
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a series of non-centrosymmetric antiperovskite and double antiperovskite borate fluorides†
Fuqiang
Chen
,
Hongping
Wu
 *,
Zhanggui
Hu
,
Jiyang
Wang
,
Yicheng
Wu
and
Hongwei
Yu
*
*,
Zhanggui
Hu
,
Jiyang
Wang
,
Yicheng
Wu
and
Hongwei
Yu
*
Tianjin Key Laboratory of Functional Crystal Materials, Institute of Functional Crystal, College of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: yuhw@email.tjut.edu.cn
First published on 18th December 2024
Abstract
Non-centrosymmetric (NCS) compounds can exhibit many symmetry-dependent functional properties, yet their rational structure design remains a great challenge. Herein, a strategy to introduce F-centered octahedra to construct a perovskite-type framework filled by π-conjugated [B2O5]4− dimers is proposed to obtain NCS compounds. The first examples of antiperovskite or double antiperovskite borate fluorides, [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5], have been successfully designed and synthesized. All three compounds exhibit a novel three-dimensional framework constructed from [F(M/Ba)4Ca2] (M = K, Rb), [FCs4Ca2] and [FBa4Ca2] octahedra, which are further filled by [B2O5]4− dimers to form antiperovskite-type structures. They all crystallize in the NCS space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m, and can exhibit moderate second harmonic generation (SHG) responses (∼0.5 × KDP@1064 nm) and short UV cut-off edges (∼190 nm), as well as suitable birefringence (Δn = 0.0405–0.0548@532 nm). This suggests their potential as UV nonlinear optical crystals.
21m, and can exhibit moderate second harmonic generation (SHG) responses (∼0.5 × KDP@1064 nm) and short UV cut-off edges (∼190 nm), as well as suitable birefringence (Δn = 0.0405–0.0548@532 nm). This suggests their potential as UV nonlinear optical crystals.
Introduction
Non-centrosymmetric (NCS) structures capable of exhibiting many symmetry-dependent properties, such as piezoelectric, ferroelectric, and second-order nonlinear optical (NLO) properties, are of the current academic and technological interest.1 In particular, NCS borates have been widely applied as ultraviolet (UV) or even deep-UV NLO crystals.2–6 However, the rational synthesis of new NCS structures remains an important ongoing challenge for chemists and experimentalists due to the weak predictability of the structure of inorganic materials. During past decades, some widely-used strategies for increasing the odds of generating NCS structures through introducing special cations with inherent asymmetric coordination environments or asymmetric anionic groups have been put forward, such as (i) the use of cations susceptible to second-order Jahn–Teller (SOJT) distortions, i.e., high-valent d0 transition metal cations in an octahedral coordination environment (Ti4+, V5+, Nb5+, Mo6+, W6+, etc.) and cations possessing stereochemically active lone pairs (Pb2+, Sb3+, Bi3+, Se4+, Te4+, I5+, etc.);7,8 (ii) the use of d10 transition metal cations revealing highly polar displacement in the center of the coordination environment (Zn2+, Cd2+, etc.);9 and (iii) the use of anions with trigonal-planar geometry exhibiting asymmetric π-conjugated molecular orbitals ([NO3]−, [CO3]2−, [BO3]3−, etc.).10–14 In addition, it is impressive that a series of polar small molecule compounds with excellent NLO response have been successfully designed through introducing a chiral backbone to a dipolar molecule.15 However, the majority of solid-state materials still crystallize in centrosymmetric (CS) structures because the locally asymmetric basic building units (BBUs) are more likely packed in structures in an antiparallel manner that can increase the local electroneutrality and is more energetically favorable according to Pauling's second rule.16 Therefore, exploring new strategies for controlling the orientation of acentric BBUs in structures will be essential for the design of new NCS compounds.The perovskite template is considered the most important and typical inorganic structural template. Its structural stability can be well predicted by the tolerance factor t (t = (rA + rX)/2(rB + rX)).17 With perovskite as the template, over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 perovskite-type structures have been designed and synthesized in many different fields, such as catalysis, photovoltaics, luminescence, etc.18,19 Beyond the perovskite template, we have noticed that antiperovskites are also a new interesting materials class, especially for the design of NCS structures. First, structurally, antiperovskite structures are isomorphic to perovskite ones. Therefore their structural stability can also be predicted by the tolerance factor t. Next, the structural frameworks of antiperovskites are generally constructed from modifiable ionic octahedra, and the rigid anion groups are filled in the voids of octahedral ionic frameworks.20 Thus, the orientation and distortion of the anion groups are easier to control or modulate in antiperovskite structures. Based on this, a series of NCS antiperovskite structures have been designed and synthesized in recent research, including M3X[B6O10] (M = K, Na/Rb; X = Cl, Br),21 Sr3[CO3][SnOSe3],22 Ae3Q[GeOQ3] (Ae = Ba, Sr; Q = S, Se),23etc. Clearly, these compounds all exhibit intriguing symmetry-dependent NLO properties. In particular, K3Cl[B6O10] and K3Br[B6O10] have been widely studied as promising UV NLO crystals.
000 perovskite-type structures have been designed and synthesized in many different fields, such as catalysis, photovoltaics, luminescence, etc.18,19 Beyond the perovskite template, we have noticed that antiperovskites are also a new interesting materials class, especially for the design of NCS structures. First, structurally, antiperovskite structures are isomorphic to perovskite ones. Therefore their structural stability can also be predicted by the tolerance factor t. Next, the structural frameworks of antiperovskites are generally constructed from modifiable ionic octahedra, and the rigid anion groups are filled in the voids of octahedral ionic frameworks.20 Thus, the orientation and distortion of the anion groups are easier to control or modulate in antiperovskite structures. Based on this, a series of NCS antiperovskite structures have been designed and synthesized in recent research, including M3X[B6O10] (M = K, Na/Rb; X = Cl, Br),21 Sr3[CO3][SnOSe3],22 Ae3Q[GeOQ3] (Ae = Ba, Sr; Q = S, Se),23etc. Clearly, these compounds all exhibit intriguing symmetry-dependent NLO properties. In particular, K3Cl[B6O10] and K3Br[B6O10] have been widely studied as promising UV NLO crystals.
In this research, we are also interested in the NCS antiperovskite borates because of their potential applications as UV or even deep-UV NLO crystals. As NLO crystals, a basic structural requirement, besides those of NCS structures, is that materials must be optically anisotropic to achieve phase-matching,24i.e. materials must crystallize in a non-cubic system. Therefore, mixed-cation-coordinated octahedra will be the best choices. Meanwhile, we have realized that borate-fluorides have more advantages than borate-chlorides/bromides for NLO crystals.25 However, to date there have been few perovskite or antiperovskite borate-fluorides reported. Therefore, we chose the F− anion as the center of octahedra of antiperovskite structures. With this in mind, we introduced F-centered [F(M/Ba)4Ca2] (M = K, Rb, Cs) octahedra to construct the antiperovskite structural framework. Then, in order to further increase the anisotropy of the structures, we filled the voids of the octahedral framework with π-conjugated [B2O5]4− groups. By doing this, two new NCS antiperovskite borate-fluorides, [(M/Ba)2Ca]F[B2O5] (M = K, Rb), and one double antiperovskite borate-fluoride, [CsBaCa]F[B2O5], were successfully designed and synthesized for the first time. Unexpectedly, the structural change from antiperovskite to double antiperovskite is caused by simple cation substitution, which is certainly exciting and worth studying. Therefore, in this communication, we will focus on finding the reasons for the structural transformation, hoping to provide ideas for the design of new antiperovskite structures. We will also report their syntheses, structures, and functional properties as well as first-principles calculation.
Results and discussion
Polycrystalline samples of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] were all synthesized via conventional solid-state reactions. Stoichiometric MF (M = K, Rb, Cs), BaCO3, CaCO3 and H3BO3 were thoroughly mixed and preheated at 300 °C for 24 h to decompose carbonates and boric acid. Then, the temperature was gradually increased to 700 °C and held for 72 h with several intermediate grindings and mixings. By doing this, the pure polycrystalline samples of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] can be obtained, and their purity can be confirmed by powder X-ray diffraction (PXRD) (Fig. S1†). For these polycrystalline samples, the thermal behaviors were studied by thermogravimetric (TG) and differential scanning calorimetry (DSC) measurements (Fig. S2a–c†). Clearly, for the three compounds, the heating DSC curves show sharp endothermic peaks at 1000 °C, 988 °C and 904 °C, accompanied by obvious weight loss in the TG curves. This suggests [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] melt incongruently. Furthermore, the PXRD measurements show the residue after TG/DSC is mainly Ba2Ca(BO3)2 (PDF #01-085-2268) and Ca3(BO3)2 (PDF #01-070-0868) (Fig. S2d–f†). This indicates [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] decomposed before melting. Therefore, a suitable flux is necessary for their single-crystal growth. With 2B2O3:0.65M2O (M = K, Rb, Cs) as the self-flux, single crystals (yields, about 80%, 85% and 90% based on Ba for [(K/Ba)2Ca]F[B2O5], [(Rb/Ba)2Ca]F[B2O5] and [CsBaCa]F[B2O5], respectively) were also obtained in the open air around 780 °C, 750 °C and 720 °C for K-, Rb- and Cs-based compounds, respectively. The crystallographic data are listed in Table 1. The related crystal data, including selected bond lengths and atomic coordinate equivalent isotropic displacement parameters, are listed in Tables S2–S6 in the ESI.†| Empirical formula | [(K/Ba)2Ca]F[B2O5] | [(Rb/Ba)2Ca]F[B2O5] | [CsBaCa]F[B2O5] |
|---|---|---|---|
| a R 1 = ∑‖Fo| – |Fc‖/∑|Fo| and wR2 = [∑w(Fo2 − Fc2)2/∑wFo4]1/2 for Fo2 > 2σ(Fo2). | |||
| Formula weight | 337.14 | 383.51 | 430.95 |
| Crystal system | Tetragonal | Tetragonal | Tetragonal |
| Space group |
P21m (113) |
P21m (113) |
P21m (113) |
| a (Å) | 8.7276(1) | 8.7713(3) | 8.8668(4) |
| c (Å) | 4.2840(9) | 4.3173(3) | 8.7669(4) |
| Z | 2 | 2 | 4 |
| Volume (Å3) | 326.32(1) | 332.15(3) | 689.26(7) |
| F (000) | 308 | 344 | 760 |
| Completeness (%) | 100.0% | 100.0% | 98.8% |
| Absolute structure parameter | 0.18(1) | 0.17(5) | 0.31(8) |
| Goodness-of-fit on F2 | 1.076 | 1.088 | 1.085 |
| Final R indices [(I > 2σ(I)]a | R 1 = 0.0401 wR2 = 0.1018 | R 1 = 0.0375 wR2 = 0.1016 | R 1 = 0.0455 wR2 = 0.1382 |
| R indices (all data)a | R 1 = 0.0506 wR2 = 0.1105 | R 1 = 0.0434 wR2 = 0.1082 | R 1 = 0.0504 wR2 = 0.1491 |
The single-crystal XRD shows [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] all crystallize in the NCS space group P21m. Remarkably, [(Rb/Ba)2Ca]F[B2O5] and [(K/Ba)2Ca]F[B2O5] are iso-structural, but they are not iso-structural with [CsBaCa]F[B2O5]. Therefore, only the structures of [(Rb/Ba)2Ca]F[B2O5] and [CsBaCa]F[B2O5] will be described in detail.
For [(Rb/Ba)2Ca]F[B2O5], the asymmetric unit contains one unique Rb/Ba (the Rb+ cations and the Ba2+ cations are disordered and occupy the same 4e Wyckoff position with 50% and 50% occupation, respectively), one Ca (Wyckoff position 2b), one B (Wyckoff position 4e), one F (Wyckoff position 2a) and two O (Wyckoff position 2c and 2f) atom(s). Each B atom is coordinated with three O atoms forming BO3 triangles, and two BO3 triangles further condensed into [B2O5]4− dimers (Fig. 1b) with B–O distances in the range of 1.352(17)–1.378(14) Å. The F− anion is surrounded by four Rb/Ba and two Ca cations to form the [F(K/Ba)4Ca2] octahedron (Fig. 1a) with Rb+/Ba2+ cations occupying four equator positions and Ca2+ cations occupying the apical position. The Rb/Ba–F and Ca–F distances are 3.1745(2) Å and 2.15865(15) Å, respectively. In the structure, the [F(Rb/Ba)4Ca2] octahedra connect with each other through corner-sharing to form the [F(Rb/Ba)2Ca]∞ octahedral framework (Fig. 1c). The [B2O5]4− dimers are fixed into the spaces of the [F(Rb/Ba)2Ca]∞ octahedral framework (Fig. 1d) by the Rb/Ba–O bonds with distances ranging from 2.899(14) to 3.329(11) Å and all Ca–O distances are identical at 2.311(10) Å. Clearly, [(Rb/Ba)2Ca]F[B2O5] exhibits an antiperovskite structure with the [B2O5]4− and F− anions, and Rb+/Ba2+ and Ca2+ cations occupying A-, B- and X-sites of the perovskite, respectively. Its crystal structure is also analogous with the antiperovskite aluminate fluoride, Sr3F[AlO4].26 As shown in Fig. 1e and f, the positions of [AlO4]5− anions are occupied by the isolated [B2O5]4− groups and the positions of Sr2+ cations are occupied by Rb+/Ba2+ and Ca2+ cations. Furthermore, the calculations of bond valence indicate that the coordination numbers of the corresponding atoms are reasonable (Tables S1 and S2†).
 | ||
| Fig. 1 (a) The [F(Rb/Ba)4Ca2] F-centered octahedra; (b) the B–O skeleton in [(Rb/Ba)2Ca]F[B2O5]; (c) the three-dimensional [F(Rb/Ba)2Ca]∞ octahedral framework; (d) the view of [(Rb/Ba)2Ca]F[B2O5] along the c-axis; (e and f) the analogy between the antiperovskite-type structure of [(Rb/Ba)2Ca]F[B2O5] and the antiperovskite structure of Sr3F[AlO4]. | ||
For [CsBaCa]F[B2O5], the asymmetric unit contains one crystallographically independent Cs (Wyckoff position 4e), one Ba (Wyckoff position 4e), one Ca (Wyckoff position 4d), two B (Wyckoff position 4e), two F (Wyckoff position 2a and 2b) and four O (Wyckoff position 2c and 8f) atom(s). All the B atoms are condensed into [B2O5]4− dimers (Fig. 2b) with B–O distances in the range of 1.317(11)–1.490(19) Å. The F(1)− and F(2)− anions are surrounded by four Ba2+ or Cs+ and two Ca2+ cations to form [F(1)Ba4Ca2] and [F(2)Cs4Ca2] octahedra, respectively (Fig. 2a). The Cs–F and Ca–F distances are in the range of 3.2012(2) to 3.1999(2) Å, and 2.1521(18) to 2.2313(18) Å, respectively. The Ba–F distances are identical, at 3.1999(6) Å. In the structure, the [FBa4Ca2] and [FCs4Ca2] octahedra connect with each other through corner-sharing to form the [FCsBaCa]∞ octahedral framework (Fig. 2c) filled by the [B(1)2O5]4− and [B(2)2O5]4− dimers to form the antiperovskite structure (Fig. 2d). Clearly, this structure has two different anionic groups at the A site, which is similar to the double antiperovskite Li6NBrBr2 (Fig. 2f) with two different anions (N and Br) at the B-site. Hence, the structure can also be written as the double antiperovskite formula [CsBaCa]2F2[B(1)2O5][B(2)2O5]. [CsBaCa]F[B2O5] is also the first reported double antiperovskite borate-fluoride. Furthermore, for the Cs–O, Ba–O and Ca–O bonds, the bond distances range from 3.029(11) to 3.444(10) Å, 2.765(8) to 2.972(9) Å and 2.320(9) to 2.345(9) Å, respectively. These bond distances are consistent with those reported in other compounds.11a,27 Bond valence sum (BVS) calculations are shown in Table S3.† The BVS indicates that the coordination numbers of corresponding atoms are reasonable.
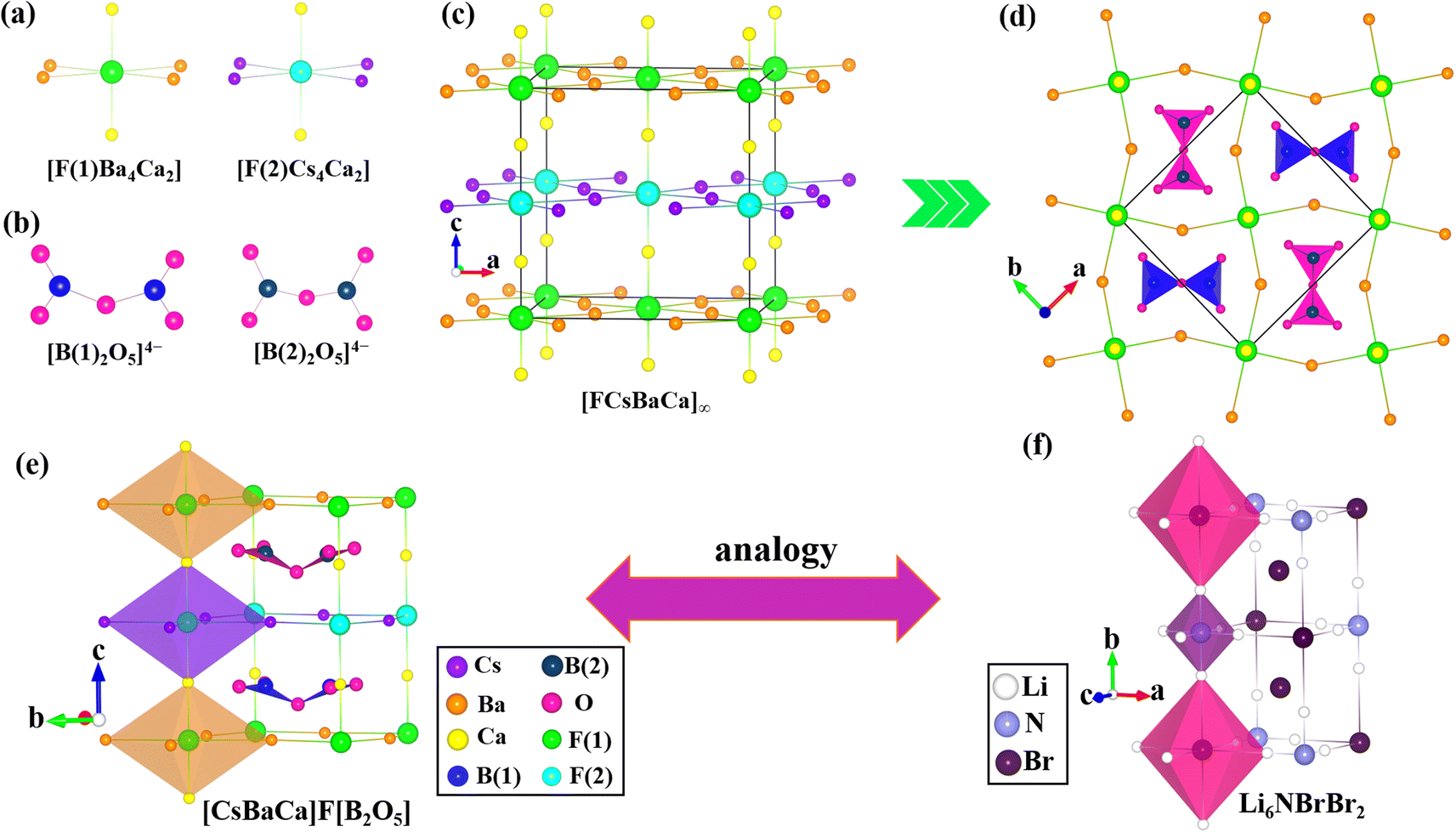 | ||
| Fig. 2 (a) The [FBa4Ca2] and [FCs4Ca2] F-centered octahedra; (b) the B–O skeleton in [CsBaCa]F[B2O5]; (c) the three-dimensional [FCsBaCa]∞ octahedral framework; (d) the view of [CsBaCa]F[B2O5] along the c-axis; (e and f) the analogy between the double antiperovskite-type structure of [CsBaCa]F[B2O5] and the double antiperovskite structure of Li6NBrBr2. | ||
From the chemical formulae, it can be seen that on going from [(Rb/Ba)2Ca]F[B2O5] to [CsBaCa]F[B2O5], the substitution of Rb+ by Cs+ cations results in a different configuration of F-based octahedra and the structure transforms from the antiperovskite to the double antiperovskite structure. For their lattice parameters, the length the of crystallographic c-axis of [CsBaCa]F[B2O5] is almost twice that of [(Rb/Ba)2Ca]F[B2O5], and the remaining lengths are nearly equal in the two structures (Table 1). This difference may be considered an effect of the cation sizes. As the cation radius on the X sites (equator positions) changes from Rb+ (1.69 Å) to Cs+ (1.85 Å),28 it will inevitably lead to an increase in the volume of the [F(M/Ba)Ca]∞ framework, which is equivalent to an increase in the effective radius of the anion F− at the B site. According to the Goldschmidt tolerance factor t = (rA + rX)/2(rB + rX), where rA, rB and rX are the effective radii of ions/groups at the A, B and X sites, in order to maintain the stability of the structure, the effective radius of the anionic group [B2O5]4− located at the A site also needs to be increased. That is to say, the [B2O5]4− dimers will be stretched. That results in an increase of the bond distances between the B atoms and the bridging O atom. As shown in Fig. 3a, the distances between the B atoms and the bridging O atom in [CsBaCa]F[B2O5] clearly increased to 1.45(2) and 1.49(19) Å (Table S7†). This means that the bridging O obtains less electrovalence from the B atoms, that is, the O atom needs to obtain more electrovalence from the cation at the X sites to ensure that its valence state is within the normal range. Therefore, the bridging O atoms need to bond with Cs+ cations with short Cs–O distances (3.029(11) Å) or directly coordinate with higher valence Ba2+ cations to increase the BVS (Fig. 3b). Thanks to the different coordination manners of bridging O atoms, the Cs+ and Ba2+ cations on the X sites no longer occupy the 4e Wyckoff position in the disordered manner of Rb+/Ba2+ in [(Rb/Ba)2Ca]F[B2O5], but instead occupy the 4e Wyckoff position separately. This coordination mode results in the formation of different F-based octahedra in the c-axis direction, further resulting in different cavities filled by two kinds of [B2O5]4− dimers (Fig. 3c). We also noticed that with the substitution of Cs+ for Rb+, the dihedral angle of the [B2O5]4− (the angle between two BO3 groups) decreased from 52.176° to 47.878° and 39.667° due to the stretching of the [B2O5]4− dimers.
 | ||
| Fig. 3 The evolution from the antiperovskite structure [(Rb/Ba)2Ca]F[B2O5] to the double antiperovskite [CsBaCa]F[B2O5]. (a) The stretching of the [B2O5]4− dimer leads to an increase in the distances between the bridging O atom and B atoms; (b) the bridging O atoms coordinate with Cs and Ba, instead of coordinating with disordered Rb/Ba in the [(Rb/Ba)2Ca]F[B2O5] structure; (c) the A site of the [(Rb/Ba)2Ca]F[B2O5] and [CsBaCa]F[B2O5] structures is occupied by one and two different sized [B2O5]4− dimers, respectively; (d) the dihedral angle of the [B2O5]4− (the angle between two BO3 units) decreased from 52.176° to 47.878° and 39.667°. | ||
In addition, it is clear that the octahedral frameworks for [(Rb/Ba)2Ca]F[B2O5] to [CsBaCa]F[B2O5] are distorted as can be seen from the F-M/Ba–F (M = Rb, Cs) bond angles (Table 2), which are much lower than 180°.29 According to Glazer's notation,30 their tilt systems can all be written as a0a0c+. Additionally, their octahedral rotation angles φ along the c-axis are 12.34° (for [(Rb/Ba)2Ca]F[B2O5]) (Fig. S3a†), and 11.57° (for [CsBaCa]F[B2O5]) (Fig. S3b†). Clearly, when going from antiperovskite [(Rb/Ba)2Ca]F[B2O5] to double antiperovskite [CsBaCa]F[B2O5], the octahedral rotation angles are slightly reduced. These indicate that on going from antiperovskite [(Rb/Ba)2Ca]F[B2O5] to double antiperovskite [CsBaCa]F[B2O5], the strain can be further alleviated. To further evaluate the stability of [(M/Ba)2Ca]F[B2O5] (M = K,Rb) and [CsBaCa]F[B2O5], the bond strain index (BSI) and global instability index (GII) were also calculated (Table 2).31 Generally, BSI and GII values imply the strain caused by electronic-induced and lattice-induced strains, respectively. The BSI values of the three title compounds are larger than 0.05 vu, indicating that they are all strained. In addition, the GII values are 0.223 vu, 0.260 vu, and 0.116 vu for K-, Rb-, and Cs-based compounds, respectively. According to Salinas-Sanchez's report,31b GII values of around 0.2 are considered highly strained and unstable and 0.1 is considered stable, which indicates that when the structure transforms from the antiperovskite to the double antiperovskite, the electronic-induced and lattice-induced strains can be released with a decrease in the BSI and GII values. As a result, the structure changes from antiperovskite [(Rb/Ba)2Ca]F[B2O5] to double antiperovskite [CsBaCa]F[B2O5] with the substitution of Rb+ by Cs+ cations.
| Compound | BVS | BSI (vu) | GII (vu) | Angle of F–M/Ba–F (°) | ||
|---|---|---|---|---|---|---|
| M0.5Ba0.5 | Cs and Ba | B | ||||
| [(K/Ba)2Ca]F[B2O5] | 1.32 | 3.25 | 0.102 | 0.223 | 154.20 | |
| (Rb/Ba)2Ca]F[B2O5] | 1.26 | 3.01 | 0.111 | 0.260 | 155.32 | |
| [CsBaCa]F[B2O5] | 0.98 and 1.87 | 2.97–3.03 | 0.084 | 0.116 | 156.64 | |
Furthermore, the existence of the [B2O5]4− groups was also confirmed by infrared spectra. As shown in Fig. 4a, the peaks in the range of 1348 cm−1 to 1207 cm−1 for the three compounds can be assigned to the asymmetric stretching of [BO3]3− units. The peaks around 1054–1106 cm−1 may be associated with the stretching vibration of B–O–B in [B2O5]4− groups. The weak absorption bands are assigned to the asymmetric stretching of [BO3]3− units. The peaks around 1054–1106 cm−1 may be associated with the stretching vibration of B–O–B in [B2O5]4− groups. The weak absorption bands at 1010–808 cm−1 can be attributed to the [BO3]3− symmetric stretching vibrations. In addition, the absorption bands around 748–596 cm−1 may be caused by the out-of-plane bending of the [BO3]3− groups. All of these are consistent with other reported borates.27,32
 | ||
| Fig. 4 (a) The infrared spectra of the title compounds; the UV-Vis diffuse reflectance spectra and optical band gaps of [(K/Ba)2Ca]F[B2O5] (b), [(Rb/Ba)2Ca]F[B2O5] (c), and [CsBaCa]F[B2O5] (d). | ||
The UV-vis-NIR diffuse reflectance spectra of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] are shown in Fig. 4b–d. It is clear that the three compounds exhibit wide UV transparent regions with UV cut-off edges around 190 nm. Based on the Kubelka–Munk function,33a the reflectance was converted into absorption. Extrapolating the linear part of the rising curve of absorption to zero, band-gaps of 5.68, 6.05, and 5.56 eV were obtained for [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5], respectively.
Since [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] all crystallize in NCS structures, their SHG responses were also measured by the Kurtz and Perry technique.33b Plots of the SHG intensity versus particle sizes for ground polycrystalline samples of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] are shown in Fig. 5a. Detailed features of those curves indicate that all three crystals are phase-matchable (PM). The comparison of the SHG signal produced by the [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] polycrystalline samples and the KDP samples in the same particle sizes (180–200 μm) reveals that they exhibit moderate SHG responses, ∼0.50, 0.44, and 0.39 × KDP, respectively (Fig. 5b). Clearly, these SHG responses are smaller than expected. According to the anionic group theory,33c the relatively small SHG responses should be mainly attributed to the non-aligned arrangement of the [B2O5]4− groups. In order to better understand the effect of orientation of [B2O5]4− groups on SHG responses, we calculated their dipole moments in the unit cell with a simple bond-valence approach.33d As shown in Table S8,† the dipole moments generated by the [B2O5]4− groups are largely cancelled in the unit cell. In addition, it should be noted that from Cs-, to Rb- to K-homologues, the SHG responses gradually increase. In order to better understand these, the number density of [B2O5]4− and the empirical ‘flexibility index’ F were also calculated (Table S9†).33e The calculation results indicate that the number density and F of [B2O5]4− groups in [(K/Ba)2Ca]F[B2O5] are the highest, followed by [(Rb/Ba)2Ca]F[B2O5] and [CsBaCa]F[B2O5], which is consistent with the experimental results of SHG responses.
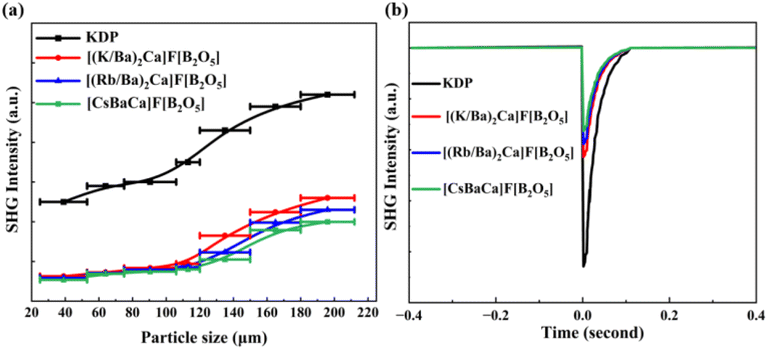 | ||
| Fig. 5 (a) The particle size dependence of SHG intensities for the title compounds and KDP as a reference; (b) the SHG signals for the three title compounds and KDP for exact particle sizes. | ||
The birefringence is also important for materials to achieve PM in a wide spectral region, so the birefringence of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] was also measured through cross polarization microscopy based on the formula R = Δn × d, where R, Δn, and d represent the optical path difference, birefringence, and thickness, respectively.33f Fig. S4† shows the original interference colour and the thickness measured on a Bruker single crystal diffractometer. The observed interference colours in cross-polarized light were second-order green for the title compounds, and matching with the Michal–Levy chart, the retardations (R values) were found to be 800 nm. The crystal thicknesses were found to be 17.1, 17.0, and 19.2 μm for [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5], respectively. Therefore, the birefringence values were determined to be 0.046, 0.052 and 0.041, respectively. Clearly, the birefringence is consistent with the calculated values based on first principles calculations (Fig. 6), and is comparable with other UV borate NLO crystals, such as LiB3O5 (0.0427@532 nm),2c CsLiB6O10 (0.050@532 nm)3c and Ba3Mg3(BO3)3F3 (0.045@532 nm),34 indicating the potential as UV NLO crystals.
 | ||
| Fig. 6 Calculated birefringence for [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5]. | ||
In order to further understand the origin of the optical and NLO properties of the title compounds, their electronic structures were analysed by first principles calculations based on density functional theory (DFT).35 The results show that [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] are all indirect band-gap compounds with calculated band gaps of 3.86, 5.55, and 3.95 eV, respectively (Fig. S5a–c†). The smaller calculated band-gaps than experimental ones can be attributed to the underestimation of the DFT method.36 The partial densities of states (PDOS) projected on the constituent atoms of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] are given in Fig. S5d–f.† Clearly, for [(M/Ba)2Ca]F[B2O5] (M = K, Rb), the top of the valence band (VB) in the region of −8 eV to the Fermi level is dominated by O 2p, B 2p, and F 2p orbitals with partial contributions of B 2s, and the upper two peaks of the VB between 0 and −2 eV mainly consist of O 2p and B 2p orbitals. The conduction band (CB) above the Fermi level is dominated by O 2p and B 2p orbitals with partial contributions of Ba 5d and Ca 3d orbitals, and the bottom of the CB mostly consists of O 2p and B 2p orbitals. For [CsBaCa]F[B2O5], the valence band maximum (VBM) is also mostly from the O 2p, B 2p and F 2p orbitals, with a small amount of O 2s and B 2s orbitals. The conduction band minimum (CBM) is mainly dominated by the O 2p and B 2p orbitals, with a small amount of Ca 3d and Ba 5d orbitals. It is obvious that the electronic states of the B atom are fully overlapped with the O atom in the energy region near the Fermi level, showing the strong interactions of B–O in the three compounds. These results indicate that the [B2O5]4− group makes the main contribution to the optical properties of [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5].
Based on the electronic structures, the SHG coefficients were also calculated. Since [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] crystallize in the space group P21m, belonging to the point group 2m, they all have the only independent non-zero SHG coefficient, d36, which can be calculated as 0.192, 0.185, and 0.181 pm V−1 for [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5], respectively. This is basically consistent with the experimental values. Remarkably, as [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] crystallize in the tetragonal system, the calculation equation of effective NLO coefficients (deff) for type-I PM for them will be ‘deff = d14sin2θcos2ψ’, where θ and ψ are the angle between the wave vector of fundamental frequency light and the optical axis and the projection of the phase matching direction on the a–b plane, respectively. This means [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] would have larger effective NLO coefficients when they are used in the UV spectral regions, especially when the wavelengths are close to their PM limit where θ is approaching 90°. Therefore, [(M/Ba)2Ca]F[B2O5] (M = K, Rb) and [CsBaCa]F[B2O5] can exhibit larger SHG responses in the UV regions.
Conclusions
In summary, through rationally choosing an antiperovskite structure as a template, two new antiperovskite borate fluorides, [(M/Ba)2Ca]F[B2O5] (M = K, Rb), and the first double antiperovskite borate fluoride, [CsBaCa]F[B2O5], have been successfully designed and synthesized. Interestingly, with the change of cation size, the structure undergoes an evolution from an antiperovskite structure to a double antiperovskite structure. Moreover, all the structures crystallize in the NCS space group P21m. The physiochemical property measurements show they all exhibit wide UV transparent regions with short UV cut-off edges (∼190 nm) and suitable birefringence (0.0405, 0.0548, and 0.0414@532 nm for Cs-, Rb- and K-based compounds), as well as moderate SHG responses (∼0.5 × KDP@1064 nm). In particular, they crystallize in the tetragonal point group 2m, so they can exhibit larger SHG responses in the UV region, close to their PM limit. This suggests their potential as UV NLO crystals, and indicates that using an antiperovskite structural template is an effective way to design new inorganic NCS solids.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2379650–2379652 has been deposited at the CCDC.Author contributions
F. Q. C. performed the experiments, data analysis, theoretical calculations, and paper writing. H. W. Y. designed and supervised the experiments. H. P. W. provided major revisions of the manuscript. Z. G. H. supervised the optical experiments. J. Y. W. and Y. C. W. helped the analyses of the crystallization process and the data. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22071179, 52322202, 52172006) and Tianjin Municipal Science and Technology Bureau (Grant No. 21JCJQJC00090 and 20JCJQJC00060).Notes and references
- (a) P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS; (b) K. M. Ok, E. O. Chi and P. S. Halasyamani, Chem. Soc. Rev., 2006, 35, 710–717 RSC.
- (a) C. T. Chen, B. C. Wu, A. D. Jiang and G. M. You, Sci. Sin., Ser. B, 1985, 28, 235 Search PubMed; (b) C. T. Chen and G. Z. Liu, Annu. Rev. Mater. Sci., 1986, 16, 136 CrossRef; (c) C. T. Chen, Y. C. Wu, A. D. Jiang, B. C. Wu, G. M. You, R. K. Li and S. J. Lin, J. Opt. Soc. Am. B, 1989, 6, 616 CrossRef CAS.
- (a) Y. Wu, T. Sasaki, S. Nakai, A. Yokotani, H. Tang and C. Chen, Appl. Phys. Lett., 1993, 62, 2614 CrossRef CAS; (b) D. A. Keszler, A. Akella, K. I. Schaoers and T. Alekel III, Mater. Res. Soc. Symp. Proc., 1994, 329, 15 CrossRef CAS; (c) Y. Mori, I. Kuroda, S. Nakajima, T. Sasaki and S. Nakai, Appl. Phys. Lett., 1995, 67, 1818 CrossRef CAS.
- (a) P. Becker, Adv. Mater., 1998, 10, 979 CrossRef CAS; (b) N. Ye, W. R. Zeng, J. Jiang, B. C. Wu, C. T. Chen, B. H. Feng and X. L. Zhang, J. Opt. Soc. Am. B, 2000, 17, 764 CrossRef CAS; (c) H. W. Huang, J. Y. Yao, Z. S. Lin, X. Y. Wang, R. He, W. J Yao, N. X. Zhai and C. T. Chen, Angew. Chem., Int. Ed., 2011, 50, 9141 CrossRef CAS PubMed.
- (a) S. C. Wang, N. Ye, W. Li and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8779 CrossRef CAS PubMed; (b) S. C. Wang and N. Ye, J. Am. Chem. Soc., 2011, 133, 11458 CrossRef CAS PubMed; (c) H. W. Yu, S. L. Pan, H. P. Wu, W. W. Zhao, F. F. Zhang, H. Y. Li and Z. H. Yang, J. Mater. Chem., 2012, 22, 2105 RSC; (d) H. W. Huang, J. Y. Yao, Z. S. Lin, X. Y. Wang, R. He, W. J. Yao, N. X. Zhai and C. Y. Chen, Chem. Mater., 2011, 23, 5457 CrossRef CAS.
- (a) Y. Z. Huang, L. M. Wu, X. T. Wu, L. H. Li, L. Chen and Y. F. Zhang, J. Am. Chem. Soc., 2010, 132, 12788 CrossRef CAS PubMed; (b) H. Yang, C. L. Hu, J. L. Song and J. G. Mao, RSC Adv., 2014, 4, 45258 RSC; (c) M. R. D. Mutailipu, K. R. Poeppelmeier and S. L. Pan, Chem. Rev., 2021, 121, 1130–1202 CrossRef CAS PubMed.
- (a) U. Opik and M. H. L. Pryce, Proc. R. Soc. London, Ser. A, 1957, 238, 425–447 CrossRef CAS; (b) R. F. W. Bader, Can. J. Chem., 1962, 40, 1164–1175 CrossRef CAS; (c) R. G. Pearson, J. Am. Chem. Soc., 1969, 91, 4947–4955 CrossRef CAS.
- (a) R. G. Pearson, J. Mol. Struct.:THEOCHEM, 1983, 103, 25–34 CrossRef; (b) R. A. Wheeler, M. H. Whangbo, T. Hughbanks, R. Hoffmann, J. K. Burdett and T. A. Albright, J. Am. Chem. Soc., 1986, 108, 2222–2236 CrossRef CAS PubMed; (c) S. E. Bang, Z. Pan, Y. H. Kim, D. W. Lee and K. M. Ok, J. Solid State Chem., 2013, 208, 65–70 CrossRef CAS.
- (a) H. L. Jiang, S. P. Huang, Y. Fan, J. G. Mao and W. D. Cheng, Chem.–Eur., J., 2008, 14, 1972–1981 CrossRef CAS PubMed; (b) H. L. Jiang, F. Kong, Y. Fan and J. G. Mao, Inorg. Chem., 2008, 47, 7430–7437 CrossRef CAS PubMed; (c) Y. C. Hao, X. Xu, F. Kong, J. L. Song and J. G. Mao, CrystEngComm, 2014, 16, 7689–7695 RSC; (d) H. W. Yu, H. P. Wu, S. L. Pan, Z. H. Yang, X. L. Hou, X. Su, Q. Jing, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2014, 136, 1264–1267 CrossRef CAS PubMed.
- (a) R. H. Cong, T. Yang, F. H. Liao, Y. X. Wang, Z. S. Lin and J. H. Lin, Mater. Res. Bull., 2012, 47, 2573–2578 CrossRef CAS; (b) J. L. Song, C. L. Hu, X. Xu, F. Kong and J. G. Mao, Angew. Chem., Int. Ed., 2015, 54, 3679–3682 CrossRef CAS PubMed; (c) S. G. Zhao, Y. Yang, Y. G. Shen, B. Q. Zhao, L. N. Li, C. M. Ji, Z. Y. Wu, D. Q. Yuan, Z. S. Lin, M. C. Hong and J. H. Luo, Angew. Chem., Int. Ed., 2017, 129, 555–559 CrossRef; (d) Y. X. Song, M. Luo, C. S. Lin and N. Ye, Chem. Mater., 2017, 29, 896–903 CrossRef CAS; (e) C. Wu, X. X. Jiang, Z. J. Wang, L. Lin, Z. S. Lin, Z. P. Huang, X. F. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3464–3468 CrossRef CAS PubMed.
- (a) G. H. Zou, N. Ye, L. Huang and X. S. Lin, J. Am. Chem. Soc., 2011, 133, 20001–20007 CrossRef CAS PubMed; (b) L. Kang, S. Y. Luo, H. W. Huang, N. Ye, Z. S. Lin, J. G. Qin and C. T. Chen, J. Phys. Chem. C, 2013, 117, 25684–25692 CrossRef CAS; (c) T. T. Tran, J. H. He, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2015, 137, 10504–10507 CrossRef CAS PubMed; (d) G. Peng, C. S. Lin and N. Ye, J. Am. Chem. Soc., 2020, 142, 20542–20546 CrossRef CAS PubMed; (e) X. M. Liu, L. Kang, P. F. Gong and Z. S. Lin, Angew. Chem., Int. Ed., 2021, 60, 13574–13578 CrossRef CAS PubMed; (f) L. Xiong, L. M. Wu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 25063–25067 CrossRef CAS PubMed.
- (a) B. C. Wu, D. Y. Tang, N. Ye and C. T. Chen, Opt. Mater., 1996, 5, 105 CrossRef CAS; (b) S. G. Zhao, P. F. Gong, L. Bai, X. Xu, S. Q. Zhang, Z. H. Sun, Z. S. Lin, M. C. Hong, C. T. Chen and J. H. Luo, Nat. Commun., 2014, 5, 4019 CrossRef CAS PubMed; (c) S. G. Zhao, P. F. Gong, S. Y. Luo, S. J. Liu, L. Li, M. Adnan, T. Khan, M. C. Hong, Z. S. Lin and J. H. Luo, J. Am. Chem. Soc., 2015, 137, 2207–2210 CrossRef CAS PubMed; (d) G. H. Zou, C. S. Lin, H. Jo, G. Nam, T. S. You and K. M. Ok, Angew. Chem., Int. Ed., 2016, 55, 12078–12082 CrossRef CAS PubMed; (e) H. W. Yu, N. Z. Koocher, J. M. Rondinelli and P. S. Halasyamani, Angew. Chem., Int. Ed., 2018, 57, 6100–6103 CrossRef CAS PubMed.
- (a) X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS PubMed; (b) G. Q. Shi, Y. Wang, F. F. Zhang, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS PubMed; (c) Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 2150–2154 CrossRef CAS PubMed; (d) C. Wu, X. X. Jiang, L. Lin, W. Y. Dan, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 27151–27157 CrossRef CAS PubMed; (e) H. T. Qiu, F. M. Li, Z. Li, Z. H. Yang, S. L. Pan and M. R. D. Mutailipu, J. Am. Chem. Soc., 2023, 145, 24401–24407 CrossRef CAS PubMed; (f) S. Liu, X. X. Jiang, L. Qi, Y. L. Hu, K. N. Duanmu, C. Wu, Z. S. Lin, Z. P. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202318107 CrossRef PubMed.
- (a) R. Q. Liu, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Chem. Mater., 2021, 33, 4240–4246 CrossRef CAS; (b) J. J. Zhou, Y. Y. Liu, H. P. Wu, H. W. Yu, Z. S. Lin, Z. G. Hu, J. Y. Wang and Y. C. Wu, Angew. Chem., Int. Ed., 2020, 59, 19006–19010 CrossRef CAS PubMed; (c) H. P. Wu, Z. J. Wei, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, Angew. Chem., Int. Ed., 2024, 63, e202406318 CrossRef CAS PubMed.
- (a) S. De, D. Asthana, C. Thirmal, S. K. Keshri, R. K. Ghosh, G. Hundal, R. Kumar, S. Singh, R. Chatterjeec and P. Mukhopadhyay, Chem. Sci., 2023, 14, 2547–2552 RSC; (b) S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. Malla Reddy, Science, 2021, 373, 321 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010–1026 CrossRef CAS.
- M. R. Filip and F. Giustino, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5397–5402 CrossRef CAS PubMed.
- (a) M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981 CrossRef CAS PubMed; (b) C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2016, 28, 5778 CrossRef CAS PubMed; (c) P. Gao, M. Graetzel and M. K. Nazeeruddin, Energy Environ. Sci., 2014, 7, 2448 RSC; (d) M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506 CrossRef CAS; (e) B. V. Lotsch, Angew. Chem., Int. Ed., 2014, 53, 635 CrossRef CAS PubMed.
- (a) X. T. Li, M. Kepenekian, L. D. Li, H. Dong, C. C. Stoumpos, R. Seshadri, C. Katan, P. J. Guo, J. Even and M. G. Kanatzidis, J. Am. Chem. Soc., 2022, 144, 3902 CrossRef CAS PubMed; (b) L. M. Kong, Y. Q. Sun, B. Zhao, K. Y. Ji, J. Feng, J. C. Dong, Y. Z. Wang, Z. R. Liu, S. B. Maqbool, Y. G. Li, Y. G. Yang, L. J. Dai, W. Lee, C. Cho, S. D. Stranks, R. H. Friend, N. Wang, N. C. Greenham and X. Y. Yang, Nature, 2024, 631, 73–79 CrossRef CAS PubMed; (c) H. N. Liu, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu, P. S. Halasyamani and H. W. Yu, ACS Mater. Lett., 2023, 5, 155–161 CrossRef CAS.
- Y. G. Wang, H. Zhang, J. L. Zhu, X. J. LU, S. Li, R. Q. Zou and Y. S. Zhao, Adv. Mater., 2020, 32, e1905007 CrossRef PubMed.
- (a) H. P. Wu, S. L. Pan, K. R. Poeppelmeier, H. Y. Li, D. Z. Jia, Z. H. Chen, X. Y. Fan, Y. Yang, J. M. Rondinelli and H. S. Luo, J. Am. Chem. Soc., 2011, 133, 7786–7790 CrossRef CAS PubMed; (b) M. Zhang, X. Su, S. L. Pan, Z. Wang, H. Zhang, Z. H. Yang, B. B. Zhang, L. Y. Dong, Y. Wang, F. F. Zhang and Y. Yang, J. Phys. Chem. C, 2014, 118, 11849–11856 CrossRef CAS; (c) Z. H. Chen, S. L. Pan, X. Y. Dong, Z. H. Yang, M. Zhang and X. Su, Inorg. Chim. Acta, 2013, 406, 205–210 CrossRef CAS; (d) C. Y. Bai, H. W. Yu, S. J. Han, S. L. Pan, B. B. Zhang, Y. Wang, H. P. Wu and Z. H. Yang, Inorg. Chem., 2014, 53, 11213–11220 CrossRef CAS PubMed.
- J. K. Wang, Y. S. Cheng, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, Angew. Chem., Int. Ed., 2022, 61, e202201616 CrossRef CAS PubMed.
- S. X. Cui, H. P. Wu, Z. G. Hu, J. Y. Wang, Y. C. Wu and H. W. Yu, Adv. Sci., 2023, 10, e2204755 CrossRef PubMed.
- P. S. Halasyamani and J. M Rondinelli, Nat. Commun., 2018, 9, 2972 CrossRef PubMed.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed.
- R. Green, M. Avdeev and T. Vogt, J. Solid State Chem., 2015, 228, 1–8 CrossRef CAS.
- S. S. Li, X. M. Liu, H. P. Wu, Z. F. Song, H. W. Yu, Z. S. Lin, Z. G. Hu, J. Y. Wang and Y. C. Wu, Chem. Sci., 2021, 12, 13897–13901 RSC.
- R. D. Shannon, Acta Cryst., 1976, A32, 751–767 CrossRef CAS.
- T. K. Mandal, A. M. Abakumov, M. V. Lobanov, M. Croft, V. V. Poltavets and M. Greenblatt, Chem. Mater., 2008, 20, 4653–4660 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater. , 1972, 28, 3384 CrossRef CAS.
- (a) C. Preiser, J. Lösel, I. D. Brown, M. Kunz and A. Skowron, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater. , 1999, 55, 698 CrossRef PubMed; (b) A. Salinas-Sanchez, J. L. Garcia-Muñoz, J. Rodriguez-Carvajal, R. Saez-Puche and J. L. Martinez, J. Solid State Chem., 1992, 100, 201–211 CrossRef CAS.
- (a) M. E. Gao, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Sci. China: Chem., 2021, 64, 1184–1191 CrossRef CAS; (b) M. M. Ding, J. J. Xu, H. P. Wu, H. W. Yu, Z. G. Hu, J. Y. Wang and Y. C. Wu, Dalton Trans., 2020, 49, 12184–12188 RSC; (c) X. Meng, M. Xia and R. Li, New J. Chem., 2019, 43, 11469–11472 RSC.
- (a) J. Tauc, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS; (b) S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS; (c) C. T. Chen, Y. C. Wu and R. K. Li, Int. Rev. Phys. Chem., 1989, 8, 65–91 Search PubMed; (d) P. A. Maggard, T. S. Nault, C. L. Stern and K. R. Poeppelmeier, J. Solid State Chem., 2003, 175, 27–33 CrossRef CAS; (e) X. X. Jiang, S. G. Zhao, Z. S. Lin, J. H. Luo, P. D. Bristowe, X. G. Guand and C. T. Chen, J. Mater. Chem. C, 2014, 2, 530 RSC; (f) B. E. Sørensen, Eur. J. Mineral., 2013, 25, 5–10 CrossRef.
- M. R. D. Mutailipu, M. Zhang, H. P. Wu, Z. H. Yang, Y. H. Shen, J. L. Sun and S. L. Pan, Nat. Commun., 2018, 9, 3089 CrossRef PubMed.
- L. Kang, F. Liang, X. X. Jiang, Z. S. Lin and C. T. Chen, Acc. Chem. Res., 2020, 53, 209–217 CrossRef CAS PubMed.
- R. W. Godby, M. Schlüter and L. J. Sham, Phys. Rev. Lett., 1986, 56, 2415–2418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2379650, 2379651 and 2379652 for [CsBaCa]F[B2O5], [(Rb/Ba)2Ca]F[B2O5] and [(K/Ba)2Ca]F[B2O5], respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05747c |
| This journal is © The Royal Society of Chemistry 2025 |