Synthesis of the C45–C53 tetrahydropyran domain of norhalichondrins and the C14–C22 tetrahydrofuran domain of the halichondrin family†
Gowravaram
Sabitha
*a,
Gajangi
Chandrashekhar
a,
Jhillu Singh
Yadav
a,
Kavitha
Rachineni
b and
Bharatam
Jagadeesh
b
aDivision of Natural Products Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 607, India. E-mail: gowravaramsr@yahoo.com; Fax: +91-40-27160512
bNuclear Magnetic Resonance Division, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 607, India
First published on 4th September 2012
Abstract
Sequential asymmetric α-aminoxylation of aldehydes and Horner–Wadsworth–Emmons olefination has been used as the key reaction for the synthesis of the C45–C53 tetrahydropyran domain of norhalichondrins and C14–C22 tetrahydrofuran domain of halichondrin family.
The halichondrins are a family of novel antimitotic polyether macrolides isolated from Halichondria okadai Kadota,1,2 collected on the coast of Aburatsubo in the Miura Peninsula which is to the south of Tokyo. Since the isolation of norhalichondrin A (Fig. 1) in 1985, there has been substantial interest from chemists and biologists in the halichondrin family of compounds. Halichondrins have attracted significant scientific attention3 because of their complex molecular architectures and their remarkable in vitro and in vivo antitumor activities.4 Recently we have disclosed the synthesis of the C38–C54 spiroketal segment of halichondrin B from our group.5 In continuation of our interest in the halichondrin family of compounds and our recent interest in the use of asymmetric α-aminoxylation reaction,6 we herein report the synthesis of the C45–C53 fully functionalized tetrahydropyran- and tetrahydrofuran containing domains of the halichondrin family utilizing the sequential asymmetric α-aminoxylation of aldehydes and Horner–Wadsworth–Emmons (HWE) olefination reaction as the key step. Phillips et al.3a,c used functionalized furfural as the starting material for the preparation of the THP domain and ketoester via diazoketone intermediate for the THF domain respectively, whereas Kishi et al.3b used 2-deoxy-L-arabinose diethyl thioacetal 4,5-acetonide as the starting material for the preparation of the THF domain in 13 steps.
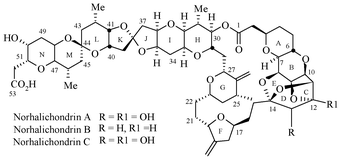 | ||
| Fig. 1 | ||
Retrosynthetic analysis for the C45–C53 tetrahydropyran containing domain 1 is depicted in Scheme 1. We envisaged that this tetrahydropyran domain 1 could be obtained via hydrolysis of the acetonide group in compound 2 followed by oxa-Michael cyclization reaction. Precursor 2 could be obtained from aldehyde 3 using asymmetric aminoxylation and HWE reaction. The aldehyde 3 in turn could be made from epoxy alcohol 4via Gillman reaction and other functional group transformations. Epoxy alcohol 4 itself could be accessed from the known secondary allylic alcohol 57 by chain elongation with Wittig ylide, DIBAL-H reduction followed by Sharpless epoxidation.
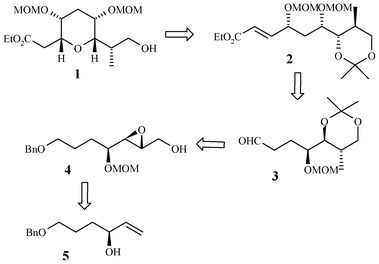 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Our synthesis of the C45–C53 tetrahydropyran domain 1 of the norhalichondrins commenced with the known secondary allylic alcohol 57 (Scheme 2). Accordingly, the secondary hydroxyl group in 5 was protected as methoxymethyl (MOM) ether to afford 6 in 85% yield. Next, compound 6 was subjected to one pot dihydroxylation, oxidative cleavage of diol (OsO4/NaIO4/THF/H2O) to furnish the corresponding aldehyde, which on chain elongation using two carbon Wittig ylides gave α,β-unsaturated ester 7. DIBAL-H reduction of the ester carbonyl group sets up the allyl alcohol 8 in 82% yield. Sharpless asymmetric epoxidation employing D-(–)-diisopropyl tartrate and TBHP furnished the desired epoxy alcohol 4 in 80% yield Regioselective opening of epoxide with lithium dimethylcuprate gave 1,3-diol compound 9 in 78% yield after removing the minor 1,2-diol compound by treatment with NaIO4. Diol 9 was masked as an acetonide 10 using 2,2-dimethoxy propane and catalytic PPTS and then debenzylation with Li in liq NH3 in dry THF provided primary alcohol 11, which was oxidized to the corresponding aldehyde 3 using IBX in DMSO/CH2Cl2. The sequential α-aminoxylation-olefination on aldehyde 3 catalyzed by 40 mol% L-proline using nitroso benzene in DMSO at rt followed by in situ Horner–Wadsworth–Emmons olefination with triethylphosphono acetate and Cs2CO3 as base resulted in the formation of aminoxyolefinic ester, followed by cleavage of the O–N bond using Cu(OAc)2 in ethanol at room temperature gave the γ-hydroxy α,β-unsaturated ester 12 with 91% de. The absolute stereochemistry of the new chiral center formed in compound 12 was confirmed at the later stage by NOE studies of compound 1 (see Fig. 2). The secondary hydroxy group was protected as a MOM ether to afford 2 in 80% yield. Hydrolysis of acetonide group under PPTS/MeOH conditions led to the free diol 13, which on exposure to TBAF in dry THF produced the desired product, the C45–C53 tetrahydropyran domain 1 of norhalichondrins with the required syn relationship via intramolecular oxa-Michael reaction.
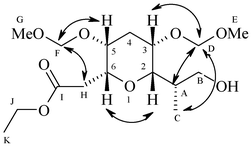 | ||
| Fig. 2 Chemical structure of 1. | ||
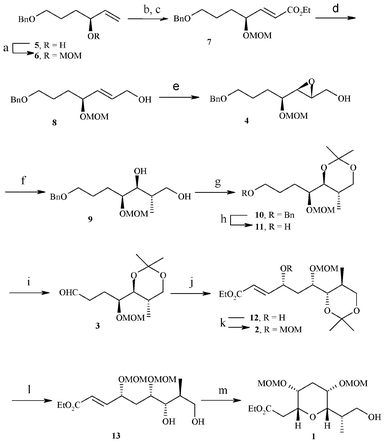 | ||
Scheme 2 reagents: a) MOMCl, N,N-diisopropylethyl amine, anhydrous CH2Cl2, 0 °C–r.t, 5 h, 85%; b) i) OsO4, NMO, Acetone:H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 48 h, 90%; c) i) NaIO4, THF:H2O (4:1), 30 min; ii) PPh3 = CHCO2Et, benzene, r.t., 3 h, (80% from 2 steps); d) DIBAL-H, anhydrous CH2Cl2, 0 °C–r.t., 1 h, 82%; e) (−)-DIPT, Ti(iOPr)4, TBHP, anhydrous CH2Cl2, −20 °C, 36 h, 80%; f) i) Me2CuLi, anhydrous Et2O, −40 °C, 3.5 h; ii) NaIO4, THF:H2O (4:1), 0 °C–r.t., 30 min, 78%. g) 2,2-DMP, PPTS, anhydrous CH2Cl2, 0 °C–r.t., 3 h, 85%; h) Li, liq NH3, anhydrous THF, −78 °C, 30 min, 82%; i) IBX, anhydrous CH2Cl2, anhydrous DMSO, 0 °C–r.t., 12 h, 80%; j) i) PhNO, L-Proline (40 mol %), triethyl phosphono acetate, Cs2CO3, anhydrous DMSO, r.t., 1 h, ii) Cu(OAc)2, EtOH, r.t., 12 h, 57%, 91%de; k) MOMCl, N,N-diisopropylethyl amine, DMAP, anhydrous CH2Cl2, 0 °C–r.t., 7 h, 80%; l) PPTS, MeOH, r.t., 5 h, 85%; m) TBAF, anhydrous THF, r.t, 3 days, 75%. :1), 48 h, 90%; c) i) NaIO4, THF:H2O (4:1), 30 min; ii) PPh3 = CHCO2Et, benzene, r.t., 3 h, (80% from 2 steps); d) DIBAL-H, anhydrous CH2Cl2, 0 °C–r.t., 1 h, 82%; e) (−)-DIPT, Ti(iOPr)4, TBHP, anhydrous CH2Cl2, −20 °C, 36 h, 80%; f) i) Me2CuLi, anhydrous Et2O, −40 °C, 3.5 h; ii) NaIO4, THF:H2O (4:1), 0 °C–r.t., 30 min, 78%. g) 2,2-DMP, PPTS, anhydrous CH2Cl2, 0 °C–r.t., 3 h, 85%; h) Li, liq NH3, anhydrous THF, −78 °C, 30 min, 82%; i) IBX, anhydrous CH2Cl2, anhydrous DMSO, 0 °C–r.t., 12 h, 80%; j) i) PhNO, L-Proline (40 mol %), triethyl phosphono acetate, Cs2CO3, anhydrous DMSO, r.t., 1 h, ii) Cu(OAc)2, EtOH, r.t., 12 h, 57%, 91%de; k) MOMCl, N,N-diisopropylethyl amine, DMAP, anhydrous CH2Cl2, 0 °C–r.t., 7 h, 80%; l) PPTS, MeOH, r.t., 5 h, 85%; m) TBAF, anhydrous THF, r.t, 3 days, 75%. | ||
The structure of compound 1 was analyzed in pure CDCl3 using 2D NMR techniques such as gDQCOSY, HSQC and NOESY. The experiential 3JHH couplings H5–H6 = 4.0 Hz (H5 equatorial and H6 axial) and H2–H3 = 1.9 Hz (H2 axial and H3 equatorial) are allowing to fix the axial and equatorial positions of the protons. Furthermore, observed NOE cross peaks between HF–HH, HF–H5, H3–HD, HA–HD, HC–HD and H2–H6 suggest the following structure for compound 1 (Fig. 2 and 3).
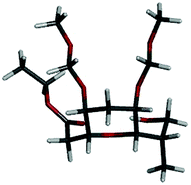 | ||
| Fig. 3 Energy-minimized diagram for 1. | ||
Retrosynthetic analysis for the C14–C22 tetrahydrofuran domain 14 of the halichondrin family is presented in Scheme 3.
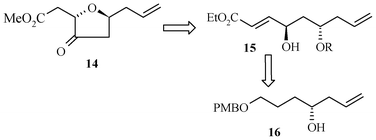 | ||
| Scheme 3 Retrosynthetic analysis. | ||
The synthesis of the C14–C22 tetrahydrofuran domain 14 of the halichondrin family starts from the known homoallyl alcohol 168 (Scheme 4), which was eventually protected as TBS ether 17. After deprotection of the PMB group, the resulting alcohol 18 was oxidized to an aldehyde 19. The desired hydroxy unsaturated ester 20 was obtained using an aminoxylation reaction. Thus, treatment of aldehyde 19 with nitrosobenzene in the presence of 40 mol% of L-proline in DMSO at room temperature followed by in situ Horner–Wadsworth–Emmons olefination with triethylphosphono acetate and Cs2CO3 as base furnished an aminoxyolefinic ester; cleavage of the O–N bond using Cu(OAc)2 in ethanol gave the γ-hydroxy α,β-unsaturated ester 20 in 52% yield with 98% de. The absolute stereochemistry of the new chiral center formed in compound 20 was confirmed at a later stage by NOE studies of compound 21 (see Fig. 4). TBS deprotection with TBAF followed by cyclization using Triton B in MeOH9 for 48 h furnished the required anti 2,5-tetrahydrofuran 21 in 78% yield, where transesterification has taken place. The oxidation of the hydroxy group using IBX provided the desired C14–C22 tetrahydrofuran domain 14 of the halichondrin family in 70% yield.
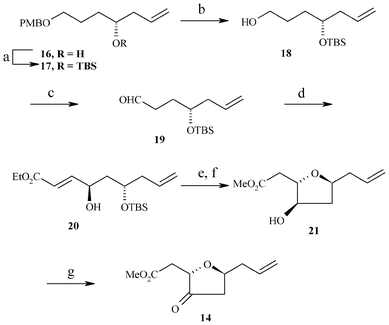 | ||
| Scheme 4 Reagents: a) TBSCI, imidazole, anhydrous CH2Cl2, 0 °C, 2 h, 92% b) DDQ, CH2Cl2:H2O (9:1), 0 °C, 30 min., 78%; c) IBX, anhydrous CH2Cl2, anhydrous DMSO, 0 °C–r.t., 10 h, 82%; d) i) PhNO, L-Proline (40 mol %), triethyl phosphonoacetate, Cs2CO3, anhydrous DMSO, r.t., 1 h; ii) Cu(OAc)2, EtOH, r.t., 12 h, 52%, 98% de; e) TBAF, anhydrous THF, 0 °C–r.t, 30 min, 80%; f) Triton-B, MeOH, r.t., 1 h, 78%; g) IBX, anhydrous CH2Cl2, anhydrous DMSO, 0 °C–r.t., 2 days, 70%. | ||
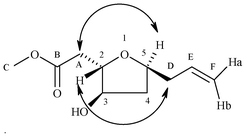 | ||
| Fig. 4 Chemical structure of 21. | ||
The structure of compound 21 was analyzed in pure CDCl3, by using 1D- 1H and 2D 1H NMR techniques such as gDQCOSY, HSQC and NOESY. Observed NOE cross peaks between H5–HA, H2–HD indicating H2 and H5 are anti to each other suggest the indicataed structure for compound 21 (Fig. 4 & 5).
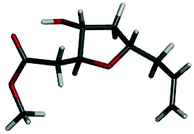 | ||
| Fig. 5 Energy-minimized diagram for 21. | ||
In conclusion, synthesis of the C45–C53 tetrahydropyran domain of norhalichondrins and the C14–C22 tetrahydrofuran domain of the halichondrin family has been achieved in 13 linear steps with a overall 5.07% yield and in 7 linear steps with a overall 13.3% yield featuring MacMillan aminoxylation and HWE followed by oxa-Michael as the key reaction steps. Our group is engaged toward the synthesis of halichondrin and norhalichondrin and the results will be published in due course.
Acknowledgements
G.C thanks UGC, New Delhi for the award of fellowship.References
- (a) D. Uemura, K. Takahashi, T. Yamamoto, C. Katayama, J. Tanaka, Y. Okumura and Y. Hirata, J. Am. Chem. Soc., 1985, 107, 4796–4798 CrossRef CAS; (b) Y. Hirata and D. Uemura, Pure Appl. Chem., 1986, 58, 701–710 CrossRef CAS; (c) M. Litaudon, S. J. H. Hickford, R. E. Lill, R. J. Lake, J. W. Blunt and M. H. G. Munro, J. Org. Chem., 1997, 62, 1868–1871 CrossRef CAS; (d) M. Litaudon, J. B. Hart, J. W. Blunt, R. J. Lake and M. H. G. Munro, Tetrahedron Lett., 1994, 35, 9435–9438 CrossRef CAS; (e) G. R. Pettit, R. Tan, F. Gao, M. D. Williams, D. L. Doubek, M. R. Boyd, J. M. Schmidt, J. C. Chapuis, E. Hamel, R. Bai, J. N. A. Hooper and L. P. Tackett, J. Org. Chem., 1993, 58, 2538–2543 CrossRef CAS.
- A recent publication (R. Bai, K. D. Paull, C. L. Herald, L. malspeis, G. R. Pettit and E. Hamel, J. Biol. Chem. 1991, 266, 15882–15889) implies that halichondrin B and homohalichondrin B are isolated from Axinella sponges Search PubMed.
- (a) K. L. Jackson, J. A. Henderson, H. Motoyoshi and A. J. Phillips, Angew. Chem., Int. Ed., 2009, 48, 2346–2350 CrossRef CAS; (b) T. D. Aicher, K. R. Buszek, F. G. Fang, C. J. Forsyth, S. H. Jung, Y. Kishi, M. C. Matelich, P. M. Scola, D. M. Spero and S. K. Yoon, J. Am. Chem. Soc., 1992, 114, 3162–3164 CrossRef CAS; (c) J. A. Henderson, K. L. Jackson and A. J. Phillips, Org. Lett., 2007, 9, 5299–5302 CrossRef CAS.
- (a) K. L. Jackson, J. A. Henderson and A. J. Phillips, Chem. Rev., 2009, 109, 3044–3079 CrossRef CAS.
- J. S. Yadav, C. N. Reddy and G. Sabitha, Tetrahedron Lett., 2012, 53, 2504–2507 CrossRef CAS.
- (a) G. Sabitha, G. Chandrashekhar, K. Yadagiri and J. S. Yadav, Tetrahedron Lett., 2010, 51, 3824–3826 CrossRef CAS; (b) G. Sabitha, D. V. Reddy, A. Senkara Rao and J. S. Yadav, Tetrahedron Lett., 2010, 51, 4195–4198 CrossRef CAS; (c) G. Sabitha, D. V. reddy, A. Senkara Rao and J. S. Yadav, Org. Lett., 2011, 13, 382–385 CrossRef CAS.
- J. S. Yadav, S. Joyasawal, S. K. Dutta and A. C. Kunwar, Tetrahedron Lett., 2007, 48, 5335–5340 CrossRef CAS.
- S. Takahashi, A. Kubota and T. Nakata, Angew. Chem., Int. Ed., 2002, 41, 4751–4754 CrossRef CAS.
- G. Sabitha, A. Y. Reddy, S. Nayak and J. S. Yadav, Synthesis, 2012, 44, 1657–1662 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: spectral data of all new compounds. NOE and HPLC diagrams for selected compounds. See DOI: 10.1039/c2ra21346j |
| This journal is © The Royal Society of Chemistry 2012 |