DOI:
10.1039/C6DT01568A
(Paper)
Dalton Trans., 2016,
45, 13750-13765
Synthesis and structural diversity of trivalent rare-earth metal diisopropylamide complexes†‡
Received
22nd April 2016
, Accepted 8th July 2016
First published on 8th July 2016
Abstract
A series of rare-earth metal diisopropylamide complexes has been obtained via salt metathesis employing LnCl3(THF)x and lithium (LDA) or sodium diisopropylamide (NDA) in n-hexane. Reactions with AM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ln ratios ≥3 gave ate complexes (AM)Ln(NiPr2)4(THF)n (n = 1, 2; Ln = Sc, Y, La, Lu; AM = Li, Na) in good yields. For smaller rare-earth metal centres such as scandium and lutetium, a Li:Ln ratio = 2.5 accomplished ate-free tris(amido) complexes Ln(NiPr2)3(THF). The chloro-bridged dimeric derivatives [Ln(NiPr2)2(μ-Cl)(THF)]2 (Ln = Sc, Y, La, Lu) could be obtained in high yields for Li:Ln = 1.6–2. The product resulting from the Li:La = 1:1.6 reaction revealed a crystal structure containing two different molecules in the crystal lattice, [La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2. Recrystallization of the chloro-bridged dimers led to the formation of the monomeric species Ln(NiPr2)2Cl(THF)2 (Ln = Sc, Lu) and La(NiPr2)3(THF)2. The reaction of YCl3 and LDA with Li:Y = 2 in the absence of THF gave a bimetallic ate complex LiY(NiPr2)4 with a chain-like structure. For scandium, the equimolar reactions with LDA or NDA yielded crystals of tetrametallic mono(amido) species, {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 and [Sc(NiPr2)Cl2(THF)]4, respectively. Depending on the Ln(III) size, AM, and presence of a donor solvent, ate complexes (AM)Ln(NiPr2)4(THF)n show distinct dynamic behaviour as revealed by variable temperature NMR spectroscopy. The presence of weak Ln⋯CH(iPr) β-agostic interactions, as indicated by Ln–N–C angles <105°, is corroborated by DFT calculations and NBO analysis.
:Ln ratios ≥3 gave ate complexes (AM)Ln(NiPr2)4(THF)n (n = 1, 2; Ln = Sc, Y, La, Lu; AM = Li, Na) in good yields. For smaller rare-earth metal centres such as scandium and lutetium, a Li:Ln ratio = 2.5 accomplished ate-free tris(amido) complexes Ln(NiPr2)3(THF). The chloro-bridged dimeric derivatives [Ln(NiPr2)2(μ-Cl)(THF)]2 (Ln = Sc, Y, La, Lu) could be obtained in high yields for Li:Ln = 1.6–2. The product resulting from the Li:La = 1:1.6 reaction revealed a crystal structure containing two different molecules in the crystal lattice, [La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2. Recrystallization of the chloro-bridged dimers led to the formation of the monomeric species Ln(NiPr2)2Cl(THF)2 (Ln = Sc, Lu) and La(NiPr2)3(THF)2. The reaction of YCl3 and LDA with Li:Y = 2 in the absence of THF gave a bimetallic ate complex LiY(NiPr2)4 with a chain-like structure. For scandium, the equimolar reactions with LDA or NDA yielded crystals of tetrametallic mono(amido) species, {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 and [Sc(NiPr2)Cl2(THF)]4, respectively. Depending on the Ln(III) size, AM, and presence of a donor solvent, ate complexes (AM)Ln(NiPr2)4(THF)n show distinct dynamic behaviour as revealed by variable temperature NMR spectroscopy. The presence of weak Ln⋯CH(iPr) β-agostic interactions, as indicated by Ln–N–C angles <105°, is corroborated by DFT calculations and NBO analysis.
Introduction
In a “Celebration of Inorganic Lives” interview in 1999 Don Bradley mentioned about the seminal discovery of the homoleptic bis(trimethylsilyl)amide complexes Ln[N(SiMe3)2]3 “to get a three-coordinated lanthanide was incredible”.1 Indeed, the “establishment of the very lowest coordination numbers of the lanthanides” in 19722 not only triggered immense research in the field of discrete organorare-earth metal complexes but also launched the exploitation of rare-earth metal amide complexes as synthesis precursors according to amine/amido elimination reactions.3 In 1976, it was again Bradley et al. who communicated the synthesis of the first diisopropylamide complexes, Nd(NiPr2)3(THF) and Ln(NiPr2)3 (Ln = Y, Yb).4 The authors of this paper emphasized that these complexes should display enhanced reactivity (due to less sterically demanding amido ligands of increased basicity) though their enhanced thermal instability might negatively affect their broad application. Later on, the use of tailor-made amido ligands,5–8 including mixed silyl/silyl,6 silyl/alkyl,7 and silyl(phenyl) derivatives,8 has immensely progressed Ln(III) tris(amido) chemistry in the fields of structure & bonding as well as precursor development for syntheses, catalysis, and materials science (Chart 1).3,9 While archetypal Ln[N(SiMe3)2]3 adopt a trigonal pyramidal coordination geometry in the solid state,10 trigonal planar LnN3 skeletons were revealed for donor-free complexes La[N(SiMe2tBu)2]3,6 Ln[N(SiMe3)(SiMe2tBu)]3 (Ln = La, Ce),6 La[N(SiMe3)(C6H3iPr2-2,6)]3,7c and Sm[N(SiMe3)(C6F5)]3,7d featuring additional secondary Ln/C/Si/F interactions, as well as Ln(tmp)3 (tmp = 2,2,6,6-tetramethylpiperidinato).8c Though coordination of donor solvent molecules is required to accomplish monomeric tris(amido) complexes bearing less bulky amido ligands, such complexes are more prone to amido exchange, as routinely screened for Ln[N(SiHMe2)2]3(THF)x.5
 |
| | Chart 1 Monomeric non-ate Ln(III) tris(amido) complexes structurally authenticated by X-ray crystallography.5–8 | |
Crucially, alkylamide complexes such as Ln(NiPr2)3(THF)x should display even higher reactivity than Ln[N(SiHMe2)2]3(THF)x due to similarly sized amido ligands of enhanced basicity (pKa,THF = 22.6 (HN(SiHMe2)2),11 35.7 (HNiPr2)).12 However, the design and development of the carbon-analogous alkylamide complexes received only little attention. The targeted synthesis of well-defined diisopropylamide derivatives is impeded by ate complex formation and their structural elucidation by extensive iPr disordering in the solid state. As a consequence there is only a limited number of protocols applying Ln(III) diisopropylamide complexes as synthesis precursors.8a,13–15 As demonstrated by Bradley in 1976, rare-earth metal diisopropylamides can be readily synthesized from LnCl3(THF)x and alkali metal amides like lithium diisopropylamide (LDA)4 and sodium diisopropylamide (NDA). It is noteworthy that LDA features the most prominent metal amide, especially its strongly basic/weakly nucleophilic behaviour makes it the deprotonating reagent of choice in organic synthesis.16 In 1993, Aspinall et al. employed La(NiPr2)3(THF) and LiLa(NiPr2)4 as precursors for the synthesis of phosphide complexes according to protonolysis protocols and emphasized a superior reactivity when compared to La[N(SiMe3)2]3.13a Ate complexes LiLn(NiPr2)4(THF) (Ln = Y, Yb) were crystallographically examined in the following year,17 but due to severe disorder the structures could not be solved. In 1996, Evans et al. structurally characterized LiNd(NiPr2)4(THF) as a side-product of the NdCl3–LDA (1:2.9) reaction in THF and pointed out the presence of Nd⋯(H–C) β-agostic interactions as suggested by short Nd–C distances and acute Nd–N–C angles,13b,18–21 whereas the La derivative was first crystallographically authenticated in 2003 but not further characerized.21 One year earlier, Gambarotta et al. described various samarium amide complexes bearing the dicyclohexylamido ligand (NCy2), among others ate-free heteroleptic complexes [Sm(NCy2)2(THF)(μ-Cl)]2.8a Our group described the synthesis of ate-free complexes Ln(NiPr2)3(THF)x (Ln = Sc, Lu (x = 1), Y (x = 2)) via protonolysis of Ln(CH2SiMe3)3(THF)2 with isopropylamine and used them as precursors to access heteroleptic complexes supported by chelating diamido ancillary ligands.22 The atomic connectivity of Y(NiPr2)3(THF)2 could be unambiguously determined by an X-ray structure analysis but due to heavy ligand disorder in the solid state at 143 K a closer inspection of the molecular structure was infeasible.22 Similarly, we applied [Sc(NiPr2)2(THF)(μ-Cl)]2 for the synthesis of alkali-metal-free salen complexes.14a Recently, trivalent ate complexes LiCe(NCy2)4(THF) and LiCe(NiPr2)4(THF) were employed for the synthesis of homoleptic tetravalent complexes Ce(NCy2)4 and Ce(NiPr2)4 applying tandem oxidation/ligand redistribution protocols.23
In view of the limited knowledge about rare-earth metal complexes bearing the supposedly most popular amido ligand we set out to gain an in-depth understanding of simple Ln(III) diisopropylamide complexes. Our interest in trivalent diisopropylamide rare-earth metal complexes was also triggered by our recent finding that homoleptic Ce(NiPr2)4 is a useful precursor in Ce(IV) chemistry.23 On the basis of a few literature reports it was clear that the outcome of any salt metathesis protocol is sensitively affected by the stoichiometry, Ln(III) metal size, alkali metal reagent, solvent, and temperature.13,15b Moreover, compared to Ln(III)–N(SiHMe2)2 derivatives, the handling of Ln(III)–NiPr2 is aggravated by an enhanced sensitivity toward moisture and thermal instability.4,24 Here we present the targeted synthesis of a number of rare-earth metal diisopropylamide complexes according to salt metathesis routes including mono(amido), bis(amido), tris(amido), and tetra(amido) derivatives. The study not only puts main emphasis on the prevailing solid-state structures but also aims at a better understanding of the solution behaviour. And it is precisely the increased thermal instability and potential formation of product mixtures, which in some cases made it difficult to obtain satisfactory microanalytical data.
Results and discussion
As part of an initial solvent screening study, we found that the best tractable complexes, in terms of purity and yield, are obtained using aliphatics like n-hexane instead of THF as a solvent. The alkali metal precursors lithium diisopropylamide (LDA) and sodium diisopropylamide (NDA) were synthesized freshly from HNiPr2/n-BuLi25 and HNiPr2/sodium metal/isoprene, respectively.16a Depending on the ratio LnCl3(THF)x/LDA (NDA) [1:1–4] complexes 1–10 could be identified by NMR spectroscopy and X-ray structure analysis (Scheme 1). In general, separation of LiCl or NaCl from the yellowish reaction mixtures via centrifugation and concentration of the supernatant solution in a vacuum was sufficient for the formation of colourless crystals at −35 °C in good yields and quality. Accordingly, performing reactions with ≥3 equivalents of LDA or NDA, the reaction pathway A led to the isolation of ate complexes (AM)Ln(NiPr2)4(THF) (Ln = Sc (1a), Y (1b), La (1c), Lu (1d) for AM = Li and 2a–d for AM = Na). In the case of NDA, >3 equiv. also favoured the formation of bis(THF) adducts (AM)Ln(NiPr2)4(THF)2 (3a–c). This is in agreement with the Aspinall study where the ratio was [1:4], more specifically the equimolar addition of LDA to Ln(NiPr2)3(THF) (Ln = Y, La, Yb) produced the respective ate complexes 1. The [1:2.5] reaction of LnCl3(THF)x/LDA in n-hexane (pathway B) generated crystals of Ln(NiPr2)3(THF) (4) for the smaller-sized rare-earth metal centres Sc(III) (4a) and Lu(III) (4d), while only 1-type ate complexes could be identified for the larger Y(III) and La(III). For comparison, the system LnCl3(THF)x/LDA/THF (Ln = Y, La, Yb; ratio [1:2.5]) was previously reported to produce non-ate trisamido complexes Ln(NiPr2)3(THF),17 while NdCl3(THF)x/LDA/THF (ratio [1:2.9]) gave Nd(NiPr2)3(THF) contaminated with a small amount of the ate complex LiNd(NiPr2)4(THF).15a Further increase of the ratio LnCl3(THF)x/LDA to [1:1.6–2] according to the pathway C led to the formation of chloro-bridged dimeric species [Ln(NiPr2)2(μ-Cl)(THF)]2 (5a–d). For the largest metal centre La(III), isolation of pure 5c was not possible, since it has the highest tendency to form 1-type ate complexes. However, we were fortunate to isolate crystals of 5c′, which revealed [La(NiPr2)2(μ-Cl)(THF)]2 and La(NiPr2)3(THF)2, coexisting in the same crystal lattice. Interestingly, recrystallization of dimeric complexes 5 from n-pentane/THF gave monomeric La(NiPr2)3(THF)2 (6) and Ln(NiPr2)2Cl(THF)2 (Ln = Sc (7a), Lu (7b)), exclusively. Treatment of unsolvated YCl3 with two equivalents of LDA in n-hexane yielded the ate complex LiY(NiPr2)4 (8, pathway D). Finally, the [1:1] reactions of ScCl3(THF)3 with LDA or NDA in n-hexane, featuring the smallest and most Lewis-acidic Ln(III) centre, afforded complexes of composition [Sc(NiPr2)Cl2] as envisaged. The enhanced bridging tendency of two chloro ligands per Sc(III) is evidenced in tetrameric arrangements {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 (9, pathway E) and [Sc(NiPr2)Cl2(THF)]4 (10, pathway F), with LDA additionally favouring LiCl inclusion. In summary, the successive reaction of LnCl3 with LDA in n-hexane can be interpreted as shown in Scheme 2.
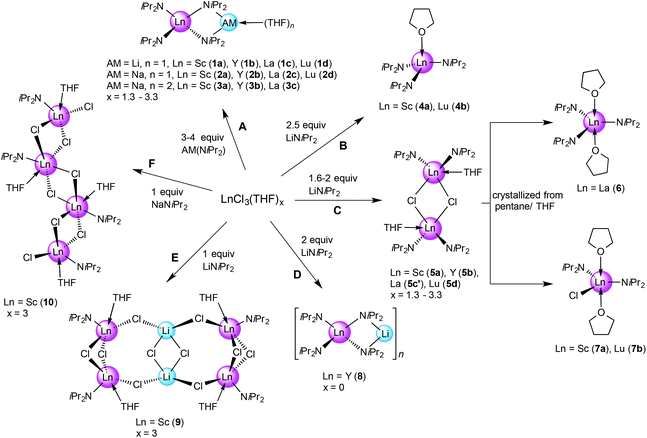 |
| | Scheme 1 Overview of the syntheses of rare-earth metal diisopropylamide complexes via salt metathesis in n-hexane. The crystal structure of 5c′ shows a 1:1 mixture of dimeric 5c and 6 in the asymmetric unit. The side products LiCl and NaCl were omitted for clarity. | |
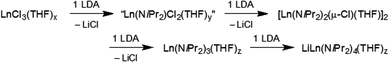 |
| | Scheme 2 Reaction sequence of LnCl3 with LDA in n-hexane. | |
The initial equimolar reaction is very slow due to the poor solubility of both Ln(III) trichloride and polymeric LDA in n-hexane.16b,23,26 The first ligand exchange product “Ln(NiPr2)Cl2(THF)]y” was isolable only for the smallest rare-earth metal centre Sc(III). Because of its enhanced solubility, the mono(amido) derivatives react more rapidly with any dissolved LDA than the trichloride does, hence favouring the formation of the di(amido) complex [Ln(NiPr2)2(μ-Cl)(THF)]2. Such dimeric complexes [Ln(NiPr2)2(μ-Cl)(THF)]2 are very stable and isolable, despite their excellent solubility in n-hexane, which facilitates further ligand exchange.14a
The enhanced reactivity of the lanthanum derivative [La(NiPr2)2(μ-Cl)(THF)]2 toward LDA most likely originates from steric unsaturation of the large La(III) centre. Isolation of pure Ln(NiPr2)3(THF)y according to a one-step synthesis, in the presence of sub-stoichiometric amounts of LDA, seems feasible only for the smaller-sized rare-earth metal centres, since already the yttrium-reaction afforded the ate complex LiY(NiPr2)4(THF). However, treatment of pre-isolated [Y(NiPr2)2(μ-Cl)(THF)]2 with 0.9 equiv. LDA gave Y(NiPr2)3(THF)y exclusively.27 The formation of pure Ln(NiPr2)3(THF)y of the large Ln(III) centres seems hardly controllable since they feature reactive intermediates en route to the thermodynamically favoured ate complexes.
Spectroscopic properties and dynamic behaviour
The compounds described in this paper have been successfully isolated as main products and recrystallized repeatedly, but in some cases the elemental analysis indicated the presence of small amounts of byproducts, the formation of which is difficult to avoid.14a,15a,17 These byproducts which are mainly protic impurities are also revealed by NMR spectroscopy. The ambient-temperature NMR spectra of complexes 1–10 are very similar, while the integration of the proton signals gave only evidence for the NiPr2/THF ratio. However, combined NMR/microanalytical data allowed for a tentative structure assignment as shown in Scheme 1. Moreover, ate complexes 1–3 were anticipated to show a distinct coordination behaviour of the amido ligands at lower temperatures. Aspinall et al. have previously reported that complexes LiLn(NiPr2)4(THF) (Ln = Y (1b), La (1c), Yb) show one set of signals for the amido ligands in ambient-temperature 1H NMR spectra, consistent with a tetrahedral symmetry at the metal centre and a highly fluxional Li(THF)+ fragment on the NMR time scale.17 On lowering the temperature to −50 °C for lanthanum complex 1c, Aspinall et al. observed decoalescence of the amido signal into two CH resonances (1:1 ratio) and three CH3 resonances (2:1:1 ratio), in agreement with two terminal amido ligands exhibiting rapid rotation about the Ln–N bonds and two bridging amido ligands featuring diastereotopic methyl groups within each iPr substituent.17 By redoing the variable temperature (VT) 1H NMR study of lanthanum complex 1c, we observed the same signal pattern with decoalescence occurring at ca. 0 °C affording three doublets at 1.43, 1.33, and 1.26 ppm and two multiplets at 3.60 and 3.51 ppm (ESI; Fig. S16‡). Surprisingly, the complex LiY(NiPr2)4(THF) (1b) with the smaller-sized yttrium centre also revealed a decoalescence temperature at ca. 0 °C (Fig. 1 and S13‡). In contrast, for the sodium ate complex NaY(NiPr2)4(THF) (2b) separation of the amido signal set did not occur until a temperature of ca. −70 °C. This might be attributed to a weaker (ionic) bonding of the Na(THF)+ fragment with the amido ligands compared to the Li(THF)+ fragment (ESI; Fig. S17‡). Moreover, the THF-free ate complex LiY(NiPr2)4 (8) revealed a dynamic behaviour distinct from LiY(NiPr2)4(THF) (1b). Separation of the amido signal set into signals for terminal and bridging ligands occurred at ca. 0 °C (CH3) and ca. −20 °C (CH), while the diastereotopy of the iPr methyl groups of the bridging amido ligands became visible only at −30 °C (Fig. 1 and S27‡). Such consecutive decoalescence behaviour can be ascribed to a distinct lithium–amido interaction in THF-free 8, which is also supported by shorter Li–N contacts compared to those in 1b (av. 2.014 versus 2.058 Å; vide infra, see Fig. 10 and Table 1). Since the formation of M⋯CH(NiPr) β-agostic bonding has been discussed at several instances in the literature we screened complexes 1–10 for the presence of such secondary interactions. While the aforementioned NMR investigations revealed highly dynamic behaviour in solution for all of the compounds even at low temperatures (e.g., 1b: 13C NMR signals at −35 °C: CH: δ = 44.9–45.9 ppm (d, 1JC–H = 125.0 Hz), CH: δ = 48.1–49.1 ppm (d, 1JC–H = 126.6 Hz), matching the coupling constants for 1JC–H (methine) observed earlier),28 the DRIFT (Diffuse Reflectance Infrared Fourier Transform) spectra are indicative of the presence of weak agostic bonds in the solid state. In addition to undisturbed C–H stretching vibrations of the methyl groups, appearing at about 2900 cm−1, additional broad but weaker ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (C–H) bands in the range of 2720 to 2680 cm−1 were detected for complexes 1–10. Such lower energy bands can be assigned to agostic and non-agostic C–H(methine) stretching vibrations.20,29 This would be in accordance with the solid-state structure of complexes 1–10 which feature asymmetric amido bonding involving distinct Ln–N–C angles and apparent close intramolecular M⋯CH contacts (vide infra). C–H(methine) stretching vibrations at even lower energy, occurring in alkali metal amides and alkoxides with heteroatom–CH bonding, have been assigned to hyperconjugation.30,31
(C–H) bands in the range of 2720 to 2680 cm−1 were detected for complexes 1–10. Such lower energy bands can be assigned to agostic and non-agostic C–H(methine) stretching vibrations.20,29 This would be in accordance with the solid-state structure of complexes 1–10 which feature asymmetric amido bonding involving distinct Ln–N–C angles and apparent close intramolecular M⋯CH contacts (vide infra). C–H(methine) stretching vibrations at even lower energy, occurring in alkali metal amides and alkoxides with heteroatom–CH bonding, have been assigned to hyperconjugation.30,31
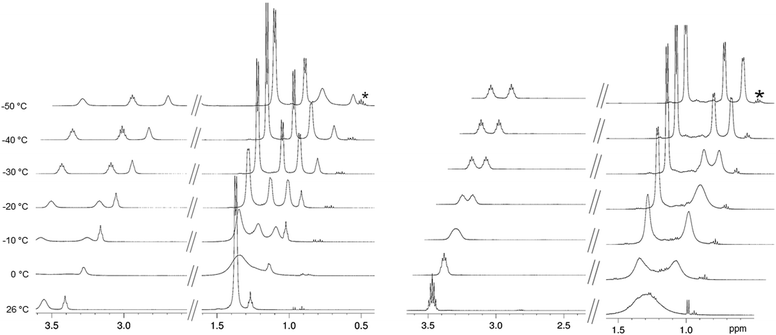 |
| | Fig. 1 VT 1H NMR spectra (500 MHz) of LiY(NiPr2)4(THF) (1b, left) and LiY(NiPr2)4 (8, right) in toluene-d8; shown are the regions of the isopropyl group and THF; * impurities. | |
Table 1 Selected bond lengths, intramolecular distances and angles for AMLn(NiPr2)4(THF) (AM = Li: Ln = Sc (1a), Y (1b), La (1c), Lu (1d); AM = Na: Ln = Sc (2a), Y (2b), Lu (2d))a
| |
1a
|
1b
|
1c
|
1d
|
2a
|
2b
|
2d
|
|
Values of molecule 1 only (a complete list of metrical parameters including those of molecule 2 are shown in the ESI).
|
|
Bond lengths (Å)
|
| Ln1–N1 |
2.176(2) |
2.315(2) |
2.461(2) |
2.260(2) |
2.170(2) |
2.307(2) |
2.258(2) |
| Ln1–N3 |
2.177(2) |
2.309(2) |
2.466(2) |
2.266(2) |
2.156(2) |
2.291(2) |
2.247(2) |
| Ln1–N2 |
2.066(2) |
2.210(2) |
2.339(2) |
2.167(2) |
2.080(2) |
2.224(2) |
2.176(2) |
| Ln1–N4 |
2.068(2) |
2.215(2) |
2.348(2) |
2.166(2) |
2.085(2) |
2.214(2) |
2.186(2) |
| AM–N1 |
2.037(2) |
2.058(3) |
2.061(4) |
2.062(4) |
2.414(2) |
2.415(2) |
2.431(2) |
| AM–N3 |
2.046(3) |
2.056(3) |
2.065(3) |
2.060(2) |
2.418(3) |
2.430(2) |
2.429(2) |
|
Interatom. distances (Å)
|
| Ln1⋯C1 |
2.918(2) |
2.967(2) |
3.034(2) |
2.956(2) |
2.935(3) |
2.992(2) |
2.968(2) |
| Ln1⋯C7 |
2.800(2) |
2.914(3) |
3.045(3) |
2.886(2) |
2.816(2) |
2.923(2) |
2.896(2) |
| Ln1⋯C16 |
2.951(2) |
3.444(2) |
3.048(2) |
2.982(2) |
2.922(3) |
2.935(2) |
2.951(3) |
| Ln1⋯C19 |
2.773(3) |
2.895(2) |
3.034(2) |
2.865(2) |
2.834(2) |
2.905(2) |
2.913(2) |
|
Bond angles (°)
|
| Ln1–N1–C1 |
104.3(2) |
100.7(2) |
97.9(2) |
102.5(2) |
105.2(2) |
102.7(2) |
103.3(2) |
| Ln1–N1–C4 |
127.6(2) |
126.2(2) |
128.6(2) |
127.8(2) |
128.1(2) |
129.0(2) |
128.1(2) |
| Ln1–N2–C7 |
103.3(2) |
103.0(2) |
104.2(2) |
103.5(2) |
103.3(2) |
102.9(2) |
103.3(2) |
| Ln1–N2–C10 |
143.9(2) |
143.1(2) |
140.6(2) |
143.1(2) |
144.0(2) |
142.9(2) |
143.4(2) |
Solid-state structures
Ate complexes AMLn(NiPr2)4(THF)n; n = 1, 2; Ln = Sc, Y, La, Lu; AM = Li, Na.
The reaction pathway A (Scheme 1) employing LnCl3(THF)x along with 3–4 equiv. LDA or NDA led to the formation of bimetallic ate complexes of the type AMLn(NiPr2)4(THF)n (n = 1, 2; Ln = Sc, Y, La, Lu; AM = Li, Na). The single crystals obtained from the 3-equiv. reactions were analyzed as the mono(THF) adducts 1a–d and 2a–d (Fig. 2 and S1–S5‡), being structurally analogous (monoclinic space group P21/c) with previously reported LiNd(NiPr2)4(THF),15a LiCe(NiPr2)4(THF),23 LiCe(NCy2)4(THF)8b,32 and LiCe[N(SiHMe2)2]4(THF).33 For comparison, the respective diphenylamido complexes tend to form separated ion pairs of the type [Li(THF)4][Ln(NPh2)4] (Ln = Er, Yb).34 Lanthanum complex 2c proved to be structurally analogous as well, but a complete crystallographic data set was not collected. The solid-state structures show two distinct molecules per asymmetric unit (Fig. S6‡). The lanthanide centres are coordinated by four nitrogen atoms in a distorted tetrahedral fashion, involving two terminal and two bridging amido ligands. The Ln–N and AM–N bonds are in the expected range (Table 1).5b,15a,23,35 Moreover, Ln–N–C angles clearly smaller than 120° were observed, which have been proposed as indicators for Ln⋯CH β-agostic interactions.20 The presence of such additional secondary interactions could not be confirmed by solution NMR spectroscopy (vide supra). In order to verify their existence and strength, DFT calculations have been carried out on 1a at the B3LYP/def2TZVP level of theory.36 The DFT-optimized geometry of 1a (henceforth denoted as 1a′) is in very good agreement with its experimentally determined structure in the solid state; in particular, the Sc–N–C bond angles deviate by not more than 1.2% from their experimental counterparts (for details, see the ESI‡). Crystal packing forces can therefore be ruled out as a cause for the asymmetric coordination observed for the amido ligands. The β-C–H bonds in the bridging amido ligands of 1a′ display identical bond lengths, whereas they differ, however slightly, for the terminal ligands: the smaller Sc–N–C angles (104.3° vs. 142.3°) are accompanied by subtly elongated β-C–H bonds (1.103 Å vs. 1.096 Å), which bring the respective H atoms in 2.502 Å proximity to Sc. The small deviations in β-C–H bond lengths suggest fairly weak agostic interactions with the Sc centre seemingly in strong contrast to the markedly different Sc–N–C bond angles. Indeed, an NBO analysis37 of 1a′ revealed that in the NLMOs (natural localized molecular orbitals) which represent the β-C–H bonds, only 1.3% of the electrons are delocalized towards Sc. It is therefore unlikely that this small electronic effect is the primary cause for the observed tilting of the amido ligands. Optimizing the geometry of a modified model system 1a′′, in which all iPr groups of the ligands are replaced by methyl groups, revealed that the sterics play the key role instead. In 1a′′, all Sc–N–C bond angles involving the terminal ligands are now larger than 120° (120.3°/127.9° and 122.3°/126.7°, respectively), and no elongation of β-C–H bonds is observed anymore (for details, see the ESI‡). The β-agostic interactions observed in these complexes are therefore a consequence rather than a cause of the asymmetric coordination mode of the amido ligands. Steric factors have also been shown to play a role in agostic amido complexes of Ti.28,38
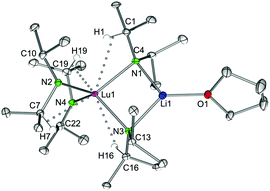 |
| | Fig. 2 Molecular structure of complexes AMLn(NiPr2)4(THF) (AM = Li: Ln = Sc (1a), Y (1b), La (1c), Lu (1d); AM = Na: Ln = Sc (2a), Y (2b), La (2c), Lu (2d)), representatively shown for LiLu(NiPr2)4(THF) (1d). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. | |
In contrast to the lithium complexes LiLn(NiPr2)4(THF) (1, 2), the sodium derivatives can accommodate a second donor molecule to yield bis(THF) adducts NaLn(NiPr2)4(THF)2 (3) (Fig. 3 and S7 and S8‡). This involves a switch to an orthorhombic crystal system (space group P212121), with one molecule in the asymmetric unit. The metrical parameters obtained from X-ray structure analyses are listed in Table 2. Similar to all complexes of the type AMLn(NiPr2)4(THF)n the geminal isopropyl groups exhibit an antiperiplanar and gauche conformation for terminal and bridging amido ligands, respectively, allowing for effective secondary interactions.
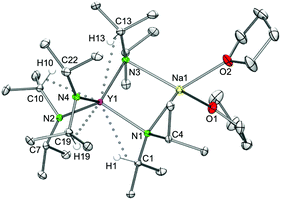 |
| | Fig. 3 Molecular structure of complexes NaLn(NiPr2)4(THF)2 (Ln = Sc (3a), Y (3b), and La (3c)), representatively shown for NaY(NiPr2)4(THF)2 (3b). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. | |
Table 2 Selected bond lengths, intramolecular distances, and angles for NaLn(NiPr2)4(THF)2 (Ln = Sc (3a), Y (3b), and La (3c))
| |
3a
|
3b
|
3c
|
|
Bond lengths (Å)
|
| Ln1–N1 |
2.157(2) |
2.293(3) |
2.465(2) |
| Ln1–N3 |
2.162(2) |
2.301(2) |
2.471(2) |
| Ln1–N2 |
2.090(2) |
2.230(2) |
2.360(2) |
| Ln1–N4 |
2.089(2) |
2.228(2) |
2.353(2) |
| Na1–N1 |
2.494(2) |
2.512(2) |
2.520(2) |
| Na1–N3 |
2.492(2) |
2.505(2) |
2.518(2) |
|
Interatom. distances (Å)
|
| Ln1⋯C1 |
2.928(3) |
2.975(2) |
2.998(5) |
| Ln1⋯C10 |
2.806(3) |
2.922(2) |
3.038(3) |
| Ln1⋯C13 |
2.910(3) |
2.955(2) |
3.518(2) |
| Ln1⋯C19 |
2.813(3) |
2.923(2) |
3.039(3) |
|
Bond angles (°)
|
| Ln1–N1–C1 |
105.8(2) |
102.1(2) |
95.8(3) |
| Ln1–N1–C4 |
128.2(2) |
129.0(2) |
125.02(18) |
| Ln1–N2–C7 |
144.3(2) |
143.8(2) |
143.11(18) |
| Ln1–N2–C10 |
102.6(2) |
102.4(3) |
102.81(16) |
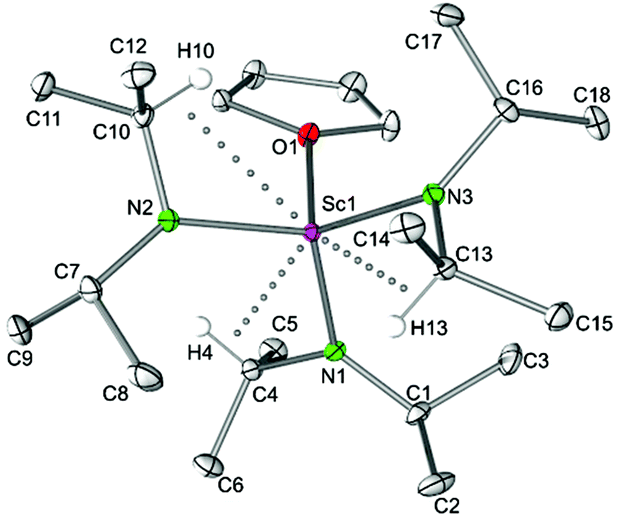
Tris(amido) complexes Ln(NiPr2)3(THF) (Ln = Sc (4a), Lu (4b)).
Following the reaction pathway B, alkali metal-free diisopropylamide complexes Ln(NiPr2)3(THF) (4) could be straightforwardly obtained for the small metal centres Sc(III) and Lu(III) by using 2.5 equivalents of LDA. The crystal structure analysis of the scandium derivative 4a confirmed the molecular composition proposed by NMR spectroscopy and the absence of lithium in the crystal lattice (Fig. 4).22 Complex 4a shows a distorted tetrahedral coordination geometry defined by the three NiPr2 ligands and one THF molecule, similar to the silylamide analogue Sc[N(SiHMe2)2]3(THF)5b and the samarium alkyl amide compound [Sm(NCy2)3(THF)]·toluene8a (Cy = cyclohexyl).
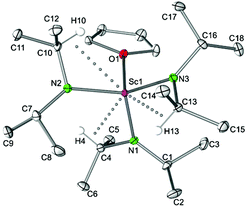 |
| | Fig. 4 Molecular structure of Sc(NiPr2)3(THF) (4a). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. Bond lengths (Å), interatomic distances (Å), and angles (°): Sc1–N1 2.035(2), Sc1–N2 2.048(2), Sc1–N3 2.047(2), Sc1–O1 2.202(3); Sc1⋯C4 2.910(2), Sc1⋯C10 2.892(2), Sc1⋯C13 2.882(3); Sc1–N1–C1 134.99(6), Sc1–N1–C4 111.21(6), Sc1–N2–C7 135.35(7), Sc1–N2–C10 109.44(6), Sc1–N3–C13 108.89(6), Sc1–N3–C16 136.18(6), N1–Sc1–N2 115.37(3), N1–Sc1–N3 113.92(3), N2–Sc1–N3 113.87(4), N1–Sc1–O1 99.09(3), N2–Sc1–O1 104.55(3), N3–Sc1–O1 108.18(3). | |
Complexes Sc(NiPr2)3(THF) (4a) and Sc[N(SiHMe2)2]3(THF) allow for direct comparison of alkyl and silylamide coordination. In the case of the diisopropylamide complex 4a, a less pronounced delocalization of the negative charge within the amido ligand is evidenced by comparatively shorter Sc–N bond lengths (av. 2.043 Å; cf., in Sc[N(SiHMe2)2]3(THF) (2.079(2), 2.063(2), 2.062(2) Å).5b As a consequence the Sc–O(THF) distances show an opposite trend (2.202(2) versus 2.181(2) Å). Both complexes exhibit asymmetrically coordinated amido ligands as evidenced by distinct Sc–N–C(Si) angles in the ranges of 109.44(6)–135.35(7)° and 104.4(1)–132.6(1)°, respectively, and secondary Sc⋯C(Si) contacts as close as 2.882(3) and 2.989(1), respectively.
Chloro-bridged complexes [Ln(NiPr2)2(THF)(μ-Cl)]2 (Ln = Sc (5a), Y (5b), Lu (5d)).
Further attempts to obtain alkali metal-free non-ate lanthanide diisopropylamide complexes by using 1.6–2 equiv. of LDA led to the formation of dimeric [Ln(NiPr2)2(THF)(μ-Cl)]2 (route C, Scheme 1: Ln = Sc (5a), Y (5b), Lu (5d); Fig. 5, S9,‡ and Table 3). Similar chloro(halo)-bridged rare-earth metal bis(trimethylsilyl)amido complexes are known, {Ln[N(SiMe3)2]2(THF)(μ-Cl)}2 (Ln = Ce, Pr, Nd Sm, Gd, Dy, Y, Yb)39 including the THF-free ytterbium derivative.19 The more relevant bis(dimethylsilyl)amido chlorides {Ln[N(SiHMe2)2]2(THF)(μ-Cl)}2 were structurally authenticated for scandium and yttrium.40 Complexes 5 crystallized from n-hexane very slowly in the form of stacked layers, which were difficult to separate and hence markedly affected the X-ray diffraction measurements. The solid-state structures revealed six independent molecules in the asymmetric unit (Fig. S10,‡ monoclinic space group Pc), accounting for lengthy data acquisitions. For this reason, only the unit cell parameters of scandium complex 5a were determined, crystallizing isomorphously to 5b–d. Analysis of the crystallographic data of the lanthanum reaction revealed two distinct molecules (denoted as 5c′).
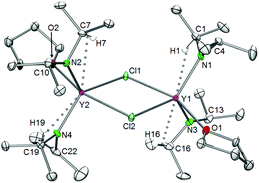 |
| | Fig. 5 Molecular structure of complexes [Ln(NiPr2)2(THF)(μ-Cl)]2 (Ln = Sc (5a), Y (5b), and Lu (5d)), representatively shown for [Y(NiPr2)2(THF)(μ-Cl)]2 (5b). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. | |
Table 3 Selected bond lengths, interatomic distances and angles for chloro-bridged lanthanide amide complexes [Ln(NiPr2)2(THF)(μ-Cl)]2 (Ln = Y (5b), La (5c′, part 1), Lu (5d))
| |
5b
|
5c′ (part 1)b |
5d
|
|
The solid-state structures of 5b and 5d contain six independent molecules each in the asymmetric unit (see the ESI). Representative metrical parameters are listed only for molecule 1.
N3 = N2; Cl2 = Cl1′; Ln2 = La1′.
|
|
Bond lengths (Å)
|
| Ln1–N1 |
2.162(4) |
2.330(2) |
2.143(7) |
| Ln1–N3 |
2.137(4) |
2.286(2) |
2.136(7) |
| Ln2–N2 |
2.141(4) |
— |
2.144(6) |
| Ln2–N4 |
2.152(4) |
— |
2.136(7) |
| Ln1–Cl1 |
2.729(1) |
2.8797(7) |
2.689(2) |
| Ln1–Cl2 |
2.698(1) |
2.8797(7) |
2.674(2) |
| Ln2–Cl1 |
2.675(2) |
2.8797(7) |
2.643(2) |
| Ln2–Cl2 |
2.717(1) |
2.8797(7) |
2.675(2) |
| Ln1–O1 |
2.385(4) |
2.601(2) |
2.321(6) |
| Ln2–O2 |
2.382(4) |
— |
2.306(6) |
|
Interatomic distances (Å)
|
| Ln2–O2 |
| Ln1⋯C1 |
2.978(5) |
3.379(3) |
2.974(9) |
| Ln1⋯C16 |
2.864(6) |
|
2.888(9) |
| Ln2⋯C7 |
2.851(5) |
3.120(4) |
2.859(8) |
| Ln2⋯C19 |
2.951(5) |
|
2.979(9) |
|
Bond angles (°)
|
| Ln1–Cl1–Ln2 |
100.5(2) |
102.80(2) |
101.7(1) |
| N1–Ln1–Cl1 |
100.5(1) |
100.07(6) |
100.7(2) |
| N1–Ln1–N3 |
115.2(2) |
119.58(9) |
114.5(3) |
| Ln1–N1–C1 |
109.0(3) |
124.24(18) |
110.5(5) |
| Ln1–N1–C4 |
136.5(4) |
122.68(16) |
136.0(6) |
| Cl1–Ln1–O1 |
156.6(1) |
158.43(5) |
156.9(2) |
In complexes 5b and 5d the rare-earth metal centres are bridged by chloro ligands building Ln2Cl2 rhombi with almost equally long edges and Ln–Cl–Ln angles larger than the respective Cl–Ln–Cl angles. The Ln(III) centres adopt a distorted trigonal bipyramidal coordination geometry, with the atoms N1, N3, and Cl2 in the equatorial positions and Cl1 and O1 arranged apically.
The deviation from the linearity of the Cl–Ln–O angles by about 23° can be explained by the bulkiness of the diisopropylamido ligands, which causes the bending of the THF ligands toward the Ln2Cl2 rhombi. The Ln–Cl bond lengths are in the range of those detected previously for similar heteroleptic complexes.39 Moreover the large difference between the Ln–N–C angles is accompanied by secondary interactions between the metal centres and β-H atoms of the diisopropyl amido groups.
For the largest rare-earth metal lanthanum, the 1.6-equiv. reaction led to single crystals consisting of two different compounds, [La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2 (5c′). Apparently, chloro-bridged [La(NiPr2)2(THF)(μ-Cl)]2 (denoted as part 1) and the bis(THF)tris(amido) complex La(NiPr2)3(THF)2 (part 2) display equally favoured products (Fig. 6). In contrast to the related complexes 5a, 5b, and 5d, the unit cell of 5c′ contains two molecules each of parts 1 and 2.
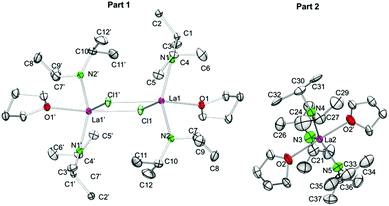 |
| | Fig. 6 Crystal structure of [La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2 (5c′). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity. The monomer (part 2) is heavily disordered and a partial disordering model is shown in the ESI (Fig. S11‡). For selected bond lengths and angles of part 1, see Table 3. | |
Useable crystal data could be collected at temperatures ≥173 K, as further temperature decrease led to crystal cracking and signal splitting. Unfortunately, 5c′ suffers from several disorders, especially in part 2 (Fig. S11‡). The metrical parameters of the chloro-bridged part 1 are in line with those of complexes 5b and 5d (Table 3). Due to the strong disorder of trigonally bipyramidally arranged part 2 the respective bond lengths and angles are not discussed here (cf., 6).
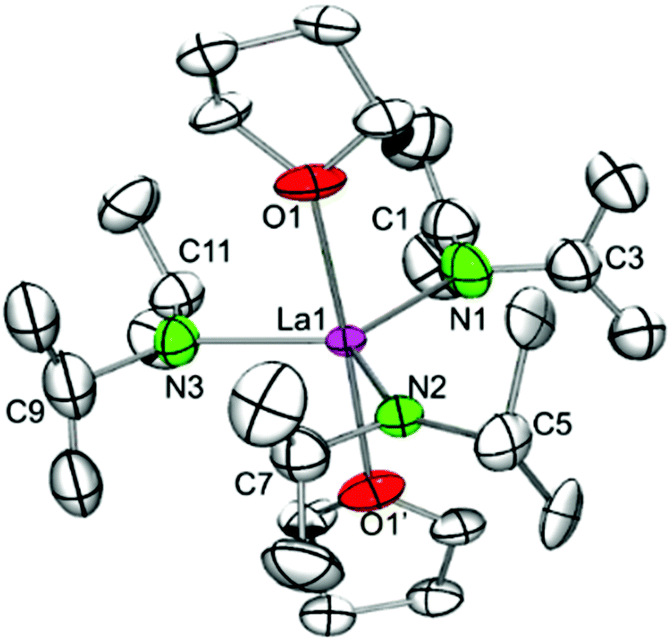
Alkali metal-free tris(amido) complex La(NiPr2)3(THF)2 (6).
Recrystallization of the mixed complex 5c′ from n-pentane/THF gave exclusively the complex La(NiPr2)3(THF)2 (6) (Scheme 1 and Fig. 7). For comparison, the isostructural bis(dimethylsilyl)amido complex La[N(SiHMe2)2]3(THF)2 was described by our group back in 1998.5c It is noteworthy that pure complexes Ln(NiPr2)3(THF)x of the smaller-sized scandium, lutetium (x = 1, 4), and yttrium (x = 2) could be obtained via protonolysis of Ln(CH2SiMe3)3(THF)2 with isopropylamine.22 The crystal system (orthorhombic space group Cmc21) and atomic connectivity of heavily distorted Y(NiPr2)3(THF)2 determined earlier could be confirmed for lanthanum complex 6. Not surprisingly, the molecular structure of 6 obtained at 150 K was disordered, but clearly revealed a regular trigonal bipyramidal coordination sphere around the lanthanum centre with three diisopropylamido ligands in the equatorial and the THF units in the axial positions (cf., 5c′, part 2). Furthermore, Ln–N–C shows only small differences in angles for La1–N1–C1 and La1–N1–C3 and almost ecliptic conformation of the geminal isopropyl groups. In contrast the geminal isopropyl units connected to N2 and N3 feature antiperiplanar conformation with one narrow and one wide La–N–C angles, which points to additional secondary interactions between β-H atoms and the metal centre. The Ln–N bond lengths of av. 2.35 Å appear slightly shorter than those in La[N(SiHMe2)2]3(THF)2 (2.395(5)–2.416(5) Å)5b corroborating the findings with complexes Sc(NiPr2)3(THF) (4a) and Sc[N(SiHMe2)2]3(THF).5b
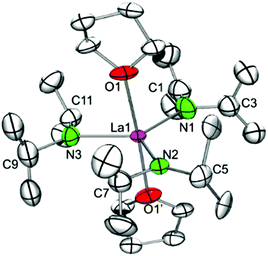 |
| | Fig. 7 Molecular structure of La(NiPr2)3(THF)2 (6). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity. The amido ligands are heavily disordered but modeling of a proper disorder failed due to lack of electron density. Bond lengths (Å), interatomic distances (Å), and angles (°): La1–N1 2.32(3), La1–N2 2.36(2), La1–N3 2.35(1), La1–O1 2.61(1); La1⋯C3 3.09(2), La1⋯C7 3.03(2), La1⋯C11 3.06(2); La1–N1–C1 138(3), La1–N1–C3 109(2), La1–N2–C5 138.1(14), La1–N2–C7 104.6(16), O1–La1–O1′ 177.1(6), N1–La1–N3 120.9(16), N2–La1–N3 119.7(12), N1–La1–N2 119.4(6), N2–La1–O1 90.1(5). | |
Alkali metal-free bis(amido) complexes Ln(NiPr2)2Cl(THF)2 (Ln = Sc (7a) and Lu (7b)).
Recrystallization of the chloro-bridged complexes 5a and 5b from n-pentane/THF mixtures gave the monomeric five-coordinate complexes Ln(NiPr2)2Cl(THF)2 (Ln = Sc (7a) and Lu (7b), Fig. 8 and S12‡). Excess of donor solvent THF disrupts the chloro bridges to afford bis(THF) adducts which are structurally similar to the binaphthylamido yttrium complex [{R-C20H12(NC5H9)2}YCl(THF)2].41
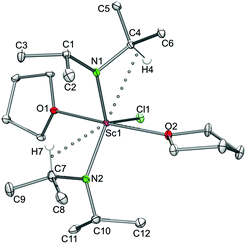 |
| | Fig. 8 Molecular structure of complexes Ln(NiPr2)2Cl(THF)2 (Ln = Sc (7a) and Lu (7b)), representatively shown for the scandium derivative. Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. Bond lengths (Å), interatomic distances (Å), and angles (°): 7a: Sc1–N1 2.039(2), Sc1–N2 2.029(2), Sc1–Cl1 2.453(2), Sc1–O1 2.245(2), Sc1–O2 2.253(2); Sc1⋯C4 2.818(2), Sc1⋯C7 2.838(2); Sc1–N1–C1 141.3(2), Sc1–N1–C4 105.3(2), O1–Sc1–O2 170.1(2), N1–Sc1–N2 123.70(4), Cl1–Sc1–N1 121.07(3), Cl1–Sc1–N2 115.19(3), Cl1–Sc1–O1 83.67(2), Cl1–Sc1–O2 86.43(2). 7b: Lu1–N1 2.145(2), Lu1–N2 2.142(2), Lu1–Cl1 2.539(2), Lu1–O1 2.325(2), Lu1–O2 2.327(2); Lu1⋯C4 2.905(2), Lu1⋯C7 2.909(2); Lu1–N1–C1 141.8(2), Lu1–N1–C4 105.0(2), O1–Lu1–O2 169.8(2), N1–Lu1–N2 122.43(6), Cl1–Lu1–N1 120.59(4), Cl1–Lu1–N2 116.93(4), Cl1–Lu1–O1 83.42(3), Cl1–Lu1–O2 86.38(3). | |
The X-ray structure analyses revealed a distorted trigonal bipyramidal environment around the metal centres, with both THF molecules located axially and the two diisopropylamido groups as well as the chloro ligand in the equatorial positions. The two oxygen atoms of both THF are slightly bent toward the chlorine atom while the diisopropylamido ligands engage in secondary Ln⋯CH interactions as indicated by distinct Ln–N–C angles. The Ln–Cl terminal bond lengths of 7a (2.453(2) Å) and 7b (2.539(2) Å) are slightly shorter than those in [{R-C20H12(NC5H9)2}YCl(THF)2],41 mainly due to the difference in metal ion radii.
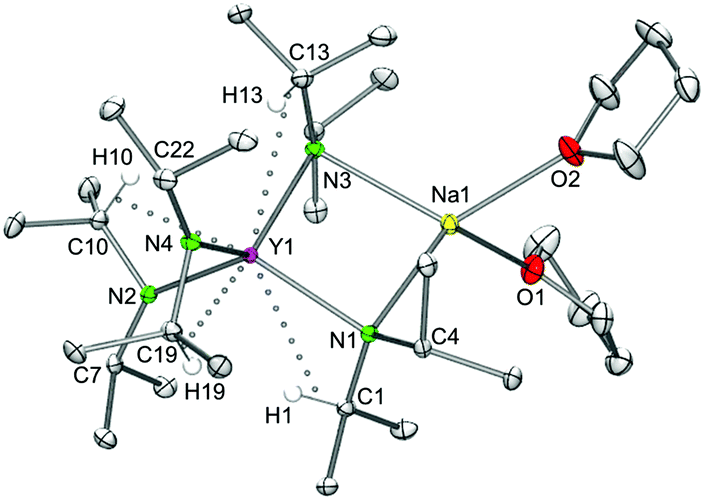
THF-free yttrium ate complex LiY(NiPr2)4 (8).
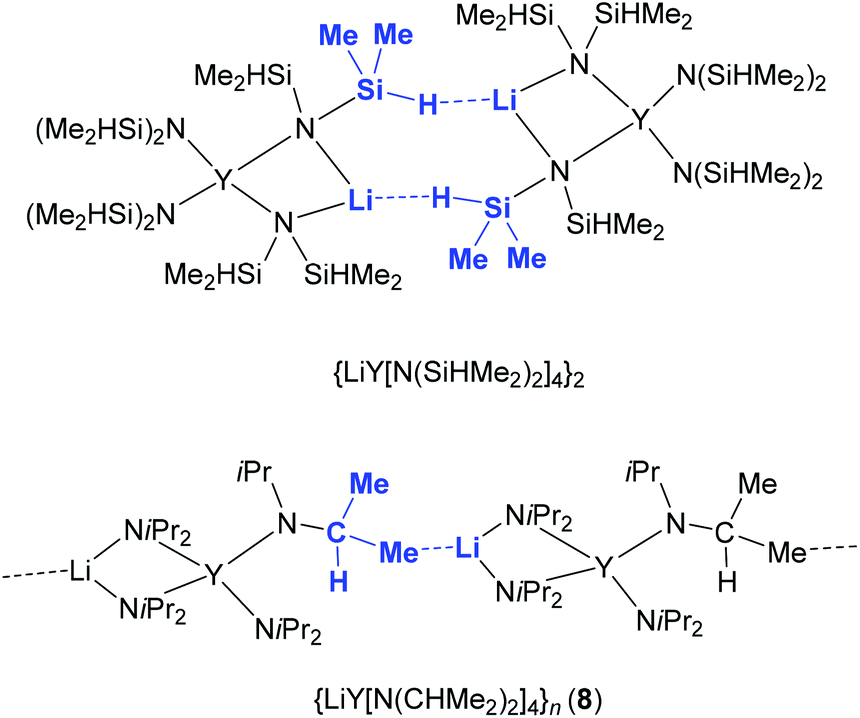
Employing donor (THF)-free YCl3 and 2 equiv. LDA according to route D (Scheme 1) led to the isolation of the ate complex LiY(NiPr2)4 (8). The X-ray structure analysis confirmed a tetrahedrally coordinated rare-earth metal centre as in complexes 1–3 (Fig. 9 and 10, space group P21/c) with metrical parameters very similar to THF-coordinated complexes LiY(NiPr2)4(THF) (1b) and NaY(NiPr2)4(THF) (2b). The overall coordination chemistry of 8 also resembles that of the anionic part of the alkyl complex {(YCH2SiMe3)x(Me3CO)1−x(μ-OCMe3)4[Li(THF)]4(μ4-Cl)}+[Y(CH2SiMe3)4]− described by Evans et al.42 Closer inspection of the solid-state structure of complex 8 revealed the occurrence of intermolecular Li⋯CH3 (iPr) contacts (Li1⋯C21′ 2.569 Å, Li⋯H21c′ 2.206 Å), leading to an infinite chain structure (Fig. 9 and 10). Similar intermolecular AM⋯CH3 interactions were previously detected in divalent bis(trimethylsilyl)amide complexes {AMSm[N(SiMe3)2]3}n, which form a chain structure (AM = Na) and a two-dimensional network (AM = K), respectively, depending on the size of the alkali metal AM.43 The chain-like solid-state arrangement of 8 is in stark contrast to that of the related bis(dimethylsilyl)amide complex {LiY[N(SiHMe2)2]4}2 (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), which assembles as dimers involving two close intermolecular Li⋯H–Si bridges (Fig. 9, Li⋯H 1.87(1) Å).44 Furthermore, the Y–N bond lengths in complex 8 are significantly shorter than those in {LiY[N(SiHMe2)2]4}2 (terminal: 2.258(2), 2.259(2); bridging: 2.386(2), 2.443(2) Å), while the Y–N–C angles in 8 differ as much as the respective Y–N–Si angles in {LiY[N(SiHMe2)2]4}2 (terminal: e.g., 98.85(9) and 135.7(1)°).44 Also in complex 8, the acute Y–N–C angles involve close intramolecular Y⋯CH secondary interactions. It is also noteworthy that already in 1993 the lanthanum derivative LiLa(NiPr2)4, obtained from La(NiPr2)3(THF) and 1 equiv. of LDA without further characterization, was employed as a suitable synthesis precursor.13a
), which assembles as dimers involving two close intermolecular Li⋯H–Si bridges (Fig. 9, Li⋯H 1.87(1) Å).44 Furthermore, the Y–N bond lengths in complex 8 are significantly shorter than those in {LiY[N(SiHMe2)2]4}2 (terminal: 2.258(2), 2.259(2); bridging: 2.386(2), 2.443(2) Å), while the Y–N–C angles in 8 differ as much as the respective Y–N–Si angles in {LiY[N(SiHMe2)2]4}2 (terminal: e.g., 98.85(9) and 135.7(1)°).44 Also in complex 8, the acute Y–N–C angles involve close intramolecular Y⋯CH secondary interactions. It is also noteworthy that already in 1993 the lanthanum derivative LiLa(NiPr2)4, obtained from La(NiPr2)3(THF) and 1 equiv. of LDA without further characterization, was employed as a suitable synthesis precursor.13a
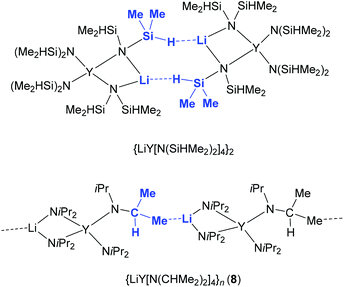 |
| | Fig. 9 Distinct intermolecular bonding detected in dimeric {LiY[N(SiHMe2)2]4}2 and polymeric [LiY(NiPr2)4]n (8). | |
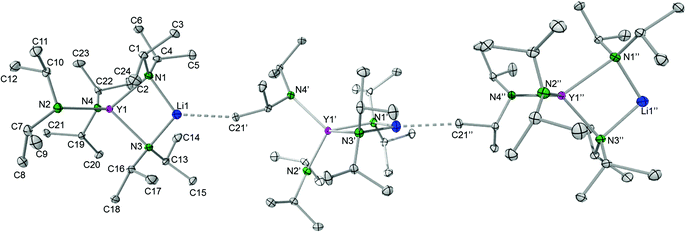 |
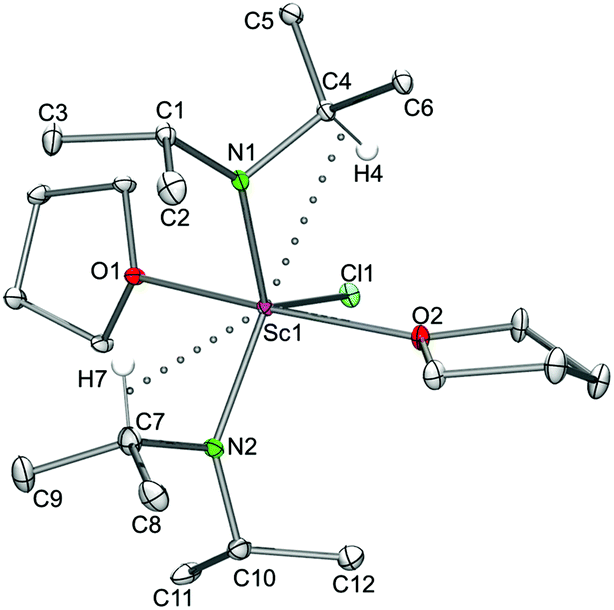
| | Fig. 10 Molecular structure of the complex LiY(NiPr2)4 (8). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity. Bond lengths (Å), interatomic distances (Å), and angles (°): Y1–N1 2.345(2), Y1–N2 2.217(2), Y1–N3 2.330(2), Y1–N4 2.226(2), Li1–N1 2.007(3), Li1–N3 2.020(3); Y1⋯C4 2.993(2), Y1⋯C10 2.931(2), Y1⋯C16 2.945(2), Y1⋯C19 2.910(2); Y1–N1–C1 123.24(8), Y1–N1–C4 100.50(8), Y1–N2–C7 142.74(9), Y1–N2–C10 103.29(9), N1–Y1–N3 84.99(5), N1–Li1–N3 103.3(2). | |
Scandium complexes {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 (9) and [Sc(NiPr2)Cl2(THF)]4 (10).
Depending on the use of LDA or NDA the [1:1] reactions with ScCl3(THF)3 in n-hexane led to the Sc4 complexes {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 (9) and [Sc(NiPr2)Cl2(THF)]4 (10) (Scheme 1, routes E and F), with LDA favouring LiCl inclusion. Similarly, the ScCl3(THF)3/NaN(SiMe3)2/THF reaction yielded alkali-metal-free Sc[N(SiMe3)2]Cl2(THF)245 while the LnCl3/LiN(SiMe3)2/THF reaction gave Ln2Li2 ate-complexes [{Ln[N(SiMe3)2](THF)(μ-Cl)2Li(THF)2}(μ-Cl)]2 (Ln = Sm, Eu, Yb).46 The Sc4Li2 ate-complex 9 is composed of two [Sc(NiPr2)(μ-Cl)(THF)]2 units, which are connected to a central Li2Cl2 rhombus via a total of four bridging chloro ligands (Fig. 11). The scandium centres adopt an almost square pyramidal coordination geometry with the THF and three chloro ligands in the basal as well as the diisopropylamido group in the apical positions. The ten chloro ligands constitute four Sc–Cl–Sc, four Sc–Cl–Li, and two Li–Cl–Li bridges, the latter two creating a heavily distorted tetrahedral environment around the lithium atoms. The geminal isopropyl groups display an antiperiplanar conformation and distinct Sc–N–C angles pointing toward β-H agostic interactions with scandium.
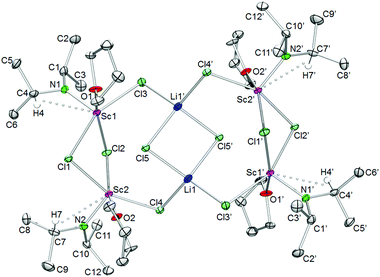 |
| | Fig. 11 Molecular structure of {[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 (9). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms, except those showing close metal contacts, and co-crystallized n-hexane (one molecule per asymmetric unit) are omitted for clarity. Bond lengths (Å), interatomic distances (Å), and angles (°): Sc1–N1 1.961(3), Sc2–N2 1.97(2), Sc1–Cl1 2.583(2), Sc2–Cl1 2.569(2), Sc1–Cl3 2.486(2), Sc2–Cl4 2.857(4), Sc1–O1 2.175(3), Sc2–O2 2.180(2), Li1–Cl3 2.294(7), Li1–Cl5 2.381(7); Sc1⋯C4 2.857(5), Sc2⋯C7 2.880(6); Sc1–N1–C1 134.7(3), Sc1–N1–C4 111.7(2), Cl1–Sc1–Cl2 80.82(4), Cl1–Sc1–Cl3 155.5(2), Cl3–Li1′–Cl4′ 112.1(3), Cl1–Sc1–N1 105.8(2), Cl5–Li1–Cl5′ 96.5(2), Li1–Cl5–Li1′ 83.5(2). | |
The tetrameric complex [Sc(NiPr2)Cl2(THF)]4 (10) features a Sc4 zigzag arrangement with two distinct scandium coordination environments (Fig. 12). The peripheral scandium atoms are coordinated in a distorted square pyramidal fashion with the amido nitrogen atom in the apical position and the THF, one terminal (Cl3) and two bridging chloro ligands (Cl1 and Cl4) forming the base. The inner scandium atoms show a distorted octahedral coordination with one bridging chloro ligand and the amido ligand in the apical and three bridging chloro ligands and one THF ligand in the equatorial positions. All scandium atoms are connected via two chloro bridges. The central Sc2–Cl2–Sc2′–Cl2′ ring is planar whereas the outer Sc2–Cl1–Sc2–Cl4 rings are not. Other alkali metal-free mixed amido/chloro rare-earth metal complexes with an amido:Ln ratio <2 feature above-mentioned monomeric Sc[N(SiMe3)2]Cl2(THF)2,45 trimeric Nd3[N(SiHMe2)2]5Cl4(THF)2,40 and donor-free pentameric Ce5[N(SiHMe2)2]8Cl7.47
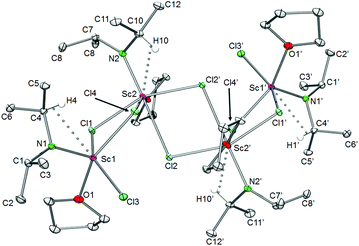 |
| | Fig. 12 Molecular structure of [Sc(NiPr2)Cl2(THF)]4 (10). Non-hydrogen atoms are represented by atomic displacement ellipsoids at the 30% level. Hydrogen atoms are omitted for clarity, except those showing close metal contacts. Bond lengths (Å), interatomic distances (Å), and angles (°): Sc1–N1 1.9584(14), Sc2–N2 1.9824(14), Sc1–Cl1 2.5886(5), Sc1–Cl3 2.3830(5), Sc1–Cl4 2.5426(5), Sc2–Cl2 2.5150(5), Sc2–Cl2′ 2.759(5); Sc1–O1 2.1813(12), Sc2–O2 2.1516(12), Sc1⋯C4 2.7857(17), Sc2⋯C10 2.8053(17); N1–Sc1–Cl1 109.58(4), N1–Sc1–Cl3 105.82(4), N2–Sc2–Cl4 97.83(4), N2–Sc2–Cl2′ 103.22(4), N2–Sc2–Cl2 177.63(5). Atoms O2 and O2′ are concealed by Sc1 and Sc1′. | |
Conclusions
Depending on the size of the rare-earth metal centre, careful choice of the synthesis conditions and molar ratios of the precursors gives access to a number of mono, bis, tris, and tetra(diisopropylamido) derivatives. Ate complexes of the type (AM)Ln(NiPr2)4(THF)n (AM = Li, Na; n = 1, 2) and chloro-bridged complexes [Ln(NiPr2)2(μ-Cl)(THF)]2 can be obtained for the entire Ln size range in good to very good yields, applying salt metathesis reactions in n-hexane and AM(NiPr2):LnCl3(THF)x ratios ≥3 and 1.6–2, respectively. In particular, the latter heteroleptic derivatives including the newly designed monomeric Ln(NiPr2)2Cl(THF)2 (Ln = Sc, Lu) might be useful precursors for subsequent salt metathesis or amine elimination reactions. For the smallest rare-earth metal centres scandium and lutetium, the synthesis of ate-free tris(amido) complexes Ln(NiPr2)3(THF) is easily accomplished using a Li:Ln ratio of 2.5. Although the accessibility of mono(amido) derivatives has been probed for the smallest rare-earth metal centre scandium only, ate-complex-free derivatives seem feasible as shown for the tetrametallic complex [Sc(NiPr2)Cl2(THF)]4. Donor (THF)-free diisopropylamide complexes can be accessed when applying unsolvated LnCl3 and n-hexane as a solvent, as shown for the isolation of LiY(NiPr2)4. Interestingly, LiY(NiPr2)4 not only shows enhanced mobility of the Li cation in solution compared to LiY(NiPr2)4(THF), but also features secondary intermolecular interactions in the solid-state distinct from LiY[N(SiHMe2)2]4. While the latter assembles as dimers involving two close intermolecular Li⋯H–Si bridges, LiY(NiPr2)4 adopts an infinite linear chain structure with single intermolecular Li⋯CH3 (iPr) contacts. Variable temperature NMR studies also show that the mobility of the alkali metal cation in ate complexes (AM)Ln(NiPr2)4(THF)n (AM = Li, Na) seems less affected by the size of the rare-earth metal centre than the size of AM (Na+ > Li+). Moreover, the formation of intramolecular Ln⋯CH(NiPr) β-agostic interactions seems less pronounced than in the corresponding bis(dimethylsilyl)amide complexes. DFT calculations and NBO analysis suggest the asymmetric coordination mode of the diisopropylamido ligands observed in the complexes under study and hence steric factors are rather the cause of weak β-agostic interactions than the consequence of it.
Seemingly long overlooked, diisopropylamido derivatives of the rare-earth elements, in particular complexes Ln(NiPr2)3(THF), might emerge as useful precursors in protonolysis protocols. The foreseeable enhanced reactivity compared to Ln[N(SiHMe2)2]3(THF)x bearing similarly-sized silylamido ligands of reduced basicity, as well as the reduced nucleophilicity compared to alkyl derivatives like routinely employed Ln(CH2SiMe3)3(THF)2, thus avoiding undesired side-reactions, might open up new avenues for catalyst design. Like the ubiquitous complexes Ln[N(SiMe3)2]3, broader applications of complexes Ln(NiPr2)3(THF) will carry on Don Bradley's legacy of fundamental organoamide chemistry.
Experimental section
General considerations
All operations were performed with rigorous exclusion of air and water, using standard Schlenk, high-vacuum, and glovebox techniques (MB Braun MB150B-G-I; <1 ppm O2, <1 ppm H2O). n-Hexane, n-pentane, and THF were purified by using Grubbs columns (MBraun SPS, Solvent Purification System) and stored in a glovebox. Benzene-d6 (99.5%) was received from Deutero GmbH and toluene-d8 (99.5%) from Euriso-top. All deuterated solvents were dried over NaK alloy for a minimum of 48 h, and filtered through a filter pipette (Whatman) before use. n-Butyllithium (2.5 M in n-hexane) and diisopropylamine (99.95%) were obtained from Sigma-Aldrich. Anhydrous LnCl3 were purchased from ABCR and converted to LnCl3(THF)xvia Soxhlet extraction. LDA25 and NDA16a were synthesized according to literature procedures. CHN elemental analyses were performed on an Elementar Vario MICRO cube. Although some carbon analyses are outside the range viewed as establishing analytical purity, they are provided to illustrate the best values obtained to date. The deviations are possibly due to the instability of the isopropylamide moieties at ambient temperature and salt contaminations.4,22,27 NMR spectra were recorded by using J. Young valve NMR tubes and obtained on Bruker AVII+400 (1H: 400.11 MHz, 13C: 100.61 MHz) and on Bruker AVII+500 (1H: 500.13 MHz, 13C: 125.76 MHz) spectrometers. 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. IR spectra were recorded on a Nicolet Impact 410 FTIR spectrometer using a DRIFT chamber with dry KBr/sample mixtures and KBr windows. For the latter the collected data were converted using the Kubelka–Munk refinement.
Syntheses
LiLn(NiPr2)4(THF) (1).
All of these complexes were synthesized in the same manner using 1 equiv. of LnCl3(THF)x and 3 equiv. of LDA in n-hexane. The synthesis of 1a is described in detail.
LiSc(NiPr2)4(THF) (1a).
To a suspension of 0.511 g ScCl3(THF)3 (1.39 mmol) in 10 ml n-hexane was added under vigorous stirring 0.440 g LDA (4.11 mmol). After stirring overnight at ambient temperature the colour of the suspension changed from white to yellow. The LiCl formed was separated by centrifugation and subsequent filtration of the supernatant. The solution was concentrated in vacuo and the product purified by crystallization from n-hexane at −35 °C, yielding 0.361 g (0.688 mmol, 50%) of the crystalline product. 1H NMR (toluene-d8, 500 MHz, 26 °C): δ = 3.91 (m, 4H, THF), 3.66 (sep, 8H, 3J = 6.5 Hz, CH), 1.38 (d, 48H, 3J = 6.4 Hz, CH3), 1.33 (m, 4H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 47.10 (CH), 27.1 (CH3), 25.0 (THF) ppm. IR (DRIFT): 2965 (vs), 2948 (vs), 2920 (s), 2855 (s), 2834 (m), 2705 (w), 2602 (w), 1470 (m), 1410 (m), 1365 (w), 1358 (m), 1320 (w), 1166 (vs), 1107 (m), 1037 (m), 995 (m), 913 (m), 794 (m), 623 (w), 512 (m), 437 (s) cm−1. Elemental analysis (%) calcd for C28H64LiN4OSc (524.73 g mol−1): C 64.09, H 12.29, N 10.68. Found: C 62.79, H 12.08, N 10.27.
LiY(NiPr2)4(THF) (1b).
YCl3(THF)3.3 (0.343 g, 0.793 mmol) and LDA (0.257 g, 2.40 mmol) yielded 0.225 g (0.396 mmol, 50%) of the crystalline product. 1H NMR (toluene-d8, 500 MHz, 26 °C): δ = 3.56 (broad m, 8H, CH), 3.41 (4H, THF), 1.37 (d, 48H, 3J = 6.15 Hz, CH3), 1.27 (4H, THF) ppm. 1H NMR (toluene-d8, 500 MHz, −33 °C): δ = 3.72 (m, 4H, 3J = 6.2 Hz, CH), 3.38 (sep, 4H, 3J = 6.1 Hz, CH), 3.21 (m, 4H, THF), 1.52 (d, 24H, 3J = 6.18 Hz, CH3 (terminal)), 1.34 (d, 12H, 3J = 6.16 Hz, CH3 (bridging)), 1.22 (d, 12H, 3J = 6.15 Hz, CH3 (bridging)), 1.08 (m, 4H, THF) ppm. 13C{1H} NMR (toluene-d8, 125.76 MHz, 26 °C): δ = 69.54 (THF), 46.16 (CH), 28.50 (CH3), 26.12 (THF) ppm. 13C NMR (toluene-d8, 125.76 MHz, −35 °C): 68.1 (t, 1JC–H = 150 Hz, THF), 48.6 (d, 1JC–H = 126.7 Hz, CH), 45.4 (d, 1JC–H = 125 Hz, CH), 29.3 (d, CH3 (bridging), 1JC–H = 123.3 Hz), 27.8 (d, 1JC–H = 126.5 Hz, CH3 (terminal)), 26.4–25.1 (d + t CH3 (bridging) + THF). IR (DRIFT): 2948 (vs), 2918 (vs), 2852 (s), 2817 (m), 2672 (m), 2602 (w), 1461 (m), 1407 (w), 1351, (m), 1323 (w), 1182 (vs), 1162 (ws), 1105 (m), 1037 (m), 915 (m), 779 (m), 606 (w), 526 (m), 416 (m) cm−1. Elemental analysis (%) calcd for C28H64LiN4OY (568.68 g mol−1): C 59.14, H 11.36, N 9.85. Found: C 57.43, H 11.11, N 9.61.
LiLa(NiPr2)4(THF) (1c).
LaCl3(THF)1.3 (0.50 g, 1.47 mmol) and LDA (0.473 g, 4.42 mmol) yielded 0.434 g (0.701 mmol, 48%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.58 (sep, 8H, CH, 3J = 6.3 Hz), 3.31 (m, 4H, THF), 1.40 (d, 48H, CH3, 3J = 5.2 Hz), 1.17 (m, 4H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.57 (THF), 48.60 (CH), 27.22 (CH3), 25.20 (THF) ppm. IR (DRIFT): 2965 (vs), 2913 (s), 2855 (s), 2663 (w), 1463 (m), 1374 (m), 1335 (m), 1164 (s), 1103 (m), 1039 (w), 997 (vw), 904 (m), 782 (w), 510 (m), 420 (w) cm−1. Elemental analysis (%) calcd for C28H64LiN4OLa (618.68 g mol−1): C 54.36, H 10.43, N 9.06. Found: C 53.19, H 9.81, N 8.93.
LiLu(NiPr2)4(THF) (1d).
LuCl3(THF)2.9 (0.611 g, 1.25 mmol) and LDA (0.40 g, 3.73 mmol) yielded 0.325 g (0.497 mmol, 40%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.66 (sep, 12H, CH, 3J = 6.4 Hz, overlapping with THF), 1.40 (d, 48H, CH3), 1.15 (m, 4H, THF). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 47.34 (CH), 27.03 (CH3), 25.03 (THF) ppm. IR (DRIFT): 2962 (vs), 2943 (vs), 2929 (s), 2859 (m), 2681 (w), 2601 (vw), 1466 (w), 1412 (w), 1351 (m), 1177 (s), 1164 (ws), 1107 (w), 1039 (m), 1002 (w), 915 (m), 784 (m), 613 (w), 536 (w), 412 (w) cm−1. Elemental analysis (%) calcd for C28H64LiN4OLu (654.74 g mol−1): C 51.36, H 9.85, N 8.56. Found: C 50.29, H 9.39, N 7.95.
NaLn(NiPr2)4(THF) (2).
All of these complexes were synthesized in the same manner using 1 equiv. of LnCl3(THF)x and 3 equiv. of NDA. The synthesis of 2a is described in detail.
NaSc(NiPr2)4(THF) (2a).
To a suspension of 44.7 mg ScCl3(THF)3 (0.12 mmol) in 7 ml n-hexane was added 45 mg NDA (0.36 mmol) under vigorous stirring. After stirring overnight at ambient temperature the colour of the suspension changed from white to slightly yellow. The NaCl formed was separated by centrifugation and subsequent filtration of the supernatant. The solution was concentrated in vacuo and the product purified by crystallization from n-hexane at −35 °C, yielding 36 mg (0.067 mmol, 55%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.66 (sep, 8H, CH, 3J = 6.3 Hz), 3.41 (m, 4H, THF), 1.38 (d, 48H, CH3, 3J = 6.4 Hz), 1.29 (m, 4H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 46.2 (CH), 27.0 (CH3), 25.0 (THF) ppm. IR (DRIFT): 2969 (vs), 2872 (m), 1604 (vw), 1467 (w), 1388 (m), 1178 (m), 993 (vw), 911 (vw), 786 (w), 692 (w), 616 (w), 509 (m), 433 (m) cm−1. Elemental analysis (%) calcd for C28H64N4NaOSc (540.79 g mol−1): C 62.19, H 11.93, N 10.36. Found: C 61.12, H 12.53, N 9.85.
NaY(NiPr2)4(THF) (2b).
YCl3(THF)3.3 (0.375 g, 0.87 mmol) and NDA (0.322 g, 2.62 mmol) yielded 0.326 g (0.557 mmol, 64%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.66 (sep, 8H, CH, 3J = 6.37 Hz), 3.41 (broad, 4H, THF), 1.38 (d, 48H, CH3, 3J = 6.37 Hz), 1.29 (m, 4H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 67.8 (THF), 46.8 (CH), 27.7 (CH3), 25.3 (THF) ppm. IR (DRIFT): 2965 (vs), 2942 (vs), 2916 (s), 2855 (s), 2809 (m), 2682 (w), 2589 (vw), 1461 (m), 1424 (w), 1404 (w), 1370 (m), 1362 (m), 1348 (m), 1179 (s), 1163 (s), 1109 (m), 1099 (m), 1052 (w), 1038 (w), 1003 (m), 910 (m), 785 (m), 777 (w), 608 (vw), 521 (m), 507 (w), 414 (m) cm−1. Elemental analysis (%) calcd for C28H64N4NaOY (584.74 g mol−1): C 57.51, H 11.03, N 9.58. Found: C 56.42, H 11.64, N 9.11.
NaLa(NiPr2)4(THF) (2c).
LaCl3(THF)2.9 (0.30 g, 0.88 mmol) and NDA (0.326 g, 2.65 mmol) yielded 0.301 g (0.474 mmol, 54%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.56 (sep, 8H, CH, 3J = 6.31 Hz), 3.41 (m, 4H, THF), 1.34–1.31 (52H: d, CH3, 3J = 6.27 Hz; m, THF). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.0 (THF), 47.5 (CH), 27.5 (CH3), 23.7 (THF) ppm. IR (DRIFT): 2944 (vs), 2853 (s), 2662 (vw), 2579 (vw), 1460 (m), 1349 (m), 1311 (w), 1161 (vs), 1095 (w), 1041 (w), 999 (m), 906 (m), 781 (m), 514 (w), 402 (w) cm−1. Elemental analysis (%) calcd for C28H64N4NaOLa (634.74 g mol−1): C 52.98, H 10.16, N 8.83. Found: C 50.89, H 10.57, N 7.87. Better microanalytical data could not be obtained.
NaLu(NiPr2)4(THF) (2d).
LuCl3(THF)2.9 (0.613 g, 1.25 mmol) and NDA (0.462 g, 3.75 mmol) yielded 0.394 g (0.587 mmol, 47%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.71 (sep, 8H, 3J = 6.23 Hz, CH), 3.32 (m, 4H, THF), 1.36 (d, 48H, 3J = 6.47 Hz, CH3), 1.25 (m, 4H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.1 (THF), 46.8 (CH), 27.9 (CH3), 25.5 (THF) ppm. IR (DRIFT): 2966 (vs), 2944 (vs), 2915 (s), 2856 (s), 2811 (w), 2690 (vw), 2591 (vw), 1462 (w), 1400 (w), 1378 (w), 1351 (m), 1319 (w), 1179 (s), 1162 (s), 1147 (m), 1110 (m), 1093 (m), 1040 (m), 1001 (m), 912 (m), 895 (w), 787 (m), 613 (vw), 525 (w), 507 (w), 437 (w), 408 (w) cm−1. Elemental analysis (%) calcd for C28H64N4NaOLu (670.79 g mol−1): C 50.13, H 9.62, N 8.35. Found: C 49.30, H 9.19, N 7.70.
NaLn(NiPr2)4(THF)2 (3).
All of these complexes were synthesized in the same manner using 1 equiv. of LnCl3(THF)x and 3–4 equiv. of NDA. Representatively, the synthesis of 3a is described in detail.
NaSc(NiPr2)4(THF)2 (3a).
To a suspension of 0.361 g ScCl3(THF)3 (0.979 mmol) in 10 ml n-hexane was added under vigorous stirring 0.491 g NDA (3.916 mmol). After stirring for 72 h at ambient temperature the colour of the suspension changed from white to deep yellow. The NaCl formed was separated by centrifugation and subsequent filtration of the supernatant. The solution was concentrated in vacuo and the product purified by crystallization from n-hexane at −35 °C, yielding 0.103 g (0.168 mmol, 17%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.81 (sep, 8H, CH, 3J = 6.4 Hz), 3.5 (m, 8H, THF), 1.37 and 1.33 (56H: m, THF; d, CH3, 3J = 6.3 Hz). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.1 (THF), 47.1 (CH), 27.5 (CH3), 25.5 (THF) ppm. IR (DRIFT): 2966 (vs), 2942 (vs), 2919 (s), 2853 (m), 2815 (w), 2595 (vw), 1463 (w), 1400 (w), 1379 (w), 1369 (m), 1352 (m), 1318 (w), 1176 (s), 1161 (vw), 1144 (m), 1008 (m), 1039 (w), 995 (m), 910 (m), 838 (vw), 829 (vw), 781 (m), 616 (vw), 528 (w), 508 (w), 481 (vw), 434 (m) cm−1. Elemental analysis (%) calcd for C32H72N4NaO2Sc (612.90 g mol−1): C 62.71, H 11.84, N 9.14. Found: C 61.54; H 12.35; N 8.78.
NaY(NiPr2)4(THF)2 (3b).
YCl3(THF)3.3 (0.396 g, 0.914 mmol) and NDA (0.450 g, 3.654 mmol) yielded 0.249 g (0.379 mmol, 42%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.67 (sep, 8H, 3J = 6.24 Hz, CH), 3.37 (m, 7H, THF), 1.39 (d, 48H, 3J = 6.38 Hz, CH3), 1.28 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.1 (THF), 47.0 (CH), 27.9 (CH3), 25.5 (THF) ppm. IR (DRIFT): 2967 (vs), 2875 (s), 2685 (w), 2587 (vw), 2808 (w), 1659 (vw), 1607 (w), 1460 (w), 1377 (m), 1173 (m), 1146 (w), 1094 (w), 1053 (vw), 1001 (w), 913 (w), 782 (w), 687 (w), 606 (vw), 507 (w), 412 (w) cm−1. Elemental analysis (%) calcd for C28H64N4NaOY (656.84 g mol−1): C 58.51, H 11.05, N 8.53. Found: C 57.82, H 11.64, N 8.49.
NaLa(NiPr2)4(THF)2 (3c).
LaCl3(THF)2.9 (0.30 g, 0.88 mmol) and NDA (0.434 g, 3.52 mmol) yielded 0.262 g (0.37 mmol, 42%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.55 (sep, 8H, CH, 3J = 6.26 Hz), 3.36 (m, 8H, THF), 1.34 and 1.28 (56H: d, CH3, 3J = 6.33 Hz; m, THF). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 68.0 (THF), 47.5 (CH), 27.5 (CH3), 25.5 (THF) ppm. IR (DRIFT): 2948 (vs), 2912 (vs), 2854 (s), 2804 (m), 2661 (w), 2584 (vw), 1464 (m), 1348 (m), 1316 (w), 1159 (ws), 1094 (w), 1040 (w), 1001 (m), 903 (s), 781 (m), 513 (w), 420 (w) cm−1. Elemental analysis (%) calcd for C32H72N4NaO2La (706.85 g mol−1): C 54.38, H 10.27, N 7.93. Found: C 53.89, H 9.34, N 8.43.
Ln(NiPr2)3(THF) (Ln = Sc (4a), Lu (4b)).
In addition to an alkyl elimination reaction employing Ln(CH2SiMe3)3(THF)2 and 3 equiv. isopropylamine22 these two complexes were synthesized according to two distinct salt metathesis protocols as follows: protocol 1: to a suspension of LnCl3(THF)x in 15 mL n-hexane were added under vigorous stirring in small portions 2.5 equiv. of solid LDA. After stirring for 16 h, the reaction mixture was filtered, the residue was extracted twice with 10 mL n-hexane, and the combined organic fractions were evaporated in vacuo. Crystallization from a small amount of n-hexane gave colourless crystals (yields <40%). Protocol 2: to a solution of [Ln(NiPr2)2(μ-Cl)(THF)]2 (5) in 15 mL n-hexane were added under vigorous stirring in small portions 0.9 equiv. of solid LDA. After stirring for 16 h, the reaction mixture was filtered, the residue was extracted twice with 10 mL n-hexane, and the combined organic fractions were evaporated in vacuo. Crystallization from a small amount of n-hexane gave colourless crystals (yields <50%). Complexes 4a and 4b were identified by NMR spectroscopy and elemental analysis, see ref. 22 and Fig. S24.‡
[Ln(NiPr2)2(THF)(μ-Cl)]2 (Ln = Sc (5a), Y (5b), Lu (5d)), [La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2 (5c′), La(NiPr2)3(THF)2 (6), and Ln(NiPr2)2Cl(THF)2 (Ln = Sc (7a) and Lu (7b)).
Complexes 5 were synthesized in the same manner using 1 equiv. of LnCl3(THF)x and substoichiometric amounts of 1.6 to 2 equiv. of LDA according to the literature.14a Representatively, the synthesis of 5a is described in detail.
[Sc(NiPr2)2(THF)(μ-Cl)]2 (5a).
To a suspension of 0.735 g ScCl3(THF)3 (1.99 mmol) in 15 ml n-hexane was added under vigorous stirring 0.418 g of LDA (3.902 mmol) and the reaction mixture was stirred at ambient temperature overnight. Centrifugation, subsequent filtration of the supernatant, concentrating in vacuo, and re-crystallization from n-hexane at −35 °C yielded 0.499 g (0.706 mmol, 71%) of the pure product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.78 (m, 8H, THF), 3.55 (m, 8H, CH), 1.36 (d, 48H, CH3, 3J = 6.4 Hz), 1.22 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 71.82 (THF), 47.28 (CH), 26.36 (CH3), 25.14 (THF) ppm. IR (DRIFT): 2969 (vs), 2923 (s), 2867 (s), 2701 (vw), 1457 (m), 1388 (m), 1371 (m), 1353 (m), 1169 (s), 1100 (m), 1023 (m), 999 (w), 919 (m), 873 (m), 798 (w), 675 (w), 525 (w), 490 (w), 445 (w) cm−1. Elemental analysis (%) calcd for C32H72Cl2N4O2Sc2 (705.76 g mol−1): C 54.46, H 10.28, N 7.94. Found: C 53.32, H 9.66, N 7.98.
[Y(NiPr2)2(THF)(μ-Cl)]2 (5b).
YCl3(THF)3.3 (2.183 g, 5.04 mmol) and LDA (1.026 g, 9.58 mmol) yielded 0.845 g (1.064 mmol, 42%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.86 (m, 8H, THF), 3.63 (m, 8H, 3J = 6.1 Hz, CH), 1.44 (d, 48H, 3J = 6.2 Hz, CH3), 1.29 (m, 8H, THF). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 70.95 (THF), 46.40 (CH), 27.33 (CH3), 25.07 (THF) ppm. IR (DRIFT): 2965 (vs), 2919 (s), 2859 (m), 2681 (vw), 1457 (w), 1405 (w), 1368 (w), 1350 (m), 1165 (vs), 1144 (m), 1100 (w), 1022 (m), 1004 (w), 918 (m), 872 (m), 845 (w), 522 (w), 423 (m) cm−1. Elemental analysis (%) calcd for C32H72Cl2N4O2Y2 (793.66 g mol−1): C 48.43, H 9.14, N 7.06. Found: C 48.39, H 8.09, N 7.35.
[Lu(NiPr2)2(THF)(μ-Cl)]2 (5d).
LuCl3(THF)2.9 (0.994 g, 2.03 mmol) and LDA (0.429 g, 4.00 mmol) yielded 0.483 g (0.48 mmol, 49%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.86 (m, 8H, THF), 3.59 (sep, 8H, CH, 3J = 6.34 Hz), 1.42 (d, 48H, CH3, 3J = 6.49 Hz), 1.23 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 70.95 (THF), 45.96 (CH), 26.99 (CH3), 25.07 (THF) ppm. IR (DRIFT): 2965 (vs), 2920 (s), 2860 (m), 2683 (vw), 2587 (vw), 1458 (m), 1404 (w), 1368 (w), 1354 (m), 1185 (s), 1168 (s), 1144 (m), 1116 (w), 1021 (m), 1005 (m), 919 (m), 868 (m), 796 (m), 674 (vw), 593 (vw), 524 (m), 419 (m) cm−1. Elemental analysis (%) calcd for C32H72Cl2Lu2N4O2 (965.79 g mol−1): C 39.80, H 7.51, N 5.80. Found: C 39.03, H 6.62, N 5.72.
[La(NiPr2)2(THF)(μ-Cl)]2·La(NiPr2)3(THF)2 (5c′).
LaCl3(THF)1.3 (1.334 g, 3.93 mmol) and LDA (0.689 g, 6.43 mmol) yielded 0.732 g (0.495 mmol, 38%) of the crystalline product (based on putative 5c′), which contained 6 as revealed by X-ray crystallographic analysis. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.68 (30H, CH and THF), 1.44 and 1.30 (100H, CH3 and THF). IR (DRIFT): 2964 (vs), 2854 (s), 2671 (w), 1456 (m), 1397 (m), 1373 (m), 1354 (m), 1329 (w), 1170 (vs), 1114 (m), 1100 (m), 1010 (w), 913 (s), 792 (m), 596 (vw), 522 (m), 407 (s) cm−1. Elemental analysis for 5c′ (%) calcd for C58H130Cl2La3N7O4 (1477.34 g mol−1): C 47.15, H 8.87, N 6.64. Found: C 49.43, H 9.21, N 9.21.
La(NiPr2)3(THF)2 (6).
Recrystallization of 5c′ from n-pentane/THF afforded 6. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.90 (m, 8H, THF), 3.62 (sep, 6H, CH, 3J = 6.1 Hz), 3.33 (m, 8H, THF), 1.38 (d, 36H, CH3, 3J = 6.0 Hz), 1.18 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 70.82 (THF), 47.45 (CH), 26.72 (CH3), 25.19 (THF) ppm. IR (DRIFT): 2954 (vs), 2923 (vs), 2858 (s), 2674 (w), 2591 (vw), 1458 (m), 1406 (m), 1368 (m), 1339 (w), 1277 (w), 1165 (s), 1116 (w), 913 (m), 873 (w), 843 (vw), 793 (m), 602 (vw), 521 (w), 410 (m) cm−1. Elemental analysis (%) calcd for C26H58LaN3O2 (583.66 g mol−1): C 53.50, H 10.02, N 7.20. Found: C 54.42, H 10.11, N 8.83. Better microanalytical data could not be obtained.
Sc(NiPr2)2Cl(THF)2 (7a).
Recrystallization of 5a from n-pentane/THF afforded 7a. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.65, 3.55 (2 m, 12H, CH and THF), 1.38 (m, 24H, CH3), 1.28 (m, 8H, THF). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 72.11 (THF), 47.80 (CH), 26.67 (CH3), 25.43 (THF) ppm. IR (DRIFT): 2968 (vs), 2868 (s), 2711 (vw), 1661 (vw), 1462 (m), 1376 (m), 1169 (m), 1103 (w), 1014 (w), 925 (w), 867 (w), 808 (w), 675 (w), 564 (w), 442 (w) cm−1. Elemental analysis (%) calcd for C20H44ClScN2O2 (424.98 g mol−1): C 56.52; H 10.44; N 6.59. Found: C 55.89; H 10.47; N 6.21.
Lu(NiPr2)2Cl(THF)2 (7b).
Recrystallization of 5d from n-pentane/THF afforded 7b. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.68 (m, 8H, THF), 3.54 (sep, 4H, 3J = 6.36 Hz), 1.36 (d, 24H, 3J = 6.45 Hz), 1.30 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 71.34 (THF), 46.52 (CH), 25.17 (CH3), 24.78 (THF) ppm. IR (DRIFT): 2965 (vs), 2920 (s), 2861 (m), 1507 (vw), 1459 (w), 1369 (m), 1350 (m), 1184 (s), 1167 (s), 1146 (m), 1102 (vw), 1018 (m), 920 (m), 870 (m), 797 (w), 521 (w), 516 (w) cm−1. Elemental analysis (%) calcd for C20H44ClLuN2O2 (555.0 g mol−1): C 43.28, H 7.99, N 5.05. Found: C 42.82, H 7.03, N 5.31.
LiY(NiPr2)4 (8).
To 0.319 g of anhydrous YCl3 (1.63 mmol. 1 equiv.) was added 0.350 g of solid LDA (3.26 mmol, 2.0 equiv.) and the reaction mixture was stirred vigorously for 72 h in 15 mL n-hexane. Centrifugation, subsequent filtration of the supernatant, concentrating in vacuo, and re-crystallization from n-hexane at −35 °C yielded 0.325 g (0.654 mmol, 40%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.46 (sep, 8H, CH), 1.27 (m, 48H, CH3) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 47.0 (CH), 26.76, 26.09 (CH3 bridging and terminal), 24.80 (THF) ppm. 1H NMR (toluene-d8, 500 MHz, −50 °C): δ = 3.56 (sep, 4H, CH), 3.41 (sep, 4H, CH), 1.52 (d, 24H, CH3 terminal), 1.24 (d, 12H, CH3 bridging), 1.10 (d, 12H, CH3 bridging) ppm. 13C{1H} NMR (toluene-d8, 125.76 MHz, −50 °C): δ = 48.3 (CH), 45.2 (CH), 29.1 (CH3), 27.4 (CH3), 25.5 (CH3) ppm. IR (DRIFT): 2967 (vs), 2949 (vs), 1920 (s), 2857 (s), 2683 (w), 2600 (vw), 1462 (m), 1373 (m), 1358 (m), 1317 (w), 1178 (s), 1165 (vs), 1109 (m), 1004 (m), 995 (w), 914 (m), 785 (m), 614 (vw), 532 (m), 509 (w), 419 (w) cm−1. Elemental analysis (%) calcd for C24H56LiN4Y (496.59 g mol−1): C 58.05, H 11.37, N 11.28. Found: C 56.10, H 10.94, N 10.83. Better microanalytical data could not be obtained.
{[Sc(NiPr2)Cl2(THF)]2(LiCl)}2 (9).
Compound 9 was synthesized according to the procedure used for complexes 1. ScCl3(THF)3 (0.150 g, 0.408 mmol) and LDA (0.0437 g, 0.408 mmol) yielded 56.0 mg (0.045 mmol, 44%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.65 (m, 16H, THF), 3.54 (m, 8H, CH), 1.37 (d, 48H, CH3), 1.28 (16H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 47.8 (CH), 26.8 (CH3), 25.1 (THF) ppm. IR (DRIFT): 2967 (vs), 2956 (vs), 2925 (s), 2881 (m), 1458 (m), 1389 (w), 1372 (w), 1355 (m), 1189 (m), 1162 (s), 1122 (w), 1103 (m), 1008 (s), 925 (s), 863 (s), 852 (s), 809 (w), 676 (vw), 598 (vw), 532 (m), 485 (m), 446 (m) cm−1. Elemental analysis (%) calcd for C40H88Cl10Li2N4O4Sc4 (without co-crystallized n-hexane) (1237.37 g mol−1): C 38.83; H 7.17; N 4.53. Found: C 39.25; H 6.96; N 4.61.
[Sc(NiPr2)Cl2(THF)]4 (10).
Compound 10 was synthesised according to the procedure used for complexes 2. ScCl3(THF)3 (67.2 mg, 0.18 mmol) and NDA (22.4 mg, 0.18 mmol) yielded 0.011 g (0.0096 mmol, 21%) of the crystalline product. 1H NMR (C6D6, 400 MHz, 26 °C): δ = 3.83 (m, 8H, THF), 3.69 (sep, 4H, CH), 1.43 (d, 24H, CH3, 3J = 5.36 Hz), 1.30 (m, 8H, THF) ppm. 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ = 47.0 (CH), 26.76, 26.09 (CH3 bridging and terminal), 24.80 (THF) ppm. IR (DRIFT): 2965 (vs), 2925 (s), 2869 (s), 2720 (vw), 1461 (m), 1410 (w), 1373 (w), 1357 (m), 1186 (m), 1167 (s), 1122 (w), 1101 (w), 1101 (w), 1037 (vw), 1010 (s), 925 (m), 865 (s), 809 (m), 678 (w), 598 (w), 531 (m), 492 (m), 481 (m), 447 (m) cm−1. Elemental analysis (%) calcd for C40H84Cl8N4O4Sc4 (1148.57 g mol−1): C 41.83; H 7.37; N 4.88. Found: C 41.21; H 7.95; N 4.53.
X-ray crystallography and crystal structure determinations of 1–10
Crystals were grown by standard techniques from saturated solutions using n-hexane for 1a–d, 2a–d, 3a–c, 4a, 5b, 5c′, 8, 9 and 10 at −35 °C and a n-pentane/THF solution for 6, 7a and 7b. Suitable single crystals for X-ray structure analyses were selected in a glovebox and coated with Paratone-N (Hampton) and fixed on a nylon loop. Data for all crystals except 4a were collected on a Bruker APEX DUO instrument equipped with a IμS microfocus sealed tube and QUAZAR optics for MoKα radiation (λ = 0.71073 Å). 4a was measured on a Bruker SMART APEX II instrument equipped with a fine focus sealed tube and curved graphite monochromator using MoKα radiation (λ = 0.71073 Å). The data collection strategy was determined using COSMO48 employing ω and ϕ scans. Raw data were processed using APEX48 and SAINT,48 corrections for absorption effects were applied using SADABS.48 The structures were solved by direct methods and refined against all data by full-matrix least-squares methods on F2 using SHELXT48 and Shelxle.49 All graphics were produced by employing ORTEP-350 and POV-Ray.51 Some disorder models were calculated using the program DSR.52 Further details of the refinement and crystallographic data are listed in Tables S1–S6‡ and in the CIF files. CCDC 1471665–1471684 contain all the supplementary crystallographic data for this paper.
Computational details
DFT calculations were performed with the Gaussian 09 program suite36 using the B3LYP density functional,53 along with the implemented def2-TZVP basis set.54 All geometry optimizations were performed without imposing any symmetry constraints, and the structures obtained were confirmed as true minima by calculating analytical frequencies. NBO analyses were carried out by employing the NBO 6.0 software.37
Acknowledgements
We thank the German Science Foundation (Grant AN 238/16-1) for funding, the bwGRiD project, as part of the German D-Grid initiative, for providing computing facilities at the University Tübingen, as well as the bwHPC initiative and the bwHPC-C5 project55 for computing facilities at the University of Ulm (JUSTUS cluster).
Notes and references
- P. O'Brien, Coord. Chem. Rev., 2000, 209, 35 CrossRef.
-
(a) D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Chem. Commun., 1972, 349 RSC;
(b) E. C. Alyea, D. C. Bradley and R. G. Copperthwaite, J. Chem. Soc., Dalton Trans., 1972, 1580 RSC;
(c) D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021 RSC.
- For review articles, see:
(a) D. C. Bradley and M. H. Chisholm, Acc. Chem. Res., 1976, 9, 273 CrossRef CAS;
(b)
D. H. Harris and M. F. Lappert, in Organometallics Chemisty Reviews: Organosilicon Reviews, Elsevier Scientific Publishing Company, Amsterdam, 1976, p. 13 Search PubMed;
(c)
Metal And Metalloid Amides, ed. M. F. Lappert, P. P. Power, A. R. Sanger and R. C. Srivastava, Ellis Horwood, Chichester, 1980 Search PubMed;
(d) R. Anwander, Top. Curr. Chem., 1996, 179, 33 CrossRef CAS;
(e) R. Kempe, Angew. Chem., Int. Ed., 2000, 39, 468 CrossRef CAS;
(f) W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131 CrossRef CAS;
(g)
Metal Amide Chemistry, ed. M. F. Lappert, A. V. Protchenko, P. P. Power and A. L. Seeber, John Wiley & Sons, Hoboken, 2009 Search PubMed;
(h) Y. Liang and R. Anwander, Dalton Trans., 2013, 42, 12521 RSC.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, Inorg. Nucl. Chem. Lett., 1976, 12, 735 CrossRef CAS.
-
(a) W. A. Herrmann, R. Anwander, F. C. Munck, W. Scherer, V. Dufaud, N. W. Huber and G. R. J. Artus, Z. Naturforsch., B: Chem. Sci., 1994, 49, 1789 CrossRef CAS;
(b) W. A. Herrmann, F. C. Munck, G. R. J. Artus, O. Runte and R. Anwander, Organometallics, 1997, 16, 682 CrossRef CAS;
(c) R. Anwander, O. Runte, J. Eppinger, G. Gerstberger, E. Herdtweck and M. Spiegler, J. Chem. Soc., Dalton Trans., 1998, 847 RSC.
- C. A. P. Goodwin, K. C. Joslin, S. J. Lockyer, A. Formanuik, G. A. Morris, F. Ortu, I. J. Vitorica-Yrezabal and D. P. Mills, Organometallics, 2015, 34, 2314 CrossRef CAS.
-
(a) W. S. Rees, O. Just, H. Schumann and R. Weimann, Angew. Chem., Int. Ed., 1996, 35, 419 CrossRef CAS;
(b) H. Schumann, J. Winterfeld, E. C. E. Rosenthal, H. Hemling and L. Esser, Z. Anorg. Allg. Chem., 1995, 621, 122 CrossRef CAS;
(c) C. Schädle, C. Meermann, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2010, 2841 CrossRef;
(d) D. R. Click, B. L. Scott and J. G. Watkin, Chem. Commun., 1999, 633 RSC;
(e) H. Yin, J. R. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Commun., 2014, 50, 3470 RSC;
(f) G. B. Deacon, C. M. Forsyth, P. C. Junk, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 1381 RSC.
-
(a) R. K. Minhas, Y. Ma, J.-I. Song and S. Gambarotta, Inorg. Chem., 1996, 35, 1866 CrossRef CAS;
(b) P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, Chem. Commun., 2006, 3546 RSC;
(c) B. S. Lim, A. Rahtu, J.-S. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951 CrossRef CAS PubMed;
(d) P. B. Hitchcock, Q.-G. Huang, M. F. Lappert and X.-H. Wei, J. Mater. Chem., 2004, 14, 3266 RSC;
(e) S. D. Daniel, J.-S. M. Lehn, J. D. Korp and D. M. Hoffman, Polyhedron, 2006, 25, 205 CrossRef CAS;
(f) Y. Yao, L. Mao, X. Lu, Q. Shen and J. Sun, J. Rare Earths, 2002, 20, 592 Search PubMed;
(g) H. Yin, A. J. Lewis, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 8234 CrossRef CAS PubMed;
(h) W. J. Evans, M. A. Ansari, J. W. Ziller and S. I. Khan, Inorg. Chem., 1996, 35, 5435 CrossRef CAS PubMed;
(i) G. R. Giesbrecht, J. C. Gordon, D. L. Clark, P. J. Hay, B. L. Scott and C. D. Tait, J. Am. Chem. Soc., 2004, 126, 6387 CrossRef CAS PubMed.
- For an example, see: R. L. LaDuca and P. T. Wolczanski, Inorg. Chem., 1992, 31, 1313 CrossRef.
- For structure analyses of Ln[N(SiMe3)2]3, see:
(a) Ln = Sc, Eu: J. S. Ghotra, M. B. Hursthouse and A. J. Welch, J. Chem. Soc., Chem. Commun., 1973, 669 RSC;
(b) Ln = Nd: R. A. Andersen, D. H. Templeton and A. Zalkin, Inorg. Chem., 1978, 17, 2317 CrossRef CAS;
(c) Ln = Sc (gas phase): T. Fjeldberg and R. A. Andersen, J. Mol. Struct., 1985, 129, 49 CrossRef;
(d) Ln = Ce, Pr (gas phase): T. Fjeldberg and R. A. Andersen, J. Mol. Struct., 1985, 129, 93 CrossRef CAS;
(e) Ln = Dy, Er (solid state), Lu (gas phase): [5a]; ;
(f) Ln = Y: M. Westerhausen, M. Hartmann, A. Pfitzner and W. Schwarz, Z. Anorg. Allg. Chem., 1995, 621, 837 CrossRef CAS;
(g) Ln = Ce: W. S. Rees Jr., O. Just and D. S. Van Derveer, J. Mater. Chem., 1999, 9, 249 RSC;
(h) Ln = Sc, La:
G. Gerstberger, PhD thesis, Technische Universität München, 1999;
(i) Ln = Yb: M. Niemeyer, Z. Anorg. Allg. Chem., 2002, 628, 647 CrossRef CAS;
(j) Ln = Sm: E. D. Brady, D. L. Clark, J. C. Gordon, P. J. Hay, D. W. Keogh, R. Poli, B. L. Scott and J. G. Watkin, Inorg. Chem., 2003, 42, 6682 CrossRef CAS PubMed;
(k) Ln = Sm: J. Sundermeyer, A. Khvorost and K. Harms, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1117 CAS;
(l) Ln = Tb: P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and Z. Li, Dalton Trans., 2004, 129 RSC;
(m) Ln = Lu: G. Scarel, C. Wiemer, M. Fanciulli, I. L. Fedushkin, G. K. Fukin, G. A. Domrachev, Y. Lebedinskii, A. Zenkevich and G. Pabia, Z. Anorg. Allg. Chem., 2007, 633, 2097 CrossRef CAS.
- J. Eppinger, M. Spiegler, W. Hieringer, W. A. Herrmann and R. Anwander, J. Am. Chem. Soc., 2000, 122, 3080 CrossRef CAS.
- R. R. Fraser, M. Bresse and T. S. Mansour, J. Chem. Soc., Chem. Commun., 1983, 620 RSC.
- For examples having revealed the enhanced reactivity of diisopropylamide complexes compared to silylamide derivatives, see:
(a) H. C. Aspinall, S. R. Moore and A. K. Smith, J. Chem. Soc., Dalton Trans., 1993, 993 RSC;
(b) W. J. Evans, R. Anwander, J. W. Ziller and S. I. Khan, Inorg. Chem., 1995, 34, 5927 CrossRef CAS;
(c) R. Anwander and R. Roesky, J. Chem. Soc., Dalton Trans., 1997, 137 RSC;
(d) P. N. O'Shaughnessy and P. Scott, Tetrahedron: Asymmetry, 2003, 14, 1979 CrossRef;
(e) R. H. Platel, L. M. Hodgson, A. J. P. White and C. K. Williams, Organometallics, 2007, 26, 4955 CrossRef CAS;
(f) H. M. Dietrich, K. W. Törnroos, E. Herdtweck and R. Anwander, Organometallics, 2009, 28, 6739 CrossRef CAS.
- For the use of mixed diisopropylamido/chloro derivatives, see:
(a) C. Meermann, P. Sirsch, K. W. Törnroos and R. Anwander, Dalton Trans., 2006, 1041 RSC;
(b) C. Meermann, K. W. Törnroos and R. Anwander, Inorg. Chem., 2009, 48, 2561 CrossRef CAS PubMed;
(c) S. Qayyum, G. G. Skvortsov, G. K. Fukin, A. A. Trifonov, W. P. Kretschmer, C. Döring and R. Kempe, Eur. J. Inorg. Chem., 2010, 248 CrossRef CAS.
- For the use of diisopropylamide ate complexes, see:
(a) Z. G. Wang, H. M. Sun, H. S. Yao, Q. Shen and Y. Zhang, Organometallics, 2006, 25, 4436 CrossRef CAS;
(b) Z. G. Wang, H. M. Sun, H. S. Yao, Y.-M. Yao, Q. Shen and Y. Zhang, J. Organomet. Chem., 2006, 691, 3383 CrossRef CAS;
(c) J. Zhang, H. Yao, Y. Zhang, H. Sun and Q. Shen, Organometallics, 2008, 27, 2672 CrossRef CAS;
(d) H. Yao, Y. Zhang, H. Sun and Q. Shen, Eur. J. Inorg. Chem., 2009, 1920 CrossRef CAS;
(e) D. Qin, F. Han, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2009, 5535 RSC;
(f) H. Yao, J. Zhang, Y. Zhang, H. Sun and Q. Shen, Organometallics, 2010, 29, 5841 CrossRef CAS;
(g) X. Hu, C. Lu, B. Wu, H. Ding, B. Zhao, Y. Yao and Q. Shen, J. Organomet. Chem., 2013, 732, 92 CrossRef CAS.
-
(a) D. Barr, A. J. Dawson and B. J. Wakefield, J. Chem. Soc., Chem. Commun., 1992, 204 RSC;
(b) D. B. Collum, A. J. McNeil and A. Ramirez, Angew. Chem., Int. Ed., 2007, 46, 3002 CrossRef CAS PubMed.
- H. C. Aspinall and M. R. Tillotson, Polyhedron, 1994, 13, 3229 CrossRef.
-
(a) M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS;
(b) M. Brookhart, M. L. H. Green and L.-L. Wong, Prog. Inorg. Chem., 1988, 36, 1 CrossRef CAS.
- M. Niemeyer, Z. Anorg. Allg. Chem., 2002, 628, 647 CrossRef CAS.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782 CrossRef CAS PubMed.
- W. Scherer, P. Sirsch, D. Shorokhov, M. Tafipolsky, G. S. McGrady and E. Gullo, Chem. – Eur. J., 2003, 9, 6057 CrossRef CAS PubMed.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212 CrossRef CAS.
- D. Schneider, T. Spallek, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Chem. Commun., 2014, 50, 14763 RSC.
- P. G. Williard and J. M. Salvino, J. Org. Chem., 1993, 58, 1 CrossRef CAS.
- W. Bauer and D. Seebach, Helv. Chim. Acta, 1984, 67, 1972 CrossRef CAS.
-
(a) N. D. R. Barnett, R. E. Mulvey, W. Clegg and P. A. O'Neil, J. Am. Chem. Soc., 1991, 113, 8187 CrossRef CAS;
(b) R. Neufeld, M. John and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 6994 CrossRef CAS PubMed;
(c) K. Bergander, R. He, N. Chandrakumar, O. Eppers and H. Günther, Tetrahedron, 1994, 50, 5861 CrossRef CAS;
(d) Y. J. Kim, M. P. Bernstein, A. S. G. Roth, F. E. Romesberg, P. G. Williard, D. J. Fuller, A. T. Harrison and D. B. Collum, J. Org. Chem., 1991, 56, 4435 CrossRef CAS.
-
F. Estler, PhD thesis, Technische Universität München, 2003.
- W. Scherer, D. J. Wolstenholme, V. Herz, G. Eickerling, A. Brück, P. Benndorf and P. W. Roesky, Angew. Chem., Int. Ed., 2010, 49, 2242 CrossRef CAS PubMed.
- D. M. Barnhart, D. L. Clark, J. C. Gordon, J. C. Huffman, J. G. Watkin and B. D. Zwick, J. Am. Chem. Soc., 1993, 115, 8461 CrossRef CAS.
- L. Lochmann and J. Trekoval, J. Organomet. Chem., 1979, 179, 123 CrossRef CAS.
- D. C. McKean, Chem. Soc. Rev., 1978, 7, 399 RSC.
- M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, Z. Li and A. V. Protchenko, Dalton Trans., 2010, 6780 RSC.
- D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Chem. – Eur. J., 2014, 20, 4426 CrossRef CAS PubMed.
-
(a) W.-K. Wong, L. Zhang, F. Xue and T. C. W. Mak, Polyhedron, 1997, 16, 2013 CrossRef CAS;
(b) W.-K. Wong, U. Zhang, F. Xue and T. C. W. Mak, Polyhedron, 1997, 16, 345 CrossRef CAS;
(c) Y. Yao, X. Lu, Q. Shen and K. Yu, J. Chem. Crystallogr., 2004, 34, 275 CrossRef CAS.
- D. Barr, W. Clegg, S. M. Hodgson, R. E. Mulvey, D. Reed, R. Snaith and D. S. Wright, J. Chem. Soc., Chem. Commun., 1988, 367 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, University of Wisconsin, Madison, WI, USA, 2013 Search PubMed.
- C. W. Spicer, C. F. Lovitt and G. S. Girolami, Organometallics, 2012, 31, 4894 CrossRef CAS.
-
(a) M. Karl, G. Seybert, W. Massa, S. Agarwal, A. Greiner and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 1405 CrossRef CAS;
(b) D. J. Berg and R. A. L. Gendron, Can. J. Chem., 2000, 78, 454 CrossRef CAS;
(c) P. B. Hitchcock, A. G. Hulkes and M. F. Lappert, Inorg. Chem., 2004, 43, 1031 CrossRef CAS PubMed;
(d) H. C. Aspinall, D. C. Bradley, M. B. Hursthouse, K. D. Sales, N. P. C. Walker and B. Hussain, J. Chem. Soc., Dalton Trans., 1989, 623 RSC.
- C. Meermann, K. W. Törnroos, W. Nerdal and R. Anwander, Angew. Chem., Int. Ed., 2007, 46, 6508 CrossRef CAS PubMed.
- Y. Chapurina, R. Guillot, D. Lyubov, A. Trifonov, J. Hannedouche and E. Schulz, Dalton Trans., 2013, 42, 507 RSC.
- W. J. Evans, J. L. Shreeve, R. N. R. Broomhall-Dillard and J. W. Ziller, J. Organomet. Chem., 1995, 501, 7 CrossRef CAS.
- W. J. Evans, M. A. Johnston, R. D. Clark, R. Anwander and J. W. Ziller, Polyhedron, 2001, 20, 2483 CrossRef CAS.
- C. Meermann, G. Gerstberger, M. Spiegler, K. W. Törnroos and R. Anwander, Eur. J. Inorg. Chem., 2008, 2014 CrossRef CAS.
- M. Karl, G. Seybert, W. Massa and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 375 CrossRef CAS.
- H.-X. Li, Q.-F. Xu, J.-X. Chen, M.-L. Cheng, Y. Zhang, W.-H. Zhang, J.-P. Lang and Q. Shen, J. Organomet. Chem., 2004, 689, 3438 CrossRef CAS.
- A. R. Crozier, A. M. Bienfait, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Chem. Commun., 2013, 49, 87 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
-
POV-Ray v. 3.6, Persistence of Vision Pty. Ltd, Williamstown, Victoria, Australia, 2004. http://www.povray.org/ Search PubMed.
- D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933 CAS.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS;
(c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
-
(a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC;
(b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- bwHPC and bwHPC-C5 (http://www.bwhpc-c5.de) funded by the Ministry of Science, Research and the Arts Baden-Württemberg (MWK) and the Germany Research Foundation (DFG).
Footnotes |
| † In memory of Donald C. Bradley, protagonist in rare-earth metal amide and alkoxide chemistry. |
| ‡ Electronic supplementary information (ESI) available: NMR, X-ray crystallographic and computational data. CCDC 1471665–1471684. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01568a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence