
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-mediated reactions of vinylferrocene: toward redox auxiliaries†
Alan A.
Wiles
a,
Xiaolu
Zhang
a,
Brian
Fitzpatrick
a,
De-Liang
Long
a,
Stuart A.
Macgregor
b and
Graeme
Cooke
*a
aGlasgow Centre for Physical Organic Chemistry, WestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: graeme.cooke@glasgow.ac.uk
bInstitute of Chemical Sciences, School of Engineering & Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
First published on 11th April 2016
Abstract
Chemical redox reactions have been exploited to transform unreactive vinylferrocene into a powerful dienophile for the Diels–Alder reaction and reactive substrate for thiol addition reactions upon conversion to its ferrocenium state. We have further investigated the ability of these reactions to facilitate redox-auxiliary-like reactivity by further hydrogenolyisis of the Diels–Alder adduct to the corresponding cyclopentane derivative.
Introduction
The fascinating structural and redox properties of ferrocene (Fc) have ensured that this metallocene has become a ubiquitous redox-active building block for a range of applications spanning materials science, catalysis and medicine.1 Arguably the most important property of this unit is its ability to reversibly undergo a one-electron chemical or electrochemical oxidation to transform the electron rich Fc to the electron deficient ferrocenium (Fc+) species. The propensity of this unit to undergo redox umpolung of this type has ensured that Fc and its derivatives have found a range of applications including a switchable motif for host–guest complexation,2 sol–gel3 and micelle4 actuation, biological recognition,5 recyclable catalysts6 and switchable dyes.7The Diels–Alder (DA) and conjugate addition (CA) reactions are two synthetically important reactions of alkenes, which generally require facilitation through a strongly electron withdrawing unit attached to the alkene. Therefore, reactions of this type are attractive vehicles for developing redox umpolung controlled transformations of appropriately functionalised redox active alkenes. Vinylferrocene (1) is a particularly attractive candidate for development of this type due to its commercial availability and its ability to undergo reversible oxidation, thereby transforming the electron donating Fc moiety to the more electron withdrawing Fc+ state. The alkene moiety of 1 is electron rich due to the electron releasing nature of Fc unit. Indeed, the Alfrey–Price e parameter (a semi-empirical measure of electron richness of the double bond) is −2.1 for vinylferrocene which predicts a reactivity worse than 4-(N,N-dimethylamino)styrene (−1.37), a derivative which is likely to be a poor dienophile for DA reactions and electrophile for CA.8 However, conversion to the Fc+ species should reverse the reactivity of 1 providing a powerful dienophile and reactive alkene for CA.
Here, we report the synthesis of compounds 2, 3 and 4 using redox umpolung-like reactions to transform 1 from an unreactive species for DA and thiol addition to a reactive ferrocenium-based reagent 1+ upon the addition of a chemical oxidant (Scheme 1). DA reactions of ferrocene containing dienophiles have been reported,9 however, they have been largely restricted to the reaction of β-acceptor-substituted vinylferrocenes10 or acrolylferrocenes11 with complementary dienes or by inverse electron demand reactions.12 In addition, compound 5 has previously been reported using a DA reaction of cyclopentadiene (CP) and the fulvalene13 form of α-carbocation derived from FcCH(OH)CH3. In other studies, cyclopentadienols have been isolated as the major products resulting from the oxygen-mediated decomposition of Fc+ species in the presence a triazoline-based trapping agent.14 In an interesting article, Gleixner et al. have shown that a single electron chemical oxidation of 1 leads to extensive decomposition of the starting material and produces a small amount of a Fc derivative that was tentatively assigned as cyclopentadiene-functionalized derivative 6, possibly through a Friedel Crafts-like process between a cationic Fc derivative (resulting from the oxidation of 1) and a vinylferrocene unit.15 The CA of nucleophiles to appropriately functionalised Fc derivatives have been reported.16 Nucleophilic thiol substitution reactions has been reported for ferrocenyl alcohols in the presence of the single electron oxidant cerium ammonium nitrate via a highly-delocalized ferrocenyl carbocation intermediate.17 Furthermore, radical-mediated thiol–ene reactions have recently been reported for 1, which afforded either the α- or β-hydrothiolated product depending upon the nature of the thiol and whether the radical reaction was thermally or photochemically initiated.18
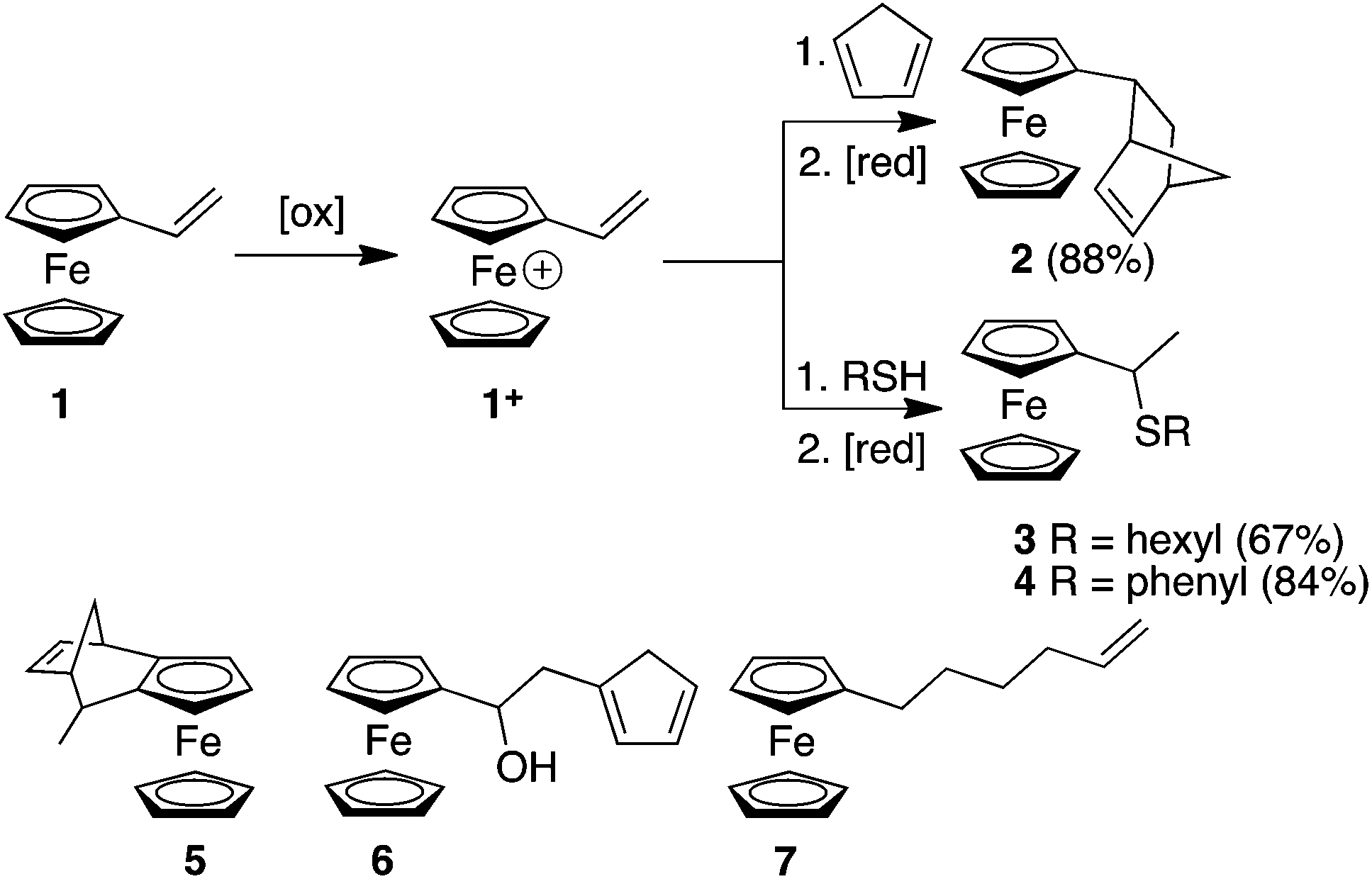 | ||
| Scheme 1 Redox umpolung-mediated reactions of vinylferrocene and the structures of derivatives 5, 6 and 7. | ||
Results and discussion
Initially we investigated the DA reactions of 1 and freshly-distilled CP. To bring about the conversion of Fc to Fc+ we used FeCl3 as the oxidant and ascorbic acid to reduce the Fc+ species back to the neutral Fc derivative. The addition of the oxidant to a solution of 1 immediately gave rise to a blue/green solution indicating the formation of 1+. Table S1 (ESI†) summarises some of the method development reactions undertaken. Of the reaction conditions investigated, the conditions described in entry 9 reliably gave the best yield of compound 2. It is noteworthy that excess CP should be added to a solution of 1 at −40 °C before the oxidant is added. Unfortunately, this DA adduct was oil-like which prevented X-ray structure determination. However, solution-based NMR experiments were consistent with the endo-structure predominating (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, endo:exo) (ESI†).
:1, endo:exo) (ESI†).
We next investigated the reaction of 1+ with hexanethiol and thiophenol. In both cases, the reactions progressed effectively, producing good yields of thiol-addition compounds 3 and 419 following conversion of the Fc+ state to the corresponding Fc derivative by treatment with ascorbic acid (ESI†). Interestingly, in all cases examined, NMR spectroscopy indicated that the sulfide unit is attached to the α-carbon, rather than the β-carbon of the alkene, which would be expected if the reactions proceeded via a CA-type reaction. The presence of the doublet for the hydrogen atoms of the methyl group is particularly diagnostic of α-addition. We have obtained the X-ray crystal structure of the thiol addition product 4 which corroborates our NMR data by clearly showing that it is the α-carbon that is functionalised (Fig. 1).20 Furthermore, the centrosymmetric P21/c space group indicates that the compound is obtained as a mixture of enantiomers.
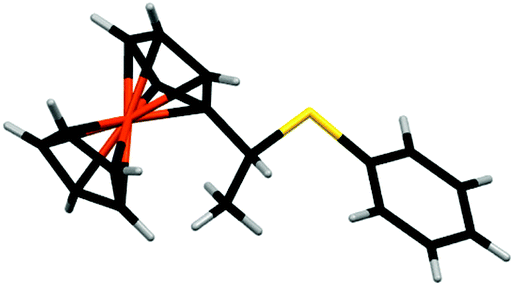 | ||
| Fig. 1 X-Ray crystal structure of 4. Colour scheme: Fe orange, S yellow, C black, H grey. | ||
In control experiments, we have investigated the DA and thiol-addition reactions of 1 without the addition of the FeCl3 as oxidant. In both cases, unreacted starting materials were recovered. Likewise, when reactions (including addition of the oxidant) were repeated with Fc derivative 7, where the vinyl group is no longer in conjugation with the cyclopentadienyl moiety of Fc, only starting materials were obtained. Thus, our experiments indicate the importance of the alkene being conjugated to the metallocene skeleton and argues against the FeCl3 merely activating the thiol component of the reaction for radical addition, or acting solely as a Lewis acid catalyst for the DA reaction.21
We have repeated the reaction described by Watts and co-workers13 to synthesise compound 5 and found in DCM at room temperature and in THF at 55 °C (to mimic our conditions) that compound 5 was produced, and that there was no evidence for the formation of compound 2. Thus these experiments argue against the possibility of a fulvalene-like structure being implicated in the DA reaction presented in this article (Fig. 2).22 Interestingly, compound 5 was observed as a minor side product using our conditions, only when the reaction was performed at room temperature. We have also investigated the reaction of the carbocation derived from FcCH(OH)CH3 with thiophenol which provided compound 4 in 65% yield.
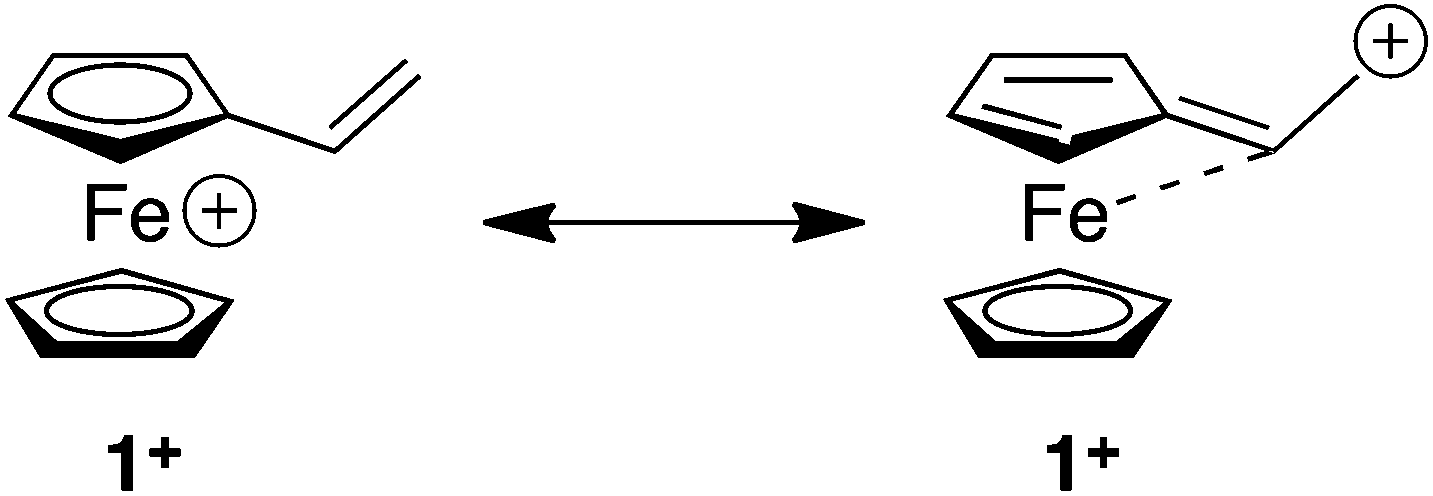 | ||
| Fig. 2 Possible fulvalene-like structure of 1+. | ||
In order to shed light on the enhanced reactivity of 1+, we have employed density functional theory (DFT) calculations23 to probe the molecular and electronic structure of this species, alongside that of 1, the neutral vinylferrocene precursor (see Fig. 3). The optimised geometries indicate little difference in the structure of the vinylcyclopentadienyl moiety within 1 and 1+. Moreover, no evidence for a fulvalene-like structure is observed with no significant bond alternation apparent in the substituted cyclopentadienyl moiety. Instead the major structural changes occur around the metal centre, with elongation of the Fe–C2a/C2b bonds and, in particular, the Fe–C1 bond (see Fig. 3 for details and the labelling scheme employed). Similar changes are computed in the unsubstituted cyclopentadienyl ring meaning that θ, the angle between the best fit planes through the two C5 rings, increases from 0.2° in 1 (i.e. essentially co-planar) to 8.8° in 1+.
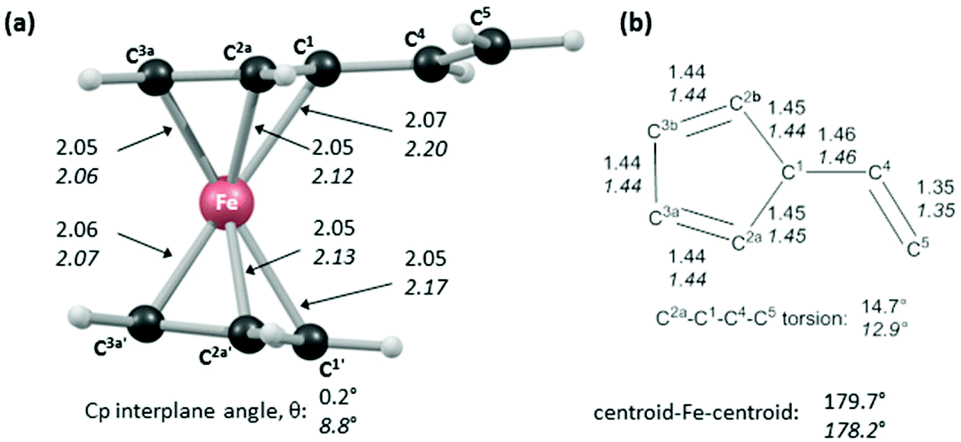 | ||
| Fig. 3 BP86-optimised structural data (Å, °) for 1 and 1+; (a) computed structure of 1+ with selected data for 1+ in italics and for 1 in plain text; average values for near-symmetry equivalent Fe–C distances are provided; (b) structure of the vinylcyclopentadienyl moiety; θ is the angle between the best fit planes through the two C5 rings. | ||
These computed structures suggest the 1e oxidation of 1 is primarily localized on iron and involves a formal Fe2+/Fe3+ couple. This is supported by a natural population analysis which indicates a large spin density of 1.08 on the Fe centre in 1+ (the next largest spin density is 0.04 on the terminal vinyl carbon, C5). The computed natural charge on the Fe centre also drops from −0.30 in 1 to +0.69 in 1+. Inspection of the HOMO of 1 reveals it is dominated by the Fe dx2−y2 orbital with some bonding character with the C1/C1′ positions (see Fig. 4). Depopulation of this orbital is therefore consistent with the computed structural changes that occur upon oxidation of 1.
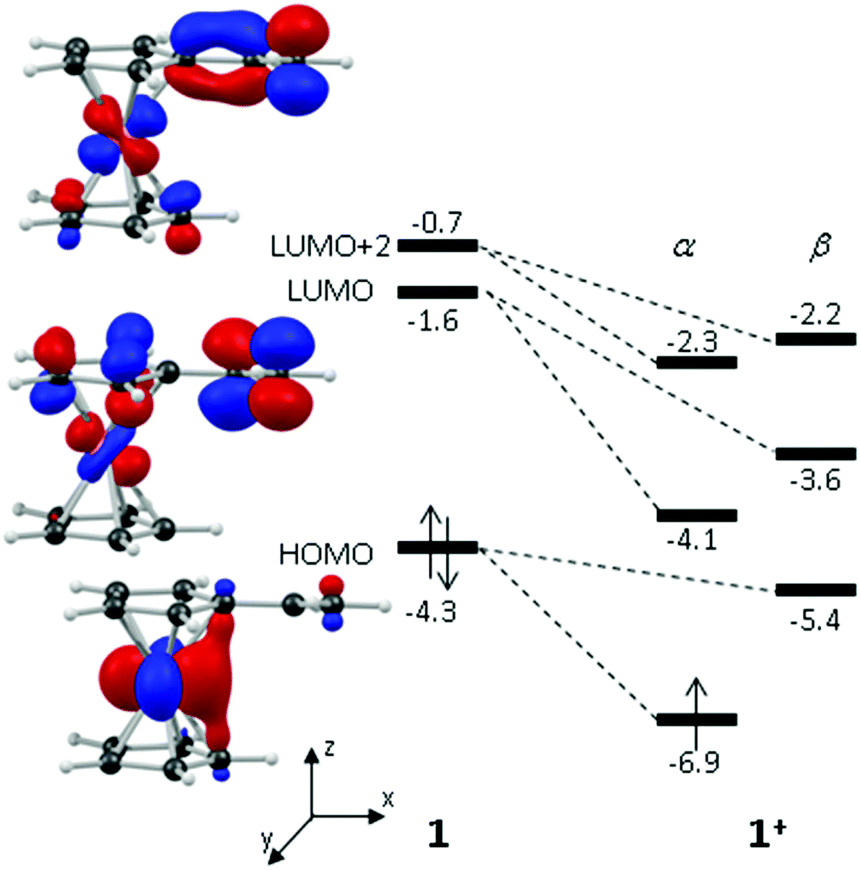 | ||
| Fig. 4 Key frontier molecular orbitals of 1 with computed energies (eV) for 1 and for the equivalent orbitals of 1+; energies computed with the BP86 functional in THF solvent (PCM approach); orbitals plotted with a maximum contour value of 0.06 au. | ||
Of the low-lying unoccupied orbitals of 1 both the LUMO and LUMO+2 have significant π* character on the vinyl substituent and hence will be important in DA reactivity. Comparison with the equivalent orbitals of 1+ revealed only minor differences in the make-up of these orbitals (see ESI†), consistent with the minimal geometric changes computed within the vinylcyclopentadienyl ligand upon oxidation. However, oxidation does result in a significant stabilisation of these orbitals of between 1.5 to 2.5 eV, and this would be consistent with the enhanced DA reactivity observed in 1+. Accordingly, the computed barriers for the reaction with cyclopentadiene to give the endo DA product reduces from 20.3 kcal mol−1 with 1 to 10.3 kcal mol−1 for 1+ while both reactions are exergonic, by 8.9 kcal mol−1 and 11.3 kcal mol−1, respectively. Fig. 5 gives details of the computed transition state structures and indicates that TS1+ shows the greater asynchronicity, with C4⋯C6 being almost 1 Å longer than C5⋯C7 in TS1+, whereas in TS1 this difference is only 0.5 Å. Similar transition states were located for the exo addition, but these were found to be less accessible by ca. 0.5–1 kcal mol−1 for 1+ which is consistent with the observed endo:exo preference.
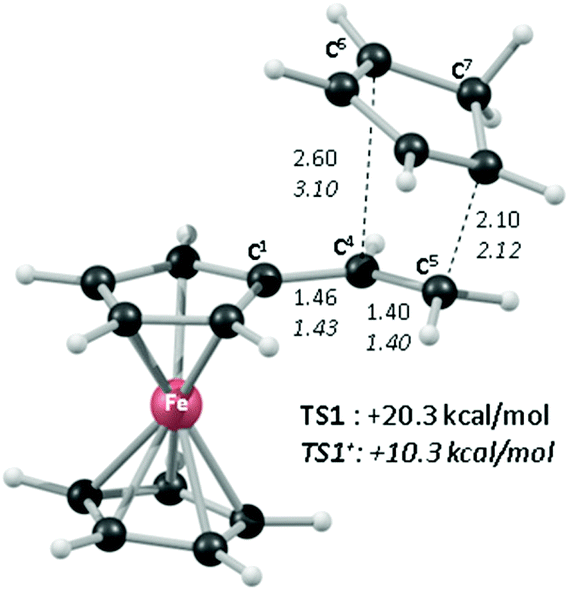 | ||
| Fig. 5 BP86-optimised transition states for the DA reactions of 1 and 1+ with C5H6 (endo selectivity); selected distances are in Å and free energies (kcal mol−1) are at the BP86-D3(THF) level and are quoted relative to the separated reactants set to 0.0 kcal mol−1 in each case. | ||
We next turned our attention to whether we could extend our methodology to develop Fc-based redox auxiliaries. In particular, whether we could couple the redox-mediated umpolung reactions with hydrogenolysis of the Fc moiety to remove the iron-containing “auxiliary” moiety to liberate the corresponding functionalised cyclopentane derivative (Scheme 2).24 To investigate the viability of this chemistry, in line with previously reported protocols for the hydrogenolyis of Fc, the alkene unit of the norbornene moiety was first reduced using Pd/C and H2 to afford compound 8 in quantitative yield. Further hydrogenolysis of 8 in the presence of trifluoroacetic acid (TFA) furnished the corresponding alkane 9 in near-quantitative yield (by GC-MS, ESI†).25
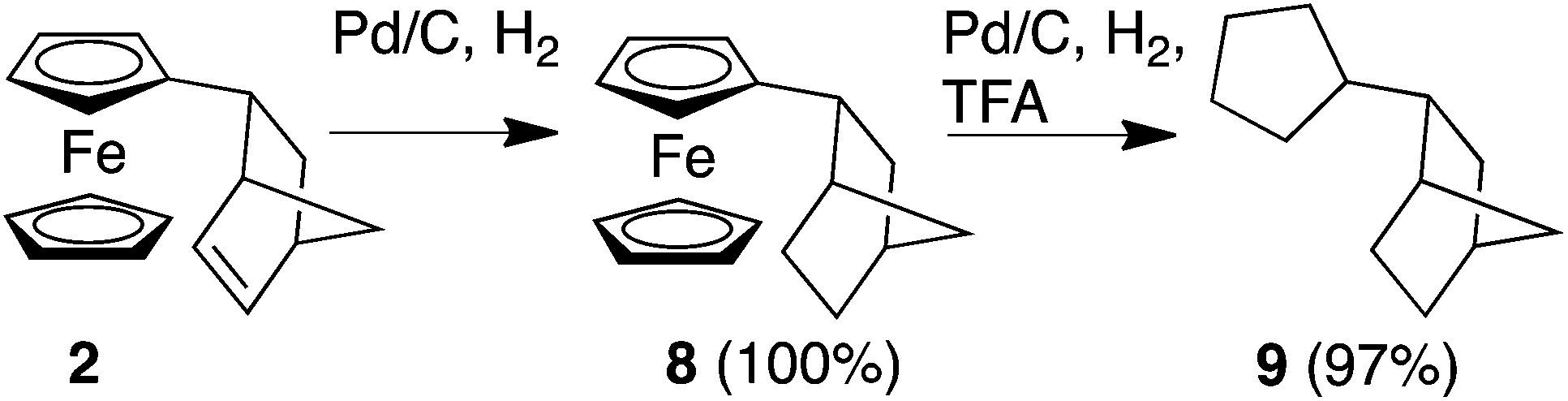 | ||
| Scheme 2 Conversion of 2 to its corresponding cyclopentane derivative 9. | ||
In conclusion, we have developed redox-umpolung chemistry to undertake DA and thiol-addition reactions of oxidised vinylferrocene. Methodologies have been developed to furnish functionalised Fc derivatives in good yield, which should open up new methods for the chemical and electrochemical functionalisation of surfaces and synthetic and biological macromolecules. Moreover, the redox-umpolung chemistry combined with the reductive cleavage of the Fc moiety described here, provides synthetic protocols to furnish functionalised cyclopentanes, which may find application in the synthesis of complex natural products such as prostaglandins.26
Experimental section
General
Dry solvents were obtained using an Innovative Technology Inc. Pure Solv 400-5-MD solvent purification system (activated alumina columns). Reactions were monitored using thin layer chromatography using SiO2-coated aluminium plates. Flash column chromatography was performed using Silica gel 40–63 μm 60 Å. Melting points were recorded on a SMP-10 Stuart Scientific melting point apparatus and are uncorrected. NMR experiments were performed on either Bruker Avance 500, Bruker Avance 400 spectrometer. All, spectra and were processed using the Mestrelab research iNMR© package (for MacOS X®).GC-MS was performed on a Shimadzu GC-2010 gas chromatograph coupled to a Shimadzu GCMS-QP2010S gas chromatograph-mass spectrometer, fitted with a 30 m Zebron ZB-5MS column. Column oven temperature: 50.0 °C; injection temperature: 250 °C; injection mode: split; split ratio: 20; pressure: 65.2 kPa; column flow: 1.16 mL min−1. GC program: 50 °C hold 3.00 min, ramp rate 10, max temperature 250 °C hold 2 min, total time 25 min. MS program: start time: 3 min; end time: 25 min, scan speed: 666; start m/z: 40.00 end m/z: 350.00.
:1, endo:exo); endo-6-ferrocene-bicyclo[2.2.1]hept-2-ene. δH (500 MHz, CDCl3) 1.13 (1H, ddd, J 11.6, 4.5, 2.6, CH), 1.39 (1H, br. d, J 8.1, CH), 1.45 (1H, m, J 8.1, CH), 2.14 (1H, ddd, J 11.6, 9.1, 3.7, CH), 2.77 (1H, br. m, CH), 2.86 (1H, br. m, CH), 3.07 (1H, ddd, J 9.1, 4.5, 4.0, CH), 3.81 (1H, m, Cp), 3.98 (1H, m, Cp), 4.01 (1H, m, Cp), 4.08 (1H, m, Cp), 4.1 (5H, s, Cp′), 5.74 (1H, dd, J 5.7, 3.0, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.19 (1H, dd, J 5.7, 2.9, CH); δC (125 MHz, CDCl3) 34.0 (CH2), 38.7 (CH), 43.1 (CH), 49.3 (CH), 50.5 (CH2), 67.0 (Cp), 67.1 (Cp), 67.2 (Cp), 68.4 (Cp), 92.5 (Cp), 68.7 (Cp), 133.5 (CH), 137.0 (CH); m/z (CI+) 278.0757 [M]+ (C17H1856Fe requires 278.0758).
CH), 6.19 (1H, dd, J 5.7, 2.9, CH); δC (125 MHz, CDCl3) 34.0 (CH2), 38.7 (CH), 43.1 (CH), 49.3 (CH), 50.5 (CH2), 67.0 (Cp), 67.1 (Cp), 67.2 (Cp), 68.4 (Cp), 92.5 (Cp), 68.7 (Cp), 133.5 (CH), 137.0 (CH); m/z (CI+) 278.0757 [M]+ (C17H1856Fe requires 278.0758).
General procedure for thiol additions
Compound 1 (0.47 mmol) was dissolved in dry THF (5 mL) and cooled to −40 °C. FeCl3 (0.52 mmol) was then added to the reaction mixture, and after 5 minutes the thiol (0.52 mmol) was also added. The reaction mixture was allowed to warm to room temperature and was left to stir at room temperature overnight. After this time a mixture of L-ascorbic acid (127 mg, 0.72 mmol) in water (10 mL) was added to the reaction mixture. The crude product was then extracted with DCM (3 × 20 mL) and washed with brine (30 mL). The combined organic extracts were dried over MgSO4, filtered and the solvent was removed under reduced pressure.:petroleum ether; 5:95) to yield the title compound as a brown oil (53 mg, 67%); δH (500 MHz, CDCl3) 1.88 (3H, t, J 7.0, CH3), 1.25–1.37 (6H, m, CH2), 1.68–1.54 (2H, m, CH2), 1.65 (3H, d, J 6.9, CH3), 2.44 (2H, td, J 7.4, 2.1, CH2), 3.72 (1H, q, J 6.9, CH), 4.11 (2H, t, J 1.9, Cp), 4.14 (6H, s, Cp and Cp′), 4.17 (1H, q, J 1.7, Cp); δC (125 MHz, CDCl3) 14.1 (CH3), 21.7 (CH3), 22.7 (CH2), 28.9 (CH2), 29.9 (CH2), 31.2 (CH2), 31.6 (CH2), 39.2 (CH), 66.0 (Cp), 67.6 (Cp), 67.7 (Cp), 68.0 (Cp), 68.8 (Cp), 92.2 (Cp); m/z (EI+) 330.1108 [M]+ (C18H2656FeS requires 330.1105).
:petroleum ether; 1:10) to yield the title compound as an orange solid (128 mg, 84%): m.p. 89–90 °C; δH (500 MHz, CDCl3) 1.56 (3H, d, J = 6.9, CH3), 3.95–3.96 (1H, m, CH), 4.01–4.03 (1H, m, Cp), 4.04–4.06 (1H, m, Cp), 4.08 (7H, s, Cp′ and Cp), 7.16–7.21 (3H, m, Ph), 7.27–7.29 (2H, m, Ph); δC (125 MHz, CDCl3) 21.3 (CH3), 43.7 (CH), 66.1 (Cp), 67.6 (Cp), 67.7 (Cp), 67.9 (Cp), 68.6 (Cp), 90.8 (Cp), 127.2 (Ph), 128.7 (2 × Ph), 133.1 (2 × Ph), 135.3 (Ph); m/z (ESI+) 345.0358 [M + Na]+ (C18H1856FeNaS requires 345.0371).
CH2), 5.02 (1H, dd, J 17.1, 2.0, CCH2), 5.82 (1H, ddt, J 17.1, 10.2, 6.7, CCH); δC (125 MHz, CDCl3) 29.0 (CH2), 29.6 (CH2), 30.7 (CH2), 33.8 (CH2), 33.8 (CH2), 67.2 (Cp), 68.2 (Cp), 68.6 (Cp), 89.5 (Cp), 114.5 (CCH2), 139.1 (CCH); m/z (ESI+) 268.0907 [M]+ (C16H2056Fe requires 268.0909).
:1, R:S). δH (500 MHz, CDCl3) 1.11–1.26 (3H, m, 3 × CH), 1.28–1.32 (1H, m, CH), 1.36 (1H, m, CH), 1.5 (2H, m, 2 × CH), 1.96 (1H, dddd, J 15.0, 11.5, 4.0, 2.0, CH), 2.04 (1H, br. t, CH), 2.24 (1H, br. t, CH), 2.93 (1H, dddd, J 11.5, 5.6, 4.0, 1.8, CH), 4.02–4.04 (1H, m, Cp), 4.07–4.09 (3H, m, Cp), 4.10 (5H, s, Cp′); δC (125 MHz, CDCl3) 23.4 (CH2), 30.2 (CH2), 36.0 (CH2), 37.5 (CH), 40.7 (CH2), 41.4 (CH), 43.7 (CH), 66.8 (Cp), 67.2 (Cp), 67.7 (Cp), 68.4 (Cp), 69.2 (Cp), 90.9 (Cp); m/z (EI+) 280.0916 [M]+ (C17H2056Fe requires 280.0915).
Acknowledgements
We thank Dr Andrew Macdonell for undertaking preliminary experiments. We thank the EPSRC (EP/E036244/1) for funding this research.Notes and references
- (a) Ferrocenes: Compounds, Properties and Applications, ed. E. S. Phillips, Nova Science Publishers Inc., 2011 Search PubMed; (b) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley and Sons, 2008 Search PubMed; (c) Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed.
- For representative examples see: (a) V. Balzani, J. Becher, A. Credi, M. B. Nielsen, F. M. Raymo, J. F. Stoddart, A. M. Talarico and M. Venturi, J. Org. Chem., 2000, 65, 1947–1956 CrossRef CAS PubMed; (b) D. Sobransingh and A. E. Kaifer, Org. Lett., 2006, 8, 3247–3250 CrossRef CAS PubMed; (c) L. Yang, A. Gomez-Casado, J. F. Young, H. D. Nguyen, J. Cabanas-Danés, J. Huskens, L. Brunsveld and P. Jonkheijm, J. Am. Chem. Soc., 2012, 134, 19199–19206 CrossRef CAS PubMed; (d) J. Guo, C. Yuan, M. Guo, L. Wang and F. Yan, Chem. Sci., 2014, 5, 3261–3266 RSC; (e) Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed; (f) Q. Yan, A. Feng, H. Zhang, Y. Yin and J. Yuan, Polym. Chem., 2013, 4, 1216–1220 RSC; (g) T. Matsue, D. H. Evans, T. Osa and N. Kobayashi, J. Am. Chem. Soc., 1985, 107, 3411–3417 CrossRef CAS.
- For representative examples see: (a) J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508–2511 CrossRef CAS; (b) M. A. Hempenius, C. Cirmi, F. L. Savio, J. Song and G. J. Vancso, Macromol. Rapid Commun., 2010, 32, 772–783 CrossRef PubMed; (c) M. Nakahata, Y. Takashima, Y. Harada and A. Harada, Nat. Commun., 2011, 2, 511, DOI:10.1038/ncomms1521; (d) C. Yuan, J. Guo, M. Tan, M. Guo, L. Qiu and F. Yan, ACS Macro Lett., 2014, 3, 271–275 CrossRef CAS.
- T. Saji, K. Hoshino and S. Aoyagui, J. Am. Chem. Soc., 1985, 107, 6865–6868 CrossRef CAS.
- For representative examples see: (a) M. E. Hays, C. M. Jewell, D. M. Lynn and N. L. Abbott, Langmuir, 2007, 23, 5609–5614 CrossRef CAS PubMed; (b) N. L. Abbott, C. M. Jewell, M. E. Hays, Y. Kondo and D. M. Lynn, J. Am. Chem. Soc., 2005, 127, 11567–11577 CrossRef PubMed; (c) B. S. Aytar, J. P. E. Muller, S. Golan, Y. Kondo, Y. Talmon, N. L. Abbott and D. M. Lynn, J. Colloid Interface Sci., 2012, 287, 56–64 CrossRef PubMed; (d) B. S. Aytar, J. P. E. Muller, Y. Kondo, Y. Talmon, N. L. Abbott and D. M. Lynn, J. Am. Chem. Soc., 2013, 135, 9111–9120 CrossRef CAS PubMed.
- For representative examples see: (a) C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed; (b) V. C. Gibson, N. J. Long, P. J. Oxford, A. J. P. White and D. J. Williams, Organometallics, 2006, 25, 1932–1939 CrossRef CAS; (c) M. Sűβner and H. Plenio, Angew. Chem., Int. Ed., 2005, 44, 6885–6888 CrossRef PubMed; (d) C. A. Fleckenstein and H. Plenio, Adv. Synth. Catal., 2006, 348, 1058–1062 CrossRef CAS; (e) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed; (f) P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311–314 CrossRef CAS PubMed; (g) Q. Zhang, X. Cui, L. Zhang, S. Luo, H. Wang and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 5210–5213 CrossRef CAS PubMed.
- For representative examples see: (a) L. Asselberghs, K. Clays, A. Persoons, A. M. McDonagh, M. D. Ward and J. A. McCleverty, Chem. Phys. Lett., 2003, 368, 408–411 CrossRef; (b) M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jefferey, J. A. McCleverty, M. D. Ward, I. Asselberghs, K. Clays and A. Persoons, Chem. Commun., 2001, 49–50 RSC; (c) S. Nagashima, M. Murata and H. Nishishara, Angew. Chem., Int. Ed., 2006, 45, 4298–4301 CrossRef CAS PubMed; (d) W.-Y. Wang, N.-N. Ma, S.-L. Sun and Y.-Q. Qiu, Organometallics, 2014, 33, 3341–3352 CrossRef CAS.
- C. U. Pittman, T. V. Jayaraman, R. D. Priester, S. Spencer, M. D. Rausch and D. Macomber, Macromolecules, 1981, 14, 237–241 CrossRef CAS.
- G.-N. Li, G.-Y. Chen, Z. G. Niu, B.-X. Lei and H. J. Feng, J. Heterocycl. Chem., 2014, 51, E367–E371 CrossRef CAS.
- M. Prokešová, S. Toma, A. Kennedy and G. R. Knox, Tetrahedron, 1998, 54, 9175–9186 CrossRef.
- M. Prokešová, E. Solčániová, S. Toma, K. W. Muir, A. A. Torabi and G. R. Knox, J. Org. Chem., 1996, 61, 3392–3397 CrossRef.
- F. A. Khan and S. K. Upadhyay, Synthesis, 2009, 2773–2777 CrossRef CAS.
- T. D. Turbitt and W. E. Watts, J. Chem. Soc. D, 1971, 631 RSC.
- J. Lorans, F. Pierre, L. Toupet and C. Moinet, Chem. Commun., 1997, 1279–1280 RSC.
- R. M. Gleixner, K. M. Joly, M. Tremayne, B. M. Kariuki, L. Male, D. M. Coe and L. R. Cox, Chem. – Eur. J., 2010, 16, 5769–5777 CrossRef CAS PubMed.
- H. J. Al-Najjar, A. Barakat, A. M. Al-Majid, Y. N. Mabkhot, M. Weber, H. A. Ghabbour and H.-K. Fun, Molecules, 2014, 19, 1150–1162 CrossRef PubMed.
- R. Jiang, Y. Zhang, Y.-C. Shen, X. Zhu, X.-P. Xu and S. J. Ji, Tetrahedron, 2010, 66, 4073–4078 CrossRef CAS.
- (a) I. Martínez-Montero, S. Bruńa, A. M. González-Vadillo and I. Cuadrado, Macromolecules, 2014, 47, 1301–1315 CrossRef; (b) A. Enríquez, A. M. González-Vadillo, I. Martínez-Montero, S. Bruńa, L. Leemans and I. Cuadrado, Organometallics, 2014, 33, 7307–7317 CrossRef.
- B. Misterkiewicz, C. Kajdas, M. Dominiak, J. Dabrowski and J. Wasilewski, Chem. Ind., 1981, 433–434 CAS.
- Crystal data for 4: C18H18FeS, Mr = 322.23, monoclinic system, space group P21/c, a = 19.1342(5), b = 7.59440(10), c = 10.3523(2) Å, β = 92.834(2), V = 1502.48(5) Å3. Z = 4, ρcalcd = 1.425 g cm−3; Mo Kα radiation, λ = 0.71073 Å, μ = 1.129 mm−1, T = 150 K. 23151 data (2958 unique, Rint = 0.0233, θ < 26°) were collected on an Oxford Diffraction Gemini A Ultra CCD diffractometer and were corrected for absorption (transmission 0.729–0.953). The structure was solved by direct methods and refined by full-matrix least-squares on F2 to give wR2 = 0.058, R1 = 0.025, S = 1.047 for 182 parameters. Residual electron density extrema were 0.28 and −0.35 e Å−3. CCDC number is 1439981.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- (a) G. Gokel, P. Hoffman, H. Klusacek, D. Marquarding, E. Ruch and I. Ugi, Angew. Chem., Int. Ed. Engl., 1970, 9, 64 CrossRef CAS; (b) T. Ireland, J. J. A. Perea and P. Knochel, Angew. Chem., Int. Ed., 1999, 38, 1457–1460 CrossRef CAS; (c) R. Gleiter, C. Bleiholder and F. Rominger, Organometallics, 2007, 26, 4850–4859 CrossRef CAS; (d) K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H.-J. Krüger, B. Sarkar and W. Kaim, Organometallics, 2009, 28, 4196–4209 CrossRef CAS.
- DFT calculations were run with the Gaussian 03 and 09 suite of programs. Optimisations employed the BP86 functional in combination with an SDD pseudopotential and basis set on Fe and 6-31g** basis sets on C and H. See ESI† for full details and references.
- A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894–908 CrossRef.
- (a) V. I. Sokolov, L. L. Troitskaya, N. S. Khrushcheva and O. A. Reutov, J. Organomet. Chem., 1989, 378, 227–233 CrossRef CAS; (b) F. V. Meurs, F. W. Metselaar, A. J. A. Post, J. A. A. M. V. Rossum, A. M. V. Wijk and H. V. Bekkum, J. Organomet. Chem., 1975, 84, C22–C24 CrossRef.
- S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286–3337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, X-ray crystallography and modelling data. CCDC 1439981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00875e. The underlying research data for this paper is available in accordance with EPSRC open data policy from 10.5525/gla.researchdata.297 |
| This journal is © The Royal Society of Chemistry 2016 |