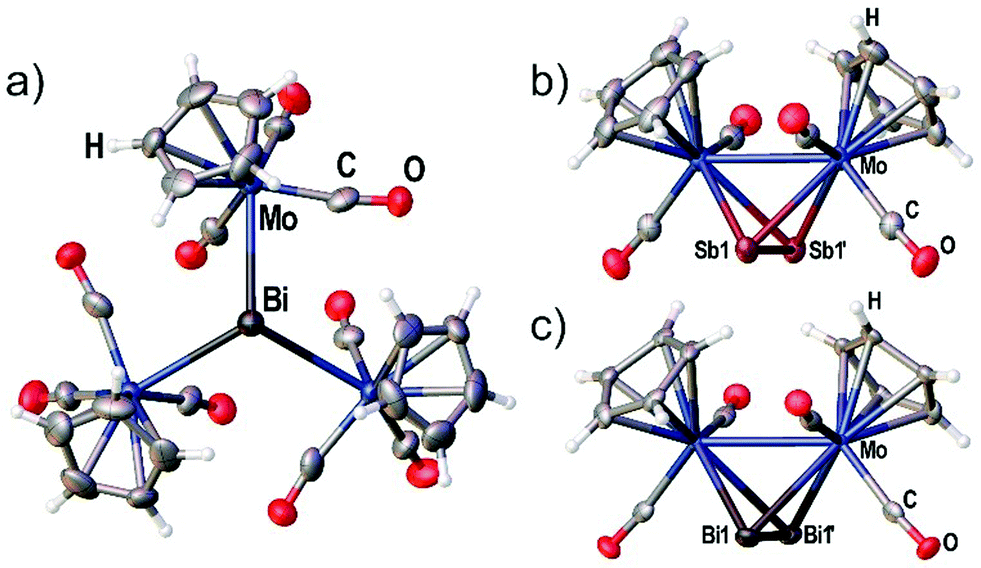 | ||
| Fig. 2 (a) Crystal structure of [{CpMo(CO)3}3Bi] (4) with viewing direction along the threefold symmetry axis. (b) Crystal structure of the Sb2 complex 3c. (c) Crystal structure of the Bi2 complex 3d. Selected bond lengths [Å] and angles [°]: 4: Mo–Bi 3.014(3), angle Mo–Bi–Mo = 113.50(5)°. 3c: Mo1–Mo1′ 3.1183(10), Sb1–Sb1′ 2.6871(7). 3d: Mo1–Mo1′ 3.1378(13), Bi1–Bi1′ 2.8580(7). Ellipsoids are drawn at 50% probability for 3c,d and 30% probability for 4. | ||
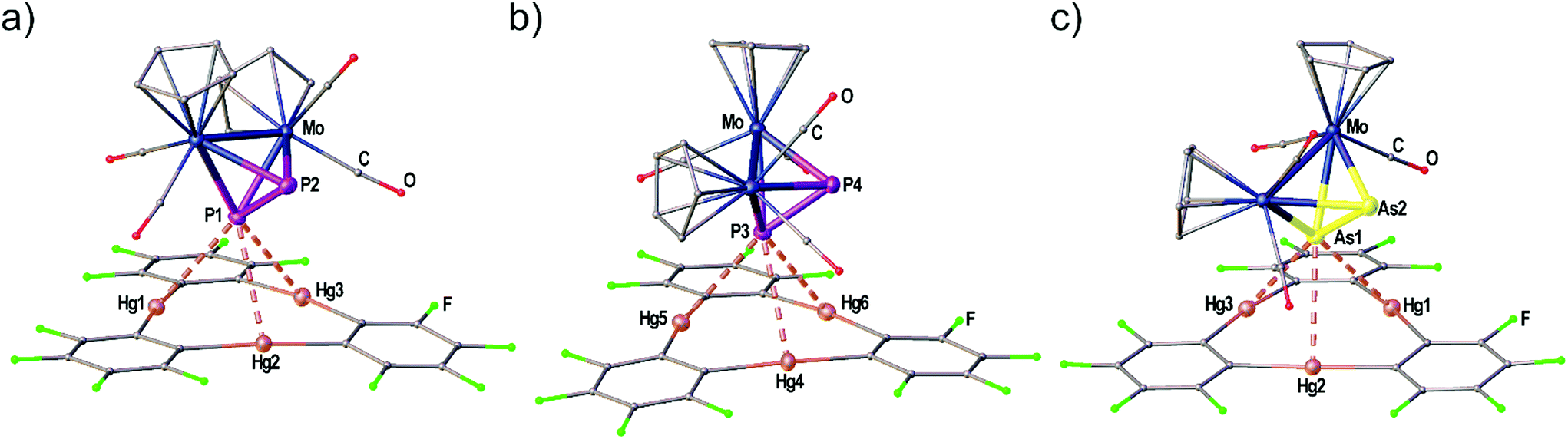 | ||
| Fig. 3 Independent molecules A (a) and B (b) found in the solid state structure of the adduct [(1)·(3a)]. (c) Crystal structure of the adduct [(1)·(3b)]. Selected bond lengths [Å]: [(1)·(3a)]: Hg1–P1 3.1040(2), Hg1–P2 3.3369(2), Hg1–P3 3.1953(2), Hg4–P3 3.3557(2), Hg5–P3 3.1609(2), Hg6–P3 3.2299(2), P1–P2 2.0883(1), P3–P4 2.0948(1). [(1)·(3b)]: Hg1–As1 3.2702(2), Hg2–As1 3.3259(1), Hg3–As1 3.3788(2), As1–As2 2.3162(1). Angles of E2 dumbbells to Hg3 plane: P1P2–Hg1Hg2Hg3 36.33(1)°, P3P4–Hg4Hg5Hg6 38.05(1)°, As1As2–Hg1Hg2Hg3 37.00(1)°. Ellipsoids are drawn at 50% probability. H atoms are omitted and C, F and O atoms are represented as small spheres for clarity. | ||
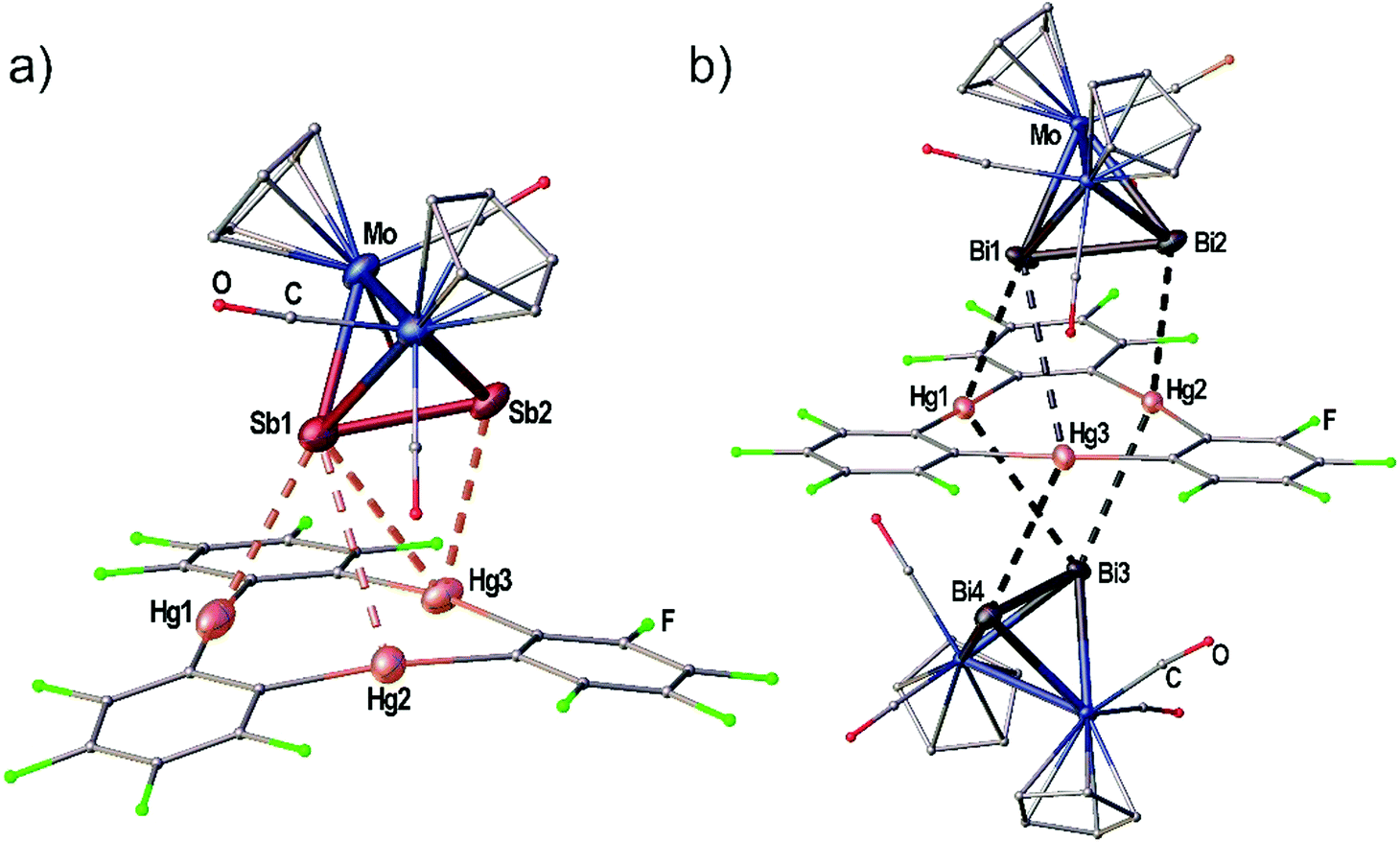 | ||
| Fig. 4 (a) Solid state structure of the adduct [(1)·(3c)]. (b) Solid state structure of the adduct [(1)·(3d)2]. Selected bond lengths [Å]: [(1)·(3c)]: Hg1–Sb1 3.4336(1), Hg2–Sb1 3.8621(1), Hg3–Sb1 3.7879(2), Hg3–Sb2 3.5222(1), Sb1–Sb2 2.6914(1). [(1)·(3d)2]: Hg1–Bi1 3.4859(1), Hg2–Bi2 3.6370(1), Hg3–Bi1 4.0481(2), Bi1–Bi2 2.8479(1), Hg1–Bi3 3.9046(1), Hg2–Bi3 3.5888(1), Hg3–Bi4 3.7827(1), Bi3–Bi4 2.8537(1). Ellipsoids are drawn at 50% probability. H atoms are omitted and C, F and O atoms are represented as small spheres for clarity. Angles E2 straight to Hg3 plane: Sb1Sb2–Hg1Hg2Hg3 7.03(1)°, Bi1Bi2–Hg1Hg2Hg3 5.72(1)°, Bi3Bi4–Hg1Hg2Hg3 2.50(1)°. | ||
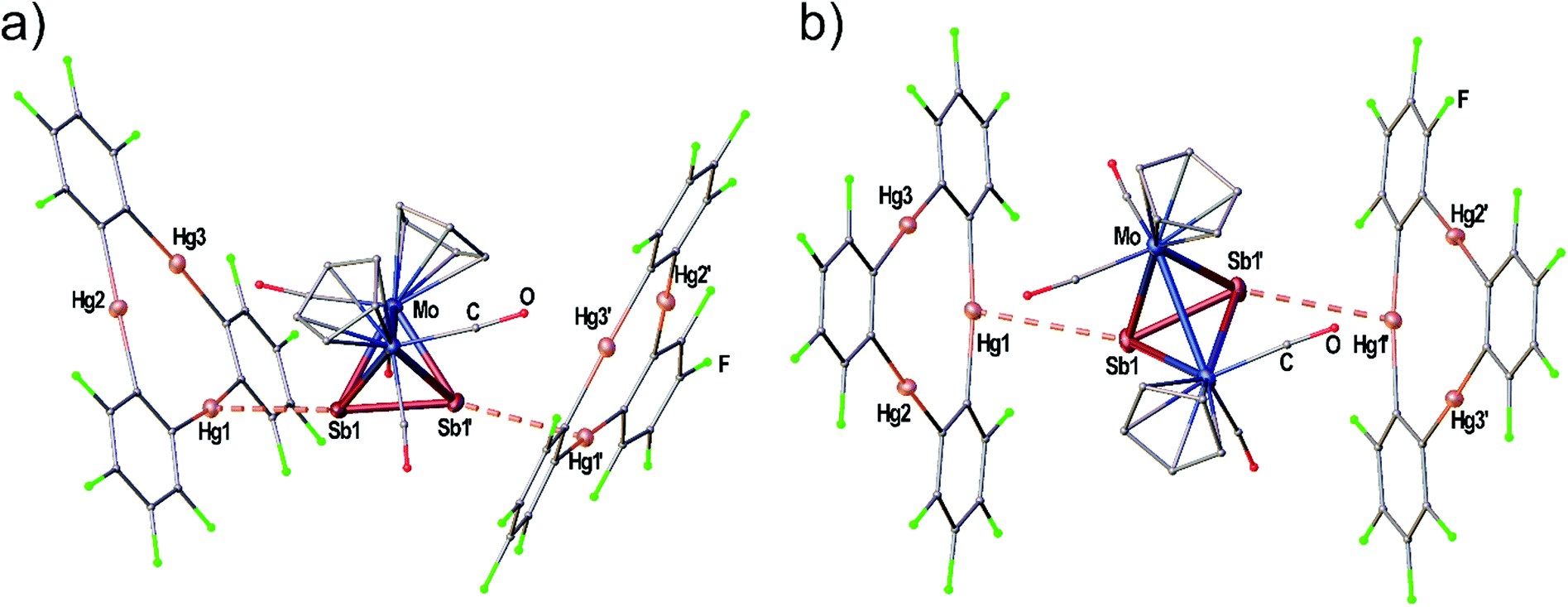 | ||
| Fig. 5 Solid state structure of the adduct [(1)2·(3c)]. (a) Viewing direction along the Mo–Mo bond. (b) Viewing direction along the crystallographic b-axis (twofold rotation axis). Selected bond lengths [Å]: Hg1–Sb1 3.5283(9), Sb1–Sb1′ 2.6863(13). Ellipsoids are drawn at 50% probability. H atoms are omitted and C, F and O atoms are represented as small spheres for clarity. | ||
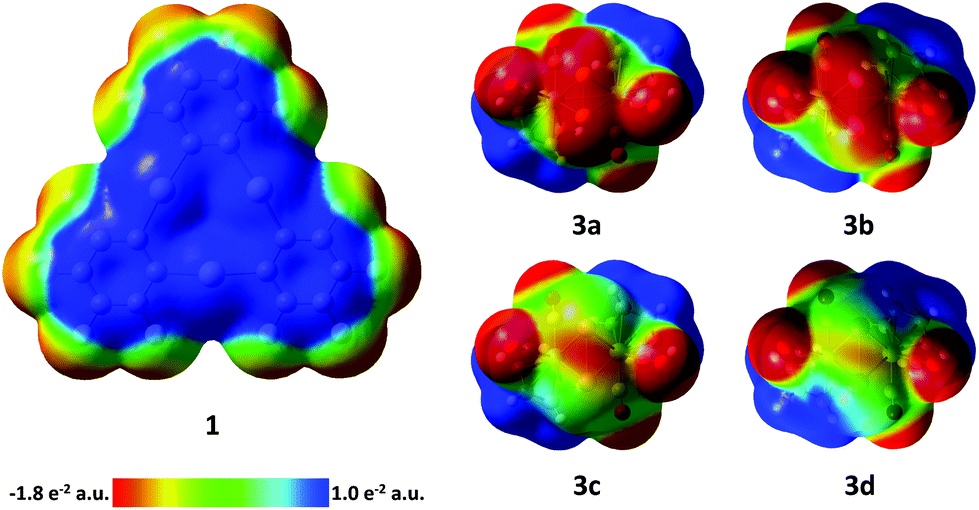 | ||
| Fig. 6 Electrostatic potential surfaces of compounds 1 and 3a–d (isovalue = 0.004). 3a–d: viewing direction along the C2 axis of the complexes. E2 bonds are in front. | ||
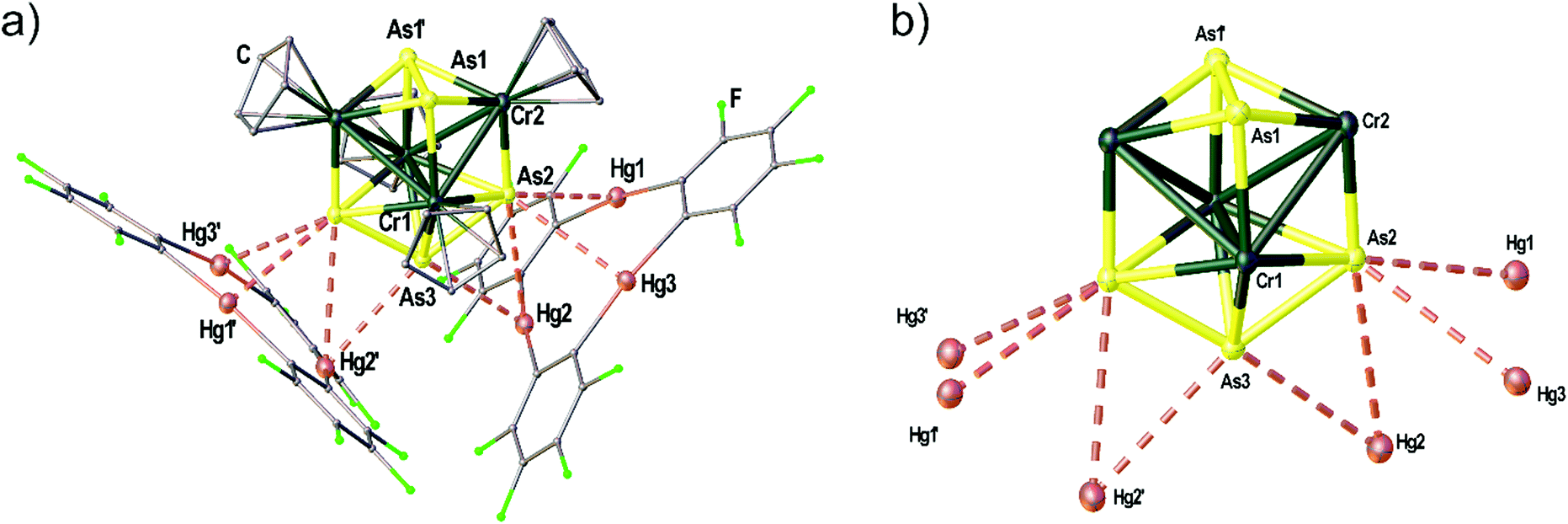 | ||
| Fig. 7 Solid state structure of the adduct [(1)2·(5)]. (a) Representation of the Cr–As cluster 5 coordinated by two molecules of 1. (b) Illustration of the central cluster core of 5 omitting the four Cp ligands on Cr and only showing the contacts to the Hg atoms of 1. Selected bond lengths [Å]: Hg1–As2 2.6863(1), Hg2–As2 3.6066(1), Hg2–As3 3.3557(1), Hg3–As2 3.3944(1), As1–As1′ 2.4922(1), As2–As3 2.4456(1), Cr1–Cr1′ 2.9075(1), Cr1–Cr2 2.9453(1), Cr1–Cr2′ 3.0134(1), Cr1–As1 2.5067(1), Cr1–As2 2.5899(1), Cr1–As2′ 2.5698(1), Cr1–As3 2.4683(1), Cr2–As1 2.4450(1), Cr2–As1′ 2.4380(1), Cr2–As2 2.3604(1); angle Hg3 plane – Hg3 plane 87.36(1)°. Ellipsoids are drawn at 50% probability. H atoms are omitted and C, F and O atoms are represented as small spheres for clarity. | ||
Crystal structure of the adduct [(1)·(2)]
Fig. 1 shows the solid state structure of the adduct [(1)·(2)]. Although the symmetry of the cyclo-P3 ligand of complex 2 would generally match the threefold symmetry of the planar Lewis acid 1 it shows a coordination of only one P atom interacting with all three Hg centers of 1. The resulting Hg–P distances (3.19349(7)–3.25262(6) Å) lie well within the sum of the vdW radii (3.6 Å). These lengths are very comparable to the Hg–P distances found for 1 and [(CpMo)2(μ,η6:η6-P6)] (Scheme 1c), but they are a bit shorter than the closest Hg–P distance that was observed for 1 and [Cp*Fe(η5-P5)] (3.2878(9) Å, Scheme 1d). The angle enclosed between the P3 plane and the Hg3 plane measures 65.43(7)°. The P–P bonds show uniform lengths of 2.144(2) Å comparable to the free complex 2.24 No Cp–Hg contacts are present.Crystal structures of the Sb2 complex 3c, the Bi2 complex 3d and the starting material [{CpMo(CO)3}3Bi] (4)
Although the synthesis of the Bi2 complex 3d was already reported in 1988 the solid state structure was only described of a derivative bearing one methyl group on each Cp ligand.25,26 During our investigations we were able to determine the solid state structures of 3d and of the starting material 4 shown in Fig. 2(a) and (c). The synthesis and X-ray structure of the analogous Sb2 complex 3c was reported27 earlier. Moreover, we could structurally characterize a new modification of 3c by X-ray analysis (see Fig. 2(b)).The new modification of 3c crystallizes in the trigonal space group P3221 with half a complex in the asymmetric unit. The complex is crystallographically completed by a twofold rotation axis. The determined bond lengths are in good agreement with the previously determined structure (Mo–Mo 3.114(1) Å, Sb–Sb 2.678(1) Å).
The Bi2 complex 3d (see Fig. 2(c)) crystallizes in the monoclinic space group C2/c with half of the complex in the asymmetric unit. The Mo–Mo bond as well as the Bi–Bi bond is bisected by the twofold rotation axis. The Bi–Bi bond length of 2.8580(7) Å is very comparable to the 2.838(1) Å found for [{(η5-C5H4Me)Mo(CO)2}2(μ,η2:η2-Bi2)].25,26
Compound 4 crystallizes in the trigonal space group P31c with the central Bi atom situated on the threefold rotation axis (see Fig. 2a) showing a Mo–Bi bond length of 3.014(3) Å. The bonding environment around Bi is best described as trigonal pyramidal with the three Mo–Bi–Mo angles summing up to 340.50(5)°.
Structural characterization of the adducts formed from 1 and the E2 complexes 3a–d
The adduct [(1)·(3a)] crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two independent formula units [(1)·(3a)] in the asymmetric unit shown in Fig. 3(a) and (b). Both molecules of 3a interact with all three Hg atoms of the Lewis acid 1via one P atom which is situated above the center of 1 while the other P atom shows no Hg⋯P contacts. The resulting Hg–P distances lie in the range of 3.10401(15)–3.3557(2) Å. The P–P bond lengths are not affected in comparison to free 3a (2.079(2) Å)28 by the coordination. The angles between the direction of the P2 dumbbell and the Hg3 plane measure 36.33(1)° (P1–P2) and 38.05(1)° (P3–P4), respectively.
with two independent formula units [(1)·(3a)] in the asymmetric unit shown in Fig. 3(a) and (b). Both molecules of 3a interact with all three Hg atoms of the Lewis acid 1via one P atom which is situated above the center of 1 while the other P atom shows no Hg⋯P contacts. The resulting Hg–P distances lie in the range of 3.10401(15)–3.3557(2) Å. The P–P bond lengths are not affected in comparison to free 3a (2.079(2) Å)28 by the coordination. The angles between the direction of the P2 dumbbell and the Hg3 plane measure 36.33(1)° (P1–P2) and 38.05(1)° (P3–P4), respectively.
When looking at the As2 complex 3b and its interaction with 1 in the solid state shown in Fig. 3(c) it can be noted that the assembly in the adduct [(1)·(3b)] is very similar to the P2 complex 3a. This observation was very surprising since we previously observed a distinctly different assembly of cyclo-P5 and cyclo-As5 ligands with 1 (see Scheme 1(d) and (e)). The compound [(1)·(3b)] crystallizes in a different unit cell (but same space group P) than [(1)·(3a)] containing only one formula unit in the asymmetric unit. The As2 complex 3b is interacting with all three Hg atoms of the Lewis acid 1via one As atom while the second As atom does not show any Hg⋯As contacts. The resulting Hg–As distances lie in the range of 3.27021(13)–3.37875(13) Å and are well within the sum of the vdW radii for Hg and As (3.7 Å). The As–As bond length is 2.31607(10) Å long which is essentially unchanged to free 3b (2.305(3) Å).29 The angle enclosed by the As2 dumbbell and the Hg3 plane (37.00(1)°) also compares very well to the analogous P2 adduct.
When the Sb2 complex 3c is allowed to react with the Lewis acid 1 two sorts of crystals (orange and red) are formed from the same solution. Single crystal X-ray diffraction analysis revealed that the orange crystals consist of the equimolar adduct of 1 and 3c [(1)·(3c)] depicted in Fig. 4a. For E = Sb, the assembly of the Mo2E2 complex is different from E = P, As. The most apparent distinction is the angle enclosed by the E2 dumbbell and the Hg3 plane in [(1)·(3c)] which measures only 7.03(1)°. Although Sb1 is also situated above the center of 1 the resulting Hg–Sb distances differ significantly. The shortest distance is observed for Hg1–Sb1 with 3.4336(1) Å while the distances to Hg2 and Hg3 are almost at the edge of vdW contacts (3.9 Å). The observed arrangement of 1 and 3c shows one additional Hg-Sb contact between Sb2 and Hg3 with 3.5222(1) Å and can be described as a side-on coordination of the Sb–Sb bond to Hg3. The Sb–Sb bond length of 2.6914(1) Å is essentially unchanged compared to free 3c (vide supra).27 X-ray analysis of the red crystals from the same reaction consist of the adduct [(1)2·(3c)] where one Sb2 complex 3c is surrounded by two molecules of the Lewis acid 1 depicted in Fig. 5(a). The Sb atom Sb1 shows only one contact to Hg1 with 3.5283(9) Å. The adduct [(1)2·(3c)] crystallizes in the monoclinic space group P2/n. The twofold rotation axis bisects the Mo–Mo bond as well as the Sb–Sb bond of 3c (see Fig. 5b). The angle enclosed by both Hg3 planes amounts to 53.45(1)°. The Sb1–Sb1′ bond length of 2.6863(13) Å is consistently unchanged compared to free 3c.
When the reactivity of the Bi2 complex 3d towards the Lewis acid 1 was investigated by using different stoichiometries, black crystals of pure 3d, colorless crystals of pure 1 and black crystals of the adduct [(1)·(3d)2] were obtained next to each other from one reaction. Although numerous crystals were mounted it was not possible to identify any adducts of 1 and 3d in different molar ratios. The solid state structure of [(1)·(3d)2] is depicted in Fig. 4(b). The compound crystallizes in the triclinic space group P with two units of 3d interacting with one molecule of the Lewis acid 1 from both sides via both Bi atoms in the asymmetric unit. The Bi–Bi dumbbells are arranged almost parallel to the Hg3 plane with enclosed angles of 2.50(1)° and 5.72(1)°, respectively. The center of 1 is not situated below one of the Bi atoms but rather the middle of the Bi–Bi bonds. The resulting Hg–Bi distances vary significantly in the range of 3.48590(8)–4.04806(13) Å. The Bi–Bi bond lengths in the adduct [(1)·(3d)2] are essentially identical to the free complex 3d (vide supra).25,26
In order to aid in the rationalization of the observed structural trends for the adducts between 3a–3d and 1, their constituent compounds were optimized by density functional theory (DFT) methods. The large computed HOMO–LUMO gaps between 1 and 3a–d of 3.06, 3.00, 2.88, and 2.74 eV, respectively, suggest that orbital-based interactions are not likely to be dominant within the pnictogen-mercury bonding found in these adducts. Instead, similar to the related adducts of 1 with the cyclo-P5 and cyclo-As5 rings of [Cp*Fe(η5-E5)] complexes,17 electrostatic and dispersion forces are likely to factor strongly in the stabilization of the observed structures. In order to probe the potential role of electrostatic forces in the observed adducts, we inspected the electrostatic potential maps of the constituent complexes shown in Fig. 6. In both 3a and 3b, the electrostatic potential surface shows accumulation of negative character both across the length of the E–E bond and at the phosphorus and arsenic termini. For the electrostatic potential map of complex 3c, negative character is preferentially accumulated at the Sb–Sb bond, with modest negative character still present at the Sb termini. Finally, in the electrostatic potential map of complex 3d, negative character is expressed almost exclusively at the Bi–Bi bond. Taken together, these results suggest that in adducts of 1 with 3a and 3b, the surfaces of the pnictogen termini are complimentary with the positive surface of the mercury centers in 1. Complex 3c appears to be a borderline case, with both residual negative character at the Sb termini and increasing preference for negative character accumulation at the Sb–Sb bond possibly accounting for the two distinctly different modes which can be observed in the two adducts of 1 and 3c. Finally, the lack of negative character at the Bi termini in complex 3d accounts for the observed side-on binding mode, as the surface of the Bi–Bi bond is most complimentary with the positive character found at the mercury surfaces in 1. In an effort to gain further insight into the nature of the E–Hg interactions, adducts [(1)·(3a)], [(1)·(3b)], [(1)2·(3c)], [(1)·(3c)], and [(1)·(3d)2] were subjected to QTAIM30 analysis at their crystal structure geometries (see ESI‡ for details). In all cases, bond critical points (BCPs) were found between the pnictogen atoms of 3a–d and the mercury atoms of 1. Additional intermolecular BCPs were found between the oxygen atoms of the carbonyl ligands of 3a–d and the mercury and carbon atoms of 1. Inspection of the features of the electron density distribution function (ρ(r)) at the intermolecular BCPs found that similar to the previously observed adducts of 1 with the cyclo-P5 and cyclo-As5 rings of [Cp*Fe(η5-E5)] complexes, electrostatic and dispersion forces predominate in the adducts of 3a–d with 1.
Structural characterization of the adduct [(1)2·(5)] and the ‘Cr4As5’ cluster 5
The Cr–As cluster 5 was characterized by chance when [(CpCr)2(μ,η5:η5-As5)]31 was reacted with 1. The reaction conducted at room temperature resulted in the formation of amorphous black powder which could not be further characterized and a few dark green crystals of the adduct [(1)2·(5)]. The solid state structure of [(1)2·(5)] is depicted in Fig. 7(a) showing the unprecedented ‘Cr4As5’ cluster 5 surrounded by two molecules of 1. The adduct [(1)2·(5)] crystallizes in the monoclinic space group C2/c with the twofold rotation axis running through As3 as well as the middle of the As1–As1′ bond. The Hg3 planes of the two molecules of 1 are arranged almost perpendicular to each other with an enclosed angle of 87.36(1)°. The assembly results in eight Hg–As distances below the sum of the vdW radii (3.7 Å). The cluster core of 5 depicted in Fig. 7(b) consists of four Cr atoms in a butterfly structure with Cr–Cr bond lengths of 2.9075(1)–3.0134(1) Å, one As2 ligand and one As3 ligand. There is a η5-bound Cp ligand on each Cr atom. Since the Lewis acid 1 generally shows the formation of weakly interacting adducts in combination with pnictogen donors14,17 (see above) without changing the geometry or composition of the reactants we assume that the unprecedented cluster 5 was an impurity of the used [(CpCr)2(μ,η5:η5-As5)] sample that could not be separated by chromatography before (most probably formed as a byproduct in the thermolytic synthesis of [(CpCr)2(μ,η5:η5-As5)]).31 The cluster could presumably only be characterized by X-ray diffraction as it crystallizes very well in the adduct with 1 due to the stabilization by several Hg⋯As contacts in the solid state. Unfortunately, 5 could not be further characterized due to the very low yield of this side-product.Conclusion
The presented results demonstrate the facile preparation of adducts starting from the planar Lewis acid 1 and polypnictogen complexes. While the interactions of the Hg atoms of 1 and the pnictogen donors are very weak resulting in dissociation in solution, several contacts below the sum of the vdW radii can be observed in the solid state. Here, the polyphosphorus complexes 2 (P3) and 3a (P2) show interaction of only one P atom with all three Hg atoms of 1 in accordance with previously described results from reactions of cyclo-P5 and cyclo-P6 complexes with 1. In the series of the E2 complexes 3a–d a significant influence of the pnictogen atom (E = P, As, Sb, Bi) is revealed. While the assembly of the As2 complex 3b is very similar to its P analogue, a different mode of aggregation was observed for the Sb2 and Bi2 complexes 3c and 3d. In case of 3c the formation of two adducts with 1 was detected. Therein, 3c shows two distinctly different coordination modes towards the planar Lewis acid 1. The Sb2 ligand either shows a side-on arrangement towards one molecule of 1 or an end-on coordination of both Sb atoms towards two independent molecules of 1. In contrast, the Bi2 complex 3d shows an almost parallel alignment of the Bi2 dumbbell situated above the center of the planar Lewis acid 1. The isolation of [(1)·(3d)2] does not only represent the first adduct of 1 with a Bi based Lewis base, but is also the first follow-up chemistry that has been described for the Bi2 complex 3d. The observed arrangements of the E2 complex series can be well rationalized by analysis of the electrostatic potential maps of the complexes 3a–d. In addition, QTAIM analysis and direct comparison to the previously described adducts of 1 and the cyclo-P5 and cyclo-As5 ligands of [Cp*Fe(η5-E5)] complexes suggest that electrostatic and dispersion forces predominate in the adducts of 3a–d with 1. Finally, the structural characterization of a new modification of the Sb2 complex [{CpMo(CO)2}2(μ,η2:η2-Sb2)] (3c), the Bi2 complex [{CpMo(CO)2}2(μ,η2:η2-Bi2)] (3d), the starting material [{CpMo(CO)3}3Bi] (4), and a novel [(CpCr)4As5] cluster 5 are presented.Acknowledgements
Manfred Zabel is acknowledged for recording the X-ray dataset for 3c. This work was financially supported by the Deutsche Forschungsgemeinschaft, the National Science Foundation (CHE-1300371) and the Welch Foundation (A-1423). Texas A&M University (Arthur E. Martell Chair of Chemistry) and the Laboratory for Molecular Simulation at Texas A&M University (computational resources) are gratefully acknowledged.Notes and references
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- M. A. Omary, R. M. Kassab, M. R. Haneline, O. Elbjeirami and F. P. Gabbaï, Inorg. Chem., 2003, 42, 2176–2178 CrossRef CAS PubMed.
- P. Sartori and A. Golloch, Chem. Ber., 1968, 101, 2004–2009 CrossRef CAS.
- M. R. Haneline, R. E. Taylor and F. P. Gabbaï, Chem. – Eur. J., 2003, 9, 5188–5193 CrossRef PubMed.
- T. J. Taylor, C. N. Burress and F. P. Gabbaï, Organometallics, 2007, 26, 5252–5263 CrossRef CAS.
- M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 8335–8336 CrossRef CAS.
- C. N. Burress, M. I. Bodine, O. Elbjeirami, J. H. Reibenspies, M. A. Omary and F. P. Gabbaï, Inorg. Chem., 2007, 46, 1388–1395 CrossRef CAS PubMed.
- M. R. Haneline, M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2002, 124, 3737–3742 CrossRef CAS PubMed.
- I. A. Tikhonova, D. A. Gribanyov, K. I. Tugashov, F. M. Dolgushin, A. S. Peregudov, D. Y. Antonov, V. I. Rosenberg and V. B. Shur, J. Organomet. Chem., 2010, 695, 1949–1952 CrossRef CAS.
- M. R. Haneline and F. P. Gabbaï, Angew. Chem., Int. Ed., 2004, 43, 5471–5474 CrossRef CAS PubMed.
- M. C. Ball, D. S. Brown, A. G. Massey and D. A. Wickens, J. Organomet. Chem., 1981, 206, 265–277 CrossRef CAS.
- M. Fleischmann, C. Heindl, M. Seidl, G. Balázs, A. V. Virovets, E. V. Peresypkina, M. Tsunoda, F. P. Gabbaï and M. Scheer, Angew. Chem., Int. Ed., 2012, 51, 9918–9921 CrossRef CAS PubMed.
- O. J. Scherer and T. Brück, Angew. Chem., Int. Ed., 1987, 26, 59–59 Search PubMed.
- O. J. Scherer, C. Blath and G. Wolmershäuser, J. Organomet. Chem., 1990, 387, C21–C24 CrossRef CAS.
- M. Fleischmann, J. S. Jones, F. P. Gabbaï and M. Scheer, Chem. Sci., 2015, 6, 132–139 RSC.
- A. J. Canty and G. B. Deacon, Inorg. Chim. Acta, 1980, 45, L225–L227 CrossRef CAS.
- S. S. Batsanov, J. Chem. Soc., Dalton Trans., 1998, 1541–1546 RSC.
- P. Pyykko and M. Straka, Phys. Chem. Chem. Phys., 2000, 2, 2489–2493 RSC.
- K. R. Flower, V. J. Howard, S. Naguthney, R. G. Pritchard, J. E. Warren and A. T. McGown, Inorg. Chem., 2002, 41, 1907–1912 CrossRef CAS PubMed.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, 2007 Search PubMed.
- It should be noted, that the CCDC take 1.55 Å as the vdW radius of Hg (taken from: A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS ), but this value is probably too small since the authors already mention the fact that it is uncertain how to estimate the vdW radius of metals in organometallic compounds.
- O. J. Scherer, H. Sitzmann and G. Wolmershäuser, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1761–1763 CrossRef.
- W. Clegg, N. A. Compton, R. J. Errington and N. C. Norman, Polyhedron, 1988, 7, 2239–2241 CrossRef CAS.
- W. Clegg, N. A. Compton, R. J. Errington, G. A. Fisher, N. C. Norman and T. B. Marder, J. Chem. Soc., Dalton Trans., 1991, 2887–2895 RSC.
- J. R. Harper and A. L. Rheingold, J. Organomet. Chem., 1990, 390, C36–C38 CrossRef CAS.
- O. J. Scherer, H. Sitzmann and G. Wolmershäuser, J. Organomet. Chem., 1984, 268, C9–C12 CrossRef CAS.
- P. J. Sullivan and A. L. Rheingold, Organometallics, 1982, 1, 1547–1549 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Cambridge University Press, Oxford, UK, 1991 Search PubMed.
- O. J. Scherer, W. Wiedemann and G. Wolmershäuser, J. Organomet. Chem., 1989, 361, C11–C14 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Max Herberhold on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Details of syntheses, characterization, X-ray crystallography and DFT calculations. CCDC 1486626–1486635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02507b |
| This journal is © The Royal Society of Chemistry 2016 |