Graphene based gene transfection†
Liangzhu
Feng
,
Shuai
Zhang
and
Zhuang
Liu
*
Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Institute of Functional Nano & Soft Materials, Soochow University, Suzhou, Jiangsu, China. E-mail: zliu@suda.edu.cn
First published on 27th January 2011
Abstract
Graphene as a star in materials research has been attracting tremendous attentions in the past few years in various fields including biomedicine. In this work, for the first time we successfully use graphene as a non-toxic nano-vehicle for efficient gene transfection. Graphene oxide (GO) is bound with cationic polymers, polyethyleneimine (PEI) with two different molecular weights at 1.2 kDa and 10 kDa, forming GO-PEI-1.2k and GO-PEG-10k complexes, respectively, both of which are stable in physiological solutions. Cellular toxicity tests reveal that our GO-PEI-10k complex exhibits significantly reduced toxicity to the treated cells compared to the bare PEI-10k polymer. The positively charged GO-PEI complexes are able to further bind with plasmid DNA (pDNA) for intracellular transfection of the enhanced green fluorescence protein (EGFP) gene in HeLa cells. While EGFP transfection with PEI-1.2k appears to be ineffective, high EGFP expression is observed using the corresponding GO-PEI-1.2k as the transfection agent. On the other hand, GO-PEI-10k shows similar EGFP transfection efficiency but lower toxicity compared with PEI-10k. Our results suggest graphene to be a novel gene delivery nano-vector with low cytotoxicity and high transfection efficiency, promising for future applications in non-viral based gene therapy.
Introduction
Graphene, two dimensional (2-D) single or few layers of sp2-bonded carbon sheets, has been extensively studied owing to its excellent physical, chemical and mechanical properties.1–4 In the past several years, intensive efforts have been devoted to explore potential biomedical applications of carbon nanotubes, a class of nanomaterials closely related to graphene, for biological sensing and detection, drug and gene delivery, as well as biomedical imaging in vitro and in vivo.5–15 The research of graphene in biomedicine has also very recently emerged as an interesting field.16–31 With all atoms exposed on its surface, graphene has an ultra-high surface area available for efficient loading of aromatic drug molecules via π–π stacking, useful for applications in drug delivery.16–18 In recent work by our group, we studied the in vivo behaviors of graphene by a fluorescent labeling method, and used polyethylene glycol (PEG) functionalized graphene for highly efficient in vivo photothermal therapy in a mouse model.19 In the past two years, chemical and biological sensing with graphene have also been widely investigated.20–28,31 It has been shown that graphene is able to strongly bind to single-strand DNA (ssDNA), however without a strong interaction with double-stranded DNA (dsDNA) molecules. Utilizing this mechanism and the effective fluorescence quenching ability of graphene, several groups have independently developed various graphene based DNA sensing platforms.24,25,28 In addition, Luet al. showed that graphene could protect oligonucleotides from enzymatic cleavage and was able to deliver ssDNA into cells.29 However, to the best of our knowledge, delivery of a gene plasmid with graphene has not yet been reported.Gene therapy to cure diseases which are difficult for traditional clinical methods has been actively pursued for decades in both the academic world and in industry. The major obstacle in this area, however, is to develop non-viral based safe and efficient gene delivery vehicles, in which nanomaterials are usually involved.10,32–34 Although much progress has been reported regarding the use of cationic polymers and various inorganic nanomaterials such as carbon nanotubes, silica nanoparticles and nanodiamonds, as gene delivery vehicles,10,32–40 a lot more effort is still demanded to develop non-toxic nano-vectors with high gene transfection efficiency for potential gene therapy. Among all materials used in gene delivery, polyethylenimine (PEI)36,38,40 has been normally recognized as the ‘golden standard’ cationic polymer in gene transfection, owing to its strong binding to nucleic acids, effective uptake by cells, and the excellent proton sponge effect that triggers the endosomal release of DNA or RNA. However, the high cytotoxicity and poor biocompatibility of PEI polymers, especially those with high molecular weights, have largely restricted their further applications in gene therapy. In this work, we try to use graphene as a novel 2-D nano-vector for gene loading and transfection. It is found that negatively charged graphene oxide (GO) is able to bind cationic PEI polymers by electrostatic interactions, forming stable GO-PEI complexes highly enriched in positively charges that allow effective loading of DNA plasmid (pDNA) via a layer-by-layer (LBL) assembly process41 (Fig. 1). These GO-PEI complexes show remarkably decreased cytotoxicities, even when a high molecular weight PEI (10 kDa) is used, much safer than the bare PEI polymers. A DNA plasmid encoding enhanced green fluorescence protein (EGFP) is used as a model system to study the GO-PEI based gene delivery, showing high transfection efficiencies.
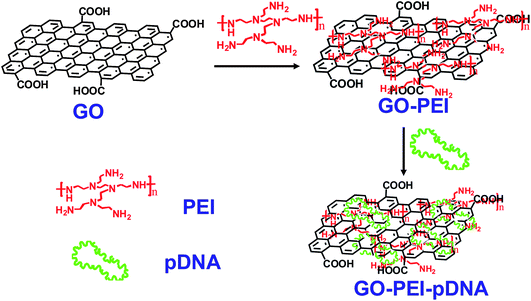 | ||
| Fig. 1 Schematic illustration showing the synthesis of GO-PEI-DNA complexes via a LBL assembly process. Firstly, graphene oxide (GO) was non-covalently functionalized by PEI polymers, forming positively charged GO-PEI complexes. Next, negatively charged pDNA was loaded on the GO-PEI complexes also by electrostatic interactions. | ||
Results and discussion
GO was synthesized using a modified Hummer's method as reported earlier.16,17 To prepare GO-PEI complexes, diluted solutions of PEI with different molecular weights (1.2 kDa, 10 kDa) were slowly added into GO solutions in water (0.1 mg mL−1) at varying GO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEI weight ratios of 1:0.1, 1:0.25, 1:0.5, 1:1, 1:2 and 1:5. Excess unbound PEI was removed by centrifugation and repeated water washing. GO-PEI-1.2k complexes at the weight ratio of 1:5 formed aggregates in water, while other GO-PEI complexes were well dispersed in water and stable for months. Interestingly, PEI coatings could enhance the stability of GO in physiological solutions. We found that various GO-PEI complexes were stable in saline (0.9% NaCl) as well as cell medium without obvious agglomeration, while naked GO aggregated in these solutions and quickly precipitated out (Fig. 2a). Atomic force microscope (AFM) images (Fig. 2b) showed that the sheet sizes of GO-PEI complexes were much smaller than that of the starting GO sample, due to the sonication step that cut graphene sheets. An increase in averaged sheet thickness from ∼1 nm for GO to 3–4 nm for GO-PEI was attributed to the binding of PEI on GO (Fig. S1, ESI†).
:PEI weight ratios of 1:0.1, 1:0.25, 1:0.5, 1:1, 1:2 and 1:5. Excess unbound PEI was removed by centrifugation and repeated water washing. GO-PEI-1.2k complexes at the weight ratio of 1:5 formed aggregates in water, while other GO-PEI complexes were well dispersed in water and stable for months. Interestingly, PEI coatings could enhance the stability of GO in physiological solutions. We found that various GO-PEI complexes were stable in saline (0.9% NaCl) as well as cell medium without obvious agglomeration, while naked GO aggregated in these solutions and quickly precipitated out (Fig. 2a). Atomic force microscope (AFM) images (Fig. 2b) showed that the sheet sizes of GO-PEI complexes were much smaller than that of the starting GO sample, due to the sonication step that cut graphene sheets. An increase in averaged sheet thickness from ∼1 nm for GO to 3–4 nm for GO-PEI was attributed to the binding of PEI on GO (Fig. S1, ESI†).
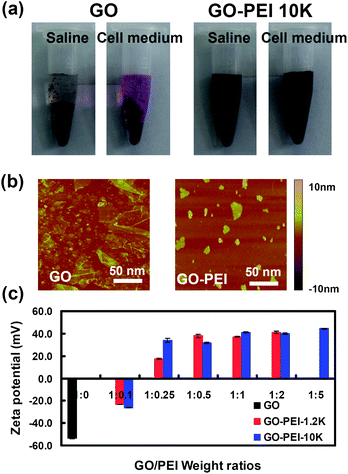 | ||
| Fig. 2 Characterizations of GO and GO-PEI complexes. (a) Photos of GO and GO-PEI-10k in saline (0.9% NaCl) and complete DMEM cell medium (with 10% serum) after centrifugation at 1,000 rpm for 10 min. GO aggregated in both physiological solutions while GO-PEI complexes were stable. (b) AFM images of GO and GO-PEI-10k. (c) Zeta potential histogram of GO and series of GO-PEI complexes at different staring GO:PEI weight ratios. Error bars were based on triplicated measurements. | ||
As-prepared GO sheets have many carboxyl groups and are highly negatively charged, allowing the binding of positively charged PEI polymers by electrostatic interactions. Zeta potentials of GO and various GO-PEI complexes were measured to determine their surface charges. We found that binding of PEI polymers dramatically increased the zeta-potential of GO at −50 mV to +30∼50 mV for GO-PEI complexes. As expected, a rise of zeta-potentials was observed with the increase of PEI weight ratios during the sample preparation (Fig. 2c). The binding of PEI on GO was also evidenced by infrared spectra (Fig. S2, ESI†) and thermogravimetric analysis (Fig. S3, ESI†). GO-PEI-1.2k at the 1:2 starting ratio and GO-PEI-10k at the 1:5 starting ratio, all with the highest zeta-potentials, were chosen and used in our following studies. The nitrogen content of these two samples was measured by elemental analysis to be 10.22% and 13.92%, corresponding to the actual GO:PEI weight ratios of 1:0.46 and 1:0.75, for GO-PEI-1.2k and GO-PEI-10k complexes, respectively.
Next, in vitro cytotoxicties of different GO-PEI complexes were evaluated with HeLa cells in comparison to bare PEI polymers by a cell viability assay (Fig. 3). Our results showed that PEI with a high molecular weight at 10 kDa appeared to be rather toxic to HeLa cells, with the half maximal inhibitory concentration (IC50) value of ∼30 mg L−1 (Fig. 3b). In marked contrast, significantly reduced toxicity was noticed for the corresponding GO-PEI-10k complex. Even at a GO-PEI-10k concentration as high as 300 mg L−1, the relative cell viability was still higher than 80%. The in vitro toxicity of GO-PEI-10k was over one order of magnitude reduced from that of PEI-10k. PEI at a low molecular weight (1.2 kDa) was much less toxic as expected (Fig. 3a). Interestingly, loading of either PEI-1.2k or PEI-10k did not increase the toxicity of GO (Fig. 3c). In fact, GO-PEI complexes afforded slightly higher relative cell viabilities compared to GO at high concentrations, likely owing to the improved stability of GO-PEI in physiological environments (Fig. 2a). Plenty of previous studies have revealed that many polycations such as PEI polymers induce cytotoxicity by binding with plasma membranes, and the rigid molecular structures (e.g. immobilized on nanoparticles) would decrease the cytotoxicity of polycations because of a lower density of cationic residues interacting with cells.36,38,40,42 The decreased cytotoxicities of GO-PEI complexes are likely attributed to a similar mechanism.
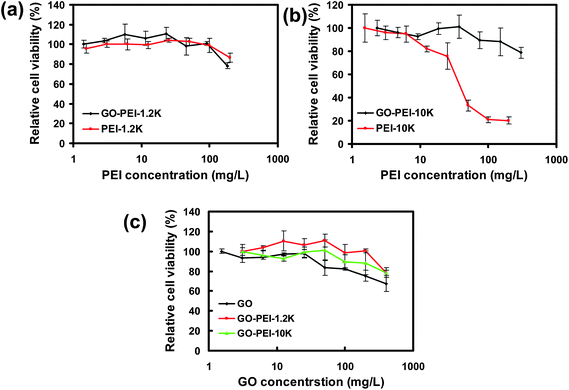 | ||
| Fig. 3 In vitro cell toxicity assay. Relative cell viability data of HeLa cells incubated with a series of concentrations of GO-PEI-1.2k and PEI-1.2k (a), GO-PEI-10k and PEI-10k (b), as well as GO and two GO-PEI complexes (c) measured by the MTT cell viability assay. The incubation time was 24 h. Error bars were based on triplicated samples. | ||
To study the pDNA loading on GO-PEI complexes, a gel electrophoresis assay was carried out after incubation of GO-PEI-1.2k and GO-PEI-10k with EGFP plasmid at different nitrogen/phosphate (N/P) ratios (Fig. 4). Significant retardation of pDNA moving in gel electrophoresis was observed when PEI and GO-PEI were mixed with pDNA at high N/P ratios. While an N/P ratio of 5 was enough to completely retard pDNA moving in electrophoresis, indicating all pDNA was bound to PEI in this circumstance, a higher N/P ratio of 10 was required for complete loading of pDNA on GO-PEI samples. This was expected as the positive charges of PEI were partially neutralized by negative charges on the GO surface. In our control experiments, plain GO showed very limited binding of the double-stranded pDNA even under high GO concentrations, consistent with earlier published results. It has been shown by others that although single-stranded DNA is able to effectively bind the graphene surface via π–π stacking, the interaction between double-stranded DNA and GO is rather weak.26,28
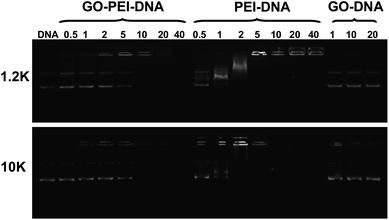 | ||
| Fig. 4 Gel retardation assay. Agarose gel electrophoresis of bare pDNA, mixtures of plain GO and pDNA, as well as GO-PEI-pDNA and PEI-pDNA at different N/P ratios. Each sample was incubated at room temperature for 1 h before electrophoresis. The amounts of GO used in the GO-pDNA samples were identical to the GO quantities in the GO-PEI-pDNA samples at N/P ratios of 1, 10 and 20. | ||
Finally, we tested the gene delivery efficiencies of GO-PEI complexes for EGFP transfection using HeLa cells. All transfection experiments were carried out at standard conditions (see method section). From the fluorescence microscope images, we found that naked EGFP pDNA and GO-pDNA failed to render appreciable EGFP expression to the treated HeLa cells (Fig. S4, ESI†). While the low molecular weight PEI-1.2k showed rather low transfection efficiency, the GO-PEI-1.2k complex was able to effectively transfect HeLa cells with EGFP at N/P ratios above 20 (Fig. 5a and b). With respect to PEI-10k, although it did offer efficient EGFP transfection at N/P ratios of 20 and 40, obvious toxicity to cells was noted at the N/P ratio of 80 (Fig. 5c). In contrast, high EGFP expression together with no obvious cell toxicity was observed when using GO-PEI-10k as the transfection vector at N/P ratios from 20 to 80 (Fig. 5d). GO-PEI with remarkably decreased cytotoxicity and high transfection efficacy could be an encouraging candidate of non-viral gene delivery vehicles.
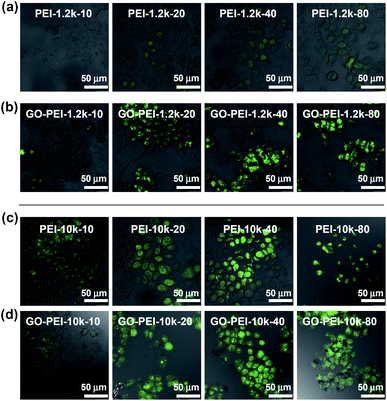 | ||
| Fig. 5 Confocal fluorescence images of EGFP transfected HeLa cells using PEI-1.2k (a), GO-PEI-1.2k (b), PEI-10k (c), and GO-PEI-10k (d) at varying N/P ratios from 10 to 80. Scale bar = 50 μm. Obvious toxicity was observed for HeLa cells after PEI-pDNA treatment at the N/P ratio of 80. | ||
Conclusions
In this work, GO was functionalized by PEI polymers by non-covalent electrostatic interactions, yielding GO-PEI complexes with strong positive charges, high stability in physiological solutions, and reduced cytotoxicity to cells. DNA plasmid could be further bound to GO-PEI for effective intracellular gene delivery as demonstrated by EGFP transfection in HeLa cells. It was found that our GO-PEI complexes exhibited improved transfection efficiency than PEI-1.2k with lower molecular weight, and lower toxicity than PEI-10k with higher molecular weight. Whether and how the structure of graphene (e.g. size, thickness) would affect the gene transfection efficiency, however, remains an important question that requires further investigation. Nevertheless, our results highlight the promise of graphene as a novel nano-carrier for safe and efficient gene transfection. Besides pDNA, small interfering RNA (siRNA) with therapeutic functions may also be delivered by our GO-PEI complexes into cancer cells for potential gene therapy, which could be further combined with graphene based chemotherapy and photothermal therapy as demonstrated earlier,17–19,24,43 for future multimodal therapies of cancer.Experiments and methods
1. Materials
Branched polyethylenimine (PEI) at molecular weights of 1.2 kDa, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), were purchased from Sigma-Aldrich. The 10 kDa branched PEI was purchased from Alfar-Aesar. Agrose (molecular biology grade) was purchased from Invitrogen. The EGFP plasmid was a gift from Dr Rui Peng's group at Soochow University. It was transformed in Escherichia coliTOP10 and amplified in Luria-Bertani (LB) medium. The plasmid was purified using an endotoxin-free plasmid kit (TianGEN, TianGEN Biotech (Beijing) CO. LTD) according to the manufacture's protocol. Purified plasmid was dissolved in Tris-EDTA (TE) buffer, and its purity and concentration were determined by ultraviolet (UV) absorbance at 260 nm and 280 nm.2. Preparation of GO-PEI complexes
Graphene oxide (GO) was prepared by a modified Hummer's method with flake expandable graphite used as the original material according to a previous protocol.16,17 For the preparation of GO-PEI complexes, we slowly added a PEI solution (1mg mL−1) to a GO solution (about 0.1 mg mL−1) in 10 min. After ultrasonication for about 10 min, the mixture was stirred overnight, washed 3–5 times with deionized (DI) water by centrifugation, and then re-dispersed in DI water. In our experiment, we obtained a series of GO-PEI complexes by mixing PEI solutions with diluted GO solutions at several GO:PEI weight ratios of 1:0.1, 1:0.25, 1:0.5, 1:1, 1:2 and 1:5.
The concentrations of GO and GO-PEI complexes were calculated by their absorbance at 230 nm recorded by a UV-Vis spectrometer (PerkinElmer) with a mass extinction coefficient measured to be 65 mg mL−1 cm−1. Elemental analyses were conducted by an Elemental Analyzer (EA1110CHNO - S Carlo - Erba). Fourier Transform Infrared Spectroscopy experiments were performed by using a spectrometer (ProStar LC240, Varian Inc). AFM images of GO and GO-PEI were taken by a MultiMode V AFM (Veeco). Zeta potentials of GO-PEI complexes were measured by Zetasizer Nano (Malvern).
3. Culture of HeLa cells
HeLa cells were cultured in Dulbecco's modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS), streptomycin (100 U mL−1) penicillin (100U mL−1), and 4 mM L-glutamine at 37 °C in a humidified 5% CO2-containing atmosphere.4. Preparation of GO-PEI-pDNA complexes and gel electrophoresis assay
Appropriate amounts of PEI, GO-PEI and GO solutions were mixed with 800 ng pDNA in 20 uL deionized water at various N/P ratios. For the mixture of GO and pDNA, the amounts of added GO were equivalent to the amounts of GO in GO-PEI-pDNA complexes at N/P ratios of 1, 10 and 20. The samples were incubated for 1 h at room temperature before they were analyzed by 0.8% agrose gel electrophoresis using a TE buffer solution as the running buffer.5. Cytotoxicity assay
The cytotoxicities of different GO-PEI complexes in comparison with GO and PEI polymers were evaluated using the standard MTT assay following the manufacture's protocol. Briefly, HeLa cells were seeded in a 96-well palate at a density of about 1 × 104cells per well. After 24 h incubation until cells were adhered to the plate, a series of concentrations of GO-PEI, GO, and PEI were added into the cell culture. After another 24 h incubation, 20 μL of MTT solution in phosphate buffered saline (PBS) was added to each well and incubated for an additional 4 h. The cell culture was discarded afterwards with 150 μL dimethyl sulfoxide (DMSO) added to each well. We then incubated the plate at 37 °C for 5 min and shocked it for another 10 min at room temperature to allow complete dissolve of formazan. Finally, the absorbance at 570 nm of each well was measured by a micro-plate reader (Model 680 Bio-RAD) to determine the relative cell viability.6. EGFP DNA transfection
HeLa cells were added to 24-well plates at a density of about 1 × 105cells per well 24 h before transfection. We diluted 400 ng EGFP plasmid in 200 μL serum-free DMEM medium, and diluted appropriate amounts of GO-PEI complexes in 200 μL serum-free DMEM medium. After 10 s vortexing, the two solutions were mixed and incubated for another 90 min at room temperature before being added into cells. The preparations of GO-pDNA and PEI-pDNA complexes were carried out following the same protocol.All cells were washed 2 times with serum-free DMEM medium right before addition of the transfection complexes. After 4 h incubation, the medium containing different transfection complexes were removed. Cells were washed 2 times with serum-free DMEM medium before addition of 750 uL formal DMEM growth medium containing 10% FBS. Additional 48 h incubation was needed for efficient EGFP expression. Control experiment was carried out using naked plasmid DNA without any transfection agent.
7. Fluorescence microscope images
Fluorescence images of cells were taken by a laser scanning confocal fluorescence microscope (TCS SP5 Leica). An excitation wavelength of 488 nm and emission band between 500 nm and 600 nm were chosen for the fluorescence imaging of EGFP. All images were taken under the same parameters setting.Acknowledgements
This work was partially supported by a research start-up fund of Soochow University, a research grant from the National Natural Science Foundation of China (project no. 51002100), and a 973 grant (project no. 2010CB934502) from the Ministry of Science and Technology (MOST) of China. We thank Dr Rui Peng at Soochow University for providing us the EGFP plasmid.Notes and references
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- Y. Kopelevich and P. Esquinazi, Adv. Mater., 2007, 19, 4559–4563 CrossRef.
- Z. H. Ni, Y. Y. Wang, T. Yu and Z. X. Shen, Nano Res., 2010, 1, 273–291.
- A. de la Zerda, Z. A. Liu, S. Bodapati, R. Teed, S. Vaithilingam, B. T. Khuri-Yakub, X. Y. Chen, H. J. Dai and S. S. Gambhir, Nano Lett., 2010, 10, 2168–2172 CrossRef.
- Z. Liu, A. C. Fan, K. Rakhra, S. Sherlock, A. Goodwin, X. Y. Chen, Q. W. Yang, D. W. Felsher and H. J. Dai, Angew. Chem., Int. Ed., 2009, 48, 7668–7672 CrossRef CAS.
- Z. Liu, X. M. Sun, N. Nakayama-Ratchford and H. J. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- Z. Liu, S. Tabakman, K. Welsher and H. J. Dai, Nano Res., 2010, 2, 85–120.
- Z. Liu, S. M. Tabakman, Z. Chen and H. J. Dai, Nat. Protoc., 2009, 4, 1372–1382 Search PubMed.
- Z. Liu, M. Winters, M. Holodniy and H. J. Dai, Angew. Chem., Int. Ed., 2007, 46, 2023–2027 CrossRef CAS.
- Z. Liu, X. L. Li, S. M. Tabakman, K. L. Jiang, S. S. Fan and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 13540–13541 CrossRef CAS.
- K. Welsher, Z. Liu, D. Daranciang and H. Dai, Nano Lett., 2008, 8, 586–590 CrossRef CAS.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. J. Dai, Nat. Nanotechnol., 2009, 4, 773–780 CrossRef CAS.
- Z. Liu, K. Yang and S. T. Lee, J. Mater. Chem., 2011, 21, 586–598 RSC.
- X. Liu, H. Tao, K. Yang, S. Zhang, S. T. Lee and Z. Liu, Biomaterials, 2011, 32, 144–151 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- X. M. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. J. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- L. M. Zhang, J. G. Xia, Q. H. Zhao, L. W. Liu and Z. J. Zhang, Small, 2010, 6, 537–544 CrossRef CAS.
- K. Yang, S. Zhang, G. Zhang, X.X. M. Sun, S. Lee and T. Z. Liu, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS.
- H. P. Cong, J. J. He, Y. Lu and S. H. Yu, Small, 2010, 6, 169–173 CrossRef CAS.
- X. C. Dong, Y. M. Shi, W. Huang, P. Chen and L. J. Li, Adv. Mater., 2010, 22, 1649–1650 CrossRef CAS.
- H. Jang, J., Y. Kim, K., H. Kwon, M., W. Yeo, S., D. Kim, E., D. Min and H., Angew. Chem. Int. Ed., 2010, 49, 5703–5707 CrossRef CAS.
- J. H. Jung, D. S. Cheon, F. Liu, K. B. Lee and T. S. Seo, Angew. Chem., Int. Ed., 2010, 49, 5708–5711 CrossRef CAS.
- F. Liu, J. Y. Choi and T. S. Seo, Biosens. Bioelectron., 2010, 25, 2361–2365 CrossRef CAS.
- C. H. Lu, J. Li, J. J. Liu, H. H. Yang, X. Chen and G. N. Chen, Chem. A Euro. J., 2010, 16, 4889–4894 Search PubMed.
- L. A. L. Tang, J. Z. Wang and K. P. Luo, J. Am. Chem. Soc., 2010, 132, 10976–10977 CrossRef CAS.
- Y. Wang, Z. H. Li, D. H. Hu, C. T. Lin, J. H. Li and Y. H. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS.
- S. J. He, B. Song, D. Li, C. F. Zhu, W. P. Qi, Y. Q. Wen, L. H. Wang, S. P. Song, H. P. Fang and C. H. Fan, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef.
- C. H. Lu, C. L. Zhu, J. Li, J. J. Liu, X. Chen and H. H. Yang, Chem. Commun., 2010, 46, 3116–3118 RSC.
- W. B. Hu, C. Peng, W. J. Luo, M. Lv, X. M. Li, D. Li, Q. Huang and C. H. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS.
- F. Li, Y. Huang, Q. Yang, Z. T. Zhong, D. Li, L. H. Wang, S. P. Song and C. H. Fan, Nanoscale, 2010, 2, 1021–1026 RSC.
- C. Hom, J. Lu, M. Liong, H. Z. Luo, Z. X. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1185–1190 CAS.
- T. A. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS.
- X. Q. Zhang, M. Chen, R. Lam, X. Y. Xu, E. Osawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS.
- B. Chertok, A. E. David and V. C. Yang, Biomaterials, 2010, 31, 6317–6324 CrossRef CAS.
- D. Fischer, T. Bieber, Y. X. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- R. Martin, M. Alvaro, J. R. Herance and H. Garcia, ACS Nano, 2010, 4, 65–74 CrossRef.
- D. Putnam, C. A. Gentry, D. W. Pack and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1200–1205 CrossRef CAS.
- S. Son, K. Singha and W. J. Kim, Biomaterials, 2010, 31, 6344–6354 CrossRef CAS.
- J. M. Zhu, A. G. Tang, L. P. Law, M. Feng, K. M. Ho, D. K. L. Lee, F. W. Harris and P. Li, Bioconjugate Chem., 2005, 16, 139–146 CrossRef CAS.
- X. Hong, J. Li, M. J. Wang, J. J. Xu, W. Guo, J. H. Li, Y. B. Bai and T. J. Li, Chem. Mater., 2004, 16, 4022–4027 CrossRef CAS.
- S. Choksakulnimitr, S. Masuda, H. Tokuda, Y. Takakura and M. Hashida, J. Controlled Release, 1995, 34, 233–241 CrossRef CAS.
- X. Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Thickness distribution of GO and GO-PEI; IR and TGA data; and confocal images of HeLa cells treated with bare EGFP pDNA and GO + pDNA. See DOI: 10.1039/c0nr00680g |
| This journal is © The Royal Society of Chemistry 2011 |