
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic oxidation of alkylarenes and polystyrene waste to benzoic acids via a copper-based catalyst†
Enjie
Xu
,
Tianwei
Liu
,
Fuyu
Xie
,
Jianghua
He
* and
Yuetao
Zhang
 *
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, Jilin 130012, China. E-mail: hjh2015@jlu.edu.cn; ytzhang2009@jlu.edu.cn
First published on 9th December 2024
Abstract
The chemical recycling of polystyrene (PS) waste to value-added aromatic compounds is an attractive but formidable challenge due to the inertness of the C–C bonds in the polymer backbone. Here we develop a light-driven, copper-catalyzed protocol to achieve aerobic oxidation of various alkylarenes or real-life PS waste to benzoic acid and oxidized styrene oligomers. The resulting oligomers can be further transformed under heating conditions, thus achieving benzoic acid in up to 65% total yield through an integrated one-pot two-step procedure. Mechanistic studies show that the CuCl2 catalyst undergoes Ligand-to-Metal Charge Transfer (LMCT) to generate a chlorine radical, which triggers activation of the C–H bond and subsequent oxidative cleavage of C–C bonds. The practicality and scalability of this strategy are demonstrated by depolymerization of real-life PS foam on a gram scale, thus showing promising application potential in chemical recycling of PS waste.
Introduction
Polystyrene (PS), one of the most manufactured plastics in the world, exhibits wide applications in various fields, such as packaging, thermal insulation and electronics. The annual production of PS is estimated to be nearly 17 million tons per year.1 However, end-of-life PS plastics either become landfill waste or escape into the environment, which not only aggravates ecological distress but also threatens human health.2–4 Although progress has been made in the development of methods to recover end-of-life polymers,5–15 the PS recycling rate remains below 1%.16,17 As one of the most valuable all recycling methods, chemical recycling has demonstrated promising potential in the depolymerization of these post-consumer polymer products into useful chemical feedstocks.18,19 However, it remains formidably challenging to realize chemical recycling of PS waste due to the inertness of C–C bonds in the polymer backbone.20,21 Research efforts have continuously focused on the traditional thermal depolymerization of PS (Scheme 1A). For example, the chemical oxidation of PE, PP and PS to organic acids was achieved by the Co(II)/Mn(II) system at 150–210 °C,22–24 the oxidative depolymerization of PS into benzoic acid was obtained by nitric acid at 180 °C.25 Yan and co-workers reported the reductive upcycling of various aromatic plastic waste (PET, PS, PPO, and PC) to arenes through catalytic hydrogenolysis over a Ru/Nb2O5 catalyst at 320 °C and 0.5 MPa H2.26 Jiao et al. utilized organic azide to synthesize arylamines from alkylarenes and PS with the catalysis of FeCl2,27 while Liu's group developed a “degradation-upcycling” strategy for the synthesis of aryl ketones and organosulfur compounds from PS waste in the presence of equivalent AlCl3.28 Despite the efficiency of existing thermal methods, the requirement of high temperatures restricts their practical application in industry.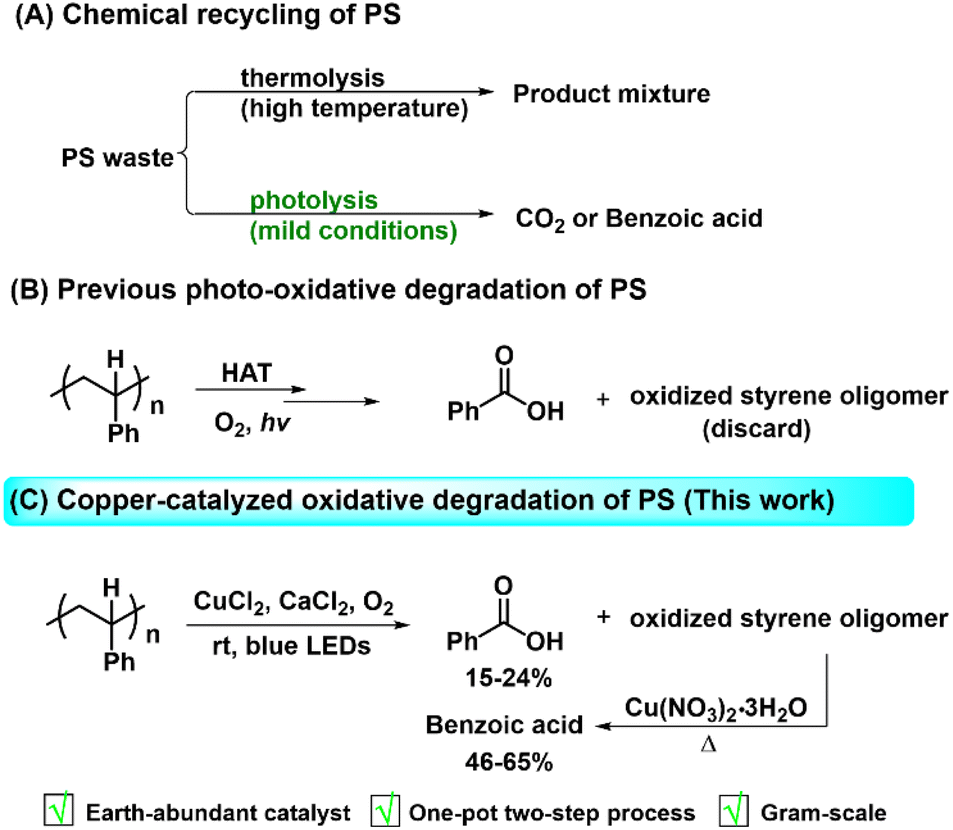 | ||
| Scheme 1 (A) Chemical recycling of PS; (B) previous studies on the photo-oxidative degradation of PS; (C) copper-catalyzed oxidative degradation of PS (this work). | ||
Compared with thermal catalysis, photocatalysis has emerged as a powerful and sustainable method that exhibits unique reactivity under mild conditions.29,30 Intense interest has been focused on the photochemical oxidation degradation of PS due to the use of visible light irradiation and the inexpensive oxidant of oxygen.31–33 The oxidative degradation of PS involves C–H bond activation and subsequent oxidation with O2 to facilitate the formation of an organic peroxide, leading to the cleavage of the C–C bond and the generation of oxidized products in the end (Scheme 1B).34 Zhu's group developed a non-selective process for photooxidation of PS plastic into CO2via the copper phthalocyanine sensitized TiO2 photocatalyst under fluorescent light,35 along with the production of CO2. Recently, progress has been made in the photocatalytic oxidative deconstruction of PS into value-added benzoic acid. It was reported that the chlorine radical generated from FeCl3 (ref. 36–39) or the catalytic system of acridinium salts and HCl40 facilitated abstraction of hydrogen atoms from the PS backbone and subsequent oxidation, resulting in formation of benzoic acid as a major product along with oligomers. It has been demonstrated that the reactive oxygen species could initiate PS degradation into benzoic acid under photo irradiation by an acid-catalyzed strategy at room temperature41,42 or heterogeneous graphitic carbon nitrides at 150 °C.43 Reisner et al. utilized fluorenone as a direct HAT agent in combination with 1 equivalent of H2SO4 as an acidic additive to realize the depolymerization of PS to benzoic acid (30%), other aromatic compounds (20%) and oligomers.44 By using an anthraquinone photocatalyst, Kokotos et al. discovered that the aerobic oxidation of PS plastics afforded benzoic acid in 25–58% yields, without requirement of acidic additive.45 Das and co-workers reported the photocatalyst system of NBS and CF3SO2Na for selective upcycling of real-life PS-based waste into benzoic acid, which can be further transformed into valuable aromatics.46 The photooxidative degradation of plastic waste (PS etc.) into commercial chemicals was achieved via V(O)(acac)2 (ref. 47) or UO2(NO3)2·6H2O.48 So far, it remains greatly challenging to obtain a single product in high yield due to the poor product selectivity and formation of the oxidized oligomer waste by the current strategies. Therefore, it is in high demand to develop a more efficient and cost-effective degradation strategy to produce a target compound from real-life plastic waste.
Earth-abundant Cu-based photocatalysts have been employed in visible light photocatalysis for production of valuable chemicals in an ecologically friendly and cost-effective manner.49–53 It is known that CuCl2 is capable of serving as a visible-light photocatalyst and the photoinduced ligand dissociation of CuCl2 complex generates CuCl and the chlorine radical through ligand-to-metal charge transfer (LMCT).54–58 The formation of the chlorine radical provides the opportunities to realize the challenging chemical transformations. For instance, Wan's group developed a CuCl2-promoted photocatalytic strategy to realize the vicinal dichlorination of alkenes and styrenes in the presence of HCl.59 Rovis's group reported the CuCl2-catalyzed alkylation of alkanes with electron-deficient olefins.60 Encouraged by these pioneering works, we also applied this photocatalytic LMCT strategy to the generation of chlorine radicals for the abstraction of the C–H bond from the polymer backbone under mild conditions. Besides, chlorine radicals are widely used as hydrogen abstraction agents due to their characteristic electrophilic selectivity towards the strong electron-rich C–H bonds (ΔHdiss(HCl) = 103 kcal mol−1),61 and are kinetically matched with the more hydridic C–H bonds of the polymer backbone.36–38 In this context, we developed a practical copper-based photocatalytic system consisting of readily available CuCl2 as a LMCT reagent, CaCl2 as a chlorine source and O2 as a terminal oxidant, which enabled the oxidative cleavage of a wide range of alkylarene and real-life PS waste to benzoic acid and oxidized styrene oligomers (Scheme 1C). The corresponding oligomers can be further transformed under heating conditions, thus achieving benzoic acid in up to 65% total yield in a one-pot two-step manner.
Experimental
Materials and methods
All reagents were used as received unless otherwise noted. CuCl2 was purchased from Energy Chemical. CaCl2 powder was purchased from Beijing Chemical Works. The methanol and MeCN (HPLC grade) were purchased from Sigma-Aldrich. Reactions were monitored by thin layer chromatography (TLC), visualizing with ultraviolet light (UV); column chromatography purifications were carried out using silica gel. The reaction mixture was analyzed by a Waters High Performance Liquid Chromatograph (HPLC) system equipped with an autosampler, a C18 column (length: 150 mm, internal diameter: 4.6 mm, 35 °C) and UV/vis detector (λ = 220 nm). CH3OH: 0.1% HCOOH in H2O was used as a mobile phase with a flow rate of 1.0 mL min−1. 1H NMR spectra was recorded on a Bruker Avance II 500 (500 MHz, 1H) instrument at room temperature (rt). Chemical shifts (δ) were reported as parts per million (ppm). The number-average molecular weight (Mn) and molecular weight distributions (PDI = Mw/Mn) of PS were measured by gel permeation chromatography (GPC) at 40 °C and a flow rate of 1 mL min−1, using THF (HPLC grade) as an eluent on a Waters 1515 instrument equipped with a Waters 4.6 × 30 mm guard column and three Waters WAT054466, WAT044226, and WAT044223 columns. The instrument was calibrated with PS standards, and chromatograms were processed with Waters Breeze2 software.General procedure for light-induced catalytic reactions
Results and discussion
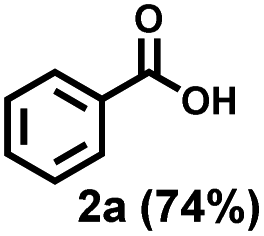
Previously, PS degradation strategies were reported to be adapted from the strategy for cleavage of C–C bonds in small molecules.44 Therefore, we initially optimized the reaction condition for the oxidative C–C bonds cleavage of propylbenzene (1u) to benzoic acid (2a) by using a copper-based catalyst in O2 atmosphere with the irradiation of blue light (Table 1). The employment of CuCl2 led to the production of 2a in 40% yield (entry 1, Table 1), whereas other non-chloride copper salts such as Cu(OAc)2·H2O, Cu(NO3)2·3H2O and CuSO4·5H2O were ineffective for this reaction in up to 24 h (entries 2–4, Table 1), highlighting the essentially important role of the chlorine radicals generated from CuCl2 for this reaction. Previously, photoactive species CuCl3− generated from the addition of exogenous chloride was found to facilitate the chlorine radical formation and C–H abstraction.60 Notably, metal chloride has a dual role in serving as a chlorine source of CuCl2 and modulating the activity of the copper-oxo intermediates (CuII-O-O and CuII-O) by the Lewis acidity of the metal ion,62 especially the latter has a predominant effect. The addition of LiCl, MgCl2 or CaCl2 can significantly enhance the yields of 2a to 67%, 72% or 75%, respectively (entries 5–7, Table 1), while there was no improvement effect exhibited by HCl, NaCl, KCl or SrCl2 due to their weak Lewis acidity63–65 (entries 8–11, Table 1). The employment of CuCl also showed high efficiency, leading the good yield of 2a in 63% (entry 12, Table 1). Literature research revealed that the bromine radicals generated from CuBr2 under photoexcitation could also abstract hydrogen atoms.66 However, using CuBr2 instead of CuCl2, drastically decreased the yield of 2a (entry 13, Table 1). The addition of bromine additives to the reactions catalyzed by CuBr2 only slightly increased the yield of 2a to 27% (entry 14, Table 1). Switching back to CaCl2 could maintain the high yields of 2a by using CuBr2 or CuBr catalyst (66% for entry 15 and 63% for entry 16, Table 1), highlighting the importance of CaCl2 in achieving 2a in high efficiency. Reducing the concentration of CuCl2 (1 mol%) or CaCl2 (10 mol%) decreased the yield of 2a to 40% or 69%, respectively (entries 17 and 18, Table 1). Irradiation of the reaction with 30 W blue LEDs furnished 2a in 63% yield whereas 71% yield of 2a was obtained in the air (entries 19 and 20, Table 1). Control experiments showed that without CuCl2, light or O2, there is no observation of 2a in 24 h, demonstrating the indispensable role of these factors for this reaction (entries 21–23, Table 1). We also screened the effectiveness of different solvents on this reaction (i.e. MeOH, THF, DCM, EtOAc, acetone and DMF) (Table S1†) and found that acetonitrile showed the best performance, probably due to the stabilization of copper complexes through the ligation of nitrile to copper.55|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] catalyst | Additive | Yield (%) | ||
| 2a | 3u | 4u | |||
| a Standard conditions: 1u (0.1 mmol), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN (1.0 mL), O2 balloon, 50 W blue LEDs, rt, 24 h. The yields were measured by HPLC using a standard curve of standard substances as reference. b MCl (40 mol%). c CuCl2 (1 mol%). d CaCl2 (10 mol%). e 30 W. f In air. g 80 °C in the dark. h In N2. | |||||
| 1 | CuCl2 | None | 40 | 14 | 9 |
| 2 | Cu(OAc)2·H2O | None | 0 | 0 | 0 |
| 3 | Cu(NO3)2·3H2O | None | 0 | 0 | 0 |
| 4 | CuSO4·5H2O | None | 0 | 0 | 0 |
| 5b | CuCl2 | LiCl | 67 | 6 | 8 |
| 6 | CuCl2 | MgCl2 | 72 | 1 | 6 |
| 7 | CuCl2 | CaCl2 | 75 | 0 | 0 |
| 8b | CuCl2 | HCl | 40 | 3 | 2 |
| 9b | CuCl2 | NaCl | 36 | 11 | 10 |
| 10b | CuCl2 | KCl | 31 | 8 | 8 |
| 11 | CuCl2 | SrCl2 | 31 | 8 | 6 |
| 12 | CuCl | CaCl2 | 63 | 3 | 0 |
| 13 | CuBr2 | None | <1 | 2 | 4 |
| 14 | CuBr2 | CaBr2 | 27 | 0 | 1 |
| 15 | CuBr2 | CaCl2 | 66 | 0 | 0 |
| 16 | CuBr | CaCl2 | 63 | 5 | 0 |
| 17c | CuCl2 | CaCl2 | 40 | 8 | 4 |
| 18d | CuCl2 | CaCl2 | 69 | 1 | 0 |
| 19e | CuCl2 | CaCl2 | 63 | 2 | 1 |
| 20f | CuCl2 | CaCl2 | 71 | 2 | 0 |
| 21 | — | CaCl2 | 0 | 0 | 0 |
| 22g | CuCl2 | CaCl2 | 0 | 0 | 0 |
| 23h | CuCl2 | CaCl2 | 0 | 0 | 0 |

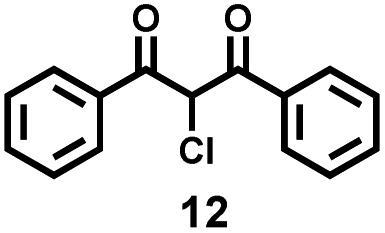
With the optimized reaction conditions in hand, we examined the generality for the deep oxidation of alkylarenes (Scheme 2). It turned out that this reaction works for methylarenes with neutral (H 1a), electron-donating (phenyl 1b and acetoxy 1c) or electron-withdrawing groups (acetyl 1d; –CN 1e; –CF31f; and –Cl 1g), furnishing the corresponding aromatic acids in good to excellent yields (68–97%). It is noted that the methylarenes with fluoride substituted at the ortho- (1h), meta- (1i), and para-positions (1j) furnished the desired products in 72–73% yields. Prolonging the reaction time to 72 h led to the smooth oxidization of 4-OH substituted methylarene (1k) into 4-hydroxybenzoic acid (2k) in 72% yield, leaving the hydroxyl group for post modifications.67 Unfortunately, this strategy was not applicable to the NH2-substituted methylarene (1l), probably due to the deactivation of the catalyst resulting from the coordination of NH2 groups with copper. A control experiment was performed by using propylbenzene as substrates, adding one equiv. of aniline completely quenched the reaction, thus confirming the above assumption (Scheme S1a†). By using the alkylarenes with secondary benzylic C–H bonds as substrates, we further examined this photocatalytic oxidative cleavage of C–C bond (1m–1x). The ethylbenzene derivatives (1m–1o) produced the corresponding acetophenones (3m–3o) as major products and benzoic acid as minor products. Moreover, there was no conversion of acetophenone under photocatalytic conditions, while the oxidative cleavage of other ketone substrates into aromatic acids occurred (Scheme S2†) as acetophenone is more difficult to transform into the enol form than other ketone intermediates.36,68 Remarkably, the strategy can be applied to the phenylethanol derivatives (1p–1t), delivering the corresponding benzoic acids in moderate to high yields (59–81%). The propylbenzene with a substituent on the benzene ring (1u–1x) produced the corresponding benzoic acids in 68–79% yields, except 1x with the 4-OH group only afforded 2k in 30% yield. The low efficiency might result from the inhibition of free radicals by the phenol group.
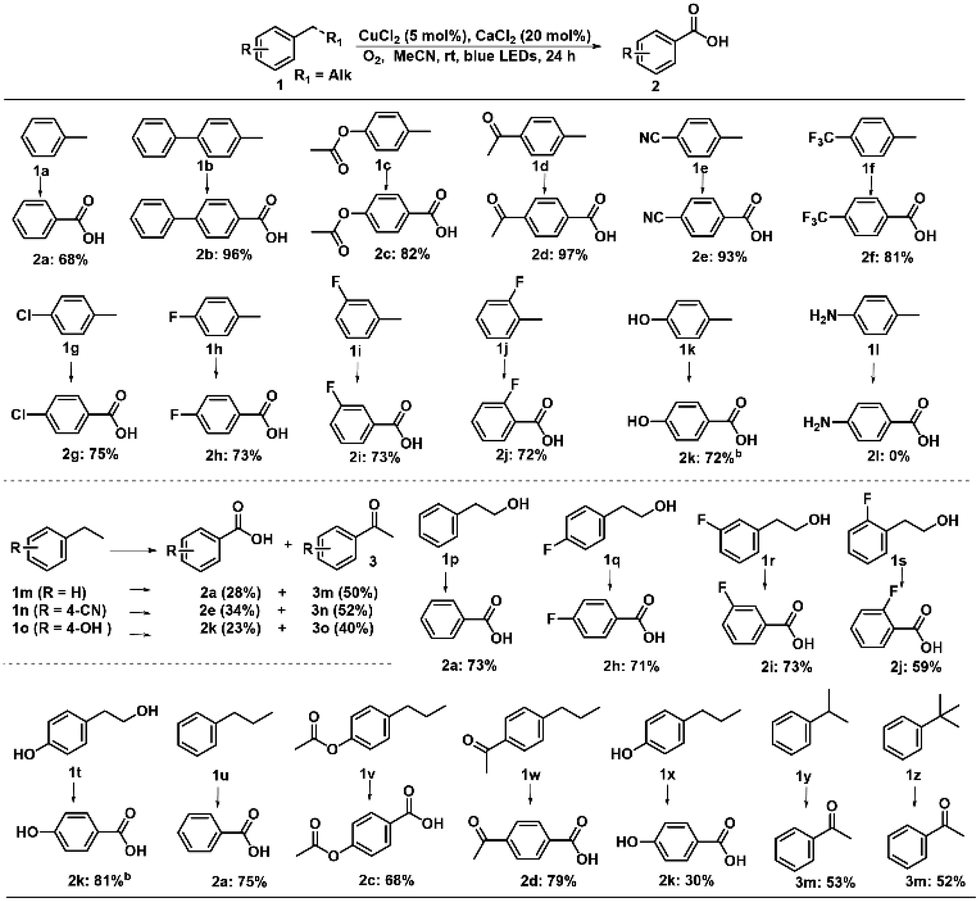 | ||
| Scheme 2 Substrate scope for the photooxidation of alkylarenes. aReaction conditions: substrate (0.1 mmol), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN (1.0 mL), O2 balloon, blue LEDs, rt, 24 h. The yields were measured by HPLC using a standard curve of standards as a reference. b72 h. | ||
To verify this assumption, we conducted a control experiment by using propylbenzene as a substrate and found that one equiv. of phenol led to the conversion of propylbenzene drastically decreasing to 49%, furnishing propiophenone (3u, 10%), 1-phenylpropan-2-one (4u, 5%) and benzaldehyde (7, 10%), rather than benzoic acid (Scheme S1b†). The cumene (1y) and tert-butylbenzene (1z) generated acetophenones in 53% and 52% yields, respectively. All these results clearly demonstrated the good versatility of this photocatalytic strategy towards the oxidative cleavage of C–C bonds.
Encouraged by such highly efficient CuCl2-catalyzed photooxidative transformation of alkylarenes into benzoic acids, we further investigated degradation of commercial PS (Mn = 87 kg mol−1, PDI = 3.29; Fig. S3†). Due to the poor solubility of PS in MeCN, we did not detect any small molecular products (Table S2†). In order to improve the solubility of PS in solvent, we investigated the cosolvent effects and found that in MeCN/benzene, high molecular weight PS could be efficiently degraded to small molecular aromatics (benzoic acid (2a) in 22% yield; 1,3-diphenylpropanetrione (5) in 3% yield; phenyl glyoxylic acid (6) in 6% yield, Scheme 3), the oxidized oligomers (Mn = 0.11 kg mol−1, PDI = 3.81; Fig. S3†) along with unidentified products in 24 h (Fig. S4†), respectively. Small amounts of aromatic products were detected with the cosolvents of acetone, THF, EtOAc or DCM (Table S2†).
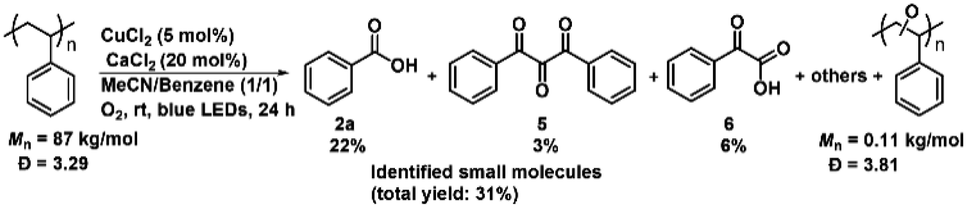 | ||
| Scheme 3 Results for the PS photooxidative degradation by CuCl2. Reaction conditions: commercial PS (0.1 mmol, based on the amounts of styrene monomer), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN/benzene (0.6 mL), O2 balloon, blue LEDs, rt, 24 h. The yields were measured by HPLC using a standard curve of standards as reference. | ||
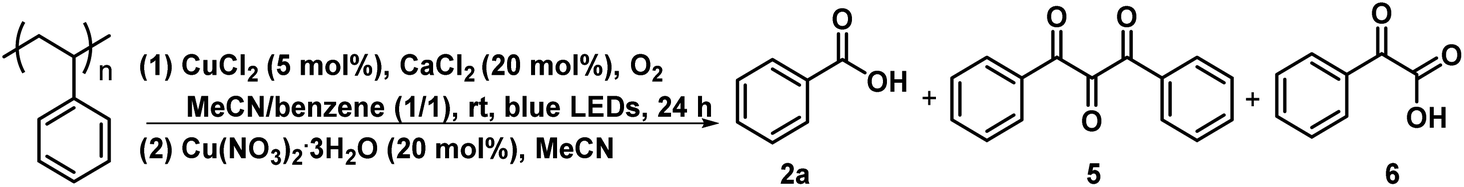
Control experiments showed that in the presence of CuCl2/CaCl2 and under irradiation of blue LEDs, PS could be degraded to 2a in 22% yield (entry 1, Table 2). Similar results were obtained with the prolonged reaction time to 36 h or heating the reaction mixture obtained from the photocatalytic first step at 120 °C for 12 h as the second step (entries 2 and 3, Table 2). The addition of Cu(NO3)2·3H2O after the first step and irradiation during the second step still did not show any improvement, while the high temperature (120 °C)69 instead of irradiation during the second step enhanced the yield of 2a to 37% after 12 h, further up to 42% for 24 h (entries 4–6, Table 2). Replacing CuCl2 with Cu(NO3)2·3H2O in the first step only produced 2a in 1% yield through such a two-step one-pot process (entry 7, Table 2), suggesting the critically important role of CuCl2 in the photocatalytic degradation of PS. Moreover, heating the reaction at 140 °C for 24 h can further increase the yield of 2a to 65%, while the higher temperature (160 °C) led to a yield decrease to 51% (entries 8 and 9, Table 2). After adding Cu(NO3)2·3H2O to CuCl2/CaCl2, the mixture was irradiated for 24 h and then heated at 140 °C for 24 h, only furnishing 2a in <1% yield, even the irradiating the reaction at 140 °C still could not produce 2a (entries 10 and 11, Table 2). These results demonstrated that the presence of Cu(NO3)2·3H2O in the first step would inhibit the reaction. Therefore, the reaction conditions were optimized as the following: CuCl2/CaCl2 as catalysts under irradiation for 24 h in the first step and Cu(NO3)2·3H2O at 140 °C for 24 h in the second step.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | First step | Second step | Yieldb (%) | ||||||
| Cu-catalyst | hν | Time (h) | Catalyst | T (°C) | Time (h) | 2a | 5 | 6 | |
| a Reaction condition: first step: commercial PS (0.1 mmol, based on the mole amount of the styrene monomer), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN/benzene (1/1, 0.6 mL), O2 balloon, blue LEDs, rt. Second step: Cu(NO3)2·3H2O (20 mol%), MeCN (1 mL) was added to the mixture obtained after first step. b The yields were measured by HPLC using standard curve of standards as reference. c Second step, blue LEDs. d Cu(NO3)2·3H2O/CaCl2 in first step. e CuCl2/Cu(NO3)2·3H2O/CaCl2 in first step. f CuCl2/Cu(NO3)2·3H2O/CaCl2 for one-step under blue-LED irradiation at 140 °C. | |||||||||
| 1 | CuCl2/CaCl2 | hν | 24 | — | — | — | 22 | 3 | 6 |
| 2 | CuCl2/CaCl2 | hv | 36 | — | — | — | 23 | 2 | 6 |
| 3 | CuCl2/CaCl2 | hν | 24 | — | 120 | 12 | 22 | 3 | 5 |
| 4c | CuCl2/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | hν | 12 | 22 | 1 | 4 |
| 5 | CuCl2/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | 120 | 12 | 37 | 0 | 3 |
| 6 | CuCl2/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | 120 | 24 | 42 | 0 | 2 |
| 7d | Cu(NO3)2·3H2O/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | 120 | 12 | 1 | 0 | 4 |
| 8 | CuCl2/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | 140 | 24 | 65 | 0 | 0 |
| 9 | CuCl2/CaCl2 | hν | 24 | Cu(NO3)2·3H2O | 160 | 24 | 51 | 0 | 0 |
| 10e | CuCl2/Cu(NO3)2·3H2O/CaCl2 | hν | 24 | — | 140 | 24 | <1 | 0 | 4 |
| 11f | CuCl2/Cu(NO3)2·3H2O/CaCl2 | hν | — | — | 140 | 24 | <1 | 0 | 0 |
Mechanistic studies
 | ||
| Fig. 1 The time-dependent UV-vis absorption spectra obtained for (A) the mixture of CuCl2/CaCl2 and (B) the mixture of CuCl2/CaCl2 with PS. | ||
To verify the generation of alkoxy radicals from the homolysis of Cu(II)-alkylperoxy complexes, we selected the secondary propylbenzene (1u) as a substrate to detect the direct cleavage product of the C–C bond, which is different from the previously reported carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) products via the aerobic oxidation of secondary sp3 carbon (–CH2–).36,44 Therefore, the time-on-course oxidation process of 1u by CuCl2 was monitored by HPLC (Fig. 2). In the first 2 h, benzoic acid (2a), propiophenone (3u), 1-phenylpropan-2-one (4u) and benzaldehyde (7) could be detected. The amounts of 3u, 4u, and 7 then decreased to zero over time, while the yield of 2a gradually increased to be the only product ultimately. It should be noted that benzaldehyde (7) might be formed during the β-scission of alkoxy radicals generated from the homolysis of O–O bond in the peroxy moiety of the Cu(II)-alkylperoxy complex, which is consistent with the observation for the CuII-OH complex in time-dependent UV-vis spectra (Fig. 1B). Besides, 3u and 7 were formed after abstracting the benzylic C–H from propylbenzene, while the formation of 4u confirmed the abstraction of the aliphatic C–H bond.
O) products via the aerobic oxidation of secondary sp3 carbon (–CH2–).36,44 Therefore, the time-on-course oxidation process of 1u by CuCl2 was monitored by HPLC (Fig. 2). In the first 2 h, benzoic acid (2a), propiophenone (3u), 1-phenylpropan-2-one (4u) and benzaldehyde (7) could be detected. The amounts of 3u, 4u, and 7 then decreased to zero over time, while the yield of 2a gradually increased to be the only product ultimately. It should be noted that benzaldehyde (7) might be formed during the β-scission of alkoxy radicals generated from the homolysis of O–O bond in the peroxy moiety of the Cu(II)-alkylperoxy complex, which is consistent with the observation for the CuII-OH complex in time-dependent UV-vis spectra (Fig. 1B). Besides, 3u and 7 were formed after abstracting the benzylic C–H from propylbenzene, while the formation of 4u confirmed the abstraction of the aliphatic C–H bond.
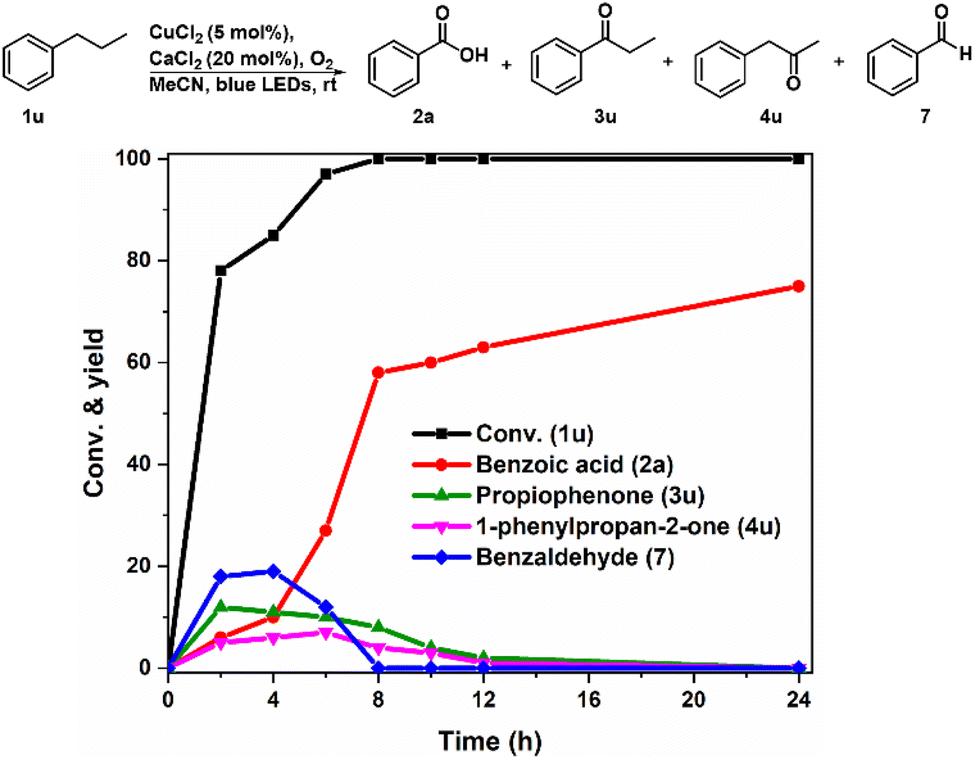 | ||
| Fig. 2 Time-on-course process of propylbenzene (1u) photocatalyzed by CuCl2. | ||
 | ||
| Fig. 3 The time-evolution experiments of CuCl2-catalyzed PS photooxidative degradation (black: Mn of PS; red: yield of 2a; blue: yield of 6; pink: yield of 5; and green: total yield). | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
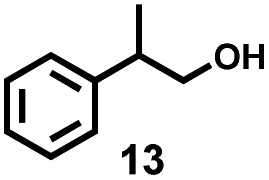
| Entry | Substrate | Conv. | Main products (yield, %) | Entry | Substrate | Conv. | Main products (yield, %) |
| a Reaction conditions: substrate (0.1 mmol), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN (1.0 mL), O2 balloon, blue LEDs, rt, 24 h; conversion and yields were measured by HPLC using a standard curve of standards as reference. b Blue LEDs, 48 h. c Substrate (0.1 mmol), Cu(NO3)2·3H2O (20 mol%), MeCN (1.0 mL), 140 °C, 24 h. | |||||||
| 1 |

|
100% |

|
7 |

|
34% |

|
| 2 |

|
100% |

|
8c |
|
100% |
|
| 3b |
|
100% |

|
9 |
|
100% |

|
| 4 |

|
100% |

|
10 |

|
100% |

|
| 5 |
|
100% |
|
11 |

|
26% |

|
| 6 |

|
100% |

|
12c |
|
100% |

|

 | ||
| Scheme 4 Possible reaction mechanism proposed for the oxidative C–C bond cleavage of 1,3-diphenylpropane (8). | ||
On the basis of the above-described experimental details and previous CuCl2-catalyzed photochemical studies,54 a plausible reaction mechanism was proposed, as depicted in Scheme 5. First, the chloride of CaCl2 was coordinated with the CuIICl2 (A) to form photoactive CuIICl3− (B), which can be confirmed by UV-vis spectroscopy (Fig. S5†). Subsequently, the irradiation of CuIICl3− (B) would undergo the LMCT process to generate a reactive chlorine radical (Cl˙) and [CuICl2−].58–60 Due to the stronger bond dissociation (BDE = 103 kcal mol−1) compared with that of C–H bond, the Cl˙ could abstract a hydrogen atom from PS (C) to generate a carbon-centred radical D along with HCl. Radical D was then captured by O2 to form a peroxy radical intermediate E, which was reduced by CuICl2− to generate the Cl2Cu(II)-alkylperoxy complex (F).71 Followed by the O–O bond homolysis of the peroxy moiety, complex F was transformed into an alkoxy radical G and generate Cl2CuIIO˙.70,72,73 The Cl2CuIIO˙ complex may abstract a hydrogen from PS other than MeCN solvent70 because the bond dissociation energy of the benzylic C–H bond (BDE = −90 kcal mol−1) is lower than that of the Cα–H bond (BDE = 96 kcal mol−1)74 of MeCN, thus transforming C into radical D and form [Cl2CuII-OH] complex. Its combination with HCl would close the photocatalytic cycle, along with the release of water. For the selectivity of abstraction of tertiary benzylic C–H or secondary aliphatic C–H via Cl˙, we propose two plausible reaction pathways for PS degradation (Scheme 5a and b). Upon the abstraction of tertiary benzylic C–H in PS and subsequent oxidation with O2 (Scheme 5a), the alkoxy radical G was generated through the repeated O–O bond homolysis, followed by rapid β-chain cleavage to form an aromatic ketone chain end (H) and a primary alkyl radical chain end (I). For aromatic ketone chain end H, the chlorine radical would abstract a Cβ–H bond adjacent to the ketone to generate a benzylic radical, which was then transformed into the alkoxy radical Jvia a repeated oxidation process.34 The β-chain scission of J produced the aromatic ketone chain end K and α-acyl radical L. The α-acyl radical L was oxidized into an oxygen-centred radical by O2, which subsequently underwent β-scission and oxidation, furnishing benzoic acid ultimately.39 The primary alkyl radical chain end (I) proceeded with the following routes for production of small molecules: First, the chain end I could be oxidized to the primary alcohol chain end (M) or aldehyde chain end (O). The Cα–H on the alcohol of M can be abstracted by a chlorine radical to form intermediate N, which would be rapidly oxidized to aldehyde chain end (O).39 The acyl hydrogen on the aldehyde of O could be abstracted to form an acyl radical chain end (P) through another HAT. P could be decarbonylated to a secondary benzylic radical chain end (Q) or partially oxidized to a carboxylic acid moieties chain end (R). The chain end Q was trapped by O2 to form alkoxy radical chain end (S) through repeated O–O bond homolysis, followed by the formation of a ketone chain end Kvia HAT or benzoic acid via β-chain scission and oxidation processes.
 | ||
| Scheme 5 Possible mechanism proposed for PS degradation through (a) benzylic C–H abstraction or (b) aliphatic C–H abstraction. | ||
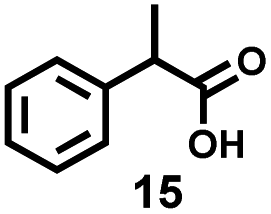
Alternatively, another degradation mechanism occurred through the abstraction of secondary aliphatic C–H bond (Scheme 5b). The reaction involving the secondary C–H abstraction and subsequent oxidation with O2 would generate the secondary alkoxy radical on the PS backbone (G1), followed by rapid β-chain cleavage to form aldehyde chain end (O) and benzylic radical chain end (Q).39 Besides, the secondary alkoxy radical G1 could also undergo HAT process to produce ketones on the PS backbone (T),44 which would subsequently be oxidized to generate the acyl radical chain end (P) and aromatic ketone chain end (H). These chain ends (O, Q, P, and H) would go through similar processes as above to generate benzoic acid and carboxylic acid moieties chain end (R). On the basis of the above-described small molecule study, low conversion of the carboxylic acid moieties chain end (R) occurred during photocatalytic process. The fact that full consumption of 2-phenylpropionic acid (15) and improved product yield of benzoic acid can be obtained by Cu(NO3)2·3H2O under heating conditions (entry 12, Table 3), which also confirmed that the integrated two-step strategy can produce benzoic acid with high efficiency.
With the understanding of the reaction mechanism and optimized reaction conditions at hand, we applied this integrated two-step strategy to a plethora of real-life plastic waste to examine the generality of this strategy (Scheme 6). Initially, the “one step” photocatalytic protocol showed somewhat different effects on the degradation of synthetic PS with different molecular weights, furnishing benzoic acid in 20–22% yields. It is known that polymer additives are typically mixed into real-life PS plastics during their manufacturing. We are very glad to see that this photocatalytic method displayed excellent compatibility with a wide variety of real-life PS waste ranging from food boxes, coffee lids, forks, bowls, cups, spoons, and foam to laboratory weighing boats and Petri dishes, delivering benzoic acid in 15–24% yields. Furthermore, the integrated two-step strategy was adopted to deconstruct PS waste as shown in the “two step”. Gratifyingly, the two-step method works well for the degradation of all PS materials, drastically increasing the yield of benzoic acid to 46–65%. There are many additives in commercial polystyrenes, so we selected several widely used polystyrene additives to further investigate the tolerance of our system, such as antioxidants (e.g. 2,6-di-tert-butyl-p-cresol (BHT) and propyl gallate), ultraviolet absorbers (e.g. phenyl salicylate and 2-(2-hydroxy-5-methylphenyl)benzotriazole) and 2,4-dihydroxybenzophenone, foaming agents e.g. azodicarbonamide and 2,2-azobis(2-methylpropionitrile) and diethyl azodicarboxylate. It turned out that the employment of 0.5 wt% additives did not affect the 2a yields obtained by our one step photocatalytic protocol and the two-step method, while increasing to 5 wt% additives led to a slight decrease in the 2a yields, except that 2-(2-hydroxy-5-methylphenyl)benzotriazole showed drastically decreased reactivity via a two-step method (Tables S3 and S4†). These results clearly demonstrated the universality and tolerance of the oxidative strategy for recycling PS waste. We also performed a gram-scale degradation of PS waste and obtained benzoic acid in 50% yield (0.57 g after acid–base wash and extractions without further purification), demonstrating the practicality and scalability of this integrated two-step strategy in PS degradation (Scheme 7).
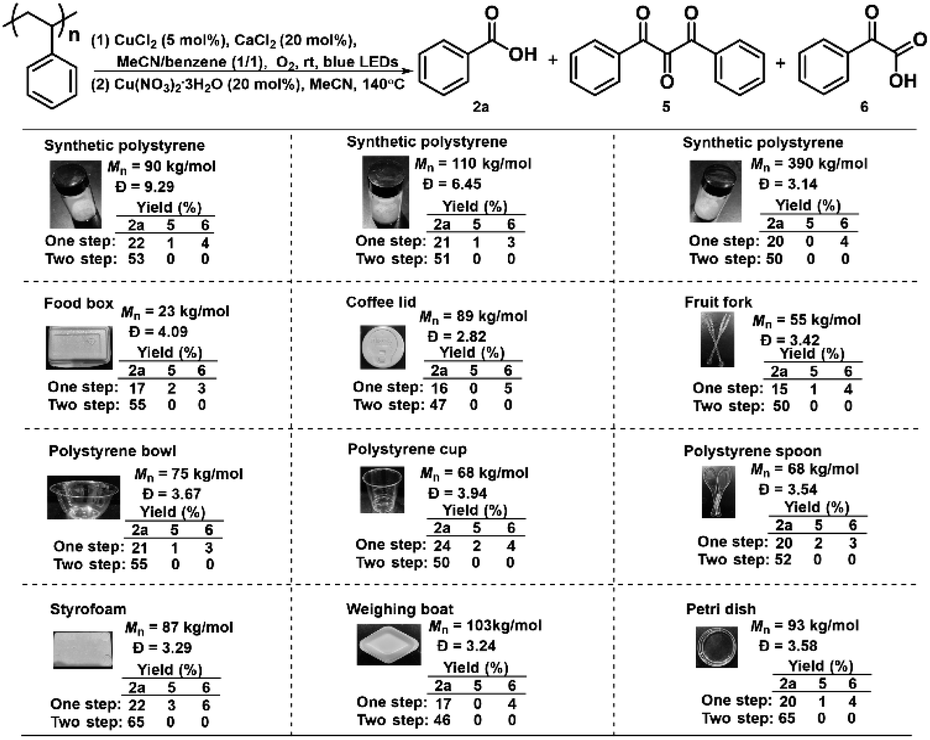 | ||
| Scheme 6 The degradation of real-life plastic waste. Reaction condition: first step: commercial PS (0.1 mmol, based on the mole amount of the styrene monomer), CuCl2 (5 mol%), CaCl2 (20 mol%), MeCN/Benzene (0.6 mL), O2 balloon, blue LED (50 W), rt, 24 h. Second step: Cu(NO3)2·3H2O (20 mol%), MeCN (1 mL) was added to the mixture obtained after first step, 140 °C, 24 h. The average values were obtained from the HPLC yields of two parallel PS degradation experiments. | ||
 | ||
| Scheme 7 The gram-scale PS degradation. | ||
Conclusions
In summary, we developed a highly efficient photocatalytic strategy for the oxidative cleavage of alkylarenes and degradation of real-life PS waste into benzoic acid and oxidized styrene oligomers, by using commercially available CuCl2 and CaCl2 as catalysts in O2 atmosphere at room temperature. The addition of Cu(NO3)2·3H2O and heating the reaction can further convert the oxidized styrene oligomers to benzoic acid, thus achieving up to 65% total yield of benzoic acid. The practicality and scalability of such a two-step protocol can be verified by the degradation of real-life PS foam on the gram scale, thus showing the great application potentials of this strategy in the recycling of plastic waste into valuable chemicals in the future.Data availability
The data supporting the findings of this article have been uploaded as the ESI,† and include full experimental details, NMR and HPLC spectra, and GPC traces.Author contributions
E. X. and Y. Z. conceived and designed the experiments. E. X. performed the experiments and characterizations. E. X., T. L., F. X., J. H., and Y. Z. analyzed the data and wrote the manuscript with input from all other authors. Y. Z. directed the projects.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22225104, 22071077, 21975102, 22471092).Notes and references
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Joule, 2021, 5, 673–686 CrossRef CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- H. Ran, S. Zhang, W. Ni and Y. Jing, Chem. Sci., 2024, 15, 795–831 RSC.
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522 CrossRef CAS PubMed.
- M. Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912–2923 CAS.
- Z. Ouyang, Y. Yang, C. Zhang, S. Zhu, L. Qin, W. Wang, D. He, Y. Zhou, H. Luo and F. Qin, J. Mater. Chem. A, 2021, 9, 13402–13441 RSC.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- D. Kwon, Nature, 2023, 616, 234 CrossRef CAS PubMed.
- L. Gan, Z. W. Dong, H. F. Xu, H. D. Lv, G. X. Liu, F. Zhang and Z. Huang, CCS Chem., 2024, 6, 313 CrossRef CAS.
- B. Zhao, H. Tan, J. Yang, X. Zhang, Z. Yu, H. Sun, J. Wei, X. Zhao, Y. Zhang, L. Chen, D. Yang, J. Deng, Y. Fu, Z. Huang and N. Jiao, The Innovation, 2024, 5, 100586 CrossRef CAS PubMed.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef PubMed.
- C. Marquez, C. Martin, N. Linares and D. De Vos, Mater. Horiz., 2023, 10, 1625–1640 RSC.
- J. C. Capricho, K. Prasad, N. Hameed, M. Nikzad and N. Salim, Polymers, 2022, 14, 5010 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A. C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- B. Singh and N. Sharma, Polym. Degrad. Stab., 2008, 93, 561–584 CrossRef CAS.
- W. Partenheimer, Catal. Today, 2003, 81, 117–135 CrossRef CAS.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- C. Rabot, Y. Chen, S. Y. Lin, B. Miller, Y. M. Chiang, C. E. Oakley, B. R. Oakley, C. C. C. Wang and T. J. Williams, J. Am. Chem. Soc., 2023, 145, 5222–5230 CrossRef CAS PubMed.
- X. Luo, J. Zhan, Q. Mei and S. Zhang, Green Chem., 2023, 25, 6717–6727 RSC.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed.
- C. Qin, T. Shen, C. H. Tang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 6971–6975 CrossRef CAS PubMed.
- N. E. Munyaneza, C. Posada, Z. Xu, V. De Altin Popiolek, G. Paddock, C. McKee and G. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307042 CrossRef CAS PubMed.
- J. Kou, C. Lu, J. Wang, Y. Chen, Z. Xu and R. S. Varma, Chem. Rev., 2017, 117, 1445–1514 CrossRef CAS PubMed.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- F. Eisenreich, Angew. Chem., Int. Ed., 2023, 62, e202301303 CrossRef CAS PubMed.
- Y. Zhang, M. Y. Qi, Z. R. Tang and Y. J. Xu, ACS Catal., 2023, 13, 3575–3590 CrossRef CAS.
- E. Skolia, O. G. Mountanea and C. G. Kokotos, Trends Chem., 2023, 5, 116–120 CrossRef CAS.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- J. Shang, M. Chai and Y. Zhu, Environ. Sci. Technol., 2003, 37, 4494–4499 CrossRef CAS PubMed.
- M. Wang, J. Wen, Y. Huang and P. Hu, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS PubMed.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, ACS Catal., 2023, 13, 10968–10975 CrossRef CAS.
- A. Ong, Z. C. Wong, K. L. O. Chin, W. W. Loh, M. H. Chua, S. J. Ang and J. Y. C. Lim, Chem. Sci., 2024, 15, 1061–1067 RSC.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- S. Xu, S. Liu, W. Song and N. Zheng, Green Chem., 2024, 26, 1363–1369 RSC.
- R. Cao, M. Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- N. F. Nikitas, E. Skolia, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Green Chem., 2023, 25, 4750–4759 RSC.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Chem, 2022, 8, 2472–2484 CAS.
- C. Li, X. Y. Kong, M. Lyu, X. T. Tay, M. Đokić, K. F. Chin, C. T. Yang, E. K. X. Lee, J. Zhang, C. Y. Tham, W. X. Chan, W. J. Lee, T. T. Lim, A. Goto, M. B. Sullivan and H. S. Soo, Chem, 2023, 9, 2683–2700 CAS.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, 450 CrossRef PubMed.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed.
- Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850–15858 CrossRef CAS PubMed.
- V. P. Charpe, A. Sagadevan and K. C. Hwang, Green Chem., 2020, 22, 4426–4432 RSC.
- A. Hossain, A. Vidyasagar, C. Eichinger, C. Lankes, J. Phan, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2018, 57, 8288–8292 CrossRef CAS PubMed.
- A. S. Mereshchenko, P. K. Olshin, A. M. Karimov, M. Y. Skripkin, K. A. Burkov, Y. S. Tveryanovich and A. N. Tarnovsky, Chem. Phys. Lett., 2014, 615, 105–110 CrossRef CAS.
- C. Meng, K. Yang, X. Fu and R. Yuan, ACS Catal., 2015, 5, 3760–3766 CrossRef CAS.
- K. Takaki, J. Yamamoto, K. Komeyama, T. Kawabata and K. Takehira, Bull. Chem. Soc. Jpn., 2004, 77, 2251–2255 CrossRef CAS.
- K. Takaki, J. Yamamoto, Y. Matsushita, H. Morii, T. Shishido and K. Takehira, Bull. Chem. Soc. Jpn., 2003, 76, 393–398 CrossRef CAS.
- J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121–2127 CrossRef CAS.
- P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Angew. Chem., Int. Ed., 2020, 59, 23603–23608 CrossRef CAS PubMed.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1594 CrossRef CAS PubMed.
- X. P. Zhang, A. Chandra, Y. M. Lee, R. Cao, K. Ray and W. Nam, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- C. García-Sancho, I. Fúnez-Núñez, R. Moreno-Tost, J. Santamaría-González, E. Pérez-Inestrosa, J. L. G. Fierro and P. Maireles-Torres, Appl. Catal., B, 2017, 206, 617–625 CrossRef.
- C. Lin, H. Wu, V. Wang, J. Huang, F. Cao, W. Zhuang, Y. Lu, J. Chen, H. Jia and P. Ouyang, Ind. Eng. Chem. Res., 2020, 59, 4358–4366 CrossRef CAS.
- C. Lin, C. Chai, Y. Li, J. Chen, Y. Lu, H. Wu, L. Zhao, F. Cao, K. Chen, P. Wei and P. Ouyang, Green Chem., 2021, 23, 2058–2068 RSC.
- P. G. David and P. A. C. da Silva, Bull. Chem. Soc. Jpn., 1985, 58, 3566–3569 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- L. Lopez, L. Troisi, S. M. K. Rashid and A. P. Schaap, Tetrahedron Lett., 1989, 30, 485–488 CrossRef CAS.
- H. Liu, M. Wang, H. Li, N. Luo, S. Xu and F. Wang, J. Catal., 2017, 346, 170–179 CrossRef CAS.
- T. Tano, M. Z. Ertem, S. Yamaguchi, A. Kunishita, H. Sugimoto, N. Fujieda, T. Ogura, C. J. Cramer and S. Itoh, Dalton Trans., 2011, 40, 10326–10336 RSC.
- H. R. Lucas, L. Li, A. A. N. Sarjeant, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 3230–3245 CrossRef CAS PubMed.
- A. Kunishita, H. Ishimaru, S. Nakashima, T. Ogura and S. Itoh, J. Am. Chem. Soc., 2008, 130, 4244–4245 CrossRef CAS PubMed.
- B. Kim, D. Jeong and J. Cho, Chem. Commun., 2017, 53, 9328–9331 RSC.
- L. Tanwar, J. Börgel and T. Ritter, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03269a |
| This journal is © The Royal Society of Chemistry 2025 |