Progress in redox flow batteries, remaining challenges and their applications in energy storage
Puiki
Leung
a,
Xiaohong
Li
*a,
Carlos
Ponce de León
*a,
Leonard
Berlouis
b,
C. T. John
Low
a and
Frank C.
Walsh
ab
aElectrochemical Engineering Laboratory, Energy Technology Research Group, Faculty of Engineering and the Environment, University of Southampton, Highfield, Southampton, SO17 1BJ, United Kingdom. E-mail: Xh.Li@soton.ac.uk; capla@soton.ac.uk
bWestCHEM, Department of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, United Kingdom
First published on 7th September 2012
Abstract
Redox flow batteries, which have been developed over the last 40 years, are used to store energy on the medium to large scale, particularly in applications such as load levelling, power quality control and facilitating renewable energy deployment. Various electrode materials and cell chemistries have been proposed; some of the successful systems have been demonstrated on a large-scale in the range of 10 kW–10 MW. Enhanced performance is attributable to the improvements in electrodes, separator materials and an increasing awareness of cell design. This comprehensive review provides a summary of the overall development of redox flow battery technology, including proposed chemistries, cell components and recent applications. Remaining challenges and directions for further research are highlighted.
 Puiki Leung | Puiki Leung received a BEng in Mechanical Engineering and a PhD in Electrochemical Engineering for studies on the development of a zinc-cerium redox flow battery from the University of Southampton, UK. Currently, he is a postdoctoral research fellow at the Hong Kong University of Science & Technology where he is working on the vanadium redox flow battery and collaborating with industry on bipolar plates and catalyst supports for fuel cell and battery applications. His research interests include functional materials for energy conversion and storage applications. He has published 6 peer-reviewed journal papers. |
 Xiaohong Li | Xiaohong Li received a BSc in Organic Chemistry (Lanzhou University, China), an MSc in Analytical Chemistry (Lanzhou University) and a PhD in Physical Chemistry (Lanzhou University & Peking University). Following experience as an engineer/project manager for PetroChina Ltd, since 2003, she has worked in the University of Southampton and is now a Senior Consulting Engineer in the Research Institute for Industry in the Faculty of Engineering & the Environment. Her research interests include flow batteries, water electrolysers, and nanostructured materials (for electrocatalysis, optoelectronics, thermoelectric devices and sensors). |
 Carlos Ponce de León | Carlos Ponce de León is a Senior Lecturer in the Faculty of Engineering and the Environment at the University of Southampton, UK. He holds a BSc and an MSc in chemistry from the Autonomous Metropolitan University, México, a PhD in electrochemistry and electrochemical engineering from the University of Southampton (1994) and an MBA from Bath University (2004). His research interests include electrochemical techniques, metal ion removal, the characterization of novel electrode materials, electrochemical strategies for pollution control and redox flow cells for energy conversion. |
 Leonard Berlouis | Leonard Berlouis is a Reader in Physical Chemistry in the Department of Pure and Applied Chemistry at the University of Strathclyde, Scotland. He obtained his PhD from the University of Southampton in 1982 for studies on ac impedance characterisation of flow-through porous electrodes. He moved to the University of Strathclyde in 1991 and has extended early work involving optical techniques for the characterization of solid/electrolyte interfaces. He has been an active researcher in redox flow battery systems for the last five years working in close collaboration with Plurion Ltd., Glenrothes, (Scotland) as well as colleagues from the University of Southampton. |
 C. T. John Low | Chee Tong John Low was born in Kuala Lumpur, Malaysia, 1980. He studied at Taylor's College, Malaysia (1999) then obtained a BEng degree in Chemical & Bioprocess Engineering including a one-year placement in industry, from the University of Bath, UK (2003) and a PhD in Electrochemical Engineering (2007) working with Prof. Frank Walsh on the controlled electrodeposition of metal alloys at rotating cylinder electrodes. His research interests are in nanostructured electrode materials for energy systems, electrochemical engineering for energy materials and reactor design together with the engineering scale-up of nanostructured electrodes. |
 Frank C. Walsh | Frank Walsh is a Professor in Electrochemical Engineering and has served as the Deputy Head (Enterprise) and Director of the Research Institute for Industry in the Faculty of Engineering & the Environment at the University of Southampton, UK. His research output spans the areas of energy conversion, electroactive nanomaterials, coating technology, electrochemical monitoring and sensors, corrosion, surface finishing and electrochemical process engineering. He was awarded the Breyer Medal of the Royal Australian Chemical Institute (2000) for international contributions to electrochemical engineering and energy and was awarded the NACE Fellow honour (2010) for international, lifelong contributions to electrochemical corrosion science and engineering. |
1. Introduction
Global energy demand is climbing rapidly due to population growth and continuing industrialization. At the same time, global CO2 emissions must be drastically reduced to restrict global warming. Renewable energy is expected to provide a central solution to our need for a sustainable fuel. The USA and many other countries have announced plans in the use of clean energy, which are accelerating the development and application of renewable energies.1–5 Major challenges presented by renewable energies, such as fluctuations in output, unavailability (e.g. sunlight in the night) and unpredictability (e.g. wind power), limit their popularity. As a solution to these problems, energy storage technologies are attracting attention.Electrical energy storage technologies can be classified into three categories, namely flywheels and supercapacitors, geological storage technologies and battery storage technologies. Flywheels and supercapacitors are low-energy, high-power storage systems used for power management (e.g., frequency regulation). These technologies are not considered to have wide applications in transmission due to materials limitations. Geological storage technologies include pumped hydro and compressed air energy storage. Both technologies are large-scale, high-energy, high-power systems, which are capable of providing significant reserves. However, they require special terrain and have large capital investment and maintenance costs. Battery storage technologies include lead-acid, lithium-ion, sodium-based batteries and redox flow batteries (RFBs). These technologies provide a potential solution to large-scale energy storage and act as a buffer between intermittent electricity production and customer requirements for the instant delivery of electricity power. For large-scale energy storage in the range of 10 kW–10 MW, RFBs have cost, mobility, flexibility, depth of discharge, rapid response, and safety advantages over lithium ion and sodium sulphur batteries.6
RFBs are potentially an efficient energy storage technology which converts and stores electrical energy into chemical energy and releases it in a controlled fashion when required, providing an alternative solution to the problems of balancing power generation and consumption, load levelling and facilitating renewable energy deployment. A typical individual RFB system (see Fig. 1a) consists of two external reservoirs which store soluble electroactive electrolytes, two electrodes, a membrane separator and a flow circulation system. The battery generates reduction and oxidation (redox) species between two active materials to store and release energy, respectively. Increasing the concentration of the electroactive species and/or the volume of the electrolytes increases the energy storage capacity of a RFB which is a great advantage over other energy storage battery systems.
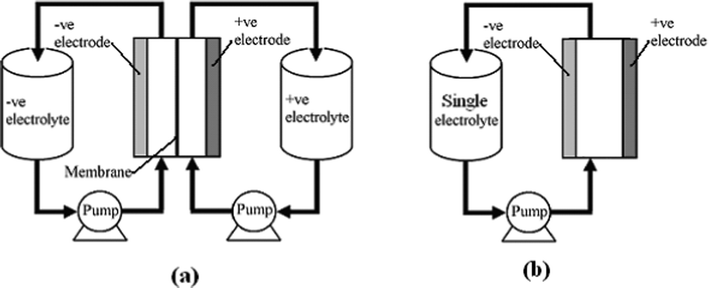 | ||
| Fig. 1 The principle of (a) a divided and (b) an undivided (membraneless) redox flow battery. | ||
The last 40 years have witnessed many developments in the field of RFBs, as evidenced by the increasing number of publications and active collaboration between research and commercial partners. Several reviews have been published, reflecting the growth in the field of RFBs. The earlier reviews by Bartolozzi7 and Ponce de León et al.8 discussed historical and fundamental aspects of RFBs, respectively. The reviews by Yang et al.9 and Skyllas-Kazacos et al.10 critically discussed technical, economic and environmental aspects of RFBs and assessed their commercial penetration in the grid. Zhang et al.11 reviewed the progress and challenges in ion exchange membranes for the all-vanadium RFB. A review in terms of financial and policy aspects of the all-vanadium RFB together with mathematical modelling of various RFBs was given by Walsh et al.12 More recently, Weber et al.13 considered the transport and kinetic phenomena in an RFB and further reviewed electrode/cell modelling and design.
This review provides a comprehensive summary of the development of RFB technologies covering advances of this area, including various proposed chemistries, materials for cell components, design considerations and modelling, and recent applications in load levelling, power quality control, integration with renewable energy sources, and electric vehicles. We also discuss remaining challenges and directions for future development.
2. Properties of redox flow batteries
In an RFB, soluble redox couples in the electrolyte(s) are used to store and release energy during the battery charge and discharge, respectively. Fig. 1a illustrates that, in a typical redox flow cell, positive and negative electrodes are separated by an ion exchange membrane. The membrane allows ion transport and helps prevent mixing of the two half-cell electrolytes, which are stored in separate tanks and circulated through the battery using pumps. Fig. 1b shows a membrane-free (undivided) system which is currently at an early stage of development.14–17Practical applications tend to require high currents and voltages. To meet this, a number of unit cells can be stacked in electrical series to increase the voltage and the stacks can be electrically connected in parallel to yield high currents. In order to reduce weight, volume and cost, bipolar electrodes are often used. In most reactor designs the cell stacks are fed by distributing the electrolyte to each cell through a manifold. An example of a redox bipolar electrode flow battery stack having four unit cells is shown in Fig. 2.8
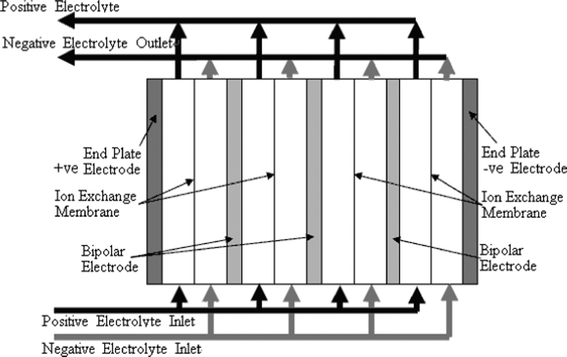 | ||
| Fig. 2 A stack design for a four cell stack with bipolar connection of electrodes.8 | ||
Table 1 highlights the differences between static batteries, RFBs and fuel cells as energy conversion devices.18–20 Flow batteries store energy in the form of reduced and oxidized electroactive species in the electrolyte while conventional static batteries store energy within the electrode structure. The electrochemical reactions involved in a flow battery should be reversible during charge and discharge. Whilst in a fuel cell the reactants, stored externally to the cell, are consumed to produce electricity, the electrolytes (reactants) of the RFB are converted and always remain within the system.
| Electrochemical device | Site of reactants/products | Electrolyte conditions | Separator |
|---|---|---|---|
| Static battery | Active electrode material | Static and held within cell | Microporous polymer separator |
| Redox flow battery | Aqueous electrolytes in reservoirs | Electrolyte recirculates through the cell | Ion-exchange membrane (cationic/anionic) or not necessary |
| Fuel cell | Gaseous or liquid fuel (anode) plus air (cathode) | Solid polymer or ceramic usually acts as solid electrolyte within cell | Ion-exchange membrane polymer or ceramic |
In comparison to conventional lead-acid batteries, which have been widely used for transportation, RFBs have cost, performance, mobility and flexibility advantages. Due to their pre-fabricated, modular design and long-life performance, the construction and maintenance of RFBs would tend to be of the lowest cost among all other energy storage systems. Furthermore, in most cases, flow batteries can be discharged completely without any damage to the electrodes or the electrolyte. 8–10
The power and energy capacity of RFBs can be easily varied and hence, the flexibility of the energy storage is enhanced. Scale-up can be achieved by increasing the electrode size or by adding more electrodes in each stack with either monopolar or bipolar connections. The power is determined by the numbers of cells in the stack and by the number and size of the electrodes. The energy storage capacity depends on the concentration of electroactive species and the volume of the electrolytes.
3. Timeline for the development of redox flow batteries
Fig. 3 is a timeline showing the history of the development of RFBs. Early developments were carried out by the National Aeronautics and Space Administration (NASA) in the 1970s for bulk storage of electrical energy, load levelling, and to facilitate the use of intermittent energy sources such as photovoltaic cells and wind turbines.21–32 A variety of redox couples were studied by NASA but their early focus was on the iron-chromium RFB. From 1973 to 1982 a 1 kW/13 kWh iron-chromium RFB system was designed, fabricated and tested. This system validated the many desirable characteristics claimed for flow batteries but NASA scientists encountered two problems with this system: (a) poor electrochemical reversibility of chromium and (b) the cross-contamination of active species. Hence, in 1981, NASA halted the system-level's effort and redirected the emphasis on fundamental studies of RFBs. This technology was further developed in Japan, as a part of the Moonlight Project. 10 kW and 60 kW system prototypes were manufactured and tested during 1984 to 1989.33,34 Iron-chromium technology is currently under redevelopment for energy storage in wireless telecom applications by Deeya Energy® in Silicon Valley, USA.35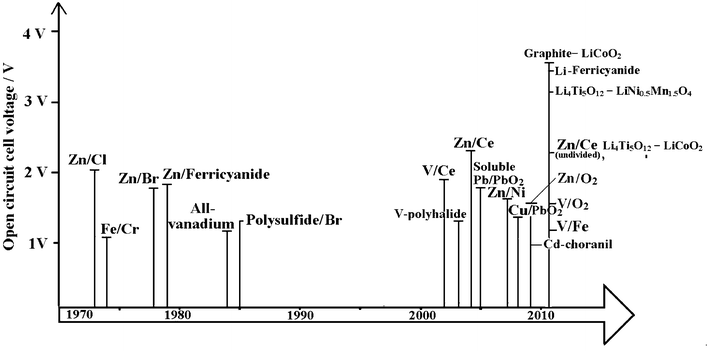 | ||
| Fig. 3 The timeline of the development of redox flow batteries over the past 40 years. | ||
In parallel with the development of the iron-chromium RFB, zinc-chlorine and zinc-bromine RFBs were developed in the USA.33,36 10 kW and 100 kW zinc-chlorine RFBs were tested and a 2 MW/6 MWh system was designed as an electric utility demonstration unit. This was viewed as a complicated system, however, and no funds had been made available to build a pilot plant to evaluate its design. A zinc-bromine RFB was developed for load levelling applications in the mid-1970s to early 1980s. Scale-up and demonstration of 3, 10 and 20 kWh submodules were attained in 1983. In 1990, a 1 MW/4 MWh zinc-bromine RFB system was installed in Japan. 36 At present, the zinc–bromine RFB is being developed by ZBB Energy Corporation, RedFlow Ltd., and Premium Power. The battery modules are expandable from 50 kWh to 500 kWh and are available for commercial and utility applications.
Vanadium-based electrolytes have been prominent during the development of RFBs.8–13 In 1977 the evaluation of the V2+/V3+ and V4+/V5+ redox couples was carried out by NASA. Since 1985, successful demonstrations and significant developments in the vanadium redox flow battery (VRFB) have been attributed to Skyllas-Kazacos et al.37–68 at the University of New South Wales, Australia. From 1993 to present, the VRFB has been commercialized for various applications, such as load-levelling, power quality control and renewable coupling. More than 20 large-scale plants have been installed globally by manufacturers.69 The considerable progress of the VRFB has been assisted by improvements in electrode47–50,53–55 and membrane materials.11,52,56,58,59,66,68,70
Bromine-polysulphide RFB systems were introduced by Remick and Ang in 198471 and developed by Regenesys® Technologies (UK) at Innogy plc between 1991 and 2004.72–76 A 1 MW pilot plant at Abershaw power state in South Wales was demonstrated and successfully tested. The construction of 15 MW/120 MWh demonstration plants started at Little Barford in the early 2000s.73,74 The bromine polysulfide technology has been one of the largest scaled-up RFB systems, driven by the moderate cost of uncatalysed electrodes and especially the low-cost of the electrolyte components which are available from multiple sources on a tonnage scale. Full production-scale modular cell stacks were proven on a pilot plant but commercial scale was ceased due to changes of the RWE holding company policy. Regenesys technology was acquired by VRB Power Systems Inc. in 2004 and was subsequently acquired by Prudent Energy (China) in 2009.
Several membrane-free (undivided) flow batteries, such as soluble lead-acid,14,77–85 zinc-nickel15,86–88 and zinc-cerium types17,89 have been developed since 2004. These batteries utilize a single electrolyte and therefore do not require an ion-exchange membrane and have advantages of lower costs and greater simplicity over other flow battery systems.77 Among these undivided systems, some were reported to have excellent round-trip DC energy efficiency of >80% and a cycle life of more than 1000 cycles.15 More recently, two types of lithium flow batteries storing energy in so-called ‘flowable’ electrode materials90,91 and an aqueous positive electrolyte92,93 have been introduced and these batteries were reported to have a discharge cell voltage of 3.8 V and energy density per electrolyte volume of 397 Wh dm−3, which are attractive for electric vehicle applications.
4. Types of redox flow batteries
Redox flow batteries may be classified into three groups according to the phases of the electroactive species presented in the system namely, a) all liquid phases, where chemical energy is stored in the electrolyte, b) all solid phases, where chemical energy is stored in an active material on the electrode plates and c) hybrid redox flow batteries. In the first group both redox couples have reactants/products dissolved in the liquid electrolytes and in the second group both redox couples involve solid species during the charge process, whilst hybrid RFBs have redox couples which involve solid species or gaseous species at one half-cell during the charge process. Fig. 4 illustrates the electrode reactions of three typical examples: (a) all vanadium, (b) lead-lead dioxide and (c) hybrid zinc-cerium RFBs.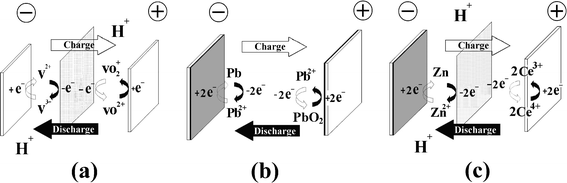 | ||
| Fig. 4 Types of redox flow battery, according to the nature of energy storage. Energy is stored (a) in the electrolytes, (b) in the active material within the electrodes and (c) hybrid (in both electrode and electrolyte phases). | ||
4.1 Chemical energy stored in the electrolyte
Table 2 summarises the operating parameters and performance of 13 RFB systems.32,44,45,94–106 The energy storage capacity can be increased by simply using a larger volume of electrolytes since chemical energy is stored in the electrolyte. All of these batteries require ion exchange membranes or separators to divide the anode and cathode compartments while allowing ion transport to maintain the electrical neutrality in the cell.| System | Operational parameters | Performance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Redox couples | Electrode reactions on charge Negative Positive |
E°OCV/V vs. SHE | Electrode materials Negative/Positive | Electrolytes Negative/Positive | Membrane | j/mA cm−2 | T/°C | E OCV /V | % Energy efficiency |
| a SHE: standard hydrogen electrode; OCV: open-circuit voltage; SCE: saturated calomel electrode; SOC: state of charge; AE: anion exchange; CE: cation exchange; NG: not given. | |||||||||
| Fe/Cr32 | Cr3+ + e− → Cr2+ | 1.18 | carbon or graphite felt + Au-Pb catalyst/carbon or graphite felt | 1 M CrCl3 in 3 M HCl/ | AE-VBC/DMAEMA | 30 | 25 (unmixed reactants) | 1.05 | 86 |
| E 0 = −0.41 V | |||||||||
| Fe2+ − e− → Fe3+ | 1 M FeCl2 in 3 M HCl | 80 | 65 (mixed reactants) | 0.95 (50% SOC) | 87 | ||||
| E 0 = +0.77 V | |||||||||
| All-Vanadium44,45 | V3+ + e− → V2+ | 1.26 | graphite impregnated polyethylene plate | 2 M VOSO4 in 2 M H2SO4/ | CE-Polystyrene sulphonic acid membrane | 30 | 35 | 1.35 (50% SOC) | 83 |
| E 0 = −0.26 V | |||||||||
| VO2+ + H2O − e− → VO2+ + 2H+ | 2 M VOSO4 in 2 M H2SO4 | ||||||||
| E 0 = +1.00 V | |||||||||
| V/polyhalide94 | VCl3 + e− → VCl2 + Cl− | 1.30 | graphite felt/graphite felt | 1 M VCl3 in 1.5 M HCl/ | CE-Nafion® 112 | 20 | NG | 1.30 (50% SOC) | 66 |
| E 0 = −0.50 V vs. SCE | |||||||||
| 2Br− + Cl− → ClBr2− + 2e− | 1 M NaBr in 1.5 M HCl | ||||||||
| E 0 = + 0.80 V vs. SCE | |||||||||
| V/Fe95 | V3+ + e− → V2+ | 1.02 | graphite felt/graphite felt | 2 M FeCl2 in 1.5 M HCl/ | CE-Nafion® | 50 | 25 | 1.02 | 80 |
| E 0 = −0.26 V | |||||||||
| Fe2+ − e− → Fe3+ | 2M VCl3 in 3M HCl | ||||||||
| E 0 = + 0.77 V | |||||||||
| V/Ce96 | V3+ + e− → V2+ | 1.93 | carbon fiber/carbon fiber | 0.5 M V3+ in 1 M H2SO4/ | AE-Asahi vycor glass | 22 | 40 | 1.87 (100% SOC) | 68 |
| E 0 = −0.26 V | |||||||||
| Ce3+ − e− → Ce4+ | 0.5 M Ce3+ in 1 M H2SO4 | ||||||||
| E 0 = + 1.67 V | |||||||||
| V/Mn97 | V3+ + e− → V2+ | 1.77 | graphite/graphite | 0.3 M V3+ in 5 M H2SO4/ 0.3 M Mn2+ in 5 MH2SO4 | CE-Nafion® 117 | 20 | 25 | NG | 63 |
| E 0 = −0.26 V | |||||||||
| Mn2+ − e− → Mn3+ | |||||||||
| E 0 = + 1.51 V | |||||||||
| Poly-Sulfide/Br298 | S42− + 2e− → 2S22− | 1.36 | Co-graphite felt/graphite felt | 1.3 M Na2S4/ 4.0 M NaBr | CE-Nafion® 117 | 40 | 26 | NG | 80 |
| E 0 = −0.27 V | |||||||||
| 3Br− − 2e− → Br3− | |||||||||
| E 0 = +1.09 V | |||||||||
| All-neptunium99-101 | Np4+ + e− → Np3+ | 1.29 | plastic formed carbon/plastic formed carbon | Single electrolyte: 0.05 M neptunium in 1 M HNO3 | AE-Asahi Kasei | 70 | 25 | 1.2∼1.3 | 99 (model predicted) |
| E 0 = 0.15 V | |||||||||
| NpO2+ − e− → NpO22+ | |||||||||
| E 0 = 1.14 V | |||||||||
| All-uranium102 | U4+ + e− → U3+ | 0.68 | NG | 0.4 M U(IV) β-diketonate complex/ | NG | 70 | NG | 1.0 | 98 (predicted) |
| E 0 = −0.52 V | |||||||||
| UO2+ − e− → UO22+ | 0.8 M U(V) β-diketonate complex | ||||||||
| E 0 = + 0.16 V | |||||||||
| All Cr-EDTA103 | CrIII-EDTA + e− → CrII-EDTA | NG | graphite felt/graphite felt | 0.05 M Cr(III)-EDTA in 1 M CH3COONa/ | CE-Nafion® 450 | 0.13 | 25 | 2.11 | NG |
| CrIII-EDTA − 3e− → CrVI-EDTA | 0.1 M Cr(III)-EDTA in 1 M CH3COONa | ||||||||
| Non-aqueous Ru-acetylacetonate104 | RuIII(acac)3 + e− → RuII(acac)3− | NG | graphite felt/graphite felt | Single electrolyte: 0.02 M Ru(acac)3 in 0.1 M TEABF4/CH3CN | AE-Neosepta AHA | 7(charge) 2 (discharge) | 25 | 1.75 | 52 (voltage) |
| RuIII(acac)3 − e− → RuIV(acac)3+ | |||||||||
| Non-aqueous V-acetylacetonate105 | VIII(acac)3 + e− → VII(acac)3− | NG | graphite/graphite | Single electrolyte: 0.01 M V(acac)3 in 0.5 M TEABF4/CH3CN | AE-AMI-7001 | 0.14 (charge) 0.014 (discharge) | 25 | 2.2 | 47 (coulombic) |
| VIII(acac)3 − e− → VIV(acac)3+ | |||||||||
| Non-aqueous Cr-acetylacetonate106 | CrIII(acac)3 + e− → CrII(acac)3− | NG | graphite/graphite | Single electrolyte: 0.05 M Cr(acac)3 in 0.5 M TEABF4/CH3CN | AE-Neosepta AHA | 0.14 (charge) 0.014 (discharge) | 25 | 3.4 | 21∼22 |
| CrIII(acac)3 − e− → CrIV(acac)3+ | |||||||||
Negative electrode.
 | (1) |
Positive electrode.
 | (2) |
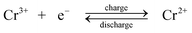
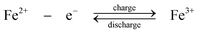
During charge, Cr3+ is reduced to Cr2+ at the negative electrode while Fe2+ is oxidized to Fe3+ at the positive electrode. During discharge the reverse process occurs. An ion-exchange membrane separates the two flowing reactant solutions and prevents the cross-mixing of the reactive cations. The standard cell voltage of this battery is 1.18 V.
Since the kinetic rate of reduction of Cr3+ to Cr2+ is slow on most electrodes, a catalyst is required on the chromium electrode. The catalyst must also have a high overpotential for hydrogen because its evolution tends to be a highly competitive reaction during the reduction of Cr3+ to Cr2+. The studied catalysts included gold, lead, thallium and bismuth.107–109 No catalyst is required on the iron electrode. It was revealed that the slow kinetics at the chromium electrode resulted from the presence of Cr3+ complexes such as Cr(H2O)63+ and Cr(H2O)5Cl2+ which showed a slow attainment of equilibrium between them.28,110 In order to improve the reversibility of the chromium redox reactions, porous three-dimensional electrodes, such as carbon felt, graphite foam and reticulated vitreous carbon were introduced to use as electrode materials for the chromium reaction.
During development of the iron-chromium RFB system, NASA researchers found that operation at elevated temperature (e.g. 65 °C) allowed the chromium species to become electrochemically active and improved battery performance.29,30 Operation at elevated temperature led to the change from unmixed reactants mode to mixed reactants mode in which both the negative and positive half-cells contain iron and chromium species. The new mode of operation permitted the use of membranes that, although having very poor selectivity, had the advantage of very low resistivity, around 0.5 Ω cm2. These changes in design and mode of operation of iron-chromium RFB increased the operating current density from 20–30 mA cm−2 to 60–80 mA cm−2 with energy efficiencies above 80% (see Table 2).
Despite great efforts toward the scale-up and optimization of the system,111–113 for the iron-chromium RFB there remain some technical challenges that have hindered its commercial development. The major problem is its low energy density which is reported to be less than 10 Wh kg−1.9 Also, the requirement of noble catalysts for the Cr3+/Cr2+ electrode reaction increases the system cost.
Negative electrode.
 | (3) |
Positive electrode.
 | (4) |
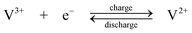
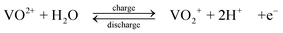
The standard open-circuit cell voltage is 1.26 V. It is reported that under actual cell conditions of using 2 M VOSO4 in 2.5 M H2SO4 electrolyte, the cell open-circuit potential is 1.35 V at 50% state-of-charge and 1.60 V in a fully charged state.45 The system can be operated over a temperature range of 10–40 °C.44,45
The VRFB was studied in detail by the group of Skyllas-Kazacos37–68 and by industrial organisations34,114 in Japan during the 1980s and 90s. Since 2002 significant research and development has been spread world-wide, especially in China,11,115–124 in the USA9,125–133 and in Europe.134,135 The advantages possessed by VRFB can be summarized as follows: (i) by employing the same element in both half-cells and utilizing four different oxidation states of vanadium in solution, in comparison to the Fe/Cr system, the VRFB eliminates the problem of cross contamination by diffusion of different redox ions across the membrane. Even if solution crossover occurs, the electrolytes can be regenerated by mixing and then electrolysis without complicated chemical treatment; (ii) it does not require a catalyst for each electrode reaction and the relatively fast kinetics of the vanadium redox couples allow high charge and voltage efficiencies to be obtained, the overall energy efficiency from an initial 71% up to 90% has been reported with a 1 kW VRFB stack; (iii) there is an extremely low rate of gas evolution during the charge rates associated with rapid charging cycles; (iv) it can be over-charged and deeply discharged, within the limits of the capacity of the electrolytes, as well as being cycled from any state of charge or discharge, without permanent damage to the cell or electrolytes; (v) a reusable electrolyte leads to a long cycle life and reduces cost of the system. These advantages and its flexibility make the VRFB a promising technology for large-scale storage of renewable energy.
For practical applications, however, the VRFB system faces several challenges. One of the major challenges is its low specific energy density (25–30 Wh kg−1) compared to other batteries for energy storage. The energy density in a VRFB depends on the concentration of vanadium species but there is a limited vanadium concentration in H2SO4 due to the precipitation of solid vanadium compounds; if the concentration is over 2 M, V2O5 precipitation occurs in the V5+ electrolyte at a temperature above 40 °C and solid vanadium oxides in V2+ or V3+ solution below 10 °C.57,60,61,64 Even with the positive effects of the additives,61,63,132,136–138 the vanadium concentration is still limited to below 2 M for most practical batteries. At such a low concentration, the VRFB system is believed to be impractical. To increase the vanadium concentration, Skyllas-Kazacos et al.57 reported that the V5+ concentration can be increased up to 3 M without any precipitation at 40 °C when a sulfuric acid concentration is higher than 5 M and, by using a concentration of 7 M SO42− as the medium, an even higher 5.4 M V5+ solution is claimed to be stable for a period of over 30 days at >50 °C, but increasing the concentration of sulfuric acid caused a decrease in solubility of VOSO4 at low temperature (<10 °C). Recently, Yang et al.129 reported that, in 10 M HCl supporting electrolytes, more than 2.3 M vanadium (V2+, V3+, VO2+, VO2+) are all stable for 15 days over a temperature range of 0–50 °C. Despite these efforts to improve the solubility of vanadium, the stability of vanadium ions is determined by a number of influential factors such as solution temperature, the concentration of supporting electrolyte, additives and state of charge. Hence low energy density still remains a major challenge for the VRFB system.
The highly oxidizing nature of V5+ is another challenge in VRFB, since it can cause degradation of the ion-exchange membrane and the positive electrode material.10,41 Early studies by Skyllas-Kazacos et al.59,68 showed that most commercially available membranes tended to deteriorate in the highly oxidizing V5+ electrolyte and could not offer a long cycle life. Only a few membranes, such as New Selemion anion exchange membrane and Nafion cation exchange membrane, were found to be chemically stable in the V5+ solution. Also, the highly oxidative activity of V5+ limits the selection of the positive electrode material to carbon or graphite felt. These limitations increase the cost of the VRFB system.
Although the VRFB has received perhaps the most attention of all RFBs and a number of successful demonstrations are scattered around the world, to date it is reported that the operational cost is about $500 kWh−1 or higher,9 obviously this technology is expensive for broad market penetration because of its high cost which is attributed to the expensive vanadium electrolyte and cell components including Nafion-based membranes and electrode materials.
In order to achieve higher cell voltage, Ce3+/Ce4+ and Mn2+/Mn3+ couples96,97 have been used as the positive electroactive species to incorporate with V2+/V3+ negative species, giving rise to vanadium-cerium and vanadium-manganese RFBs. Table 2 shows more information on these systems. According to Fang et al.,96 the vanadium-cerium RFB has advantages of high coulombic efficiency (87%), high cell voltage (1.87 V) and low self-discharge rate, but the low solubility of cerium sulfate and slow redox kinetics of the Ce3+/Ce4+ couple are the main technical issues of this system. Further research is needed to develop low-cost catalysts and high surface area electrode materials for the improvement of the kinetics of the Ce3+/Ce4+ couple.
Similarly, a vanadium-iron RFB based on mixed acid electrolytes was introduced to reduce the cost by using relatively low-cost elements Fe2+/Fe3+ in the positive electrolyte. This system was reported to have improved stability at elevated temperature and allows the use of low-cost separators.95,143 A recent investigation also demonstrated that both V4+/V5+ and Fe2+/Fe3+ can be mixed together and used as the positive redox couples of a battery. This arrangement can effectively increase the energy density by 66% compared to a typical vanadium-iron RFB.144
Negative electrode.
 | (5) |
Positive electrode.
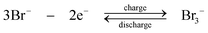 | (6) |
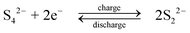
During discharge, the sulphide ion is the reducing agent and the tribromide ion the oxidizing species. In contrast to other systems using bromide salts, no complexing agent is required. The open-circuit cell voltage is reported to be 1.74 V in a fully charged state and 1.5 V at a 50% state of charge when operated at 25 °C in 1 M NaBr and 2 M Na2S4 which are separated by Nafion 125 membrane, using graphite as the positive electrode and porous nickel sulphide as the negative electrode.71
The bromine-polysulphide RFB technology was financed by National Power in UK and developed by Regenesys® Technologies between 1991 and 2004.72–76 It was scaled up at three sizes, in which 60, 120 and 200 bipolar electrodes respectively were assembled into a modular stack using a filter-press type of assembly. The nominal power ratings of the stacks were 5 kW, 20 kW, and 100 kW, respectively. A key feature of the modular technology was the use of carbon-polyolefin composite electrodes, enabling the electrodes to be welded to the polyolefin cell frames using laser-based production engineering techniques.74 A 1 MW pilot scale facility was installed and tested at the Aberthaw Power Station in South Wales (UK) with a round-trip efficiency of 60–65% and energy density of 20–30 Wh L−1.6,9
To improve the efficiency and performance of the bromine-polysulphide RFB, Zhang et al.98,145–147 have examined various electrode materials, including activated carbon, carbon/graphite felt, nickel foam and metal-coated graphite felt. They reported that using cobalt-coated graphite felt as the negative electrode and graphite felt as the positive one, the battery achieved a stable energy efficiency of up to 81% over 50 cycles (600 h) at a current density of 40 mA cm−2.
There are a number of technical challenges facing the bromine-polysulfide technology: (i) there is a risk of cross-contamination of the electrolytes due to cross-over of the ionic species through the membrane; (ii) long term transport of species across the membrane leads to the need for analytical monitoring and chemical treatment of the electrolytes, increasing cost and complexity of the process plant; (iii) the system also encountered the problem of the build-up of sulfur species (e.g. S2− and/or HS−) either on the electrodes or membrane and therefore over extended cycling there is a net loss of sulfur from the system; (iv) mixing of the electrolytes can generate heat and toxic gases such as Br2 and H2S. All of these challenges have to be fully met before a true power station scale is realised.
Recently, liquid electroactive species, such as Cr3+/Cr2+ and Cr6+/Cr3+,103 Fe3+/Fe2+,150,151 and Ce4+/Ce3+152 were reported to have higher electrochemical rate constants on complexing with triethanolamine (TEA), diethylenetriaminepentaacetate (DTPA) and ethylenediaminetetra-acetate (EDTA) ligands, respectively. This opens the possibility of using certain redox couple combinations in flow batteries, such as all chromium, vanadium-cerium and iron-bromine systems.
4.2 Chemical energy stored in active material on the electrodes
Energy can be stored as electrodeposits at the negative and positive electrodes and released by transformation or dissolution of the electrodeposits during discharge. Several flow battery systems of this type have been developed (see Table 3).14–16,157 In most of the systems proposed, a single electrolyte is circulated through the cell and no ion-exchange membrane is required. Since the systems operate without a membrane separator; it avoids the problems associated with leakage and cross-contamination of species through a membrane that have severely hampered other flow battery technologies. Moreover, this reduces the cost and design complexity of the batteries significantly. These features make them attractive for energy storage in large-scale applications.158| System | Operational parameters | Performance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Redox couples | Electrode reactions on charge Negative electrode Positive electrode |
E 0 OCV /V vs. SHEa | Electrode materials Negative/Positive | Electrolytes | j/mA cm−2 | Temp./°C | E OCV/Va | Charge/Discharge voltage /V | % Energy efficiency |
| a SHE: standard hydrogen electrode; OCV: open-circuit voltage; NG: not given. | |||||||||
| Pb/PbO277 | Pb2+ + 2e− → Pb | 1.62 | reticulated nickel foam/reticulated vitreous carbon | 1.5 M Pb(CH3SO3)2 in 0.9 CH3SO3H | 20 | 25 | 1.69 | 2.07/1.45 | 65 |
| E 0 = −0.13 V | |||||||||
| Pb2+ + 2H2O − 2e− → PbO2 + 4H+ | |||||||||
| E 0 = +1.49 V | |||||||||
| Cu/PbO216 | Cu2+ + 2e− → Cu | 1.35 | graphite/lead dioxide | 0.6 M CuSO4 in1.9 M H2SO4 | 20 | 25 | NG | 1.45/1.29 | 83 |
| E 0 = +0.34 V | |||||||||
| PbSO4 + 2H2O − 2e− → PbO2 + 4H+ + SO42− | |||||||||
| E 0 = +1.69 V | |||||||||
| Zn/Ni 15,86 | Zn(OH)42− + 2e− → Zn + 4OH− | 1.71 | cadmium-plated copper/sintered nickel hydroxide | 1 M ZnO in 10 M KOH | 10 | 25 | NG | 1.85/1.72 | 86 |
| E 0 = −1.22 V | |||||||||
| 2Ni(OH)2 + 2OH− − 2e− → 2NiOOH + 2H2O | |||||||||
| E 0 = + 0.49 V | |||||||||
| Zn/MnO2312 | ZnO + H2O + 2e− → Zn + 2OH− | NG | granular zinc/30 wt% manganese(IV) oxide | 2 M KOH | NG | 25 | NG | NG/1.1–1.2 | NG |
| Mn2O3 + 2OH− − 2e− → 2MnO2 + H2O | |||||||||
Negative electrode.
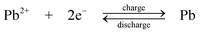 | (7) |
Positive electrode.
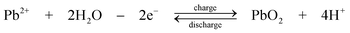 | (8) |
The chemistry is different from the traditional lead-acid battery159 in that the electrode reactions do not involve insoluble Pb(II), e.g. as lead sulphate within a paste. Operation of the laboratory flow battery gives a charge efficiency > 85%, an energy efficiency ≈ 65% and an open-circuit cell voltage of 1.69 V. This technology had been scaled up from the 2 cm2 electrode laboratory cell to a several-kW pilot plant cell containing 10 bipolar electrodes each with an area of 1000 cm2.14,77–85,160 During the development, the problems associated with dendrite growth of Pb and shedding of PbO2 led to significant loss in energy efficiency and usually resulted in the failure of the battery. Detailed study has shown that the additives such as hexadecyltrimethylammonium cation would prevent the formation of Pb dendrites on the negative electrode.80,81 It was also revealed that the phase structure of PbO2 has significant effects on cell performance. Ideally pure α-PbO2 is preferred in this system as β-PbO2 which is relatively loose causes shedding of PbO2. Experimental results suggest that, to prevent the formation of β-PbO2, the system should operate at Pb(II) concentrations > 0.3 M and elevated temperatures should be avoided.82
Due to the lead-acid's combination of low cost, high-efficiency, safety, and proven reliability, this innovative system has attracted much attention both from academics and industry. In the USA, a project (2010–2013) entitled “Grids Soluble Lead Flow Battery Technology” is being undertaken by University of California and industrial partners with funding from the US Department of Energy.161 In addition, based on the membrane-free concept, several new flow batteries were proposed.15–17,162 For example, a single flow Cu–H2SO4–PbO2 battery was reported16 with a round-trip DC energy efficiency of up to 83% and an open-circuit cell voltage of 1.35 V.
4.3 Hybrid redox flow batteries
Hybrid RFBs involve the deposition of solid species or evolution of gaseous species in one half-cell during the charging process. Zinc-chlorine and zinc-bromine are the best-known examples of this type of RFB. The operating parameters for selected hybrid systems are summarized in Table 4.18,162–190 Zinc is a typical negative electrode that has been used extensively in the batteries due to its large negative electrode potential in both acidic (−0.76 V vs. SHE) and alkaline (−1.22 V vs. SHE) aqueous media.| System | Operational parameters | Performance | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Redox couples | Electrode reactions on charge Negative Positive |
E o ocv/V vs. SHE | Electrode materials Negative/Positive | Electrolytes Negative/Positive | Membrane | j /mA cm−2 | T/°C | Charge/Discharge voltage /V | % Energy efficiency |
| a SHE: standard hydrogen electrode; OCV: open-circuit voltage; CE: cation exchange; GDE: gas diffusion electrode; NG: not given. | |||||||||
| Zn/Cl2163 | Zn2+ + 2e− → Zn | 2.12 | dense graphite/porous graphite | single electrolyte: 2 M ZnCl2 in 4 M KCl | membraneless | 22 | 25 | 2.3/1.8 | 66 |
| E 0 = −0.76 V | |||||||||
| 2Cl− − 2e− → Cl2 | |||||||||
| E 0 = +1.36 V | |||||||||
| Zn/Br2164 | Zn2+ + 2e− → Zn | 1.85 | carbon/carbon | 2 M ZnBr2 in 4 M KCl/ | CE-Nafion® 125 | 20 | 54 | 1.92/1.64 | 80 |
| E 0 = −0.76 V | |||||||||
| 2Br− − 2e− → Br2 | 0.05 M Br2 + 2 M ZnBr2 in 4 M KCl | ||||||||
| E 0 = +1.09 V | |||||||||
| Zn/Ce8,18,19 (Plurion) | Zn2+ + 2e− → Zn | 2.43 | carbon/platinised titanium | 0.6 M Ce(CH3SO3)3 + 1.3 M Zn(CH3SO3)2/ | CE Nafion® | 50–100 (charge) 5–50 (discharge) | 60 | 2.75–3.15/1.8 | NG |
| E 0 = −0.76 V | |||||||||
| 2Ce3+ − 2e− → 2Ce4+ | 0.72 M Ce(CH3SO3)3 + 0.9 M Zn(CH3SO3)2 | ||||||||
| E 0 = +1.67 V | |||||||||
| Zn/Ce17 (undivided) | Zn2+ + 2e− → Zn | 2.43 | carbon plastic/carbon felt | 1.5 M Zn(CH3SO3)2 + 0.2 M Ce(CH3SO3)3 in 0.5 M CH3SO3H | membraneless | 20 | 23 | 2.5/2.1 | 75 |
| E 0 = −0.76 V | |||||||||
| 2Ce3+ − 2e− → 2Ce4+ | |||||||||
| E 0 = +1.67 V | |||||||||
| Zn/V 313 | Zn2+ + 2e− → Zn | 1.76 | graphite felts | 1.5 M Zn(CH3SO3)2 + 0.5 M CH3SO3H | CE membrane | 40 | 25 | 2.2/1.5 | 70 |
| E 0 = −0.76 V | |||||||||
| 2V4+ − 2e− → V5+ | 3 M V(MSA) + 2.8 M CH3SO3H | ||||||||
| E 0 = +1.0 V | |||||||||
| Zn/Fe(CN)6165 | Zn2+ + 2e− → Zn | 1.12 | silver-plated iron/porous nickel | ZnO saturated in 5 M NaOH/ | CE-Nafion XR 475 | 20 (charge) 35 (discharge) | 38 | 2.0/1.5 | 74 |
| E 0 = −0.76 V | |||||||||
| [Fe(CN)6]4− − e− → [Fe(CN)6]3− | K4[Fe(CN)6] saturated in 5 M NaOH | ||||||||
| E 0 = + 0.36 V | |||||||||
| Zn/O2162 | Zn(OH)42− + 2e− → Zn + 4OH− | 1.62 | copper/GDE with catalysts MnO2-NaBiO3 & β-Ni(OH)2 | 0.7 M ZnO + 0.7 M LiOH in 7 M KOH | membraneless | 20 | 60 | 1.78/1.32 | 72 |
| E 0 = −1.22 V | |||||||||
| 2OH− − 2e− → ½O2 + H2O | |||||||||
| E 0 = +0.40 V | |||||||||
| V/O2166 | V3+ + e− → V2+ | 1.49 | carbon/titanium mesh catalyzed with Pt-Ir | 2 M V(III) in 3 M H2SO4/air | CE-Nafion® 117 | 2.4 | 80 | 1.88/1.0 | 46 |
| E 0 = −0.26 V | |||||||||
| 2H2O − 4e− → 4H+ + O2 | |||||||||
| E 0 = +1.23 V | |||||||||
| Cd/chloranil (QCl4)167 | Cd2+ + 2e− → Cd | 1.11 | copper foil/chloranil film mixed with carbon black | 0.5 M CdSO4 in 1 M (NH4)2SO4 + 0.5 M H2SO4 | membraneless | 10 | 25 | 1.18/0.97 | 82 |
| E 0 = −0.40 V | |||||||||
| QH2Cl4 − 2e− → QCl4 + 2H+ | |||||||||
| E 0 = +0.71 V | |||||||||
| Pb/Tiron168 | PbSO4 + 2e− → Pb + SO42− | 1.07 | lead/carbon felt | 3 M H2SO4/0.25 M tiron in 3 M H2SO4 | CE-Nafion® 115 | 10 | 25 | 1.1/0.9 | 82 |
| E 0 = −0.35 V | |||||||||
| RedTiron − 2e− → OxTiron | |||||||||
| E 0 = +0.72 V | |||||||||
| Li4Ti5O12/LiCoO291 | Li1−xCoO2 + xLi+ + xe− → LiCoO2 | 2.2 | 22.6 M Li4Ti5O12/51.2 M LiCoO2 suspensions | LiPF6, carbonate electrolyte, Ketjen black | Celgard 2500 micro-porous | NG | 25 | 2.5/2.34 | 88 |
| E 0 = +3.7 V vs. Li | |||||||||
| Li4+xTi5O12 − xe− → Li4xTi5O12 + xLi+ | |||||||||
| E 0 = +1.5 V vs. Li | |||||||||
| Li/Fe(CN)693 | Li+ + e− → Li | 3.40 | Li ribbon/aqueous K3Fe(CN)6 | 166 μm solid electrolyte Li1+x+yAlxTi2−xSiyP3−yO12/0.1 M K3Fe(CN)6 | Celgrade® polypropylene separator | 0.5 | 25 | 3.48–3.67/3.30–3.58 | 97–99 (coulombic efficiency) |
| E 0 = −3.04 V | |||||||||
| [Fe(CN)6]4− − e− → [Fe(CN)6]3− | |||||||||
| E 0 = +0.36 V | |||||||||
 | (9) |
 | (10) |
The reactions are highly reversible and the theoretical cell voltage is 2.12 V. During charge, the ZnCl2 electrolyte is electrolysed, yielding a zinc deposit on negative electrode and chlorine evolution on positive electrode. The chlorine gas is removed from the cell to another chamber where it is mixed with water at approximately 10 °C and chlorine hydrate (Cl2·H2O) is formed. During discharge, the cooled chlorine hydrate is passed through a heat exchanger, where it is decomposed. The chlorine-rich stream is then pumped to the positive electrode where chlorine is reduced to chloride. Simultaneously, the zinc deposit is dissolved at the negative electrode.
EDA's cell design is based on comb-like bipolar electrodes which incorporate low-cost electrode frames that can be injection-moulded to facilitate manufacture and assembly of the battery stack.170 The zinc electrodes are made of dense graphite whereas the chlorine electrodes are made of porous graphite. There is no need to use separators since the solubility of chlorine in the electrolyte is low. Hydrogen evolution occurs at the zinc electrode and the battery system includes a fluorescent lamp to convert hydrogen, in the presence of chlorine gas, to hydrogen chloride.171
In Japan, development of zinc-chlorine RFB technology was carried out within the framework of the Moonlight Project for energy storage during the 1980s.172–175 The 1 kW, 10 kW and 60 kW battery modules have been constructed and tested with energy efficiency of 71–76%, 66% and 63%, respectively. Japanese workers have made several efforts to improve the battery performance by i) developing a new method to store chlorine in a chilled organic solvent;173 ii) applying highly corrosion-resistant polyacrylonitrile-type graphite for the chlorine electrode substrate and RuO2 for the chlorine electrode catalyst;174 iii) reducing the requirement for pumping power; and iv) improving the insulation method and materials.174 These improvements allowed an 80% DC to DC energy efficiency for the 10 kW battery and 76% for the 60 kW battery system. Despite the improvements, further development of zinc-chlorine batteries was discontinued after a critical analysis of the technological, performance and economical parameters of the battery modules. This technology was judged to be too complicated for practical use and poses serious environmental hazards associated with the evolution of toxic chlorine gas.171,172
Negative electrode.
 | (11) |
Positive electrode.
 | (12) |
During charge, zinc is deposited at the negative electrode while bromide ions are oxidized to bromine at the positive electrode. During discharge, the reverse reactions occur. The theoretical cell potential is 1.82 V. The theoretical energy density is 440 Wh kg−1, while the practical one is around 65–75 Wh kg−1.10,172 In fact, the chemical species present in the electrolyte are more complex than those described in the eqn (11), (12). The bromine produced at the positive electrode during charge is in equilibrium with bromine ions and forms highly water soluble anions such as Br3− and Br5−.36,164
One of the problems with the zinc-bromine battery is the high rate of self-discharge caused by the migration of bromine ions into the zinc electrode compartment. To avoid this process, a microporous separator or an ion-exchange membrane is required to separate the positive and negative half-cells. In addition, it is essential to use complexing agents to contain and store bromine. The complexing agents associate with bromine ions to form an emulsion, which is insoluble in water, has different density from water and travels with the electrolyte to the storage tank where it is separated by gravity. The commonly used complex agents are quaternary ammonium salts such as N-methyl-N-ethyl-morpholinium bromide and N-methyl-N-ethyl-prolidinium bromide.36,176,177
In order to optimise the zinc-bromine battery, various mathematical models have been used to describe the system.178–181 The problems with the zinc-bromine battery include high cost electrodes, material corrosion, dendrite formation during zinc deposition on charge, high self-discharge rates, unsatisfactory energy efficiency and relatively low cycle life. Another disadvantage of this system is that the Zn/Zn2+ couple reacts faster than the Br2/Br− couple causing polarization and eventually battery failure. To overcome this, a high surface area carbon electrode on the cathode side is normally used however, the active surface area of the carbon eventually decreases and oxidation of the carbon coating occurs.
Despite the drawbacks of this system, the zinc-bromine battery has been one of the most developed, commercially scaled-up flow battery systems since 1970s because of its high energy density (65–75 Wh kg−1), reasonably high cell voltage (1.82 V), and high energy efficiency (80%), high degree of reversibility, and abundant low cost reactants. Exxon's ZBB system utilized carbon plastic electrodes in a bipolar stack design and important technology improvements included shunt current protection, use of low-cost microporous separators, and insert injection moulding of electrodes and separators. Scale-up and demonstration of 3, 10 and 20 kWh sub-modules were attained in 1983.
In Japan, the zinc-bromine RFB, as one of the advanced battery systems within the framework of the Moonlight Project, was developed by Meidensha Electric Co. from the 1980s to the early 1990s. Development proceeded smoothly through 1 kW, 10 kW and 50 kW battery modules with acceptable energy efficiency and cycle life. In 1990 a 1 MW/4MWh zinc-bromine RFB was installed at the Kyushu Electric Power Company in Japan. The system is composed of 24 25 kW sub-modules connected in series and completed over 1300 cycles with an overall energy efficiency of 66%.175,184
In recent work,17,89 an undivided zinc-cerium flow battery using a single electrolyte at low acid concentration was proposed with the use of a carbon felt positive electrode and a mixed electrolyte of both zinc and cerium electroactive species. In contrast to the 50 to 60 °C temperature range reported in the earlier systems, the membrane-less battery was reported to cycle at room temperature with an average round-trip DC energy efficiency of 75%. The optimum cerium(III) concentration was limited to only 0.2 mol dm−3 as although higher concentrations (particularly of Ce(IV)) could be achieved by increasing the acid concentration, this would lead to poor charge efficiency at the zinc negative electrode due to the parasitic hydrogen evolution reaction. In order to achieve larger storage capacity, further investigations are important in an attempt to increase the solubility of cerium species by modifying the ligands in other solvents and to facilitate efficient zinc electrodeposition and stability in a highly acidic medium without corrosion problems. The long term stability of the carbon felt electrode in the strongly oxidizing Ce(IV) species also needs further investigation.17,89
A dual functional zinc-air flow battery system was proposed by Wen et al. in 2008.188 Apart from storing energy, this flow battery can be used to produce organic acids, including propanoic acid, glyoxylic acid and cysteic acid from raw materials of propanol, glyoxal and cysteine, respectively.188–190Fig. 5a shows the configuration of a zinc-air flow battery using zinc regeneration electrolysis with propanol oxidation as a counter electrode reaction. This system consists of two cells connected with a zinc electrode in flowing electrolytes and used for electrolysis and discharge, respectively. During charge, propanol is oxidized to propanoic acid on the positive electrode and zinc is deposited at the negative electrode. During discharge, an air electrode where oxygen is fed from the air is employed on the positive electrode to combine with the zinc electrode, forming another cell to deliver energy. An energy efficiency of 59% was reported for this battery.
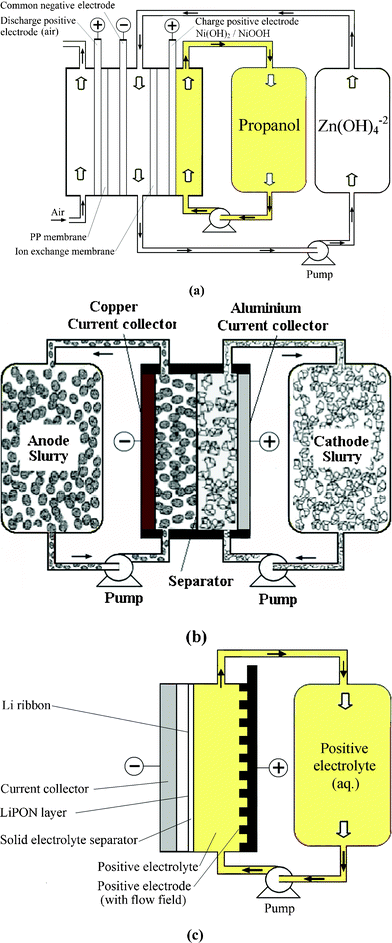 | ||
| Fig. 5 Examples showing configurations of three types of flow batteries: (a) zinc-oxygen bifunctional,188 (b) lithium flowable electrode91 and (c) lithium-aqueous ferricyanide flow batteries.93 | ||
Pan et al.162 designed a single flowing zinc-oxygen battery based on a composite oxygen electrode with one side facing the aqueous electrolyte using nano-structured Ni(OH)2 for the oxygen evolution reaction and the other side facing the atmospheric air using electrocatalytic MnO2 doped with NaBiO3 for the oxygen reduction reaction. At 20 mA cm−2 and 60 °C, the charge and discharge cell voltages of the battery are 1.78 V and 1.32 V, respectively, with an average charge efficiency of 97% and an energy efficiency of 72% over 150 cycles.
A novel type of semi-solid suspension containing ion-storing insertion “flowable electrode materials” was proposed by Chiang and Carter et al.90,91 in 2010. As shown in Fig. 5b the positive and negative suspensions are circulated through the battery and are stored in two reservoirs. A microporous polymer separator (Celgard 2500 and Tonen) was used to prevent the crossover of the negative and positive active materials. Due to the solid nature of the suspending flowable electrode material, the equivalent concentration of positive electrode suspensions such as LiCoO2 and LiNi0.5Mn1.5O4 and negative electrode suspensions such as Li4Ti5O12 and graphite are 51.2, 24.1, 22.6 and 21.4 mol dm−3, respectively, which are significantly higher than those of RFBs (<5 mol dm−3). This resulted in an increase in the energy density per electrolyte volume to approximately 10 times that of a VRFB.
The round-trip coulombic and energy efficiencies of the lithium flow battery using Li4Ti5O12 negative flowable electrode and LiCoO2 positive flowable (i.e., slurry) electrode were reported to be 98% and 88%, respectively. The architecture of using such suspension flowable active materials may be used to refuel electric vehicles by pumping out the used active materials and pumping in the fully charged one. Despite of all these advantages, further studies of the safety issues related to an LiPF6 electrolyte and microporous polymer separator, positive electrodes, particularly LiCoO2 electrode, together with the practical implications of ‘flowable’ electrodes are needed.
Taking advantage of the recent advance of solid-electrolyte lithium superionic conductive separator (Li1+x+yAlxTi2−xSiyP3−yO12), Lu and Goodenough92,93 proposed a lithium flow battery based on a solid lithium negative electrode and aqueous positive electrolyte as shown in Fig. 5c. By selecting Fe(CN)63−/Fe(CN)64− as the positive redox species, the lithium-ferricyanide flow battery gives an effective voltage between 3.33 V and 3.68 V at 0.5 mA cm−2 as shown in Fig. 6. A coulombic efficiency of more than 97% is also achieved over 1000 cycles. However, the limitation of this system is the low current density (<2.5 mA cm−2) due to the deterioration of the solid electrolyte separator. Also, the low mobility of lithium ion in the solid electrolyte separator can increase the resistance significantly. Aiming for larger power output and extended life time, the lithium ion conductivity and the chemical resistance properties of the solid electrolyte separators need further improvements.
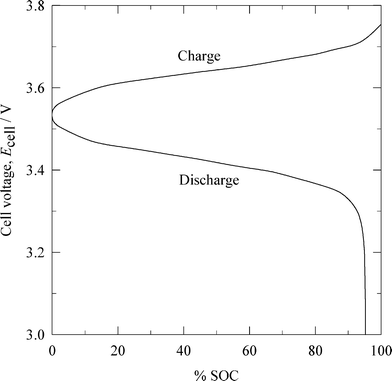 | ||
| Fig. 6 Cell voltage vs. normalized capacity characteristics of a lithium-ferricyanide flow battery during charge-discharge.92 (SOC: state of charge.) | ||
5. Design considerations and components of redox flow batteries
5.1 Constructional materials
A typical unit cell of a flow battery stack is illustrated in Fig. 7; it is composed of a positive and a negative electrode with an ion exchange membrane separating them.200 Rubber gasket seals and steel tie-bolts are normally used to compress the cell stack in order to avoid electrolyte leakages. Metallic end-plates, such as aluminum and copper, can be used as current conductors to provide electrical conductivity. Turbulence promoters can be employed in the compartment to increase the mass transport and exchange of electroactive species.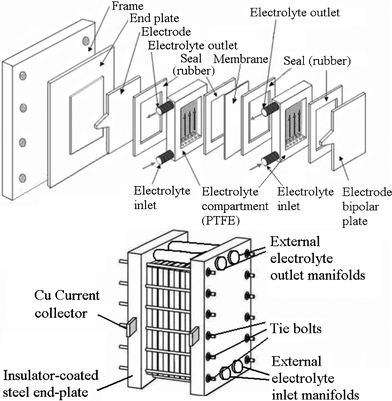 | ||
| Fig. 7 Components of a flow battery and a cell stack.200,276,277 | ||
Since the electroactive species used in RFBs are often highly oxidizing, no metallic component should be in contact with the electrolytes. Chemically resistant polymers, such as polytetrafluoroethylene (PTFE), ethylene-polypropylene-diene (EPPD), polyvinylchloride (PVC), polyvinylidene (PVDF) and acrylics (Perspex®) are the typical materials for producing the battery components (excluding the metallic end-plate and the electrode catalysts). By connecting a number of unit cells in series in a bipolar configuration to form a battery stack, a large cell voltage can be obtained. In some circumstances easily folded rubber tanks can effectively utilize the available space, such as in underground cisterns in the building.114
5.2 Electrode materials
An ideal electrode material should provide a high electrical conductivity, good mechanical properties, strong chemical resistance, be of reasonable price and have a long cycle life in highly oxidizing media. Typical electrodes used in RFBs are made of carbon-based composites or inert metallic materials. Carbon-based electrodes are more common than their metallic counterparts, as they do not undergo dissolution or formation of oxide during chemical oxidation. Metal ions from the metallic electrodes can dissolve into the electrolyte during discharge and corrosion, which could lead to unstable redox potentials and disturb the chemistry of the RFBs. If metallic electrodes are used they are generally based on noble metals, which have good electrochemical stability or catalytic property. Some metallic electrode materials with high overpotentials for gas evolution have been used to facilitate the desired reactions and avoid side reactions. The electrode materials used in various RFBs are summarized in Table 5.| Electrode material | Manufacturer | Thickness/mm | Electrode polarity | RFB system |
|---|---|---|---|---|
| Carbon based electrodes | ||||
| Carbon polymer: PVA, PVDF, HDPE184,185 | Entegris, USA | 6 | −ve | Zinc-cerium |
| Carbon felt GFA-type184 | SGL Group, Germany | 8 | +ve | Zinc-cerium |
| Graphite felt42 | Le Carbonne, France | NG | +ve & −ve | All-vanadium |
| PAN-based graphite felt98 | XinXing Carbon, China | 5 | +ve | Bromine-polysulfide |
| Cobalt coated PAN-based graphite felt98 | XinXing Carbon, China | 5 | −ve | Bromine-polysulfide |
| Reticulated vitreous carbon77 | ERG Material and Aerospace, USA | 1.5 | +ve | Soluble lead acid |
| Graphite felt bonded electrode assembly with nonconducting plastic substrate62 | FMI Graphite, USA | NG | +ve & −ve | All-vanadium |
| Carbon paper (non wet-proofed)206 | SGL Group, Germany | 0.41 | +ve & −ve | All-vanadium |
| Porous graphite163 | Union Carbide, USA | 2 | +ve & −ve | Zinc-chlorine |
| Carbon felt CH type198 | Fiber Materials, USA | 2.8 | +ve & −ve | Zinc-bromine |
| Cylindrical bed of carbon particles217 | Sutcliffe Speakman Carbons, UK | 2.5 | +ve & −ve | Bromine-polysulfide |
| Metallic electrodes | ||||
| Nickel foam146 | LuRun Material, China | 2.5 | −ve | Bromine-polysulfide |
| Cadmium-plated copper188 | NG | NG | −ve | Zinc-nickel & Zinc-air |
| Sintered nickel hydroxide15,86 | NG | NG | −ve | Zinc-nickel |
| Nickel foam77 | NG | 1.5 | +ve | Soluble lead acid |
| 3D-platinised-titanium mesh184 | Magneto GmbH, Netherlands | 2.5 | +ve | Zinc-cerium |
Fig. 8a and 8b show two typical configurations used in electrochemical flow cells; a) flow-through the electrode and b) flow-parallel to the electrode (flow-by configuration).201 Although the flow-through configuration has a more uniform concentration of electroactive species and gives an enhanced mass transport,198 it is often impractical due to the very low flow rate required 202 and the high scale-up costs.203,204 Therefore, most flow batteries reported in the literature adopt the flow-by configuration (Fig. 8b). In this configuration, the electrodes are generally made of two- or three-dimensional materials corresponding to different electrolyte flow modes as shown in Fig. 8c and 8d.98 Various three-dimensional electrodes have been used extensively in RFBs due to their large surface area, resulting in faster electrochemical reactions and lower polarization at the electrodes.
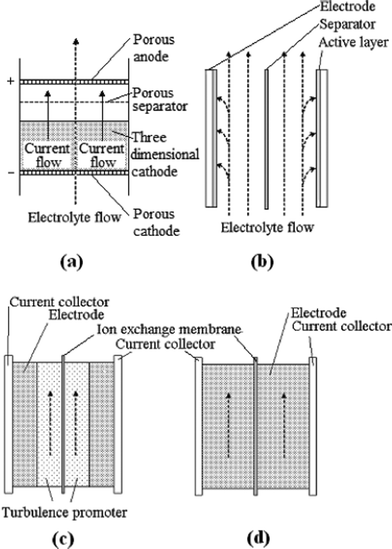 | ||
| Fig. 8 Schematics of (a) a flow-through, (b) a flow-by electrode; (c) two-dimensional and (d) three-dimensional electrode configurations for a redox flow battery98,201 | ||
Carbon polymer composite materials can be held together by polymer binders, such as polyvinylidene fluoride (PVDF), high-density polyethylene (HDPE),185 polyvinyl acetate (PVA)195 and polyolefin213 by injection moulding, which is a low-cost technique and has the advantage of producing large amounts easily. A polymer-impregnated graphite plate can be manufactured by compression moulding the expanded graphite with thermoplastic polymers. Since this moulding technique allows the use of a higher proportion of graphite than the injection moulding method, it has better electrical conductivity. Due to the thermoplastic properties of the polymer used in this technique, the mechanical properties, chemical resistance and thermal stability are superior to those produced by injection moulding. Since longer time is required to cool down the mould before each half plate can be removed, the production cycle requires longer times (typically >10 min). Therefore, the higher costs of this method could hinder its use in RFBs for large-scale applications.214,215 Conductivity of the aforementioned composite materials can be provided by the addition of conductive filler material62 or activated carbon particles.125,216 The high carbon content can lead to poor mechanical properties and some investigations have reported that carbon black filler can cause undesirable reactions, such as gas evolution and water decomposition, especially when the battery is overcharged. The oxidation of such carbon black-filler could also increase the electrical resistance.62
Two-dimensional carbon electrodes were reported to be chemically unstable for the redox reactions of V4+/V5+ (ref. 41, 43, 50, 217, and 218) and Ce3+/Ce4+ (ref. 184 and 219) due to the carbon dioxide evolution.220 Since these reactions occur at highly positive potentials and the local current density at the planar electrode surface underneath the carbon felt can be very high, physical deterioration of the electrode surface can be observed. By mechanically compressing the carbon felt onto the carbon-based current collector used in the positive electrode, the all-vanadium and the divided zinc-cerium systems can operate at a projected current density of >50 mA cm−2 with a voltage efficiency of over 75%.43,184
Due to the large volumetric surface area, typically 240–400 cm2 cm−3 and 5–70 cm2 cm−3 for the carbon felt221 and the reticulated vitreous carbon foam,222 respectively, three-dimensional carbon-based material has been used in various systems. To date, the polyarylonitrile (PAN) based carbon felt electrode is the commonest electrode material used in the all-vanadium system. The PAN based graphite felt has advantages in wide potential range, good electrochemical activity, high chemical stability and low cost. Novel modifications of graphite felt materials have been carried out.223-228 For instance, the graphite felt can be modified by treating it with sulphuric acid followed by heat treatment.48-50 During the acid treatment, surface functional groups, such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH, were formed on the electrode surface, which was found to significantly increase the chemical activity in the highly acidic media.
O and –COOH, were formed on the electrode surface, which was found to significantly increase the chemical activity in the highly acidic media.
Some researchers have further improved the catalytic properties and the conductivity of carbon felts by depositing metals on the electrode surface.48,229 For instance, Sun and Skyllas-Kazacos48 have modified carbon felt electrodes through impregnation or by ion-exchange with solutions containing ions, such as Pt4+, Pd2+, Te4+, Mn2+, In3+, Au4+ and Ir3+. In terms of the electrocatalytic activity and stability, a modified Ir3+ electrode was found to have the best performance when it was used as the positive electrode in all-vanadium system, while a large amount of hydrogen evolution was observed on Pt, Pd and Au modified electrodes. In another study, Fabjan et al.230 reported that RuO2 can improve the reaction rate and decrease the side reactions such as gaseous evolution. More recently, non-precious bismuth231 and manganese oxide (Mn3O4)232 were also found to give improved electro-catalytic activity after modification.
Reticulated vitreous carbon electrodes have also been used in zinc-bromine222 and soluble lead-acid RFBs.77 The porosity of this material is beneficial to retain the solid complex of bromide during battery charge of a zinc-bromine system,233 while the rough surface of the scraped reticulated vitreous carbon in the soluble lead-acid flow battery allows adherent deposits to be formed within the compressed foam structure.77 The use of nanostructured electrode materials, such as carbon nanotubes, graphenes and graphene oxide, has also been introduced in all-vanadium RFBs. To take advantage of the excellent electrical conductivity and mechanical properties of carbon nanotubes, several researchers234,235 suggested a graphite-carbon nanotube composite for VRFB. Despite the great advantages of the carbon nanotubes, the sole use of carbon nanotube in a vanadium reaction was found to give poor reversibility and activity compared to pure graphite. In order to benefit from both graphite and carbon nanotubes, a composite of graphite with 5 wt.% carbon nanotubes has been introduced and exhibited good reversibility and electrical conductivity. In another work, a graphene oxide nanoplatelet electrode (GONP) was suggested for VRFB by Han et al.236 This material does not require tedious synthesis procedures and has good catalytic property for both V2+/V3+ and V4+/V5+ redox reactions due to the functional groups of C–OH and COOH on the electrode surface. Polarization of the GONP can be further reduced after heat treatment at a temperature of 120 °C. Furthermore, the authors have prepared a graphene oxide nanosheets/multi-walled carbon nanotubes hybrid electrode by an electrostatic spray technique and found that the electrode shows excellent electrocatalytic redox reversibility towards VO2+/VO2+ couple, especially for the reduction from VO2+ to VO2+.237
Due to the high cost of platinum, a dimensionally stable anode (DSA) has been used as the positive electrode in all-vanadium42 and zinc-cerium flow batteries.184,196 In general, a DSA electrode is manufactured by coating a titanium substrate with a thin metal or alloy oxide selected from the following group of metals: Mn, Pt, Pd, Os, Rh, Ru and Ir. In the study of Rychcik and Skyllas-Kazacos,41 an IrO2-coated DSA electrode was reported to have good reversibility for the vanadium reactions. In order to discharge a zinc-cerium flow battery at high current densities, such as 50 mA cm−2, a three-dimensional DSA mesh stack electrode, manufactured by spot-welding three pieces of platinised titanium mesh, has been used which operates better than the two-dimensional electrode.184 As shown in Fig. 9, the high surface area of such a platinised titanium mesh stack and the carbon felts allowed a high positive half-cell potential for the reduction of Ce4+ at increased discharge current densities, while a significant potential drop was observed when two-dimensional platinised titanium, graphite and carbon polymer positive electrodes were used at current densities larger than 25 mA cm−2.
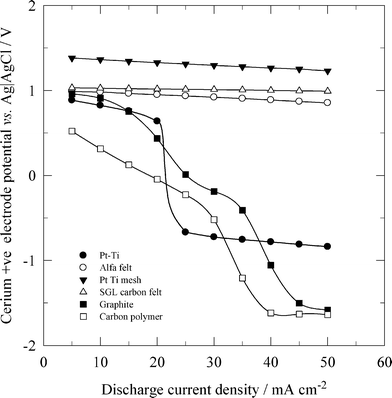 | ||
| Fig. 9 Effect of discharge current density on cerium positive electrode potentials of a zinc-cerium redox flow battery using different positive electrode materials. ● 2-D Pt-Ti, ○ Alfa Aesar carbon felt, ▼ Pt-Ti mesh stack, Δ SGL carbon felt, ■ 2-D graphite and □ 3-D carbon-polymer composite.184 | ||
Apart from the noble metals, some metals having a high overpotential for gas evolution have been used as the substrate electrodes in several RFBs. For example, zinc-nickel and zinc-air RFBs have used a high H2-overpotential cadmium-plated nickel sheet as the negative electrode to reduce the dendritic growth and to suppress hydrogen evolution effectively.86,188 Three-dimensional nickel foam is another metallic electrode, which has been used in sodium polysulfide-bromine146 and soluble lead-acid systems.78 Like other three-dimensional materials, nickel foam provides a large surface area, hence further reducing the polarization. Zhao et al.146 reported that this material is electrocatalytically active for both the negative and positive electrode reactions in a sodium polysulfide-bromine RFB. The small pores (about 150–250 μm) of nickel foam electrodes can allow the electrolyte to flow smoothly within the electrode, which further improves mass-transport at the reaction sites.
5.3 Separator materials
An ideal separator should prevent mixing of the electrolytes and the direct chemical reaction of active species or chemical products, such as electrodeposits from the positive and negative electrolytes, which can result in energy losses. Hence, permeation of the active species and the selectivity of the charge-carrying ions throughout the separator are highly important.52 Typical separators for RFB are the ion exchange membranes, which are produced from polymeric materials with pore diameters of 20 Å.239 In aqueous electrolyte, ion-exchange membranes are designed to conduct positively charged ions (i.e. H+ or Na+) or negatively charged ions (i.e. Cl−, NO3− or SO42−) by means of cationic or anionic exchange membranes, respectively. In a recent paper on lithium-ferricyanide flow battery, a solid electrolyte separator is used to conduct Li+ ions and effectively prevents the aqueous positive electrolyte from reaching the negative electrode.93 The ideal membrane for an RFB should be chemically resistant to aggressive species and pose low area resistivity to minimize the ohmic loss.240,241 Ion exchange capacity (IEC) is often used to measure the charge-carrying ions passing through the membrane and the conductivity of a membrane is determined by various factors, such as thickness, ionic group concentration, the degree of cross-linking, the size and valence of the counter ions.According to Vafiadis and Skyllas-Kazacos,142 there is no direct correlation observed amongst IEC, resistivity, diffusivity and the membrane thickness for the vanadium electrolytes. Electrolyte concentration and ion exchange groups within the membrane can have great influence on water uptake and hence the degree of swelling. With a higher degree of crosslinking, better selectivity was observed with less pronounced swelling or water transfer across the membrane. Although the transfer of vanadium ions through the membrane was found to be highly dependent on the concentration difference between the positive and negative electrolytes,120 cation- and anion-exchange membranes tend to have net volumetric transfer towards the positive and negative compartments, respectively.56 Compared to an anion exchange membrane, the cationic counterpart tends to have lower area resistance per thickness due to the higher mobility of H+ ions compared to anions, such as SO42−. This explains the fact that a cation exchange membrane is the most widely investigated membrane material for VRFB. However, the chemical stability of different ion exchange membranes could vary from each other depending on their chemical structure. In general, ion-exchange membranes based on a perfluorinated backbone, particularly Nafion cation exchange membrane (Dupont, USA), have superior chemical stability to most types of hydrocarbon membranes. However, recent studies suggest that anion exchange membranes could effectively reduce the crossover of vanadium ions in VRFB mainly due to the Donnan exclusion effect,242 which is an electrostatic repulsion between the cation groups within the membrane and the electroactive species. The development of ion-exchange membranes for vanadium RFB has been reviewed by Li et al.11 and concluded that Nafion, although expensive, is still the most used membrane but other alternatives such as modified Nafion and pore filled composite membranes with new polymers and new materials should receive more attention in the future.
As reported by Chieng,242 most of the commercial membranes have good selectively and high ionic conductivity, including Nafion® 112, 117 and 324 (Dupont, USA), K142 (Asahi Chemical, Japan), Selemion® CMV, CMS, AMV, DMV, ASS, DSV, Flemion® CMF, New Selemion® (Asahi Glass, Japan) and RAI® R1010, R4010 (Pall RAI, USA). Despite this, excessive transfer of electrolyte from one compartment to another has been observed with all these membranes. Apart from the membranes mentioned, properties and cycling performance of the other 15 commercial membranes have been evaluated by Vafiais and Skyllas-Kazacos142 for the vanadium-bromine RFB system. As summarized in Table 6, the properties of these membranes are compared with the data for the Nafion® membrane from the literature.70 Although Nafion® 117 and Hipore® are thicker than the others, smaller area resistance and larger permeation of V4+ ions have been observed. Also, higher area resistance was observed with the high IEC membranes, such as the ABT membranes, than those with a low IEC, such as Gore® and Hipore® membranes. These suggested that there is no direct correlation between thickness, IEC, resistivity and diffusivity. Among these membranes, only Nafion®, ABT3, L01854 and M04494 were reported to have acceptable performance in terms of round-trip DC energy efficiency and chemical stability. Since all other membranes degrade rapidly in the vanadium-bromine electrolyte, the cycle life has been limited to less than 40 cycles.142
| Commercial membrane | Manufacturer | Typea | Dry thickness/mm | Area resistance/Ω cm2 | V(IV) permeability/10−7 cm2 min−1 | IECa/mmol g−1 |
|---|---|---|---|---|---|---|
| a CE: cation exchange; IEC: ion exchange capacity; NG: not given. | ||||||
| Nafion® 117 | Dupont, USA | CE | 0.165 | 2.5 | 8.63 | NG |
| Gore L01854 | W.L. Gore & Associates, USA | CE | 0.03 | 0.38 | 0.36 | 0.69 |
| Gore M04494 | W.L. Gore & Associates, USA | CE | 0.04 | 0.41 | 0.96 | 1.00 |
| ABT3 | Australian Battery Tech. & Trading | CE | 0.02 | 3.24 | 0.11 | 6.01 |
| ABT4 | CE | 0.04 | 9.97 | 1.44 | 3.77 | |
| ABT5 | CE | 0.06 | 5.39 | 1.44 | 3.92 | |
| SZ | Guangzhou Delong Technologies Pry, China | CE | 0.13 | 19.03 | 2.34 | 2.5 |
| Hipore® | Asahi Kasei, Japan | Microporous separator | 0.62 | 1.4 | 148 | 1.14 |
Regarding the high cost of Nafion® membranes, some developments have focused on the modified/composite membranes based on non-Nafion® or other non-perfluorinated separators for VRFB as summarized in Table 7. Early approaches include the modification of low cost separators, such as Daramic256,257 and low-density polyethylene (LDPE)56,257,258 by grafting and sulfonation processes. Although the resulting membranes have high IEC and conductivity, the permeability of active species across the membrane is still significant. Therefore, it has been suggested that polymerizable monomers might be grafted onto the matrix of polyvinylidene fluoride (PVDF). One example is the poly(vinylidene fluoride)-graft-poly(styrene sulfonic acid) (PVDF-g-PSSA-PMAn),258 where MAn is the maleic anhydride used to reduce the irradiation dose to the membrane. The PVDF-g-PSSA-PMAn was reported to have good chemical stability and low ion permeability in vanadium electrolyte. Further increasing of the grafting degree could lead to higher water uptake, IEC and conductivity as observed by Luo et al.259 (Table 7). As with polyvinylidene fluoride, other polymer matrices, such as sulfonated poly(arylene thioether ketone) (SPTK) and sulfonated poly(tetrafluorotheylene) (SPTKK),260 have been investigated.
| Modified membrane | Supplier | Typea | Thickness/mm | Area resistance/Ω cm2 | V(IV) permeability/10−7 cm2 min−1 | IECa/mmol g−1 |
|---|---|---|---|---|---|---|
| a CE: cation exchange; AE: anion exchange; IEC: Ion Exchange Capacity; NG: not given. | ||||||
| PVDF-g-PSSA-11 259 | NG | NG | 0.151 | NG | 2.20 | 0.82 |
| PVDF-g-PSSA-22 259 | NG | NG | 0.115 | NG | 2.53 | 1.2 |
| PVDF-g-PSSA-co-PMAn 258 | Kureha Co., Japan | CE | 0.07 | NG | 0.73 | NG |
| ETFE-g-PDMAEMA 70 | Kureha Engineering Ltd, Japan & Acros Organic, USA | AE | 0.070 | 2.3 | 0.36 | NG |
| SPEEK314 | Victrex, PEEK450PF | NG | 0.100 | 1.27 | 2.432 | 1.80 |
| Nafio/SPEEK314 | Nafion: Nafion R-100 Resin PEEK: Victrex, PEEK450PF | CE | 0.100 | 1.6 | 1.928 | 1.67 |
| PSSS-composite (concentration 75 g dm−3)56 | W.R. Grace Co. & Aldrich Chemical Company Inc., USA | NG | NG | 1.09 | 4.48 | NG |
| NG | NG | 1.36 | 3.31 | NG | ||
Due to the electrostatic repulsion between the cation groups of the membrane and the soluble species, anion exchange membranes tend to have lower permeability than their cation counterpart membranes. Typical anion exchange membranes can be synthesized by chloromethylating a polymer substrate with chloromethyl ether and then quaternizing an amination reagent, such as trimethylamine.240,261-263 Early modified anion exchange membranes based on polysulfone (PSf) and polyphenylenesulfidesulfone (PPSS) have been suggested by Hwang and Ohya.264 However, the IEC of this membrane is still less than that of Nafion® 117. Therefore, some researchers70,265 suggested to graft dimethylaminoethyl methacrylate (DMAEMA), an anion monomer, onto ethylene–tetrafluoroethylene (ETFE) and PVDF films by UV-induced grafting. At 40% graft yield, the ETFE-g-PDMAEMA membrane exhibits higher IEC, lower area resistance and less pronounced permeability than those of the Nafion® 117 membrane (Table 7). Despite this, Hwang et al.240 reported that the high degree of crosslinking in anion membranes obtained by accelerated electron radiation can lead to membrane failure due to the reduced tensile stress.
By hot-pressing or immersing the substrate membranes into an aqueous Nafion® solution, multilayered composite membranes can be prepared to increase the chemical resistance. Substrate membranes are often low-cost materials with high thermal conductivity. Due to the low ion permeability, sulfonated polyether ether ketone (SPEEK) has been used as the substrate for the Nafion-SPEEK composite membrane. A thin layer of Nafion® was used to prevent degradation in the electrolyte, while diamine was used to crosslink the sulfonic groups of Nafion® ionomer with the SPEEK ionomer.266 Chen et al.267 reported that a SPEEK membrane not only exhibits high chemical stability in vanadium electrolyte, but also has a round-trip DC energy efficiency similar to that of the Nafion membrane. Many investigations also attempted to blend SPEEK membranes with PTFE,268 WO3,269 SiO2,267 sulfonated poly(ether sulfone (SPES)270 and polysulfone-2-amide-benzimidazole (PSf-ABIm)271 to reduce the permeability of vanadium ions across the membranes.
In order to enhance the battery performance, novel sandwich-type membranes, such as sulfonated poly(ether ether ketone) (SPEEK)/tungstophosphoric acid (TPA)/polypropylene (PP) and sulfonated poly(ether ether ketone) (SPEEK)/perfluorosulfonic acid (PFSA) acid (TPA)/polypropylene (PP) composite membranes have been introduced by Jia et al.,272 which consist of a layer of polypropylene (PP) membrane sandwiched by two layers of SPEEK/TPA membranes. Due to the sandwich design, the battery can operate for longer cycles even when the external layers are detached or out of function. With this membrane, improved round-trip DC energy efficiency and permeability have also been observed in an all-vanadium flow battery. PTFE reinforced SPEEK/PTFE composite membranes were reported to have low water uptake and swelling ratio compared to pristine SPEEK membranes.268 The VRFB single cell assembled with SPEEK/PTFE membranes exhibit higher coulombic efficiency and energy efficiency, and longer cycle life than that with SPEEK membranes. Other low-cost separators including sulfonated poly phenylsulfonate membrane (Radel)126,273 and sulfonated poly(arylene thioether) (SPTK)260 have also been evaluated but long-term stabilities in vanadium electrolytes need to be further investigated.
Recently, Zhang et al.122,274 have reported that polyacrylonitrile nanofiltration membranes could enhance the efficiency of VRFB, making them a more viable tool for large-scale energy storage. The idea is based on tuning the vanadium/proton selectivity via pore size exclusion, which provides a totally new concept in the design and preparation of the membrane separator. This can remove the need for the traditional restriction from the ion exchange resin and allow many more material options for RFB application. They reported that VRFB assembled with silica modified nanofiltration membranes can achieve a coulombic efficiency of 98% and an energy efficiency of 79% at a current density of 80 mA cm−2,274 which is promising for the replacement of commercial Nafion membranes at a much lower cost.
5.4 Flow distributors and turbulence promoters
Flow distributors and turbulence promoters are often employed to enhance the mass transport and promote the exchange of active species between the bulk electrolyte and the electrode surface.275 Turbulence promoters are usually in the form of insulating nets or ribs but mesh, foam or fibrous bed electrodes sometimes can also function as turbulence promoters (Fig. 10).203 Frías-Ferrer et al.200,276 have evaluated the mass transport of four types of polyvinylchloride (PVC) turbulence promoters within the rectangular channel of the filter-press reactors. The effect of such turbulence promoters was found to be more pronounced in large systems as indicated by a significant increase in the global mass transport coefficient, while contrary effects were observed in small systems as the electrolyte flow may not be fully developed due to the entrance/exit manifold effects. At increased Reynolds numbers, the decrease in mass transport coefficient was reported to have no correlation with the projected area of the open spaces or the surface blocked by the promoter strands in contact with the electrodes, but could be due to the geometry of the fibres (or filaments), such as shape and size that modify the flow regime.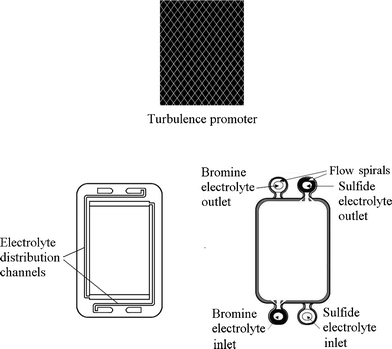 | ||
| Fig. 10 Schematics of turbulence promoter, flow frame and spiral-shaped flow paths.276,277 | ||
As shown in Fig. 10, the entrance of the electrolyte in the manifolds are often aligned to form the inlet and outlet of the electrolyte in the battery components and the change in geometry at the inlet and outlet can generate a high degree of turbulence.276 Since the electrolyte in the manifold can lead to shunt (bypass or leakage) current in a battery stack, the electrolyte flow channels in the electrode frame are often designed to be long and narrow to increase the ionic resistance of the electrolyte. In the Regenesys® system the shunt current is reduced by creating a labyrinth in the spiral-shaped electrolyte paths.277 Tsuda et al.278 suggested that flushing an inert gas bubble through the pipe can reduce the shunt current by breaking the ionic contact in the electrolyte stream, however the introduction of gas might have an impact on the mass transport, which needs to be assessed.
The reaction environment in a filter-press sodium polysulfide-bromine battery containing spiral-shaped paths in the manifolds has been investigated; the pressure drop across the compartments was higher than that in the conventional filter-press reactors, such as the FM01-LC, as the flow was restricted by the narrow spiral path in the manifold.279Fig. 11 shows that both the mass transport coefficient and pressure drop in the bromine compartment of the sodium polysulfide-bromine RFB increase significantly with mean linear flow velocity. Although the high pressure drop is often attributed to increased mass transport, it is not favourable as more powerful pumps are needed and leads to more expensive cost and power consumption.277
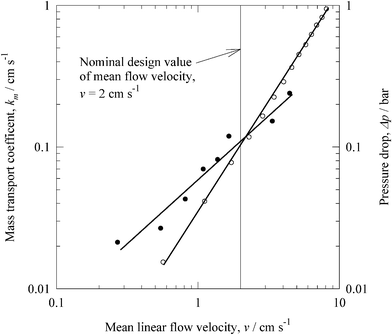 | ||
| Fig. 11 Log-plots of (a) ● mass transport coefficient and (b) ○ pressure drop vs. mean linear electrolyte velocity for the bromine compartment of the polysulfide-bromine redox flow battery. Electrolyte: 1 mol dm−3 NaBr in 0.5 mol dm−3 Na2SO4 at pH 2.277 | ||
5.5 Electrolyte flow circuit
RFB stores energy in the electroactive species, which are circulated during operation and storage tanks, pipes, pumps, and control valves are the key components. Control valves can be operated by a microprocessor and are used to control the flow rate of the electrolyte carried to and from the corresponding tanks. Alternatively, in some systems the tanks can be divided into two sealed compartments by a floatable device which allows the extraction of the spent electrolyte from one compartment while feeding the fresh electrolyte to the other compartment. A system of several taps and valves allows recharging of the tanks with fresh electrolyte without the need to remove all the spent electrolyte and flux the tanks with fresh electrolyte.246For large-scale RFBs, centrifugal pumps are installed and are controlled by variable speed motors. In general, pump efficiency is about 50%,278 which accounts for about 2–3% of the overall energy efficiency.280 At a low concentration of the electroactive species, a larger amount of electrolyte and higher flow rates are required, which can result in larger pump losses.281 Tsuda et al. suggested that the pump loss can be significant for RFBs under low current conditions, and this can be reduced by adopting intermittent pump operation. Under the intermittent pump operation, the evaluation of the energy efficiency increases significantly compared with continuous flow operation.278
The pump rates for the negative and positive electrolytes are highly dependent on the operating conditions. For instance, some researchers282,283 have developed a control system that uses the open-circuit electrode potentials and temperature measurements to determine the required pump speed. Due to the variation in electrolyte density and the viscosity at different state of charge, this method is not very accurate. Hence, other approaches including conductivity and spectrophotometric measurements have been used to monitor the state of the battery.284 The incorporation of flow meters and pressure transducers to measure the flow rates and pressures, respectively, makes the system more complicated and costly. In order to minimize the energy consumption associated with the pumps, pumps are switched off when the battery is idle and laminar electrolyte flow is used to minimize the pressure drops.285 By optimizing the electrolyte flow rates at different states of charge, the system efficiencies were able to improve by as much as 8% while maintaining the capacity.286
5.6 Modelling and simulation
Driven by the great interest in the development and applications of RFB for large-scale energy storage, the past thirty years have seen a large academic effort into the development of mathematical modeling180 to rationalize and predict battery behaviour and system performance. This work has mainly involved four RFB systems. The first involves the Zn/Br2 system which was extensively simulated in the 1980s due to the early commercial interest.178–181 Various types of models have been used to predict the cell and stack performance, including partial differential equation models to predict current and potential distributions, an algebraic model for shunt currents and associated energy losses, and ordinary differential equation models to predict the energy efficiency of the cell as a function of the state of charge. These models provided useful design criteria for improving cell performance. The second system is based on an Fe/Cr RFB which was described by Fedkiw and Watts.287 The model, based on porous electrode theory, incorporates redox reaction kinetics, mass transport, and ohmic effects as well as the parasitic hydrogen reaction which occurs in the chromium electrode. The model has been applied to the NASA Fe/Cr system. Scale-up studies of the Fe/Cr RFB based on shunt current and flow distribution analyses have been performed by Codina and Aldaz.112,113 Another simulated system is the bromine/polysulphide RFB due to Roberts et al.288,289 which was aimed at predicting the battery's technical and commercial performance, species concentration, current distribution and electrolyte deterioration for the Regenesys system. The final and most extensively modelled system is the VRFB;118,119,290–294 aspects studied include concentration, temperature, electrolyte flow rate, electrode porosity, gas evolution, self-discharge process, and open-circuit voltage. For more detailed treatments, the reader is referred to the reviews by Weber et al.13 and Kear et al.126. Applications
Since the 1970s, various RFBs have been introduced but only all-vanadium, vanadium-polyhalide, zinc-bromine, zinc-cerium and bromine-polysulfide batteries have been tested or commercialized on a large-scale. The commercial specifications of these RFBs are summarized in Table 8.6,295–298 Among the four RFBs, the VRFB has the highest efficiency and the largest life cycle, while zinc-cerium and bromide-polysulfide systems have advantages in power density and cost, respectively. Fig. 12 shows the plots of the cell voltage vs. time of the charge-discharge cycle of the four systems: all-vanadium (30 mA cm−2 for 2 h),44 zinc-bromine (15 mA cm−2 for 10 h),164 bromine-polysulfide (40 mA cm−2 for 30 min),213 and the undivided zinc-cerium system (20 mA cm−2 for 30 min).17 Up to now, only all-vanadium and zinc-bromine systems have been used in applications, such as load levelling, power quality control applications, facilitating renewable energy deployment and energy storage for electric vehicles.299 | ||
| Fig. 12 Cell voltage vs. time response during charge/discharge for all-vanadium (30 mA cm−2 for 2 h),44 zinc-bromine (15 mA cm−2 for 10 h),164 bromine-polysulfide (40 mA cm−2 for 30 mins)213 and the undivided zinc-cerium (20 mA cm−2 for 30 mins)17 redox flow batteries. | ||
| All-vanadium | Zinc-bromine | Bromine-polysulfide | Zinc-cerium | |
|---|---|---|---|---|
| Cell open-circuit voltage/V | 1.4 | 1.8 | 1.5 | 2.4 |
| Energy density/W L−1 | 16–33 | Up to 60 | 20–30 | 12–20 |
| Cycle round-trip DC energy efficiency/% | 78–80 | 65–75 | 60–75 | 63 |
| Cycle life (cycles) | >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
>2000 |
>2000 |
NG |
| Operation T/°C | 30 | 50 | 35 | 60 |
| Total system cost/$ kW−1 | 1828 | 1044 | 639 | 750 |
| Cost attribution of storage module/% | 80 | 80 | 60 | 50 |
| Typical size range/MWh | 0.5–5 | 0.01–5 | 0–120 | NG |
| Unit design life time/years | 5–10 | 5–10 | 15 | 15 |
| Stage of development | Demonstration/Commercial units | Demonstration /Commercial units | Demonstration | Demonstration |
| Major companies involved | Prudent Energy (formerly Pinnacle, VRB), Sumitomo Electric Industries, Cellstorm GmbH, Mitsubishi Chemical, Cellenium, V-fuel Pty, Ashlawn Energy and Re-fuel Tech. | ZBB, Premium Power, Kyushu Electric Power, Meidensha, Primus Power and Redflow | VRB (using Regenesys technology), further acquired by Prudent Energy | Plurion |
| Number of installations | >20 | >10 | 2 | 1 |
| Largest installation | 4 MW/6 MWh in Subaru windfarm, Hokkaido by Sumitomo Electric Industries | 1 MW in Kyushu, by Kyushu Electric Power | 15 MW in Little Barford, UK by Regenesys Technology | 2 kW–1 MW testing facility in Glenrothes, Scotland by Plurion |
6.1 Load levelling
Large-scale energy storage systems allow the use of excess power and avoid high cost power plants to cycle on and off. By employing an RFB, bottlenecks within the transmission system can be relieved; this can result in reduced power transmission losses and more reliable electrical power. The main installations of RFBs used in load-levelling and uninterruptible power supply (UPS) applications are summarized in Table 9.| System | Company | Customer | Basic specification | Application | Installation date |
|---|---|---|---|---|---|
| All-vanadium | VRB Power System | Stellenbosch University, South Africa | 250 kW/500 kWh | Load levelling and UPS | 2001 |
| Castle Valley, US | 250 kW/2 mWh | Load levelling | Feb 2004 | ||
| Sumitomo Electric Industries | Kansai Electric Power, Japan | 450 kW/900 kWh | Load levelling | 1996 | |
| Urban Ace Awaza Building, Japan | 100 kW/800 kWh | Office building load levelling | 2000 | ||
| Kansai Electric Power, Japan | 200 kW/1.6 MW | Load levelling | 2000 | ||
| Tottori Sanyo Electric, Japan | AC 3 MW × 15 s. | UPS | Apr 2001 | ||
| Kwansei Gakuin University, Japan | AC 500 kW × 10 h | Load levelling | Nov 2001 | ||
| Cesi, Italy | 42 kW × 2 h | Load levelling | Nov 2001 | ||
| Mitsubishi Chemicals | Kashima-Kita Electric Power Station, Japan | 200 kW/800 kWh | Load levelling | 1996 | |
| Premium Power | Nairn substation, Scotland | 150 kWh | Load levelling | 2009 | |
| Prudent Energy | Oxnard, California | 500 kW/3.6 MWh | Load levelling | 2011 | |
| Ashlawn Energy, V-fuel | Painesville Power Plant, Ohio | 1 MW/8 MWh | Smart-grid/load levelling | 2011 | |
| Zinc-bromine | ZBB Energy | Detroit Edison, US | 400 kWh | Load levelling | Jun 2001 |
| United Energy, Melbourne, Australia | 200 kWh | Demonstration for network storage applications | Nov 2001 | ||
| Nunawading Electrical Distribution Substation in Box Hill, Australia | 400 kWh | Load levelling | 2001 | ||
| Pacific Gas and Electric Co., US | 2 MWh | Peak power capacity | Oct 2005 | ||
| Kyushu Electric Power & Meidensha | Imajuku substation in Kyushu Electric Power, Japan | 1 MW/4 MWh | Electric-utility applications | 1990 | |
| Bromine-polysulphide- | Regenesys Technologies | Little Barford Power Station, UK | 15 MW/120 MWh | Load levelling | 2000 |
| Tennessee Valley Authority, US | 15 MW/120 MWh | Load levelling | 2001 | ||
| Zinc-cerium | Plurion | Glenrothes, Scotland | 2 kW–1MW | Testing facility | 2007 |
Early installations of RFBs started in Japan and Australia. In 1990, a 1 MW/4 MWh zinc-bromine battery has been installed in Imajuku, Fukuoka, Japan, which was under the ‘Moonlight Project’ sponsored by the Japanese government.33,36 Since 1994, more than 20 all-vanadium RFBs have been installed. The first all-vanadium flow battery was a 15 kWh installed by the University of New South Wales in a demonstration solar house in Thailand.10,300 In Japan, a number of vanadium RFBs have been installed by Sumitomo Electric Industries (SEI)301 and one has been installed by Mitsubishi Chemical in 1996.69
Innogy plc's bromine-polysulfide system (Regenesys®) focused on a multi-MW system. In the early 2000s, two 15 MW/120 MWh Regenesys® flow batteries were under construction in Little Barford, Cambridgeshire, UK and in Columbus, Mississippi, USA. On completion, these would have been the largest RFB energy storage in the world. As shown in Fig. 13a, the 15 MW plant in Mississippi comprised of 120100 kW filter-press cell stacks (Fig. 13b), each having 200 bipolar electrodes. The electrolyte tanks of such system were 30 feet tall and about 65 feet in diameter. The tank capacities for sodium bromide and sodium polysulfide were as high as 1798000 and 21578000 litres, respectively.73,74
 | ||
| Fig. 13 Photographs of (a) 15 MW bromine polysulfide (Regenesys® Technologies) under construction in Little Barford, UK in 2001. (b) Three sizes of 1.4 kW, 8.1 kW and 100 kW modular cells (S, M, XL series) developed by Regenesys Technologies.73,74 | ||
6.2 Power quality control applications
Failure of an electrical power system during power generation often leads to the collapse of the grid system, which can be costly as well as devastating. As a result, fast stabilization is necessary. RFBs are favourable in these situations as their response time to power demand can be less than 1 min and the maximum short-time overload output can be several times that of the rated capacity. This makes RFBs attractive for both voltage and frequency control.69 RFBs can be used as a battery-backed UPS system for protection against faults on the power utility transmission and distribution systems.Energy storage for the UPS system is to provide electricity during power failure, which is generally used for orderly shutdown of computer systems or switch-on of the backup generator. Since diesel generators are often difficult to switch on, longer discharge times of energy storage system are needed. Compared to sealed lead-acid batteries, RFBs have the advantage of longer discharge times. Some of the RFBs, such as those storing energy in the electrolytes, have a system capacity that can be increased easily by using more electrolyte volume and larger tanks. In addition, RFBs can be restarted from idle within 1 min by pumping electrolyte into the cell stacks.280
The installations of RFBs in UPS applications have been mainly VRFBs by SEI in Japan.301 Since 2001, SEI has installed a number of VRFBs in high-tech factories and office buildings. One prominent example is the 3 MW × 1.5 s plus a 1.5 MW × 1 h system at the liquid crystal manufacturing plant of Tottori Sanyo Electric in 2001. This system not only serves as an UPS, but also for load levelling. Other examples of load levelling and UPS back-up include the early installation of a 250 kW/500 kWh VRFB power by VRB in 2001 at Stellenbosch University for ESKOM Power Corporation, South Africa.69
6.3 Coupling with renewable energy sources
Due to the large capacity and the long discharge time, flow batteries are attractive when coupling with renewable energy sources. As the energy production from renewable energy sources are usually intermittent and are not connected to the grid, energy storage is hence important. The rapidly growing demand for energy generated by renewable energy sources has given rise to massive market opportunities.| System | Company | Customer | Basic specification | Start date |
|---|---|---|---|---|
| All-vanadium | University of South New Wales, Thai Gypsum | Solar Demonstration House, Thailand | 5 kW/12 kWh | 1994 |
| Sumitomo Electric Industries | Obayashi, Japan | DC 30 kW × 8 h | Apr 2001 | |
| VRB Power System | University of Aalborg, Denmark | 5 kW × 4 h | 2006 | |
| Risø TDU, Denmark | 15 kW × 8 h | May 2007 | ||
| Prudent Energy | Zhangbei, Heibei, China | 1 MWh | May 2011 | |
| California Public Utilities Commission | 100 kW/300 kWh | 2011 | ||
| Zinc-bromine | ZBB Energy | Australian Inland Energy, Australia | 500 kWh | Jun 2002 |
| Power Light, US | 2 × 50 kWh | Nov 2003 | ||
| CSIRO research center, Australia | 500 kWh | Dec 2009 | ||
| Redflow | University of Queensland, Australia | 12 × 120 kWh | Apr 2011 | |
| Department of Energy, US | Albuquerque, New Mexico, US | 2.8 MWh | 2011 |
| System | Company | Customer | Basic specification (nominal DC power and energy) | Start date |
|---|---|---|---|---|
| All-vanadium | VRB Power System | Hydro Tasmania, Japan | 200 kW × 4 h | Nov 2003 |
| Tapbury Management Ireland | 2 MW × 6 h | Aug 2006 | ||
| University of Aalborg, Denmark | 5 kW × 4 h | 2006 | ||
| Risø TDU, Denmark | 15 kW × 8 h | May 2007 | ||
| Prudent Energy | Zhangbei, Heibei, China | 1 MWh | May 2011 | |
| Sumitomo Electric Industries | Hokkaido Electric Power Wind Farm, Japan | 170 kW/1 MWh | 2001 | |
| Institute of Applied Energy, Japan | AC 170 kW × 6 h | Mar 2001 | ||
| J Power at Subaru Wind Farm, Hokkaido, Japan | 4 MW/6MWh | 2005 | ||
| Pinnacle VRB | Hydro Tasmania, King island, Australia | 250 kW/1MWh | 2003 | |
| Zinc-bromine | ZBB Energy | Dundalk Institute of Technology, Ireland | 125 kW/500 kWh | Dec 2008 |
In Japan and Ireland, several RFBs have been installed with wind turbines since 2000. The well-known ones include the early installation of a 170 kW/1 MWh VRFB by Sumitomo Electric Power in Hokkaido, Japan, while SEI recently installed another 4 MW/6 MWh VRFB in Hokkaido for a Subaru wind farm, being the largest installation of VRFB so far.69 Apart from SEI, the largest RFBs coupled with wind turbines are the 2 MW x 6 h VRFB installed by VRB at the Sorne Hill, Ireland in 2006.304 Although most of the large-scale RFBs coupled with wind power were installed by SEI, smaller scales of vanadium and zinc-bromine flow batteries coupled to a wind farm have been installed by manufacturers, such as Pinnacle69 and ZBB.305 In 2011, Prudent Energy installed a 1 MWh all-vanadium battery sized at a rated power 500 kW for the National Wind Power Integration Research and Test Center of China in Zhangbei, Hebei province.306
6.4 Electric vehicles
As early as 1980, a 50 kWh zinc-chlorine RFB was investigated by the Energy Development Associates (EDA) for a vehicle power system to compete with the lead-acid battery.33,307 Meanwhile, the Studiengesellschaft für Energiespeicher and Antriebssysteme (SEA, now PowerCell of Boston, GmbH) in Mürzzuschlag, Austria, has been developing zinc/bromine batteries for electric vehicles since 1983 and has produced batteries with capacities ranging between 5 and 45 kWh. A SEA 45 kWh/216 V battery has been installed in a Volkswagen bus, which the Austrian Postal Service has been using. An electric vehicle with a 15 kWh/114 V battery was designed by Hotzenblitz (Germany).36Electric vehicles with a 35 kWh zinc-bromine battery were launched by the University of California in the 1990s.308,309 In Japan, a zinc-bromine battery was installed by the Toyota Motor Corporation in an electric vehicle, namely EV-3036 and, in Australia, an electric golf-cart with a 36 V, 3.9 kWh vanadium battery prototype was demonstrated at the University of South Wales in 1994. The golf cart was under road-testing for more than two years and was reported to have a maximum speed of 19 km h−1. The total vehicle weight, including two passengers was in excess of 400 kg.94 Since energy is stored in the electrolyte, 24 h operation of an electric vehicle is possible if the electrolyte is refuelled every 4 or 5 h.310 However, the low energy density of VRFB remains the main challenge. Therefore, the recent larger specific energy density of vanadium-air166 and lithium flow batteries90,91 could be promising for longer mileages of electric vehicles but the safety conditions of using a lithium flow battery still need to be identified. Previous progress and challenges of RFBs in hybrid vehicle applications have been discussed in detail by Mohammed et al.311
7. Summary and future R&D needs
As reviewed, the key attribute of a conventional RFB (all liquid phases) is the ability to separate power and capacity where the energy storage capacity can be increased by simply using larger volumes of electrolytes while other types of RFBs each have their own unique technological advances. RFBs can have a discharge time, ranging from minutes to many hours, and most of them are capable of overload and total discharge without any risk of damage. Therefore, the combination of flexibility, rapid response, depth of discharge and mobility etc. makes RFBs an attractive candidate for electrical energy storage (EES) systems as the EES system allows for flexibility in power generation and distribution, improving the economic efficiency, and making the grid more reliable and robust. Particularly, RFBs are more attractive when coupling with the ever-growing renewable energy sources as energy production from these sources is usually intermittent and fluctuating. Yet, the rapid growing demand for energy generated from renewable energy sources has given rise to a massive market opportunity.For broad market penetration, RFB technologies, however, have to demonstrate themselves to be cost-competitive. High cost equally means high energy consumption. If an RFB technology stands at the high cost, it would be meaningless to use in coupling with renewable energy sources to store/release the energy. In 2011, the US Department of Energy set clear near-term (within 5 years) and longer-term targets for battery storage technologies including RFBs. In the former case, these need to have a system capital cost under $250 kW−1 with a system efficiency of >75% whereas in the longer term, a capital cost of $150 kWh−1 and a system efficiency of over >80% are required.2 Additionally, as a utility asset, storage technologies are required for a lifetime of at least 10 years or longer. As is already known, over the last four decades, a number of RFB systems with potential applications have been introduced and the systems, such as VRFB, zinc-bromine, and polysulfide-bromine, have been tested or commercialized on a large-scale. However, to date none of these systems could confidently be declared to reach the cost level required for the absolute practical applications. For example, the most developed VRFB system is still expensive for broad market penetration, which is evaluated at $500 kWh−1,9 because of its dependence on expensive vanadium, ionic exchange membrane, and other components. In short, the cost-competitiveness and effectiveness should be kept as one of the key objectives in R&D work for RFB technology if aiming to win in practical applications over other EES technologies.
Fundamental studies on chemistry and kinetics are necessary for many RFB systems. In principle, many redox couples can be employed in an RFB. It is, therefore, not surprising that new RFB systems are often proposed and reported with promising characteristics and voltage/energy efficiencies. These systems were usually based on small lab-cells and often the long-term cycle performance (e.g. reliability and durability) is not considered. In general, many RFB systems encountered some un-addressed problems on the road to system-scaling-up. An obvious example is that in 1981 when NASA halted the Fe/Cr system-level effort and redirected the emphasis onto fundamental studies of RFBs. Another one would be the development of bromine polysulfide RFB systems at Regenesys® Technologies (UK). In fact, the electrochemical reactions in an RFB system are usually more complicated than the balanced stoichiometric equations presented. They involve reactions at or near the electrode surface, mechanisms of charge transport and crossover in the ionic exchange membrane, and behaviour of active species in a flowing electrolyte environment. Moreover, the operational parameters, including electrolyte concentration, additives, current density, temperature etc., are all the influential factors contributing to the complexity of RFB chemistry. Hence, more research work on fundamental studies of RFBs would result in a better understanding of RFBs and well optimized RFB systems.
Electrochemical engineering and design in RFB development is another area which needs to be highlighted. Electrodes, ion-exchange membranes, cells, and stack and system design are critical to improve the performance and economy of the RFB technologies. Good electrode structure should have high electrocatalytic activity and high surface area, and should be robust and capable of low-cost and volume production. Improved ion-exchange membranes should have better selectivity, controlled solvent transport, low-cost and high stability. Further investigations on the use of bipolar electrodes in a stack are necessary to scale-up the battery and to increase the power density and energy outputs. Integration of electrochemical reactors with other devices and unit processes (e.g. optical-electrochemical sensors and combined adsorption/electrochemical treatment of soluble contaminants) is also crucial in an RFB system.
Modelling and simulation are certainly of great importance but until now it has been a low priority in RFB technologies. Excellent modelling and simulation work would bring those benefits to RFB technology, including building up approximate and detailed mathematical models to understand the effects of variations of the entire system and ancillary equipment; examining the proposed mechanism of electrochemical and chemical reactions and their kinetics against the experimental data; avoiding working in hazardous environments as many RFB systems utilize toxic species; providing simulated long-term reliability and durability test; and reducing the R&D cost.
Finally, in terms of an environmentally attractive technology, attention should be paid to the following specific areas: avoidance of hazardous chemicals and materials; limiting electrical leakage currents and preventing electrolyte escape in the large-scale systems; use of biodegradable polymers in stack construction; consideration of problems of materials degradation and corrosion of electrodes, membranes and other cell/stack components. Unfortunately, there are few publications which address the issues of environmental compatibility and energy/materials sustainability of RFB technologies.
8 Abbreviations
| AE | Anion exchange |
| CE | Cation exchange |
| DC | Direct current |
| DMAEMA | Dimethylaminoethyl methacrylate |
| DSA | Dimensionally stable anode |
| DTPA | Diethylenetriaminepentaacetate |
| EDA | Energy Development Association |
| EDTA | Ethylenediaminetetraacetate |
| EES | Electrical energy storage |
| EPPD | Ethylene-polypropylene-diene |
| ETFE | Ethylene-tetrafluoroethylene |
| GDE | Gas diffusion electrode |
| GONP | Graphene oxide nanoplatelet |
| HDPE | High-density polyethylene |
| IEC | Ion exchange capacity |
| NASA | National Aeronautics and Space Administration |
| OCV | Open-circuit voltage |
| PAN | Polyarylonitrile |
| PDDA | Polydiallydimethyl-ammonium |
| PEI | Polyethylenimine |
| PP | Polypropylene |
| PPSS | Polyphenylenesulfidesulfone |
| PSS | Polystyrene sulfonate |
| PSSA | Polystyrene sulphonic acid |
| PTFE | Polytetrafluoroethylene |
| PVA | Polyvinyl acetate |
| PVC | Polyvinyl chloride |
| PVDF | Polyvinylidene fluoride |
| RFB | Redox flow battery |
| SCE | Saturated calomel electrode |
| SEA | Studiengesellschaft für Energiespeicher and Antriebssysteme |
| SEI | Sumitomo Electric Industries |
| SHE | Standard hydrogen electrode |
| SOC | State of charge |
| SPEEK | Sulfonated poly(ether ether ketone) |
| SPTK | Sulfonated poly(arylene thioether ketone) |
| TEA | Triethanolamine |
| TPA | Tungstophosphoric acid |
| UPS | Uninterruptible power supply |
| VBC | Vinylbenzyl chloride |
| VRFB | Vanadium redox flow battery |
| ZBB | Zinc bromine battery |
Acknowledgements
This review has benefited from an invited paper “Progress & Challenges in the Development of Flow Battery Technology” delivered by Professor Frank Walsh to the First International Flow Battery Forum, IFBF2010, Vienna, 15–16, June 2010.References
- Energy Storage on the Grid Pike, Research Report, 2010 Search PubMed.
- Energy Storage Program Planning Document, US Department of Energy, Office of Electricity Delivery & Energy Reliability, 2011 Search PubMed.
- Meeting the Energy Challenge, A White Paper on Energy, Department of Trade and Industry, London, 2007 Search PubMed.
- Electricity Storage, Parliamentary office of Science and technology Report 306 London, 2008, 1–4 Search PubMed.
- The Future of Energy Storage Technologies and Policy, Royal Academy of Engineering (London) and Chinese Academy of Sciences (Beijing), 2012 Search PubMed.
- S. Eckroad, Handbook of Energy Storage for Transmission or Distribution Applications, Electric Power Research Institute Report 1007189, California, USA, 2002 Search PubMed.
- J. Bartolozzi, J. Power Sources, 1989, 27, 219–234 CrossRef CAS.
- C. Ponce de León, A. Frías-Ferrer, J. González-García, D. A. Szánto and F. C. Walsh, J. Power Sources, 2006, 160, 716–732 CrossRef CAS.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS.
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–79 CrossRef CAS.
- X. F. Li, H. M. Zhang, Z. S. Mai, H. Z. Zhang and I. Vankelecom, Energy Environ. Sci., 2011, 4, 1147–1160 RSC.
- G. Kear, A. A. Shah and F. C. Walsh, Int. J. Energy Res., 2012, 36, 1105 Search PubMed.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. H. Liu, J. Appl. Electrochem., 2011, 41, 1137–1164 CrossRef CAS.
- A. Hazza, D. Pletcher and R. Wills, Phys. Chem. Chem. Phys., 2004, 6, 1773–1778 RSC.
- J. Cheng, L. Zhang, Y. S. Yang, Y. H. Wen, G. P. Cao and X. D. Wang, Electrochem. Commun., 2007, 9, 2639–2642 Search PubMed.
- J. Q. Pan, Y. Z. Sun, J. Cheng, Y. H. Wen, Y. S. Yang and P. Y. Wan, Electrochem. Commun., 2008, 10, 1226–1229 Search PubMed.
- P. K. Leung, C. Ponce de León and F. C. Walsh, Electrochem. Commun., 2011, 13, 770–773 Search PubMed.
- R. L. Clarke, B. J. Dougherty, S. Harrison, P. J. Millington and S. Mohanta, US 2004/0202925 A1, 2004.
- R. L. Clarke, B. J. Dougherty, S. Harrison, J. P. Millington and S. Mohanta, US 2006/0063065 A1, 2005.
- ITI Energy Powerpoint of "Strengthening Scottish Bussiness Roadshow" in Lighthouse, Glasgow, 2006.
- L. H. Thaller, Electrically rechargeable redox flow cells, NASA, 1974, TM X 71540 Search PubMed.
- L. H. Thaller, US Pat. 3996064, 1976.
- L. H. Thaller, Redox flow cell energy storage systems, NASA, 1979, TM-79143, DOE/NASA/1002-79/3 Search PubMed.
- L. H. Thaller, Recent advances in redox flow cell storage systems, NASA, 1979, TM-79186, DOE/NASA/1002-79 Search PubMed.
- M. A. Hoberecht and L. H. Thaller, NASA, 1981, TM-82707, DOE/NASA/12726-13 Search PubMed.
- N. H. Hagedorn and L. H. Thaller, NASA, 1982, TM-82854, DOE/NASA/12726-16 Search PubMed.
- N. Hagedorn, M. A. Hoberecht and L. H. Thaller, NASA, 1982TM-82686, DOE/NASA/12726-11 Search PubMed.
- D. A. Johnson and M. A. Reid, NASA, 1982, TM-82913, DOE/NASA/12726-17 Search PubMed.
- J. S. Ling, NASA, 1984, TM-87074, DOE/NASA, 12726-26 Search PubMed.
- L. Swette and V. Jalan, NASA, 1984, CR-174724, DOE/NASA/0262-1 Search PubMed.
- R. F. Gahn, N. H. Hagedorn and J. A. Johnson, NASA, 1985, TM-87034, NASA, DOE, US Search PubMed.
- N. H. Hagedorn, NASA, 1984, TM-83677, DOE/NASA/12726-24 Search PubMed.
- P. C. Symons and P. C. Butler, Handbook of Batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill, 2002, vol. 37 Search PubMed.
- T. Shigematsu, SEI Technical Rev., 2011, 73, 5–13 Search PubMed.
- Deeya Energy, in Redox Flow Battery Technology, http://www.deeyaenergy.com/about/board-of-directors/deeyas-flow-battery-technology/ Search PubMed.
- P. C. Butler, P. A. Eidler, P. G. Grimes, S. E. Klassen, R. C. Miles, Handbook of Batteries, ed. D. Linden and T. B. Reddy, 3rd edn, McGraw-Hill, 2002, 39 Search PubMed.
- M. Skyllas-Kazacos, M. Rychick and R. Robins, US Pat., 4786567, 1988.
- E. Sum, M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1985, 16, 85–95 CrossRef CAS.
- E. Sum and M. Skyllas-Kazacos, J. Power Sources, 1985, 15, 179–190 CrossRef CAS.
- M. Skyllas-Kazacos, M. Rychcik, P. G. Robins, A. G. Fane and M. A. Green, J. Electrochem. Soc., 1986, 133, 1057–1058 CrossRef CAS.
- M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1987, 19, 45–54 Search PubMed.
- M. Skyllas-Kazacos and F. Grossmith, J. Electrochem. Soc., 1987, 134, 2950–2953 CrossRef CAS.
- M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1988, 22, 59–67 Search PubMed.
- M. Kazacos and M. Skyllas-Kazacos, J. Electrochem. Soc., 1989, 136, 2759–2760 CAS.
- M. Kazacos, M. Cheng and M. Skyllas-Kazacos, J. Appl. Electrochem., 1990, 20, 463–467 CrossRef CAS.
- M. Skyllas-Kazacos, D. Kasherman, D. R. Hong and M. Kazacos, J. Power Sources, 1991, 35, 399–404 CrossRef CAS.
- S. Zhong, M. Kazacos, R. P. Burford and M. Skyllas-Kazacos, J. Power Sources, 1991, 36, 29–43 CrossRef CAS.
- B. Sun and M. Skyllas-Kazacos, Electrochim. Acta, 1991, 36, 513–517 CrossRef CAS.
- B. Sun and M. Skyllas-Kazacos, Electrochim. Acta, 1992, 37, 2459–2465 CrossRef CAS.
- B. T. Sun and M. Skyllas-Kazacos, Electrochim. Acta, 1992, 37, 1253–1269 CrossRef CAS.
- S. Zhong and M. Skyllas-Kazacos, J. Power Sources, 1992, 39, 1–9 Search PubMed.
- S. C. Chieng, M. Kazacos and M. Skyllas-Kazacos, J. Power Sources, 1992, 39, 11–19 CrossRef CAS.
- S. Zhong, C. Padeste, M. Kazacos and M. Skyllas-Kazacos, J. Power Sources, 1993, 45, 29–41 CrossRef CAS.
- F. Mohammadi, P. Timbrell, S. Zhong, C. Padeste and M. Skyllas-Kazacos, J. Power Sources, 1994, 52, 61–68 Search PubMed.
- V. Haddadi-Asl, M. Kazacos and M. Skyllas-Kazacos, J. Appl. Electrochem., 1995, 25, 29–33 Search PubMed.
- T. Mohammadi and M. Skyllas-Kazacos, J. Power Sources, 1995, 56, 91–96 CrossRef CAS.
- M. Skyllas-Kazacos, C. Menictas and M. Kazacos, J. Electrochem. Soc., 1996, 143, L86–88 CrossRef CAS.
- T. Mohammadi and M. Skyllas-Kazacos, J. Power Sources, 1996, 63, 179–186 CrossRef CAS.
- T. Mohammadi and M. Skyllas-Kazacos, J. Appl. Electrochem., 1997, 27, 153–160 CrossRef CAS.
- F. Rahman and M. Skyllas-Kazacos, J. Power Sources, 1998, 72, 105–110 CrossRef CAS.
- M. Skyllas-Kazacos, C. Peng and M. Cheng, Electrochem. Solid-State Lett., 1999, 2, 121–122 Search PubMed.
- C. M. Hagg and M. Skyllas-Kazacos, J. Appl. Electrochem., 2002, 32, 1063–1069 Search PubMed.
- M. Skyllas-Kazacos and M. Kazacos, US Pat., 6562514 B1, 2003.
- F. Rahman and M. Skyllas-Kazacos, J. Power Sources, 2009, 189, 1212–1219 CrossRef CAS.
- M. Skyllas-Kazacos, G. Kazacos, G. Poon and H. Verseema, Int. J. Energy Res., 2010, 34, 182–189 Search PubMed.
- T. Suuar and M. Skyllas-Kazacos, J. Membr. Sci., 2003, 222, 249–264 CrossRef CAS.
- T. Suuar and M. Skyllas-Kazacos, J. Membr. Sci., 2003, 222, 235–247 Search PubMed.
- T. Suuar and M. Skyllas-Kazacos, J. Appl. Electrochem., 2004, 34, 137–145 CrossRef CAS.
- V-fuel Pty Ltd., Status of Energy Storage Technologies as Enabling Systems for Renewable Energy from the Sun, Wind, Waves and Tides, http://www.aph.gov.au/House/committee/Isr/renewables/submissions/sub21.pdf Search PubMed.
- J. Y. Qiu, M. Y. Li, J. F. Ni, M. L. Zhai, J. Peng, L. Xu, H. H. Zhou, J. Q. Li and G. S. Wei, J. Membr. Sci., 2007, 297, 174–180 CrossRef CAS.
- R. J. Remick and P. G. P. Ang, US 1984/4485154, 27 Nov 1984, 1984.
- R. Zito, US Pat. 5612148, 1997.
- A. Price, S. Bartley, S. Male and G. Cooley, Power Eng. J., 1999, 13, 122–129 CrossRef.
- F. C. Walsh, Pure Appl. Chem., 2001, 73, 1819–1837 Search PubMed.
- P. J. Morrissey, P. J. Mitchell and S. E. Male, Pat. Pub. No.: WO/2001/003221, 2001.
- P. J. Morrissey and N. J. Ward, GB Pat. 2374722, 2002.
- D. Pletcher and R. Wills, Phys. Chem. Chem. Phys., 2004, 6, 1779–1785 RSC.
- D. Pletcher and R. Wills, J. Power Sources, 2005, 149, 96–102 CrossRef CAS.
- A. Hazza, D. Pletcher and R. Wills, J. Power Sources, 2005, 149, 103–111 CrossRef CAS.
- D. Pletcher, H. T. Zhou, G. Kear, C. T. J. Low, F. C. Walsh and R. G. A. Wills, J. Power Sources, 2008, 180, 621–629 CrossRef CAS.
- D. Pletcher, H. T. Zhou, G. Kear, C. T. J. Low, F. C. Walsh and R. G. A. Wills, J. Power Sources, 2008, 180, 630–634 CrossRef CAS.
- X. Li, D. Pletcher and F. C. Walsh, Electrochim. Acta, 2009, 54, 4688–4695 CrossRef CAS.
- J. Collins, X. Li, D. Pletcher, R. C. Tangirala, D. S. Campbell, F. C. Walsh and C. Zhang, J. Power Sources, 2010, 195, 2975–2978 CrossRef CAS.
- R. G. A. Wills, J. Collins, D. Stratton-Campbell, C. T. J. Low, D. Pletcher and F. C. Walsh, J. Appl. Electrochem., 2010, 40, 955–965 CrossRef CAS.
- J. Collins, G. Kear, X. Li, C. T. J. Low, D. Pletcher, R. Tangirala, D. Stratton-Campbell, F. C. Walsh and C. Zhang, J. Power Sources, 2010, 195, 1731–1738 CrossRef CAS.
- L. Zhang, J. Cheng, Y. S. Yang, Y. H. Wen, X. D. Wang and G. P. Cao, J. Power Sources, 2008, 179, 381–387 Search PubMed.
- Y. Ito, M. Nyce, R. Plivelich, M. Klein, D. Steingart and S. Banerjee, J. Power Sources, 2011, 196, 2340–2345 Search PubMed.
- Y. Ito, M. Nyce, R. Plivelich, M. Klein and S. Banerjee, J. Power Sources, 2011, 196, 6583–6587 Search PubMed.
- P. K. Leung, C. Ponce de León and F. C. Walsh, Electrochim. Acta, 2012, 80, 7 Search PubMed.
- Y. M. Chiang, W. C. Carter, B. Y. Ho and M. Duduta, US 2010/0047671 A1 2010.
- M. Duduta, B. Ho, V. C. Wood, P. Limthongkul, V. E. Brunini, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2011, 1, 511–516 Search PubMed.
- Y. H. Lu, J. B. Goodenough and Y. S. Kim, J. Am. Chem. Soc., 2011, 133, 5756–5759 CrossRef CAS.
- Y. H. Lu and J. B. Goodenough, J. Mater. Chem., 2011, 21, 10113–10117 RSC.
- M. Skyllas-Kazacos, J. Power Sources, 2003, 124, 299–302 CrossRef CAS.
- W. Wang, S. Kim, B. Chen, Z. Nie, J. Zhang, G. Xia, L. Li and Z. Yang, Energy Environ. Sci., 2011, 4, 4068–4073 RSC.
- B. Fang, S. Iwasa, Y. Wei, T. Arai and M. Kumagai, Electrochim. Acta, 2002, 47, 3971–3976 CrossRef CAS.
- F. Q. Xue, Y. L. Wang, W. H. Wang and X. D. Wang, Electrochim. Acta, 2008, 53, 6636–6642 CrossRef CAS.
- H. T. Zhou, H. M. Zhang, P. Zhao and B. L. Yi, Electrochim. Acta, 2006, 51, 6304–6312 Search PubMed.
- T. Yamamura, J. Electrochem. Soc., 2005, 152, A830–A836 Search PubMed.
- K. Hasegawa, A. Kimura, T. Yamamura and Y. Shiokawa, J. Phys. Chem. Solids, 2005, 66, 593–595 CrossRef CAS.
- T. Yamamura, N. Watanabe and Y. Shiokawa, J. Alloys Compd., 2006, 408–412, 1260–1266 Search PubMed.
- T. Yamamura, Y. Shiokawa, H. Yamana and H. Moriyama, Electrochim. Acta, 2002, 48, 43–50 Search PubMed.
- C. H. Bae, E. P. L. Roberts and R. A. W. Dryfe, Electrochim. Acta, 2002, 48, 279–287 CrossRef CAS.
- M. H. Chakrabarti, E. P. L. Roberts, C. Bae and M. Saleem, Energy Convers. Manage., 2011, 52, 2501–2508 Search PubMed.
- Q. Liu, A. E. S. Sleightholme, A. A. Shinkle, Y. Li and L. T. Thompson, Electrochem. Commun., 2009, 11, 2312–2315 CrossRef CAS.
- Q. Liu, A. A. Shinkle, Y. Li, C. W. Monroe, L. T. Thompson and A. E. S. Sleightholme, Electrochem. Commun., 2010, 12, 1634–1637 CrossRef CAS.
- J. Giner and K. Cahill, US Pat. 4192910, 1980.
- D. S. Cheng and E. Hollax, J. Electrochem. Soc., 1985, 132, 269–273 Search PubMed.
- C. Wu, D. A. Scherson, E. J. Calvo, E. B. Yeager and M. A. Reid, J. Electrochem. Soc., 1986, 133, 2109–2112 Search PubMed.
- D. A. Johnson and M. A. Reid, J. Electrochem. Soc., 1985, 132, 269–273 Search PubMed.
- M. Lopez-Atalaya, G. Codina, J. R. Perez, J. L. Vazquez, A. Aldaz and M. A. Climent, J. Power Sources, 1991, 35, 225–234 Search PubMed.
- G. Codina, J. R. Perez, M. Lopez-Atalaya, J. L. Vazquez and A. Aldaz, J. Power Sources, 1994, 48, 293–302 Search PubMed.
- G. Codina and A. Aldaz, J. Appl. Electrochem., 1992, 22, 668–674 Search PubMed.
- N. Tokuda, T. Kanno, T. Hara, T. Shigematsu, Y. Tsutsui, A. Ikeuchi, T. Itou and T. Kumamoto, SEI Tech. Rev., 1998, 50, 88–94 Search PubMed.
- P. Zhao, H. Zhang, H. Zhou, J. Chen, S. Gao and B. Yi, J. Power Sources, 2006, 162, 1416–1420 CrossRef CAS.
- Q. Luo, H. Zhang, J. Chen, P. Qian and Y. Zhai, J. Membr. Sci., 2007, 311, 98–103 Search PubMed.
- M. C. Li, S. Z. Luo, Y. H. Qian, W. Q. Zhang, L. L. Jiang and J. I. Shen, J. Electrochem. Soc., 2007, 154, D567–D571 Search PubMed.
- D. J. You, H. M. Zhang and J. Chen, Electrochim. Acta, 2009, 54, 6827–6836 Search PubMed.
- D. J. You, H. M. Zhang, C. X. Sun and X. K. Ma, J. Power Sources, 2011, 196, 1578–1585 Search PubMed.
- C. X. Sun, J. Chen, H. M. Zhang, X. Han and Q. T. Luo, J. Power Sources, 2010, 195, 890–897 CrossRef CAS.
- Z. S. Mai, H. M. Zhang, X. F. Li, C. Bi and H. Dai, J. Power Sources, 2011, 196, 482–487 CrossRef CAS.
- H. Zhang, H. Zhang, X. Li, Z. Mai and J. Zhang, Energy Environ. Sci., 2011, 4, 1676–1679 RSC.
- Z. S. Mai, H. M. Zhang, X. F. Li, S. H. Xiao and H. Z. Zhang, J. Power Sources, 2011, 196, 5737–5741 Search PubMed.
- F. Xing, H. Zhang and X. Ma, J. Power Sources, 2011, 196, 10753–10757 Search PubMed.
- Y. Shao, X. Wang, M. Engelhard, C. Wang, S. Dai, J. Liu, Z. Yang and Y. Lin, J. Power Sources, 2010, 195, 4375–4379 CrossRef CAS.
- S. W. Kim, J. L. Yan, B. Schwenzer, J. L. Zhang, L. Y. Li, J. Liu, Z. G. G. Yang and M. A. Hickner, Electrochem. Commun., 2010, 12, 1650–1653 CrossRef CAS.
- M. Vijayakumar, S. D. Burton, C. Huang, L. Li, Z. Yang, G. L. Graff, J. Liu, J. Hu and M. Skyllas-Kazacos, J. Power Sources, 2010, 195, 7709–7717 CrossRef CAS.
- M. Vijayakumar, L. Li, G. Graff, J. Liu, H. Zhang, Z. Yang and J. Hu, J. Power Sources, 2011, 196, 3669–3672 CrossRef CAS.
- S. Kim, M. Vijayakumar, W. Wang, J. Zhang, B. Chen, Z. Nie, F. Chen, J. Hu, L. Li and Z. Yang, Phys. Chem. Chem. Phys., 2011, 13, 18186–18193 RSC.
- B. Schwenzer, S. Kim, M. Vijayakumar, Z. Yang and J. Liu, J. Membr. Sci., 2011, 372, 11–19 Search PubMed.
- L. Y. Li, S. W. Kim, W. Wang, M. Vijaayakumar, Z. M. Nie, B. W. Chen, J. L. Zhang, G. G. Xia, J. Z. Hu, G. Graff, J. Liu and Z. G. Yang, Adv. Energy Mater., 2011, 1, 394–400 Search PubMed.
- J. Zhang, L. Li, Z. Nie, B. Chen, M. Vijayakumar, S. Kim, W. Wang, B. Schwenzer, J. Liu and Z. Yang, J. Appl. Electrochem., 2011, 41, 1215–1221 Search PubMed.
- S. Kim, T. B. Tighe, B. Schwenzer, J. Yan, J. Zhang, J. Liu, Z. Yang and M. A. Hickner, J. Appl. Electrochem., 2011, 41, 1201–1213 CrossRef CAS.
- M. Schreiber, M. Harrer, A. Whitehead, H. Bucsich, M. Dragschitz, E. Seifert and P. Tymciw, J. Power Sources, 2011, 206, 483–489 Search PubMed.
- J. Samuel, Vanadium flow battery energy storage, International Renewable Energy Storage Conference, Berlin, 2011 Search PubMed.
- F. Chang, C. W. Hu, X. J. Liu, L. Liu and J. W. Zhang, Electrochim. Acta, 2012, 60, 334–338 Search PubMed.
- S. Li, K. L. Huang, S. Q. Liu, D. Fang, X. W. Wu, D. Lu and T. Wu, Electrochim. Acta, 2011, 56, 5483–5487 Search PubMed.
- Z. J. Jia, B. G. Wang, S. Q. Song and X. Chen, J. Electrochem. Soc., 2012, 159, A843–A847 Search PubMed.
- A. Tang, J. Bao and M. Skyllas-Kazacos, J. Power Sources, 2011, 196, 10737–10747 Search PubMed.
- M. Skyllas-Kazacos and L. Goh, J. Membr. Sci., 2012, 399–400 Search PubMed 43-48.
- M. Skyllas-Kazacos and Y. Limantari, J. Appl. Electrochem., 2004, 34, 681–685 CrossRef CAS.
- H. Vafiadis and M. Skyllas-Kazacos, J. Membr. Sci., 2006, 279, 394–402 CrossRef CAS.
- W. Wang, Z. M. Nie, B. W. Chen, F. Chen, Q. T. Luo, X. L. Wei, G. G. Xia, M. Skyllas-Kazacos, L. Y. Li and Z. G. Yang, Adv. Energy Mater., 2012, 2, 487–493 Search PubMed.
- W. Wang, L. Li, Z. Nie, B. Chen, Q. Luo, Y. Shao, X. Wei, F. Chen, G.-G. Xia and Z. Yang, J. Power Sources, 2012, 216, 99–103 Search PubMed.
- S. H. Ge, B. L. Yi and H. M. Zhang, J. Appl. Electrochem., 2004, 34, 181–185 Search PubMed.
- P. Zhao, H. M. Zhang, H. T. Zhou and B. L. Yi, Electrochim. Acta, 2005, 51, 1091–1098 Search PubMed.
- H. Zhou, H. Zhang, P. Zhao and B. Yi, Electrochem. Commun., 2006, 74, 296–298 Search PubMed.
- Y. Shiokawa, T. Yamamura and K. Shirasaki, in Energy Efficiency of an Uranium Redox-Flow Battery Evaluated By the Butler-Volmer Equation, 2006, Vol. 75, pp.137-142 Search PubMed.
- K. Shirasaki, T. Yamamura, T. Herai and Y. Shiokawa, J. Alloys Compd., 2006, 418, 217–221 Search PubMed.
- Y. H. Wen and H. M. Zhang, et al., Electrochim. Acta, 2006, 51, 3769–3775 CrossRef CAS.
- Y. H. Wen and H. M. Zhang, et al., J. Electrochem. Soc., 2006, 153, A929–A934 CrossRef CAS.
- P. Modiba and A. M. Crouch, J. Appl. Electrochem., 2008, 38, 1293–1299 Search PubMed.
- Y. Matsuda, K. Tanaka, M. Okada, Y. Takasu, M. Morita and T. Matsumura-Inoue, J. Appl. Electrochem., 1988, 18, 909–914 CrossRef CAS.
- M. H. Chakrabarti, R. A. W. Dryfe and E. P. L. Roberts, Electrochim. Acta, 2006, 52, 2189–2195 Search PubMed.
- D. Zhang, Q. Liu, X. Shi and Y. Li, J. Power Sources, 2012, 203, 201–205 CrossRef CAS.
- A. E. S. Sleightholme, A. A. Shinkle, Q. H. Liu, Y. D. Li, C. W. Monroe and L. T. Thompson, J. Power Sources, 2011, 196, 5742–5745 CrossRef CAS.
- B. D. Sawyer, G. J. Suppes, M. J. Gordon and M. G. Heidlage, J. Appl. Electrochem., 2011, 41, 543–550 Search PubMed.
- C. Lotspeich, International Conference (EESAT2002), San Francisco, 2002 Search PubMed.
- C. P. Zhang, S. M. Sharkh, X. Li, F. C. Walsh, C. N. Zhang and J. C. Jiang, Energy Convers. Manage., 2011, 52, 3391–3398 Search PubMed.
- A. Oury, A. Kirchev and Y. Bultel, Electrochim. Acta, 2012, 63, 28–36 Search PubMed.
- D. M. Keogh, Soluble Lead Flow Battery Technology, The ARPA-E Energy Innovation Summit, Washington DC, 2011 Search PubMed.
- J. Pan, L. Ji, Y. Sun, P. Wan, J. Cheng, Y. Yang and M. Fan, Electrochem. Commun., 2009, 11, 2191–2194 Search PubMed.
- J. Jorné, J. T. Kim and D. Kralik, J. Appl. Electrochem., 1979, 9, 573–579 Search PubMed.
- H. S. Lim, A. M. Lackner and R. C. Knechtli, J. Electrochem. Soc., 1977, 124, 1154–1157 Search PubMed.
- G. B. Adams, US Pat. 4180623, 25/12/, 1979.
- S. S. Hosseiny, M. Saakes and M. Wessling, Electrochem. Commun., 2010, 13, 751–754 Search PubMed.
- Y. Xu, Y. H. Wen, J. Cheng, G. P. Cao and Y. S. Yang, Electrochem. Commun., 2009, 11, 1422–1424 CrossRef CAS.
- Y. Xu, Y. H. Wen, J. Cheng, G. P. Cao and Y. S. Yang, Electrochim. Acta, 2010, 55, 715–720 Search PubMed.
- P. C. Symons, US Pat., 3809578, 1973.
- C. C. Whittlesey, G. L. Henriksen, W. B. Keryluk and M. T. Wolff, US Pat. 4518664, 1985.
- P. C. Butler and S. E. Klassen, Materials for advanced rechargeable batteries, Sandia National Laboratories, SAND93-2023, 1993 Search PubMed.
- D. Pavlov and G. PapazovM. Gerganska, Battery Energy Storage Systems, the United Nations Educational, Scientific and Cultural Organization, Regional Office for Science and Technology for Europe, Technical Report, no. 7, 1991 Search PubMed.
- T. Horie, H. Ogino, K. Fujiwara, Y. Watakabe, T. Hiramatsu and S. Kondo, Development of a 10 kW/80 kWh Zinc-Chloride Battery for Electric Power Storage Using Solvent Absorption Chlorine Storage System (Solvent Method), Proceedings of the 21st IECEC, San Diego, California, 1986, 986–991 Search PubMed.
- Y. Misawa, A. Suzuki, A. Shimizu, H. Sato, K. Ashizawa, T. Sumii and M. Kondo, Demonstration Test of a 60 kW-Class Zinc/Chloride Battery as a Power Storage System, Proceedings of the 24th IECEC, Washington D.C., 1989, 1325–1329 Search PubMed.
- T. Nakayama, Y. Sera and A. Mitsuda, The current status of development of advanced battery electric energy storage system in Japan, Proceedings of the 24th IECEC, Washington D.C., 1989, 1297–1301 Search PubMed.
- D. J. Eustace, J. Electrochem. Soc., 1980, 127, 528–532 CrossRef CAS.
- C. Ponce de leon and F. C. Walsh, ECPS00856, Zinc/bromine redox flow batteries, 2009 Search PubMed.
- T. I. Evans and R. E. White, J. Electrochem. Soc., 1987, 134, 2725–2733 Search PubMed.
- T. I. Evans and R. E. White, J. Electrochem. Soc., 1987, 134, 866–874 Search PubMed.
- G. D. Simpson and R. E. White, J. Electrochem. Soc., 1989, 136, 2137–2144 Search PubMed.
- G. D. Simpson and R. E. White, J. Electrochem. Soc., 1990, 137, 1843–1846 Search PubMed.
- R. M. Spotnitz, R. P. Kreh, J. T. Lundquist and P. J. Press, J. Appl. Electrochem., 1990, 20, 209–215 CrossRef CAS.
- G. Brodt, J. Haas, W. Hesse and H. U. Jäqer, US 2003/0141195 Al 31/07/2003, 2003.
- P. K. Leung, C. Ponce-de-Leon, C. T. J. Low, A. A. Shah and F. C. Walsh, J. Power Sources, 2011, 196, 5174–5185 Search PubMed.
- G. Nikiforidis, L. Berlouis, D. Hall and D. Hodgson, J. Power Sources, 2011, 206, 497–503 Search PubMed.
- C. Chakkaravarthy, A. K. Abdul Waheed and H. V. K. Udupa, J. Power Sources, 1981, 6, 203–228 Search PubMed.
- K. Kinoshita, Electrochemical oxygen technology, Wiley, 1992 Search PubMed.
- Y. H. Wen, J. Cheng, S. Q. Ning and Y. S. Yang, J. Power Sources, 2009, 188, 301–307 CrossRef CAS.
- Y. H. Wen, J. Cheng, P. H. Ma and Y. S. Yang, Electrochim. Acta, 2008, 53, 3514–3522 Search PubMed.
- Y. H. Wen, J. Cheng, Y. Xun, P. H. Ma and Y. S. Yang, Electrochim. Acta, 2008, 53, 6018–6023 Search PubMed.
- J. Jorne, J. Electrochem. Soc., 1982, 129, 2251–2253 Search PubMed.
- R. L. Clarke, B. Dougherty, S. Harrison, J. P. Millington and S. Mohanta, US Pat. 20040197651, 2004.
- R. L. Clarke, B. Dougherty, S. Harrison, J. P. Millington and S. Mohanta, US2004197649 (A1), 2004.
- R. P. Kreh, R. M. Spotnitz and J. T. Lundquist, J. Org. Chem., 1989, 54, 1526–1531 CrossRef CAS.
- P. K. Leung, C. Ponce de León, C. T. J. Low and F. C. Walsh, Electrochim. Acta, 2011, 56, 6536–6546 Search PubMed.
- P. K. Leung, C. Ponce-de-Leon, C. T. J. Low and F. C. Walsh, Electrochim. Acta, 2011, 56, 2145–2153 Search PubMed.
- Z. P. Xie, F. J. Xiong and D. Zhou, Energy Fuels, 2011, 25, 2399–2404 Search PubMed.
- K. Kinoshita and S. C. Leach, J. Electrochem. Soc., 1982, 129, 1993–1997 CAS.
- C. Menictas and M. Skyllas-Kazacos, J. Appl. Electrochem., 2011, 41, 1223–1232 Search PubMed.
- Á. Frías-Ferrer, J. González-García, V. Sáez, C. Ponce de León and F. C. Walsh, AIChE J., 2008, 54, 811–823 Search PubMed.
- L. Joerissen, J. Garche, C. Fabjan and G. Tomazic, J. Power Sources, 2004, 127, 98–104 CrossRef CAS.
- N. A. Hampson and A. J. S. McNeil, SPR-Electrochemistry, Royal Society of Chemistry, 1984, 9, 1–65 Search PubMed.
- F. C. Walsh, A First Course in Electrochemical Engineering, Electrochemical Consultancy, Romsey, UK, 1993 Search PubMed.
- J. A. Trainham, Electrochim. Acta, 1981, 26, 455–469 Search PubMed.
- N. Akira, K. Hiroko and N. Ken, Denki Kagaku oyobi Kogyo ButsuriKagaku, 1993, 61, 1442 Search PubMed.
- D. S. Aaron, Q. Liu, Z. Tang, G. M. Grim, A. B. Papandrew, A. Turhan, T. A. Zawodzinski and M. M. Mench, J. Power Sources, 2012, 206, 450–453 Search PubMed.
- P. Qian, H. M. Zhang, J. Chen, Y. H. Wen, Q. T. Luo, Z. Liu, D. J. You and B. L. Yi, J. Power Sources, 2008, 175, 613–620 Search PubMed.
- M. S. Yazici, D. Krassowski and J. Prakash, J. Power Sources, 2005, 141, 171–176 CrossRef CAS.
- K. Fushimi, H. Tsunakaw and K. Yonahara, US Pat. 4551267, 1985.
- G. Tomazic, US Pat. 4615108, 1986.
- C. Herscovici, A. Leo and A. Charkey, US Pat. 4758473, 1988.
- C. Herscovici, US Pat. 4920017, 1990.
- D. A. Szánto, Characterization of Electrochemical Filter-Press Reactor, PhD Thesis University of Portsmouth, UK, 1989 Search PubMed.
- K. Roβberg and V. Trapp, Graphite–based bipolar plates. Handbook of Fuel Cells, John Wiley & Sons, Ltd, 2010 Search PubMed.
- E. H. Weber, Doctor of Philosophy, Chemical Engineering, Michigan Technological University, 2001.
- G. J. W. Radford, J. Cox, R. G. A. Wills and F. C. Walsh, J. Power Sources, 2008, 185, 1499–1504 Search PubMed.
- Y. M. Zhang, Q. M. Huang, W. S. Li, H. Y. Peng and S. J. Hu, J. Inorg. Mater., 2007, 22, 1051–1056 Search PubMed.
- H. Kaneko, K. Nozaki, A. Negishi, Y. Wada, T. Aoki and M. Kamimoto, Electrochim. Acta, 1991, 36, 1191 CrossRef CAS.
- A. Paulenova, S. E. Creager, J. D. Navratil and Y. Wei, J. Power Sources, 2002, 109, 431–438 Search PubMed.
- H. J. Liu, Q. Xu, C. W. Yan and Y. L. Qiao, Electrochim. Acta, 2011, 56, 8783–8790 Search PubMed.
- J. González-Garcıa, P. Bonete, E. Exposito, V. Montiel, A. Aldaz and R. Torregrosa-Macia, J. Mater. Chem., 1999, 9, 419–426 RSC.
- J. M. Friedrich, C. Ponce-de-León, G. W. Reade and F. C. Walsh, J. Electroanal. Chem., 2004, 561, 203–217 Search PubMed.
- X. Li and K. Horita, Carbon, 2000, 38, 133–138 Search PubMed.
- M. Santiago, F. Stübera, A. Fortunyb, A. Fabregata and J. Fonta, Carbon, 2005, 43, 2134–2145 Search PubMed.
- K. Jurewicz, K. Babe, A. Źiókowskic and H. Wachowska, Electrochim. Acta, 2003, 48, 1491–1498 CrossRef CAS.
- N. S. Jacobson and D. M. Curry, Carbon, 2006, 44, 1142–1150 CrossRef CAS.
- X. G. Li, K. L. Huang, S. Q. Liu and L. Q. Chen, J. Cent. South Univ. Technol., 2007, 14, 51–56 Search PubMed.
- T. Wu, K. L. Huang, S. Q. Liu, S. X. Zhuang, D. Fang, S. Li, D. Lu and A. Q. Su, J. Solid State Electrochem., 2012, 16, 579–585 Search PubMed.
- W. H. Wang and X. D. Wang, Electrochim. Acta, 2007, 52, 6755–6762 CrossRef CAS.
- C. Fabjan, J. Garche, B. Harrer, L. Jörissen, C. Kolbeck, F. Philippi, G. Tomazic and F. Wagner, Electrochim. Acta, 2001, 47, 825–831 CrossRef CAS.
- Z. González, A. Sánchez, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Electrochem. Commun., 2011, 13, 1379–1382 Search PubMed.
- K. J. Kim, M. S. Park, J. H. Kim, U. Hwang, N. J. Lee, G. J. Jeong and Y. J. Kim, Chem. Commun., 2012, 48, 5455–5457 RSC.
- M. Mastragostino and S. Valcher, Electrochim. Acta, 1983, 28, 501–505 Search PubMed.
- H. Q. Zhu, Y. M. Zhang, L. Yue, W. S. Li, G. L. Li, D. Shu and H. Y. Chen, J. Power Sources, 2008, 184, 637–640 CrossRef CAS.
- W. Y. Li, J. G. Liu and C. W. Yan, Carbon, 2011, 49, 3463–3470 Search PubMed.
- P. X. Han, H. B. Wang, Z. H. Liu, X. Chen, W. Ma, J. H. Yao, Y. W. Zhu and G. L. Cui, Carbon, 2011, 49, 693–700 CrossRef CAS.
- P. X. Han, Y. H. Yue, Z. H. Liu, W. Xu, L. X. Zhang, H. X. Xu, S. M. Dong and G. L. Cui, Energy Environ. Sci., 2011, 4, 4710–4717 RSC.
- Y. Liu, X. Xia and H. Liu, J. Power Sources, 2004, 130, 299–305 Search PubMed.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS.
- G. J. Hwang and H. Ohya, J. Membr. Sci., 1997, 132, 55–61 CrossRef CAS.
- D. G. Oei, J. Appl. Electrochem., 1985, 15, 231–235 Search PubMed.
- S. C. Chieng, Ph.D. Thesis, University of New South Wales, Sydney, Australia, 1993.
- W. Grot, Fluorinated ionomers, ed. William Andrew Inc., 2008, Vol. 978, pp. 10 Search PubMed.
- H. Tasai, T. Horigome, N. Nozaki, H. Kaneko, A. Negishi and Y. Wada, The 31th Denchi Touron Kouengai Yousisyu, Japan, 1990, 301–302 Search PubMed.
- J. C. Woong, S. Venkataramani and S. C. Kim, Polym. Int., 2006, 55, 491–499 Search PubMed.
- J. Zeng, C. P. Jiang, Y. H. Wang, J. W. Chen, S. F. Zhu, B. J. Zhao and R. L. Wang, Electrochem. Commun., 2008, 10, 372–375 CrossRef CAS.
- S. Tan and D. Bélanger, J. Phys. Chem. B, 2005, 109, 23480–23490 CrossRef CAS.
- J. Y. Xi, Z. H. Wu, X. G. Teng, Y. T. Zhao, L. Q. Chen and X. P. Qiu, J. Mater. Chem., 2008, 18, 1232–1238 RSC.
- J. Y. Xi, Z. H. Wu, X.P. Qiu and L. Q. Chen, J. Power Sources, 2007, 166, 531–536 CrossRef CAS.
- M. Vijayakumar, B. Schwenzer, S. W. Kim, Z. G. Yang, S. Thevuthasan, J. Liu, G. L. Graff and J. Z. Hu, Solid State Nuclear Magnetic Resonance, 2012, 42, 71–80 Search PubMed.
- D. Schulte, J. Drillkens, B. Schulte and D. U. Sauer, J. Electrochem. Soc., 2010, 157, A989–A992 Search PubMed.
- X. G. Teng, Y. T. Zhao, J. Y. Xi, Z. H. Wu, X. P. Qiu and L. Q. Chen, J. Power Sources, 2009, 189, 1240–1246 CrossRef CAS.
- X. G. Teng, Y. T. Zhao, J. Y. Xi, Z. H. Wu, X. P. Qiu and L. Q. Chen, J. Membr. Sci., 2009, 341, 149–154 CrossRef CAS.
- N. F. Wang, S. Peng, D. Lu, S. Q. Liu, Y. N. Liu and K. L. Huang, J. Solid State Electrochem., 2012, 16, 1577–1584 Search PubMed.
- S. B. Sang, Q. M. Wu and K. L. Huang, J. Membr. Sci., 2007, 305, 118–124 CrossRef CAS.
- B. Tian, C. W. Yan and F. H. Wang, J. Membr. Sci., 2004, 234, 51–54 CrossRef CAS.
- T. Mohammadi and M. Skyllas-Kazacos, J. Membr. Sci., 1995, 107, 35–45 CrossRef CAS.
- J. Y. Qiu, L. Zhao, M. L. Zhai, J. F. Ni, H. H. Zhou, J. Peng, J. Q. Li and G. S. Wei, J. Power Sources, 2008, 177, 617–623 CrossRef CAS.
- X. L. Luo, Z. Z. Lu, J. Y. Xi, Z. H. Wu, W. T. Zhu, L. Q. Chen and X. P. Qiu, J. Phys. Chem. B, 2005, 109, 20310–20314 CrossRef CAS.
- D. Y. Chen, S. J. Wang, M. Xiao and Y. H. Meng, Energy Environ. Sci., 2010, 3, 622–628 RSC.
- D. B. Xing, S. H. Zhang, C. X. Yin, B. G. Zhang and X. G. Jian, J. Membr. Sci., 2010, 354, 68–73 CrossRef CAS.
- S. H. Zhang, C. X. Yin, D. B. Xing, D. L. Yang and X. G. Jian, J. Membr. Sci., 2010, 363, 243–249 CrossRef CAS.
- Y. Wang, J. Qiu, J. Peng, L. Xu, J. Li and M. Zhai, J. Membr. Sci., 2011, 376, 70–77 Search PubMed.
- G. J. Hwang and H. Ohya, J. Membr. Sci., 1998, 140, 195–203 Search PubMed.
- G. W. Hu, Y. Wang, J. Ma, J. Y. Qiu, J. Peng, J. Q. Li and M. L. Zhai, J. Membr. Sci., 2012, 407–408, 184–192 Search PubMed.
- Q. T. Luo, H. M. Zhang, J. Chen, P. Qian and Y. F. Zhai, J. Membr. Sci., 2008, 311, 98–103 CrossRef CAS.
- D. Y. Chen, S. J. Wang, M. Xiao and Y. Z. Meng, J. Power Sources, 2010, 195, 2089–2095 CrossRef CAS.
- W. P. Wei, H. M. Zhang, X. F. Li, Z. S. Mai and H. Z. Zhang, J. Power Sources, 2012, 208, 421–425 Search PubMed.
- N. F. Wang, S. Peng, H. M. Wang, Y. H. Li, S. Q. Liu and Y. N. Liu, Electrochem. Commun., 2012, 17, 30–33 Search PubMed.
- X. Ling, C. Jia, J. Liu and C. Yan, J. Membr. Sci., 2012, 415–416, 306 Search PubMed.
- X. S. Zhao, Y. Z. Fu and W. Li, RSC Adv., 2012, 2, 5554–5556 RSC.
- C. K. Jia, J. G. Liu and C. W. Yan, J. Power Sources, 2010, 195, 4380–4383 CrossRef CAS.
- D. Y. Chen, S. J. Wang, M. Xiao, D. M. Han and Y. Z. Meng, J. Power Sources, 2010, 195, 7701–7708 CrossRef CAS.
- H. Z. Zhang, H. M. Zhang, X. F. Li, Z. S. Mai and W. P. Wei, Energy Environ. Sci., 2012, 5, 6299–6303 RSC.
- S. K. Karode and A. Kumar, J. Membr. Sci., 2001, 193, 69–84 Search PubMed.
- A. Frías-Ferrer, J. Gonzalez-Garcia, V. S. E. Expósito, C. M. Sánchez-Sánchez, V. Montiel, A. Aldaz and F. C. Walsh, J. Chem. Educ., 2005, 82, 1395–1398 Search PubMed.
- A. Ponce de León, G. W. Reade, I. Whyte, S. E. Male and F. C. Walsh, Electrochim. Acta, 2007, 52, 5815–5823 Search PubMed.
- I. Tsuda, K. Kurokawa and K. Nozaki, Photovoltaic Energy Conversion, 1994, Conference Record of the Twenty Fourth IEEE Photovoltaic Specialists Conference, IEEE First World Conference Search PubMed.
- R. A. Scannell and F. C. Walsh, J. Inst. Chem. Engr. Symp. Ser., 1989, 112, 59–71 Search PubMed.
- M. Skyllas-Kazacos and C. Menictas, Telecommunications Energy Conference, INTELEC 97, 19th International, 1997, 463–471 Search PubMed.
- I. Nagashima, J. Fukui, H. Gotoh, H. Kaneko, K. Nozaki and T. Ozawa, US Pat. 4814241.
- T. Hennessy, WO/2006/076059, System and method for optimizing efficiency and power output from a vanadium redox battery energy storage system, 2006 Search PubMed.
- M. Skyllas-Kazacos, Pub. No. WO/2008/148148, 2008.
- M. Skyllas-Kazacos and M. Kazacos, J. Power Sources, 2011, 196, 8822–8827 Search PubMed.
- A. Zocchi and B. M. Broman, Pat. Appl. No.: 0176000, 2001.
- X. K. Ma, H. M. Zhang, C. X. Sun, Y. Zou and T. Zhang, J. Power Sources, 2012, 203, 153–158 Search PubMed.
- P. S. Fedkiw and R. W. Watts, J. Electrochem. Soc., 1984, 131, 701–709 Search PubMed.
- D. P. Scamman, G. W. Reade and E. P. L. Roberts, J. Power Sources, 2009, 189, 1231–1239 Search PubMed.
- D. P. Scamman, G. W. Reade and E. P. L. Roberts, J. Power Sources, 2009, 189, 1220–1230 Search PubMed.
- A. A. Shah, H. Al-Fetlawi and F. C. Walsh, Electrochim. Acta, 2010, 55, 1125–1139 Search PubMed.
- H. Al-Fetlawi, A. A. Shah and F. C. Walsh, Electrochim. Acta, 2009, 55, 78–89 Search PubMed.
- H. Al-Fetlawi, A. A. Shah and F. C. Walsh, Electrochim. Acta, 2010, 55, 3192–3205 Search PubMed.
- A. A. Shah, R. Tangirala, R. Singh, R. G. A. Wills and F. C. Walsh, J. Electrochem. Soc., 2011, 158, A671–A677 Search PubMed.
- A. A. Shah, X. Li, R. G. A. Wills and F. C. Walsh, J. Electrochem. Soc., 2010, 157, A589–A599 Search PubMed.
- EA technology, Review of electrical energy storage technologies and system of their potential for the UK, DTI report contract no.: DG/DTI/00055/00/00, URN no: 041876, 2004 Search PubMed.
- A. Gonzalez, B. Ó. Gallachóir and E. McKeogh, Final Report, Sustainable Energy Research Group, University College Cork, 2004 Search PubMed.
- I. P. Gyuk and S. Eckroad, EPRI-DOE Handbook of Energy Storage for Transmission & Distribution Applications, 1001834, Final Report, Washington DC, 2003 Search PubMed.
- R. Baxter, Energy Storage - A nontechnical guide, Electricity Storage Association, Penn Well Books, 2006 Search PubMed.
- J. Makansi and J. Abboud, Energy Storage- The Missing Link in the Electricity Value Chain, Energy Storage Council White Paper, 2002 Search PubMed.
- C. Menictas, D. R. Hong, Z. Yan, J. Wilson, M. Kazacos and M. Skyllas-Kazacos, Status of the vanadium redox flow battery development program, Proceedings of the Electrical Engineering Congress, Sydney, Australia, 1994, 1, 299 Search PubMed.
- S. Miyake and N. Tokuda, Vanadium Redox-Flow Battery for a Variety of Applications, IEEE summer conference, 2001 Search PubMed.
- B. J. Norris, P. Lex, G. J. Ball and V. Scaini, in Grid-connected solar energy storage using the zinc-bromine flow battery, 2003 Search PubMed.
- D. H. Doughty, P. C. Butler, A. A. Akhil, N. H. Clark and J. D. Boyes, Electrochem. Soc. Interface, 2010, 19, 49–53 Search PubMed.
- Tapbury Management Ltd., Energy Storage at Sorne Hill, Feasibility Study Report, 2006, http://www.seai.ie/Publications/Renewables_Publications/VRB-ESS-Energy-Storage-Rpt-Final.pdf Search PubMed.
- B. L. Norris, R. J. Parry and R. M. Hudson, in An evaluation of windfarm stabilization and load shifting using the zinc-bromine battery, (ZBB), Windpower 2002 Conference Search PubMed.
- Prudent Energy VRB® Systems, http://www.pdenergy.com/.
- D. L. Douglas and J. R. Birk, Annu. Rev. Energy, 1980, 5, 61–88 Search PubMed.
- D. H. Swan, B. Dickinson, M. Arikara and G. S. Tomazic, Proceedings of the Ninth Annual Battery Conference on Applications and Advances, 1994 Search PubMed.
- , A. H. SwanB. E. DickinsonM. P. ArikaraM. K. Prabhu, Proceedings of the Tenth Annual Battery Conference on Applications and Advances, 1995 Search PubMed.
- M. Skyllas-Kazacos in Recent Progress with the UNSW Vanadium Battery, http://www.ceic.unsw.edu.au/centers/vrb/webframe/vanart2a.htm Search PubMed.
- R. Mohammad, S. M. Sharkh and F. C. Walsh, 2009 IEEE Vehicle Power and Propulsion Conference, 2009, 551–557 Search PubMed.
- B. D. Sawyer, G. J. Suppes, M. J. Gordon and M. G. Heidlage, J. Appl. Electrochem., 2011, 41, 543–550 Search PubMed.
- C. Tang and D. Zhou, Electrochim. Acta, 2012, 65, 179–184 Search PubMed.
- Q. T. Luo, H. M. Zhang, J. Chen, D. J. You, C. X. Sun and Y. Zhang, J. Membr. Sci., 2008, 325, 553–558 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |