Thienyl-substituted BODIPYs with strong visible light-absorption and long-lived triplet excited states as organic triplet sensitizers for triplet–triplet annihilation upconversion†
Yinghui
Chen
ab,
Jianzhang
Zhao
*b,
Lijuan
Xie
a,
Huimin
Guo
b and
Qiuting
Li
b
aInstitute of Molecular Medicine, Huaqiao University, 269 Chenghua North Road, Quanzhou, 362021, P. R. China
bState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, E-208 West Campus, 2 Ling-Gong Road, Dalian, 116024, P. R. China. E-mail: zhaojzh @dlut.edu.cn; Fax: +86 (0) 411 8498 6236; Web: http://finechem.dlut.edu.cn/photochem
First published on 13th March 2012
Abstract
Thienyl-substituted BODIPY derivatives were prepared as organic triplet photosensitizers for triplet–triplet annihilation (TTA) upconversion. The photophysical properties of the sensitizers were fully studied with steady state and time-resolved spectroscopy, as well as density functional theory (DFT) calculations. Sensitizers with both 2-monothienyl substituted (BI-1) and 2,6-dithienyl substituted BODIPY cores (BI-2) were prepared. These sensitizers show strong absorption in the visible range. Interestingly, the sensitizers show large Stokes shifts (up to 86 nm) vs. small Stokes shifts (ca. 15 nm) for the normal BODIPY derivatives. DFT/TDDFT calculations show that the large Stokes shifts are due to the remarkable geometry relaxation of the sensitizers upon photoexcitation. The sensitizers show long-lived triplet excited states (up to 95.2 μs at room temperature), which is the longest T1 state lifetime of organic sensitizers used for TTA upconversion. DFT calculations indicate that the energy level T1 state does not decrease, although the absorption–emission wavelengths of the thiophene-substituted sensitizers are red-shifted compared to the unsubstituted BODIPY, which is beneficial for TTA upconversion. The organic sensitizers were used for TTA upconversion and upconversion quantum yields up to 16.5% were observed, compared to the quantum yields of 0.6% and 6.1% observed previously with organic triplet sensitizers. The efficient TTA upconversion is attributed to the enhanced triplet–triplet-energy-transfer (TTET) process, confirmed by the lifetime quenching experiments of the photosensitizers. Our results are useful for the design of new efficient organic triplet photosensitizers to replace the currently used phosphorescent transition metal complex sensitizers for TTA upconversion and other appropriate photophysical processes, such as photocatalysis, photodynamic therapy, etc.
Introduction
Upconversion has attracted much attention, due to its potential applications in photovoltaics, photocatalysis, and optical materials, etc.1–15 A few techniques are available for upconversion, such as using rare earth metal nanoparticles,5,16 two-photon absorption dyes,18 and more recently, the triplet–triplet annihilation (TTA) based upconversions.3,6 TTA upconversion is based on a combination of the triplet sensitizer and the triplet acceptor (annihilator/emitter), very often in fluid matrix.3a,6 The excitation light was harvested by the triplet sensitizer, for which firstly the singlet excited state is populated then the triplet excited state is populated via intersystem crossing (ISC).19 The excitation energy is in turn transferred to the triplet acceptor through the triplet–triplet-energy-transfer (TTET) process, by which the triplet excited state of the acceptor is populated. Then by annihilation, the singlet excited state of the acceptor will be produced and the upconverted fluorescence from the acceptor can be observed. Energy levels of the triplet sensitizer and the acceptor have to be matched with each other (for details, please refer to Scheme 2 in the later section).3a,6TTA upconversion is in particularly promising for applications, due to its advantages over other upconversion techniques, such as (1) non-coherent excitation with low power density is sufficient for sensitizing TTA upconversion. Excitation power density at a few mW cm−2 is effective, which is lower than terrestrial solar radiance (100 mW cm−2, AM1.5G); (2) high upconversion yield (ΦUC) of TTA upconversion, or more precisely, the higher overall upconversion capability, η = ε × ΦUC (ε is the molar extinction coefficients of the sensitizer and ΦUC is the upconversion quantum yield).6 The spin statistical rule predicts that the maximal upconversion quantum yield will be 11.1%.3 However, this previously thought value has been challenged by much higher upconversion quantum yields observed experimentally. For example, an upconversion quantum yield up to 16.4% has been observed.3a The triplet sensitizers used in the TTA upconversion can be optimized to show strong absorption of the excitation light. In comparison, rare earth metal upconversion materials usually show weak absorption, thus the η values of these materials are low;5,16,17 (3) the excitation and emission wavelength, or more generally, various energy levels of the components of the TTA upconversion, can be readily optimized by independent selection of the sensitizers and acceptors. Chemical modifications of the molecular structures of the sensitizers and the acceptors is feasible. This is an obvious advantage over the rare earth materials or the two photon-absorption fluorescent dyes.5,18
However, the development of TTA upconversion is facing some challenges.6 The most prominent one is to develop organic triplet photosensitizers to replace the conventional transition metal complex triplet photosensitizers. Currently transition metal complex sensitizers are used for TTA upconversion, due to the efficient ISC thus the high quantum yield of the triplet excited state of these compounds upon photoexcitation.3a,6 However, these complexes are expensive, difficult to prepare, and difficult to modify the molecular structures to optimize the energy levels.
Thus, similar to the development scenario of photosensitizers for dye-sensitized solar cells (DSCs), we propose that organic triplet sensitizers will be the next generation of sensitizers for TTA upconversion.6 However, compared to the development of the photosensitizers of DSCs, it is more challenging to develop organic triple photosensitizers for TTA upconversion, because the triplet excited state, not the quantum mechanically allowed singlet excited state, must be populated upon photoexcitation (direction S0→T1 transition is usually a strongly-forbidden process).19 Thus, some methods, such as the heavy atom effect, have to be used for the organic triplet sensitizers to facilitate the ISC process. The only two reported organic triplet sensitizers used for TTA upconversion are butanedione and 2,4,5,7-tetraiodo-6-hydroxy-3-fluorone (TIHF).20,21 However, these sensitizers show disadvantages of either short excitation wavelength or the difficulty of tuning the energy levels of the compounds by molecular structural modification.21
On the other hand, BODIPY is a versatile chromophore and its fluorescence has been extensively studied.22–25 Recently some iodo-substituted BODIPYs were studied for sensitizing singlet oxygen (1O2).26 However, to the best of our knowledge, the triplet excited state of BODIPY is rarely use for TTA upconversion.6 Recently we designed a small library of iodo-BODIPY based triplet sensitizers for TTA upconversion.13 The iodo-BODIPY triplet sensitizers show upconversion quantum yields of 5.40%. The result is promising but it is lower than that observed with Pt(II)/Pd(II) porphyrin complex sensitizers (ca. 15%).27
In order to address the low upconversion quantum yield of the iodo-BODIPY based organic triplet photosensitizers,13 herein we design thienyl-substituted BODIPY based organic triplet photosensitizers for TTA upconversion (Scheme 1). Compared to the previously reported iodo-BODIPY based sensitizers,13 these new triplet sensitizers show enhanced absorption in the visible range with prolonged triplet excited state lifetime (95.2 μs, ca. 2-fold of the previous value). An upconversion quantum yield up to 16.5% was observed, which is 3-fold higher than the previous value.13 Our result is useful for the design of new organic triplet photosensitizers for TTA upconversion and photocatalysis, etc.
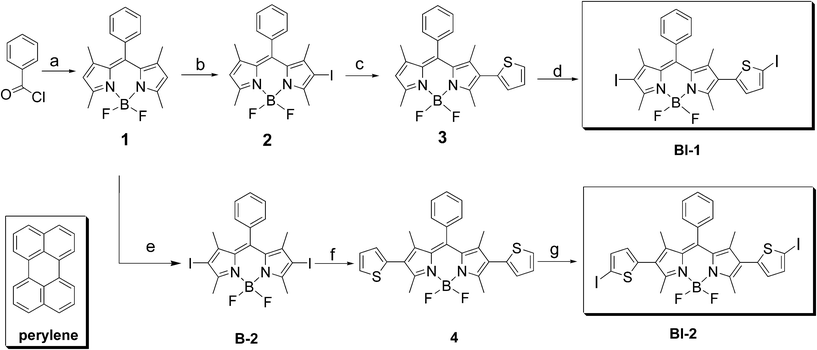 | ||
| Scheme 1 Synthesis of the thiophene-substituted BODIPY triplet sensitizers BI-1 and BI-2. The known organic triplet sensitizer B-2 and the molecular structure of the triplet acceptor perylene are also presented. (a) 2,4-Dimethylpyrrole, under Ar, overnight; TEA, BF3·OEt2, overnight; yield: 55.0%; (b) N-iodosuccinimide (NIS), CH2Cl2; yield: 45.0%; (c) thiophene-2-boronic acid, K3PO4·3H2O, Pd(OAc)2, toluene, ethanol, under Ar, 80 °C, 8 h; yield: 63.4%; (d) CHCl3, glacial acetic acid, NIS, overnight; yield: 70.0%; (e) NIS, CH2Cl2; yield: 77.0%; (f) thiophene-2-boronic acid, K3PO4·3H2O, Pd(OAc)2, toluene, ethanol, under Ar, 80 °C, 8 h; yield: 54.4%; (g) CHCl3, glacial acetic acid, NIS, overnight; yield: 50.0%. | ||
Experimental
Materials and reagents
All the chemicals used in the synthesis are analytically pure and were used as received. Solvents were dried and distilled for synthesis. Fluorescence quantum yields were measured with 1,3,5,7-tetramethyl-8-phenyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene as the standard (compound 1, Scheme 1. ΦF = 0.72 in tetrahydrofuran). NMR spectra were carried out on a 400 MHz Varian Unity Inova spectrometer. Mass spectra were recorded with a Q-TOF Micro MS spectrometer. UV-Vis spectra were carried on a HP8453 UV-visible spectrophotometer. Fluorescence spectra were recorded on a RF5301PC or 970 CRT spectrofluorometer. Fluorescence lifetimes were measured on an OB 920 luminescence lifetime spectrometer (Edinburgh Instruments, U. K.).4,4-Difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (1)
Under an argon atmosphere, benzoyl chloride (1.4 g, 11 mmol) was dissolved in CH2Cl2 (150 mL), then 2,4-dimethylpyrrole (2.0 mL, 20 mmol) was added. After the reaction mixture was stirred at room temperature overnight, triethylamine (TEA) (10 mL) and then BF3·OEt2 (10 mL) were added dropwise to the mixture in an ice-water bath, and the mixture was stirred overnight until the reaction is complete (thin-layer chromatography (TLC, CH2Cl2 as the eluent). The solvent was removed under reduced pressure. The residue was washed with saturated Na2CO3 solution (100 mL) and water (100 mL). The aqueous solutions were extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was purified by column chromatography (silica gel, CH2Cl2) to obtain a red powder (1.96 g), yield: 55.0%. 1H NMR (400 MHz, CDCl3): δ 7.49–7.47 (m, 3H), 7.29–7.26 (m, 2H), 5.98 (s, 2H), 2.56 (s, 6H), 1.37 (s, 6H).2-Iodo-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (2)
To the solution of compound 1 (200.0 mg, 0.62 mmol) in CH2Cl2 (50 mL), N-iodosuccinimide (NIS) (140.0 mg, 0.62 mmol) in CH2Cl2 (20 mL) was added dropwise and the mixture was stirred for 1 h at room temperature. After complete consumption of compound 1, the solution was evaporated under reduced pressure. The obtained residual was purified by column chromatography (silica gel, n-hexane/CH2Cl2 = 2/1, v/v) to get a bright red solid (125.6 mg), yield: 45.0%. 1H NMR (400 MHz, CDCl3): δ 7.51–7.48 (m, 3H), 7.27–7.25 (m, 2H), 6.04 (s, 1H), 2.63 (s, 3H), 2.57 (s, 3H), 1.38 (s, 6H). TOF LD+: calcd ([C19H18BF2IN2]+), m/z = 450.0576, found, m/z = 450.0535.2-(Thiephene-2-yl)-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (3)
To the solution of compound 2 (100.0 mg, 0.22 mmol) in toluene (5 mL), ethanol (5 mL), thiophene-2-boronic acid (56.3 mg, 0.44 mmol) and K3PO4·3H2O (117.2 mg, 0.44 mmol) were added under Ar atmosphere. Then palladium diacetate (Pd(CH3COO)2) (2.6 mg, 0.0132 mmol) was added. The solution was heated at 80 °C for 8 h. After complete consumption of the starting material, the solution was evaporated under reduced pressure. The residual was purified by column chromatography (silica gel, n-hexane/CH2Cl2 = 1/1, v/v) to get a red solid (56.6 mg), yield: 63.4%. M. p. 155.2–157.2 °C. 1H NMR (400 MHz, CDCl3): δ 7.50 (d, 3H, J = 5.9 Hz); 7.33–7.30 (m, 3H); 7.08 (t, 1H, J = 3.6 Hz); 6.84 (d, 1H, J = 3.40 Hz); 6.02 (s, 1H); 2.58 (s, 6H); 1.39 (d, 2H, J = 11 Hz); 13C NMR (100 MHz, CDCl3): δ 156.7, 154.3, 144.1, 142.3, 140.3, 135.2, 134.7, 132.1, 130.9, 129.4, 129.2, 128.1, 127.8, 127.3, 126.0, 121.9, 30.5, 14.9, 14.7, 13.5, 12.9. TOF HRMS ES+: Calc (C23H21BF2N2S) m/z = 406.1487, found, m/z = 406.1498.2-Iodo-6-(5-iodothiephene-2-yl)-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (BI-1)
To a round-bottomed flask (25 mL) was added compound 3 (40.6 mg), CHCl3 (10 mL), and AcOH (2 mL) under darkness. NIS (99.0 mg, 0.44 mmol) was added in one-portion, and the solution was stirred at room temperature overnight. After the addition of sat. aq. Na2SO3 solution, the mixture was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The residual was purified by column chromatography (silica gel, n-hexane–CH2Cl2 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to get a red solid (46.1 mg), yield: 70.0%. M.p. 168.0–168.9 °C. 1H NMR (400 MHz, CDCl3), δ 7.52 (d, 3H, J = 3.5 Hz), 7.29–7.28 (m, 2H), 7.21 (d, 1H, J = 3.6 Hz), 6.52 (d, 1H, J = 3.6 Hz), 2.66 (s, 6H), 2.57 (s, 6H), 1.40 (s, 6H), 1.34 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 173.7, 156.5, 155.8, 144.8, 142.3, 141.9, 140.5, 137.4, 134.9, 131.8, 131.1, 129.7, 129.6, 128.0, 126.0, 100.9, 85.6, 73.7, 79.9, 17.1, 16.2, 13.7, 13.1. TOF HRMS ES+: Calc (C23H19BF2I2N2S) m/z = 657.9419, found, m/z = 657.9424. Elemental analysis calculated (%) for C23H19BF2I2N2S+0.4(H2O): C 41.52, H 3.00, N 4.21; found: C 41.52, H 3.08, N 4.15.
:1, v/v) to get a red solid (46.1 mg), yield: 70.0%. M.p. 168.0–168.9 °C. 1H NMR (400 MHz, CDCl3), δ 7.52 (d, 3H, J = 3.5 Hz), 7.29–7.28 (m, 2H), 7.21 (d, 1H, J = 3.6 Hz), 6.52 (d, 1H, J = 3.6 Hz), 2.66 (s, 6H), 2.57 (s, 6H), 1.40 (s, 6H), 1.34 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 173.7, 156.5, 155.8, 144.8, 142.3, 141.9, 140.5, 137.4, 134.9, 131.8, 131.1, 129.7, 129.6, 128.0, 126.0, 100.9, 85.6, 73.7, 79.9, 17.1, 16.2, 13.7, 13.1. TOF HRMS ES+: Calc (C23H19BF2I2N2S) m/z = 657.9419, found, m/z = 657.9424. Elemental analysis calculated (%) for C23H19BF2I2N2S+0.4(H2O): C 41.52, H 3.00, N 4.21; found: C 41.52, H 3.08, N 4.15.
2,6-Diiodo-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (B-2)
To the solution of compound 1 (100.0 mg, 0.31 mmol) in CH2Cl2 (10 mL), NIS (140.0 mg, 0.62 mmol) was added, and the solution was stirred for 2 h. After complete consumption of compound 1, the solution was evaporated under reduced pressure. The residual was purified by column chromatography (silica gel, n-hexane–CH2Cl2 = 1:1, v/v) to get a red solid (137.5 mg), yield: 77.0%. 1H NMR (400 MHz, CDCl3): δ 7.54–7.51 (m, 3H), 7.26–7.24 (m, 2H), 2.65 (s, 6H), 1.38 (s, 6H). TOF LD−: calcd ([C19H17BF2I2N2]−), m/z = 575.9542, found, m/z = 575.9528.
2,6-Di(thiephene-2-yl)-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (4)
To the solution of compound B-2 (100.0 mg, 0.17 mmol) in mixed solvent of toluene (5 mL) and ethanol (5 mL), thiophene-2-boronic acid (111.1 mg, 0.87 mmol) and K3PO4·3H2O (181.1 mg, 0.68 mmol) were added. Then palladium diacetate (2.0 mg, 0.0102 mmol) was added under argon atmosphere. The reaction mixture was refluxed for 8 h. After complete consumption of compound B-2, the solution was evaporated under reduced pressure. The obtained residual was purified by column chromatography (silica gel, n-hexane–CH2Cl2 = 1:1, v/v) to get a red solid (45.1 mg), yield: 54.4%. M.p. > 250 °C. 1H NMR (400 MHz, CDCl3), δ 7.51 (d, 3H, J = 6.6 Hz); 7.35–7.33 (m, 4H); 7.09 (t, 2H, J = 3.7 Hz); 6.86 (d, 2H, J = 3.5 Hz); 2.60 (s, 6H); 1.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 155.5, 141.0, 129.5, 128.2, 127.9, 127.4, 126.2, 29.9, 13.7, 13.1; TOF HRMS ES+,Calc(C27H23BN2F2S2) m/z = 488.1364, found, m/z = 488.1385.
2,6-Di(5-iodothiephene-2-yl)-4,4-difluoro-1,3,5,7-tetramethyl-8-phenyl-4-bora-3a,4a-diaza-s-indacene (BI-2)
Compound 4 (48.8 mg), CHCl3 (10 mL) and AcOH (2 mL) were mixed under darkness. NIS (99.0 mg, 0.40 mmol) was added in one-portion, and the solution was stirred at room temperature overnight. After the addition of sat. aq. Na2SO3 solution, the mixture was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The residual was purified by column chromatography (silica gel, n-hexane/CH2Cl2 = 2/1, v/v) to get a red solid (46.1 mg), yield: 50.0%. M.p. 189.7–190.5 °C . 1H NMR (400 MHz, CDCl3), δ 7.50 (d, 3H, J = 3.6 Hz), 7.31–7.29 (m, 2H) 7.23–7.20 (m, 2H), 6.53 (d, 2H, J = Hz), 2.58 (6H, s), 1.36 (d, 6H, J = 4.5 Hz). 13C NMR (100 MHz, CDCl3): δ 155.9, 155.4, 155.2, 143.9, 143.3,143.1, 142.6, 141.5, 141.2, 140.7, 140.5, 137.3, 134.9, 134.8, 131.7, 131.4, 129.5, 128.0, 126.0, 125.8, 84.3, 75.9, 73.7, 28.9, 13.6, 13.3. TOF HRMS ES+, Calc (C27H21BF2I2N2S2) m/z = 739.9297, found, m/z = 739.9310. Elemental analysis calculated (%) for C27H21BF2I2N2S2+1.4(CH2Cl2)+0.1(H2O): C 39.61, H 2.81, N 3.25; found: C 39.85, H 2.85, N 3.18.Nanosecond time-resolved transient absorption spectra
The nanosecond time-resolved transient difference absorption spectra were measured on a laser flash photolysis instruments (LP920, Edinburgh Instruments, Livingston, U. K.) and recorded on a Tektronix TDS 3012B oscilloscope. The lifetime were obtained by monitoring the decay trace of the transients. All samples were deaerated with Ar for ca. 15 min before measurement and the gas flow was kept during the measurement.TTA upconversions
Diode pumped solid state laser (emission wavelength: 532 nm) was used as the excitation source for the TTA upconversions. The diameter of the laser spot is ca. 4 mm (the spot is an oval shape). The laser power was measured with photodiode detector. The output power of the DPSS laser can be adjusted continuously. For the upconversion experiments, the mixed solution of the triplet sensitizer and perylene (triplet acceptor) was degassed for at least 30 min with N2 or Ar. Then the solution was excited with laser. The upconverted fluorescence of perylene was observed with spectrofluorometer. The fluorescence of the photosensitizers was observed simultaneously. In order to suppress the scattered laser, a black box was put behind the cuvette to trap the laser beam.For the measurement of the TTET efficiency, i.e. the Stern–Volmer quenching constants, the concentration of sensitizers was fixed at 1.0 × 10−5 M, the lifetime of the sensitizers was measured with transient absorption spectroscopy upon increasing the perylene concentration.
The upconversion quantum yields were determined with the prompt fluorescence of the sensitizer B-2 as the standard, e.g. ΦUC of BI-1 was determined by comparing the upconverted fluorescence and its prompt fluorescence. The upconversion quantum yields were calculated with eqn (1), where ΦUC, Aunk, Iunk and ηunk represent the quantum yield, absorbance, integrated photoluminescence intensity and the refractive index of the samples and the solvents, respectively (eqn (1)). The equation is multiplied by factor of 2 in order to make the maximum quantum yield to be unity (100%) otherwise the maximum quantum yield will be only 50%.3a
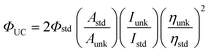 | (1) |
DFT calculations
The geometry of the molecules was optimized using density functional theory (DFT) with the B3LYP functional and 6-31G(d) basis set. The excited state related calculations were carried out with the time dependent DFT (TD-DFT) with the ground state geometry. No solvent was used for the calculation of the emission of BI-2 (base set is B3LYP/LanL2DZ/6-31G). There are no imaginary frequencies for all optimized structures. Calculations were performed with Gaussian 09W.28Results and discussions
Design and synthesis of the organic triplet sensitizers
Based on the photophysical principals of the TTA upconversion (Scheme 2, later section), the absorption of the excitation light by the photosensitizer and the lifetime of the triplet excited state of the sensitizer, are crucial to improve the upconversion quantum yield.6 Thus, we selected the thiophene moiety to be attached to the fluorophore core of BODIPY. This selection was taken with a few rationales. First, it is known that the thiophene moiety is an efficient π-conjugation linker.29 Thus we expect that the absorption of the BODIPY core can be extended to longer wavelength. Second, we used the thiophene linker between the BODIPY core and the iodine atom, instead of benzene, to ensure the effective ISC, due to the more efficient π-conjugation capability of the thiophene moiety. Isolation of the iodine from the chromophore π-core will make the ISC much less efficient.13,27 Third, the slightly-reduced ISC of the new sensitizers compared to the reported triplet sensitizers may prolong the triplet state lifetime, which is beneficial to the TTET process, a crucial step for the TTA upconversion.6,10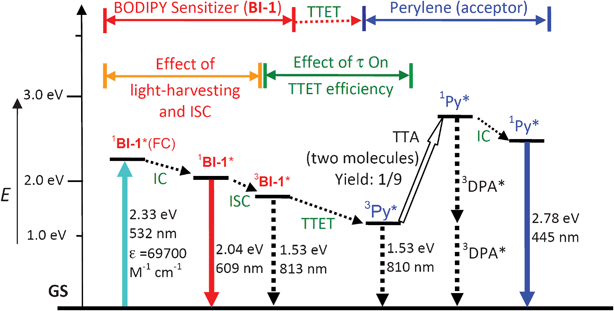 | ||
| Scheme 2 Qualitative Jablonski diagram illustrating the sensitized TTA upconversion process between BI-1 (sensitizer) and perylene (Py, acceptor). The effect of the light-harvesting ability and the triplet state lifetime of the BI-1 sensitizer on the efficiency of TTA upconversion is also shown. E is energy. GS is ground state (S0). 1BI-1*(FC) is BI-1 singlet excited state at Franck–Condon state. IC is inner conversion. ISC is intersystem crossing. 3BI-1* is the BI-1 based triplet excited state. TTET is triplet–triplet-energy-transfer. 3Py* is the triplet excited state of perylene. TTA is the triplet–triplet annihilation. 1Py* is the singlet excited state of perylene. The emission observed for the sensitizers alone is due to the 1BI-1* emissive excited state. The emission observed in the TTA upconversion is the simultaneous 1BI-1* emission (prompt fluorescence) and the 1Py* emission (upconverted fluorescence). | ||
The synthesis of the organic triplet sensitizer is summarized in Scheme 1. The BODIPY chromophore was iodated with N-iodosuccinimide (NIS). Mono- and bisiodo-substituted BODIPYs, compound 2 and B-2 were obtained in satisfying yields. Then the Suzuki cross coupling reaction with the 2-thiopheneboronic acid lead to the thiophene-substituted intermediates compounds 3 and 4. Iodation with NIS again gives BI-1 and BI-2 (Scheme 1). All the compounds were obtained with moderate to good yields.
Steady-state UV-vis absorption and fluorescence spectra
The UV-vis absorption spectra of the compounds were studied (Fig. 1). Compared to the unsubstituted BODIPY 1, which gives absorption at 504 nm (ε = 70280 M−1 cm−2),23,25 the new compounds BI-1 and BI-2 give absorption at red-shifted range at 532 nm and 526 nm, respectively. We noticed that these two new compounds show red-shifted tailing in comparison to the iodo-BODIPY without the thiophene moieties B-2. However, the molar extinction coefficient of BI-1 (ε = 69700 M−1 cm−2 at 532 nm) and BI-2 (ε = 49000 M−1 cm−2 at 526 nm) decreased sharply compared to the iodo BODIPY B-2 (ε = 87000 M−1 cm−2 at 529 nm). Red-shifted absorption is beneficial for the application in upconversions.3,6 Compared to the emission, the absorption of BI-1 and BI-2 is only slight red-shifted vs. that of compound 1. This is due to the limited π-conjugation between the BODIPY and the thienyl moieties at the ground state (S0 state).
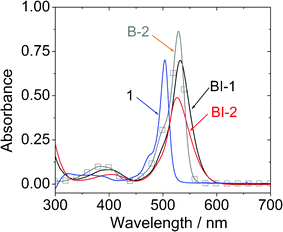 | ||
| Fig. 1 UV-vis absorption of BI-1 and BI-2. Data of B-2 and 1 (BODIPY) are presented for comparison. c = 1.0 × 10−5 M in toluene. 20 °C. | ||
The fluorescence spectra of the compounds were studied (Fig. 2 and ESI†). Red-emission was observed. We found with thiophene substitution, the emission of BI-1 (609 nm) is red-shifted compared to that of B-2 (which is at 552 nm). Notably, the Stokes shifts of the new BODIPY derivatives (77 nm) is much larger than that of the B-2 (23 nm) or the unsubstituted BODIPY (ca. 12 nm). Similar emission and large Stokes shift were observed for BI-2 (see ESI†). BODIPY derivatives with such a large Stokes shift are rare.23,25 Although the π-conjugation framework of the BODIPY core can be extended by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds or C
C bonds or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds,29a and the emission wavelength of the BODIPY derivatives can be red-shited, the Stokes shifts of the BODIPY derivatives are usually unaltered with chemical modification.23,25 With DFT calculations, we propose that the large Stokes shifts of the new derivatives are due to the increased geometry relaxation of BI-1 and BI-2 upon photoexcitation (please refer to the DFT calculations section).
C triple bonds,29a and the emission wavelength of the BODIPY derivatives can be red-shited, the Stokes shifts of the BODIPY derivatives are usually unaltered with chemical modification.23,25 With DFT calculations, we propose that the large Stokes shifts of the new derivatives are due to the increased geometry relaxation of BI-1 and BI-2 upon photoexcitation (please refer to the DFT calculations section).
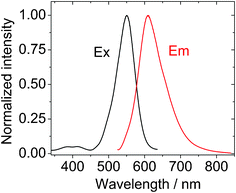 | ||
| Fig. 2 Normalized excitation spectra and emission spectra of BI-1, λex = 520 nm, c = 1.0 × 10−5 M in toluene. 20 °C. | ||
The emission of the BI-1 and BI-2 are red-shifted by 50–60 nm compared to the known compound B-2. That is, the energy levels of the singlet excited state (S1 state) of BI-1 and BI-2 are lower than that of B-2. Considering that the objective of our molecular design is to obtain the triplet photosensitizers for TTA upconversion, we don't expect the T1 state energy levels of the sensitizers to decrease, otherwise the selection of the triplet acceptor will become difficult because the T1 state energy level of the triplet acceptor has to be lower than the T1 state energy level of the triplet sensitizer. Fortunately our TTA upconversion experiments indicate that the T1 state energy level of the BI-1 and BI-2 do not decrease. DFT calculations on the energy levels of the T1 excited state of the BI-1 and BI-2 confirmed our anticipations (please see later section for detail). This uncompromised T1 state energy level is an advantage for TTA upconversion, that is, the chemical derivatisation of the BODIPY chromophore with thiophene substituents does not decrease the T1 state energy levels of the triplet sensitizers.
The photophysical properties of the compounds are summarized in Table 1. The fluorescence quantum yields of BI-1 and BI-2 are higher than that of B-2, indicating the ISC effect in BI-1 and BI-2 are less efficient than that in B-2. This result is within expectation because the iodo atoms in BI-1 and BI-2 are more far from the chromophore core than that of B-2.
| Compounds | λ abs/nmc | λ em/nm d | Δν/nm e |
ε/M−1 cm−1f |
Φ F (%)g | τ F/nsh | ΔE, T1-S0i |
Φ ISC j |
|---|---|---|---|---|---|---|---|---|
| a Measured in toluene. b Measured in acetonitrile. c λ abs (nm): absorption wavelength of first absorption maximum. d λ em (nm): emission wavelength (at the maximum intensity). e Δν: Stokes shifts. f ε: extinction coefficient. g Φ F: fluorescence quantum yields, with unsubstituted BODIPY as the standard (Φ = 72.0% in tetrahydrofuran). h τ F (ns): Fluorescence lifetimes. i The calculated energy gap between S0 and T1 state: in nm. j Intersystem crossing efficiency, calculated by Ermolev's rule. ΦISC = 1 − ΦF. k Data from ref. 13. l Not determined. | ||||||||
| BI-1 a | 532 | 609 | 77 | 69700 |
7.5 | 1.42 | 813 | 0.925 |
| BI-2 a | 526 | 612 | 86 | 49000 |
12.4 | 1.91 | 815 | 0.876 |
| B-2 b | 529 | 552 | 23 | 87000 |
2.7 l | 0.13 l | 826 k | 0.973 k |
| 3 a | 513 | 592 | 78 | 69000 |
20.6 | 3.20 | — l | 0.793 |
| 4 a | 530 | 609 | 79 | 16000 |
47.2 | 3.40 | — l | 0.528 |
| 1 a | 504 | 516 | 12 | 70280 |
85.2 | 3.40 | — l | 0.148 |
Nanosecond time-resolved transient difference absorption spectroscopy
In order to prove the population of the triplet excited states of the compounds, the nanosecond time-resolved transient difference absorption spectra of the BI-1 and BI-2 were recorded (Fig. 3).23,25 Upon pulsed 532 nm laser excitation, the transient absorption spectra of BI-1 show significant bleaching at 530 nm (Fig. 3a). This bleaching band is due to the depletion of the ground state of the BODIPY chromophore (the UV-vis absorption position). Furthermore, transient absorption bands at 340 nm, 450 nm and 700 nm, were observed for BI-1. The decay kinetics of the transient at 532 nm gives a lifetime of 94.7 μs. In order to prove that this transient is due to the triplet excited state of the BI-1, the transient spectra were measured in air-saturated solution (see ESI†) and we found the transient was significantly quenched by O2. The lifetime of the transient species decreased to about 0.5 μs. Thus we confirm that the long-lived species (τ = 94.7 μs) is due to the triplet excited state of BI-1.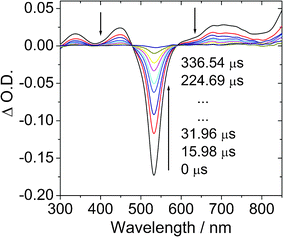 | ||
| Fig. 3 Transient difference absorption spectra of BI-1 after pulsed laser excitation (λex = 532 nm). c = 1.0 × 10−5 M in deaerated toluene. 20 °C. | ||
Under the same experimental conditions we measured the transient difference absorption of B-2 (see the ESI†). A lifetime of 56.3 μs was observed. This value is very close to the literature result of 57.1 μs.13 This control experiment proved that the lifetime of the triplet excited state of BI-1 is much longer than that of B-2.13
Furthermore, the time-resolved transient absorption spectra of BI-2 were also measured (see ESI†). Significant bleaching at 530 nm was observed. Transient absorption bands at 350 nm and 440 nm were observed. However, the transient in the range 600 nm–900 nm was different from that of BI-1.
The lifetime of the triplet excited state of BI-2 was determined as 95.2 μs. The control experiment under air atmosphere was also carried out and the lifetime was greatly reduced to about 0.5 μs. Thus, the lifetime of the triplet excited states of BI-1 and BI-2 are much longer than that of B-2.13 The longer triplet excited state lifetime of the triplet sensitizer is beneficial for the improvement of the TTA upconversion quantum yield because the crucial TTET step is more efficient with the long-lived triplet excited state of the sensitizer.6
Rationalization of the photophysical properties by DFT calculations
The BODIPY derivatives will be used for TTA upconversion, therefore the T1 excited state energy levels of these compounds are important.6 Thus the T1 state energy levels were calculated by the DFT methods (Table 2). We found that the T1 state energy levels of BI-1 and BI-2 are 1.53 eV (813 nm) and 1.52 eV (815 nm), respectively. These values are slightly higher than the T1 state energy level of B-2 (1.50 eV, 826 nm).13| Compounds | Transitionsa | TDDFT//B3LYP/6-31G(d) | |||
|---|---|---|---|---|---|
| T1/S0 energy gap | f b | Composition | CIc | ||
| a Only the selected low-lying excited states are presented. b Oscillator strength. No spin-orbital coupling was considered in the calculation thus the oscillators are zero. c The CI coefficients are in absolute values. | |||||
| BI-1 | S0→T1 | 1.53 eV (813 nm) | 0.0000 | H-2→L | 0.1091 |
| H-1→L | 0.1973 | ||||
| H → L | 0.6760 | ||||
| BI-2 | S0→T1 | 1.52 eV (815 nm) | 0.0000 | H-2→L | 0.2499 |
| H → L | 0.6661 | ||||
This is not our initial expectation because the thiophene conjugation linkers are attached to the BODIPY core and we envisaged decreased energy levels for the excited states of the sensitizers. Indeed we observed red-shifted absorption and especially much red-shifted fluorescence emission for BI-1 and BI-2 compared to that of B-2.13 Thus we expected that the T1 excited state energy levels of BI-1 and BI-2 may be decreased compared to that of B-2.
In order to rationalize the discrepancy, the spin density surfaces of the compounds BI-1 and BI-2 were analyzed (Fig. 4). We found that the spin density surface of BI-1 is localized on the BODIPY core, the thienyl moiety makes only a small contribution to the spin density surface. Similar density surface was found for BI-2. These results suggest that the thienyl moieties in BI-1 and BI-2 are not involved in the triplet excited states of BI-1 and BI-2. Therefore, the energy levels of T1 excited states of BI-1and BI-2 do not decrease compared to that of B-2.13
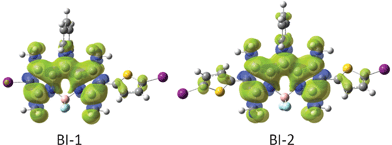 | ||
| Fig. 4 Spin density surfaces of the BODIPY based triplet sensitizers BI-1 and BI-2 (the iodo atoms are purple and sulfur atoms are yellow). Calculated by DFT at the B3LYP/LanL2DZ level using Gaussian 09W. | ||
This observation is interesting for the design of organic triplet photosensitizers for TTA upconversion because the T1 state energy level does not decrease with the structural modification of the triplet photosensitizer, despite the red-shifted UV-vis absorption and the fluorescence emission (these red-shifted emissions are indications of the decrease of the energy level of the singlet excited state of the triplet sensitizers). A decreased T1 state energy level will make the selection of the appropriate triplet acceptor difficult and the anti-Stokes shift smaller. All these consequences are detrimental to the TTA upconversion.3a,6
Since the TDDFT calculations show that it is mainly HOMO and LUMO that were involved in the triplet excited states (Table 2), thus the HOMO and LUMO isosurfaces of the triplet state of BI-1 and BI-2 were presented (Fig. 5). For BI-1, the HOMO is distributed on the BODIPY core and the thienyl moiety. However, the LUMO is localized on the BODIPY core, the thienyl moiety makes no contribution to LUMO. Similar MOs were found for BI-2. We found that the phenyl substituent at the 8-position (meso-) of the BODIPY does not contribute to the HOMO or LUMO, which is in agreement with the known photophysical properties of BODIPY.23,24
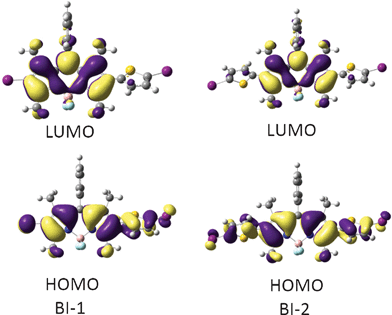 | ||
| Fig. 5 Frontier molecular orbitals of the organic triplet photosensitizers of BI-1 (left) and BI-2 (right) at the optimized triplet state geometry. Calculated by DFT at the B3LYP/GENECP/LanL2DZ level using Gaussian 09W. | ||
Origin of the large Stokes shifts of the triplet photosensitizers: geometry relaxation upon photoexcitation
The sensitizers BI-1 and BI-2 are fluorescent (Fig. 2 and Table 1) due to the radiative decay of the singlet excited states, in other words, the non-efficient ISC effect (otherwise the fluorescence will be quenched completely). The most prominent photophysical property of these dyes is the large Stokes shift (77 nm and 86 nm, Table 1). These large Stokes shifts are remarkable because usually very small Stokes shifts are observed for BODIPY and its derivatives.23,24a For example, styryl or acetylide substituents have been connected to the BODIPY core and red-shifted absorption–emission have been observed, but the Stokes shifts are still very small (ca. 10 nm).30,31 Herein we propose that the Stokes shifts of the BODIPY derivatives can be increased by inducing geometry relaxation to the rigid molecular framework of BODIPY.24b With DFT calculations, we proved that this strategy is working for the present triplet sensitizers BI-1 and BI-2. Remarkable geometry relaxation occurred, that is, the dihedral angle between the thiophene and the BODIPY core changed significantly upon excitation.Firstly the ground state geometry of BI-1 was optimized. The BODIPY core takes a coplanar geometry. The dihedral angle between the thienyl moiety and the BODIPY core is −57.2°. Furthermore, the phenyl substituent at the meso-position takes a dihedral angle 80.7° to the BODIPY core. HOMO is spread to the thiophene moiety, but not the phenyl moiety. The excitation energy of the transitions was calculated based on the optimized S0 state geometry (Franck–Condon rule, vertical excitation).19 The absorption band at the maximal wavelength was calculated as 483 nm (S0→S1 transition). This value is not far from the experimental value of 532 nm.
In order to study the fluorescence emission, the geometry of the S1 excited state was optimized by the TDDFT method (Fig. 6).
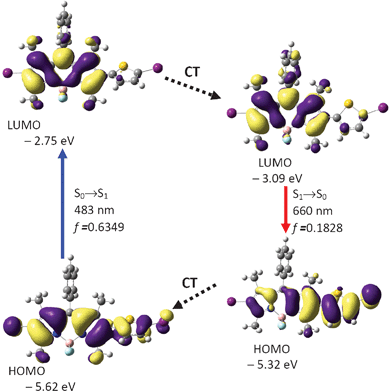 | ||
| Fig. 6 Calculation on the absorption and emission of BI-1 and rationalization of the large Stokes shift: the frontier molecular orbitals (MOs) involved in the vertical excitation (i.e. UV-vis absorption, the left column) and emission (the right column). The vertical excitation related calculations are based on the optimized ground state geometry (S0 state), the emission related calculations are based on the optimized excited state geometry (S1 state). Note the energy levels of the HOMO and LUMO at S1 state are different from that at the S0 state (which indicates geometry relaxation). CT stands for conformation transformation. Excitation and radiative processes are marked as solid lines and the non-radiative processes are marked by dotted lines. Toluene was used as the solvent for the calculations of the absorptions. At B3LYP/6-31G(d)/level using Gaussian 09W. | ||
Interestingly, the geometry of BI-1 at the excited state is drastically different from the geometry of the ground state. For example, the dihedral angle between the thiophene moiety and the BODIPY moiety is −25.9°, which is much smaller than that at the S0 state geometry (−57.2°). This result indicates that the molecule takes a more coplanar geometry at the S1 excited state. This large geometry relaxation upon photoexcitation is responsible for the large Stokes shift of BI-1.24b The emission wavelength was calculated as 660 nm, which is close to the experimental result at 609 nm (Fig. 2). We noticed the energy level of the HOMO and the LUMO are different from that at the ground state. This is mainly due to the geometry relaxation (Fig. 6).
Similar calculations were carried out for BI-2 (Fig. 7 and Table 3). The optimized S0 state geometry of BI-2 indicates a twisted conformation. The dihedral angles between the BODIPY core and the thiophene units are −56.8° and −57.0°, respectively. The HOMO is distributed on the BODIPY core and the thiophene units. However, the LUMO is more localized on the BODIPY core. The excitation was calculated as 494 nm (experimental result is 526 nm. Fig. 2 and Table 1). In order to study the fluorescence emission of the compound, the S1 state geometry was optimized (Fig. 8). The dihedral angles between the two thiophene moieties and BODIPY core are −45.9° and −22.2°, respectively. Our results are giving a clue about how to design a fluorophore that show large Stokes shift, that is, to increase the geometry relaxation of the fluorophore upon excitation.24b We believe this design rationale is useful for preparation of new fluorophores that show large Stokes shift.
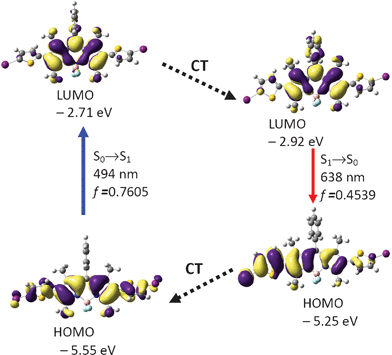 | ||
| Fig. 7 Rationalization of the absorption–emission and the large Stokes shift of BI-2: the frontier molecular orbitals (MOs) involved in the vertical excitation (i.e. UV-vis absorption, the left column) and emission (the right column) of BI-2. The vertical excitation related calculations are based on the optimized ground state geometry (S0 state), the emission related calculations were based on the optimized excited state geometry (S1 state). Note the energy levels of the HOMO and LUMO at S1 state are different from that at the S0 state (which indicates geometry relaxation). CT stands for conformation transformation. Excitation and radiative processes are marked as solid lines and the non-radiative processes are marked by dotted lines. Toluene was used as the solvent. At the B3LYP/6-31G(d)/level using Gaussian 09W. | ||
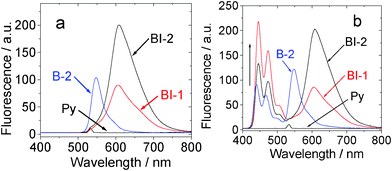 | ||
| Fig. 8 Upconversion with the organic triplet sensitizers. (a) Emission of compounds BI-1, BI-2, and the acceptor (perylene, simplified as Py) in toluene. (b) Upconversion of the perylene emission with compound BI-1 and BI-2 (1.0 × 10−5 mol L−1, both were added with 1.3 equiv of acceptor) as the sensitizer in toluene. (λex = 532 nm, the excitation power was 5.2 mW). Data of B-2 was presented for comparison. 20 °C. | ||
| Compounds | Electronic transitionsa | TDDFT//B3LYP/6-31G(d) | ||||
|---|---|---|---|---|---|---|
| Excitation energy | f b | Compositionc | CId | |||
| a Only selected excited states were presented. The numbers in parentheses are the excitation energy in wavelength. b Oscillator strength. c H stands for HOMO and L stands for LUMO. Only the main configurations are presented. d Coefficient of the wavefunction for each excitations. The CI coefficients are in absolute values. e The calculation is based on the optimized ground state geometry. f The calculation is based on the optimized S1 state geometry. | ||||||
| BI-1 | Absorptione | S0→S1 | 2.57 eV (483 nm) | 0.6349 | H-1→L | 0.1992 |
| H→ L | 0.6711 | |||||
| S0→S2 | 2.99 eV (415 nm) | 0.2688 | H-2→L | 0.1051 | ||
| H-1→L | 0.6708 | |||||
| H→ L | 0.1881 | |||||
| Emissionf | S1→S0 | 1.88 eV (660 nm) | 0.1828 | H-1→L | 0.6943 | |
| BI-2 | Absorptione | S0→S1 | 2.51 eV (494 nm) | 0.7605 | H-2→L | 0.1909 |
| H→ L | 0.6788 | |||||
| S0→S3 | 3.08 eV (402 nm) | 0.4445 | H-2→L | 0.6754 | ||
| H→ L | 0.1945 | |||||
| Emissionf | S1→S0 | 1.94 eV (638 nm) | 0.4539 | H→ L | 0.6995 | |
Triplet–triplet annihilation upconversion
Since we have shown that the long-lived triplet excited states of BI-1 and BI-2 can be populated upon photoexcitation, efficient ISC (low fluorescence quantum yields were observed for BI-1 and BI-2, Table 1), both compounds show strong absorption of visible light and the DFT calculations predicted energy level of T1 excited state at 1.53 eV (810 nm), thus, these compounds were used as triplet photosensitizers for TTA upconversion.3,6 Considering the energy level of the T1 excited states of the compounds BI-1 and BI-2, perylene was used as the triplet acceptor (energy level of the T1 excited state is 810 nm).3a,6The compounds were excited with 532 nm laser (continuous wave, CW) (Fig. 8a). BI-2 gives strong emission than that of BI-1 and B-2. This is due to the larger fluorescence quantum yields of BI-2, as well as the larger molar extinction coefficients at the excitation wavelength (ε = 69700, 46100 and 82400 M−1 cm−1 for BI-1, BI-2 and B-2 at 532 nm, respectively).
Next, the triplet acceptor perylene was added in the solution of BI-1, BI-2 or B-2. Then the mixed solution, for example, BI-1–perylene, was excited with a 532 nm laser. Besides the fluorescence emission of sensitizer BI-1, blue-shifted emission in the range 445 nm–518 nm was observed (Fig. 8b). This emission band is superimposable to the fluorescence emission of perylene (see ESI†). Excitation of the sensitizer BI-1 or perylene alone at 532 nm did not produce this emission band at 445 nm–518 nm. Thus, the emission in the range 445 nm–518 nm for the BI-1–perylene mixed solution is due to the TTA upconversion. Similar results were observed for BI-2 and B-2. It should be noted that the fluorescence of the sensitizers was not quenched in the TTA upconversion (Fig. 8b). This is reasonable since the TTET process occurs between the triplet excited states of the photosensitizers and the acceptor. The singlet excited state of the sensitizers are not involved in the photophysical processes of the TTA upconversion.3a,6,13
The upconversion quantum yields of BI-1 and BI-2 were determined as 16.5% and 10.3%, respectively. Notably, these values are 3-fold of that reported for iodo-BODIPY triplet sensitizers.13 The upconversion quantum yields are the highest values for organic triplet sensitizers. These values are comparable to those achieved with phosphorescent Pt(II)/Pd(II) porphyrin complexes.3,6 Considering the strong absorption of visible light by the sensitizers, the overall upconversion capabilities (η) of the sensitizers are significant.6 For example, the η values of BI-1 and BI-2 are 10210 M−1 cm−1 and 5052 M−1 cm−1, respectively. These values are 2 or 3-fold that of B-2.13
TTA upconversion is strongly dependent on the experimental conditions, such as the concentration of the acceptors (certainly the concentration of sensitizers).6 Thus the relationship between the upconversion intensity and the concentration of the acceptor was studied (Fig. 9). For BI-1, the upconversion intensity increased along with the addition of perylene. The upconversion reaches a plateau with an acceptor–sensitizer molar ratio of 1.23:1. A similar result was found for BI-2 (Fig. 9c). The acceptor–sensitizer molar ratio is 0.7:1 when the upconversion reaches a plateau. It should be noted that a much higher acceptor–sensitizer molar ratio was used in the literature (4:1–10:1).32,33 The smaller acceptor–sensitizer molar ratio with BI-1 and BI-2 indicated an efficient TTET process, otherwise higher concentration of acceptor is required to maximize the TTA upconversion intensity. This efficient TTA upconversion is attributed to the long-lived triplet excited state of the sensitizers.6,10
![Acceptor concentration dependency of the upconverted perylene emission with BI-1 (a) and (b), and BI-2 (c) and (d) as the sensitizers. c [sensitizer] = 1.0 × 10−5 M in toluene (λex = 532 nm, the excitation power was 5.2 mW). 20 °C.](/image/article/2012/RA/c2ra01064j/c2ra01064j-f9.gif) | ||
| Fig. 9 Acceptor concentration dependency of the upconverted perylene emission with BI-1 (a) and (b), and BI-2 (c) and (d) as the sensitizers. c [sensitizer] = 1.0 × 10−5 M in toluene (λex = 532 nm, the excitation power was 5.2 mW). 20 °C. | ||
Interestingly, the fluorescence emission of BI-1 was not quenched with increasing the perylene concentration (Fig. 9). This is in agreement with the TTA upconversion mechanism, that is, the triplet excited state, instead of the singlet excited state of the sensitizer, is involved in the TTET process and thus the TTA upconversion. This is also a demonstration that dark triplet excited state can be used for TTA upconversion.11,12,13,20,21,34
The excitation power-dependency of the upconversions with BI-1 and BI-2 was also studied (Fig. 10). Previously a quadratic relationship was usually observed for the TTA upconversion.3a However, it was proposed that linear relationship between the upconversion intensity and the excitation power will be observed if the TTET and the TTA processes are efficient.4 Our recent results with iodo-BODIPY organic triplet sensitizers confirmed this anticipatation.13
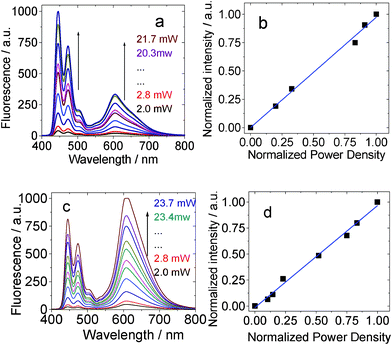 | ||
| Fig. 10 Excitation power dependency of the upconverted perylene emission with (a) and (b) BI-1 as sensitizer in toluene. λex = 532 nm. 1.0 × 10−5 M, 1.3 equiv of acceptor. BI-2 as the sensitizer (c) and (d) (1.0 × 10−5 M, 1.3 equiv of acceptor). In toluene. λex = 532 nm. 20 °C. | ||
The TTET process between the triplet sensitizers (BI-1, BI-2 and B-2) and the acceptor perylene can be quantitatively studied by the quenching of the triplet excited state of the sensitizers. Herein the quenching effect was studied by the lifetime changes, monitored by the time-resolved transient difference absorption spectra (the triplet excited state of these organic triplet photosensitizers are non-phosphorescent) (Fig. 11).13
![Stern–Volmer plots generated from triplet excited state lifetime (τT) quenching of BI-1, BI-2, and B-2 measured as a function of perylene concentration. Measured with the nanosecond time-resolved transient absorption. c [sensitizers] = 1.0 × 10−5 M. In deaerated toluene. 20 °C.](/image/article/2012/RA/c2ra01064j/c2ra01064j-f11.gif) | ||
| Fig. 11 Stern–Volmer plots generated from triplet excited state lifetime (τT) quenching of BI-1, BI-2, and B-2 measured as a function of perylene concentration. Measured with the nanosecond time-resolved transient absorption. c [sensitizers] = 1.0 × 10−5 M. In deaerated toluene. 20 °C. | ||
The Stern–Volmer plots give the quenching constants of 2.74 × 105 M−1, 3.96 × 105 M−1 and 2.13 × 105 M−1 for BI-1, BI-2 and B-2, respectively (Table 4). Thus the TTET process of the new sensitizers BI-1 and BI-2 are more efficient than that of B-2. A more efficient triplet quenching effect for BI-1 and BI-2 is partially due to the longer triplet excited state lifetime than that of B-2.6
The upconversions with the organic triplet photosensitizers are clearly visible by eye (Fig. 12). For example, with 532 nm laser excitation, the emission of B-2 is green (Fig. 12a). For BI-1 and BI-2, however, the emission is orange/red.
![(a) Photographs of the emission of sensitizers alone and (b) the upconversion (mixture of the sensitizer and acceptor). (c) CIE diagram of the emission of sensitizers alone and (d) in the presence of perylene (upconversion). c [sensitizers] = 1.0 × 10−5 M, c [perylene] = 1.3 × 10−5 M. λex =532 nm. In toluene. 20 °C.](/image/article/2012/RA/c2ra01064j/c2ra01064j-f12.gif) | ||
| Fig. 12 (a) Photographs of the emission of sensitizers alone and (b) the upconversion (mixture of the sensitizer and acceptor). (c) CIE diagram of the emission of sensitizers alone and (d) in the presence of perylene (upconversion). c [sensitizers] = 1.0 × 10−5 M, c [perylene] = 1.3 × 10−5 M. λex =532 nm. In toluene. 20 °C. | ||
In the presence of triplet acceptor perylene, significant upconverted blue emission of the perylene was observed (Fig. 12a). For example, the emission of B-2–perylene is blue with 532 nm laser excitation. For BI-2, the upconversion solution shows a pink color (due to the upconverted blue emission and the red-emission of the sensitizer). For BI-1, blue emission was observed. The different color of the upconversion with BI-1 and BI-2 are due to the different relative intensity of the blue/red emission bands (Fig. 8).
The emission color changes from excitation of the sensitizers alone and to the upconversion were quantified with CIE coordinates (Fig. 12 c and d). For the sensitizers alone, the CIE coordinates are (0.57, 0.40), (0.60, 0.38) and (0.37, 0.58) for BI-1, BI-2 and B-2, respectively. In the presence of perylene, the CIE coordinates of the solutions changed to (0.30, 0.21), (0.44, 0.29) and (0.28, 0.36), respectively. Due to the tunability of the emission color of the upconversion systems, novel luminescence materials with different emission color can be obtained by changing the molar ratio of the sensitizer/acceptor.
The mechanism of the TTA upconversion with the organic triplet photosensitizers can be demonstrated in Scheme 2. Firstly the singlet excited state of the sensitizer was populated upon excitation (usually direct S0→S1 transition is quantum mechanically allowed), then the triplet excited state (3π–π*) of the sensitizer was populated via the ISC process (direct S0→T1 transition is quantum mechanically forbidden). Note the ISC is not 100% efficient thus fluorescence of the sensitizer was observed (radiative decay from the singlet excited state). The excitation energy (harvested by the sensitizer) was transferred to acceptor via the TTET process. The efficiency of the TTET process is dependent on the lifetime of the triplet excited state of the sensitizer, as well as the energy level of the triplet states of the sensitizer and the acceptor. The singlet excited state of the sensitizer will be populate by the TTA, with the direct possibility of 11.1%.35,36 The overall production of the singlet excited state by TTA is most probably higher because the quintet state produced by the TTA will decay to a triplet excited state, with the probability of 92%, the triplet state will be involved in the TTA again to gain a singlet excited state.3a Radiative decay of the singlet excited state of acceptor will produce the upconverted fluorescence.
Note the crucial properties of the triplet photosensitizer for TTA upconversion is the light-harvesting ability of the sensitizer, the efficiency to produce the triplet excited state (ISC efficiency) and the lifetime of the triplet excited state of the sensitizer (Scheme 2).6 The new thiophene-substituted BODIPY derivatives are ideal sensitizers for TTA upconversion. The energy levels of the triplet excited states of the photosensitizer and the acceptor may be also important for the TTET and thus the TTA upconversion efficiency.
Conclusions
In summary, we prepared thienyl iodo-BODIPY derivatives as organic triplet photosensitizers for triplet–triplet annihilation (TTA) upconversion. The photophysical properties of the sensitizers were studied with steady state and time-resolved spectroscopy, as well as DFT calculations. The sensitizers show red-shifted absorption compared to the unsubstituted BODIPY. Interestingly the sensitizers show large Stokes shifts up to 86 nm, vs. ca. 20 nm for normal BODIPY derivatives. Geometry relaxation upon photoexcitation was proposed to be responsible for the large Stokes shifts based on the DFT calculations. The photosensitizers show long-lived triplet excited states (τT is up to 95.2 μs), which is the longest for organic triplet photosensitizers used for TTA upconversion. The absorption–emission wavelength of the thiophene-substituted sensitizers are red-shifted compared to the unsubstituted BODIPY, but the energy level T1 state does not decrease, which is beneficial for TTA upconversion. The organic sensitizers were used for TTA upconversion and upconversion quantum yields up to 16.5% were observed, which is improved by 3-fold compared to the previous organic triplet sensitizers. The efficient TTA upconversion with the new organic triplet sensitizers is attributed to the efficient triplet–triplet-energy-transfer (TTET) process, confirmed by lifetime quenching experiments. Our results are useful for the design of efficient organic triplet sensitizers for TTA upconversion, to replace the currently used phosphorescent transition metal complex sensitizers. Organic triplet photosensitizers are more cost-efficient, environmental benign and can be readily derivatized to optimize the energy levels of the excited states to improve the upconversion performance. These compounds can also be used for other photophysical processes that required triplet excited states to initiate, such as photodynamic therapy (PDT), etc.Acknowledgements
We thank the NSFC (20972024, 21073028 and 21103015), the Royal Society (UK) and NSFC (China-UK Cost-Share Science Networks, 21011130154), the Fundamental Research Funds for the Central Universities (DUT10ZD212 and DUT11LK19), Ministry of Education (SRFDP-200801410004 and NCET-08-0077), the State Key Laboratory of Fine Chemicals (KF0802 and KF0901) and Dalian University of Technology for financial support.References
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Mullen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef CAS.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155122 CrossRef.
- (a) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS; (b) R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC.
- Y. Y. Cheng, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66–71 RSC.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834–7841 RSC.
- Y. Liu, W. Wu, J. Zhao, X. Zhang and H. Guo, Dalton Trans., 2011, 40, 9085–9089 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953–5963 RSC.
- S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626–1629 CrossRef CAS.
- (a) J. Sun, W. Wu, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2011, 3165–3173 CrossRef CAS; (b) J. Sun, J. Zhao, H. Guo and W. Wu, Chem. Commun., 2012 10.1039/c2cc16690a.
- S. Ji, H. Guo, W. Wu, W. Wu and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 8283–8286 CrossRef CAS.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS.
- (a) W. Wu, J. Sun, S. Ji, W. Wu, J. Zhao and H. Guo, Dalton Trans., 2011, 40, 11550–11561 RSC; (b) W. Wu, J. Zhao, H. Guo, J. Sun, S. Ji and Z. Wang, Chem.–Eur. J., 2012, 18, 1961–1968 CrossRef CAS; (c) W. Wu, S. Ji, W. Wu, J. Shao, H. Guo, T. D. James, J. Zhao, Chem. Eur. J., 2012 DOI:10.1002/chem.201101377.
- (a) Y. Liu, Q. Li, J. Zhao and H. Guo, RSC Adv., 2012, 2, 1061–1067 RSC; (b) Q. Li, H. Guo, L. Ma, W. Wu, Y. Liu and J. Zhao, J. Mater. Chem., 2012, 22, 5319 RSC.
- W. Wu, L. Yao, T. Yang, R. Yin, F. Li and Y. Yu, J. Am. Chem. Soc., 2011, 133, 15810–15813 CrossRef CAS.
- I. Etchart, M. Bérard, M. Laroche, A. Huignard, I. Hernández, W. P. Gillin, R. J. Curry and A. K. Cheetham, Chem. Commun., 2011, 47, 6263–6265 RSC.
- H. M. Kim, B. R. Cho. Kim and H. M. Cho, Acc. Chem. Res., 2009, 42, 863–872 CrossRef CAS.
- N. J. Turro, V. Ramamurthy, J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction; University Science Books: Sausalito, CA, 2009 Search PubMed.
- T. N. Singh-Rachford and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5912–5917 CrossRef CAS.
- H. C. Chen, C. Y. Hung, K. H. Wang, H. L. Chen, W. S. Fann, F. C. Chien, P. Chen, T. J. Chow, C. P. Hsu and S. S. Sun, Chem. Commun., 2009, 4064–4066 RSC.
- A. B. Descalzo, H. J. Xu, Z. L. Xue, K. Hoffmann, Z. Shen, M. G. Weller, X. Z. You and K. Rurack, Org. Lett., 2008, 10, 1581–1584 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- (a) A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11, 4124–4131 RSC; (b) Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192–2206 CrossRef CAS.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC.
- (a) T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS; (b) N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2010, 12, 5720–5723 CrossRef CAS; (c) Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS; (d) S. H. Lim, C. Thivierge, P. Nowak-Sliwinska, J. Han, H. van den Bergh, G. Wagnières, K. Burgess and H. B. Lee, J. Med. Chem., 2010, 53, 2865–2874 CrossRef CAS; (e) B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC; (f) A. D. Quartarolo, N. Russo and E. Sicilia, Chem.–Eur. J., 2006, 12, 6797–6803 CrossRef CAS; (g) S. G. Awuah, J. Polreis, V. Biradar and Y. You, Org. Lett., 2011, 13, 3884–3887 CrossRef CAS.
- T. N. Singh-Rachford, A. Haefele, R. Ziessel and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 16164–16165 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) M. Baruah, W. Qin, C. Flors, J. Hofkens, R. A. L. Vallée, D. Beljonne, M. V. Auweraer, W. M. D. Borggraeve and N. Boens, J. Phys. Chem. A, 2006, 110, 5998–6009 CrossRef CAS; (b) C.-Y. Chen, J.-G. Chen, S.-J. Wu, J.-Y. Li, C.-G. Wu and K.-C. Ho, Angew. Chem., Int. Ed., 2008, 47, 7342–7345 CrossRef CAS; (c) W.-H. Liu, I-C. Wu, C.-H. Lai, C.-H. Lai, P.-T. Chou, Y.-T. Li, C.-L. Chen, Y.-Y. Hsu and Y. Chi, Chem. Commun., 2008, 5152–5154 RSC; (d) N. Koumura, Z.-S. Wang, M. Miyashita, Y. Uemura, H. Sekiguchi, Y. Cui, A. Mori, S. Mori and K. Hara, J. Mater. Chem., 2009, 19, 4829–4836 RSC; (e) Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 RSC; (f) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS.
- T. Cheng, Y. Xu, S. Zhang, W. Zhu, X. Qian and L. Duan, J. Am. Chem. Soc., 2008, 130, 16160–16161 CrossRef CAS.
- J. Shao, H. Guo, S. Ji and J. Zhao, Biosens. Bioelectron., 2011, 26, 3012–3017 CrossRef CAS.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
- P. Du and R. Eisenberg, Chem. Sci., 2010, 1, 502–506 RSC.
- L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011, 4527–4533 CrossRef CAS.
- J. Saltiel, G. R. March, W. K. Smothers, S. A. Stout and J. L. Charlton, J. Am. Chem. Soc., 1981, 103, 7159–7164 CrossRef CAS.
- P. P. Levin, Dokl. Phys. Chem., 2003, 388, 10–12 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: More structural characterization data and Z-matrix of the compounds for DFT calculations. See DOI: 10.1039/c2ra01064j/. |
| This journal is © The Royal Society of Chemistry 2012 |