
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive activity–stability correlation study of tantalum-doped tin oxide as a support for iridium oxide in low loading water electrolysis cell anodes†
Ignacio
Jiménez-Morales‡
*,
Jacques
Rozière
,
Deborah
Jones
and
Sara
Cavaliere
 *
*
ICGM, University of Montpellier, CNRS, ENSCM, 34095 Montpellier Cedex 5, France. E-mail: ijimenezm@usal.es; sara.cavaliere@umontpellier.fr
First published on 7th April 2025
Abstract
A systematic study on the impact of the treatment temperature of IrOx supported onto doped-tin oxide (1 at% Ta–SnO2 and 10 at% Sb–SnO2) fibres led to electrocatalysts with high oxygen evolution reaction activity and resistance to degradation. The electrolytic performance was comparable to that of unsupported commercial IrO2 with seven times higher loading.
1. Introduction
Sustainable and clean energy sources are needed to avoid the use of fossil fuels and limit environment pollution and global warming.1 Solar and wind renewable energy sources suffer from intermittency and uneven distribution; therefore, they require the development of efficient energy storage and conversion technologies. Hydrogen is a clean energy carrier enabling the storage of electrical energy with high specific weight energy density, and its further conversion via fuel cells occurs without carbon emissions. Green hydrogen can be produced by proton exchange membrane water electrolysis (PEMWE) coupled with renewable sources, which leads to high purity, efficiency, production rate, safety as well as rapid control response capabilities.2,3 PEMWE can operate at high current densities and moderate temperatures reducing the operational costs, and the polymer electrolyte membrane used has low gas crossover, allowing operation under a wide range of power inputs. In addition, this process uses smaller mass-volume characteristics, no corrosive electrolyte is involved and low maintenance is needed.2–4 In contrast, one of the foremost issues in PEMWE is the scarcity and high cost of the constituting electrode materials. In particular, unsupported platinum group metals (PGMs) and/or oxides of iridium or ruthenium with a high loading due to low catalyst utilisation are used to electrocatalyse the anodic oxygen evolution reaction (OER).5 IrO2 is the most used OER catalyst due to its high corrosion resistance, and it shows only a slightly lower activity than RuO2, which is less stable at high cell voltages.5,6 The use of binary IrO2–RuO2 solid solutions can partially mitigate the instability of RuO.7–9 Their activity can be enhanced by producing nanostructured or alloyed metal oxides employing different synthetic routes.10–14 A promising approach to reduce the PGM loading at the anode is the utilisation of support materials to uniformly disperse the electrocatalyst and increase its efficiency.15–18 Furthermore, the support can promote activity and stability, providing corrosion resistance in the harsh PEMWE conditions. For this reason, mainly metal oxides such as TiO2, Ta2O5 (ref. 19 and 20) and SnO2 are used21–25 that can also interact with the electrocatalyst through electron transfer, affecting its activity. Catalyst–support interactions26–30 modify the catalyst activity through alteration in the electron density around the active sites, as identified by XPS, XRD or XAS analyses. Tin oxide has been extensively used as an electrocatalyst support in the oxygen reduction reaction (ORR) and OER due to its high resistance to corrosion. To enhance its intrinsically low electrical conductivity, aliovalent ions such as niobium, antimony and tantalum ions were introduced in its structure in atomic percent (n doping).24,28,31–36 Its chemical structure, size, morphology and textural properties can be tuned depending on the synthetic route and the doping agent used.The crystallinity of the IrO2 electrocatalyst plays a crucial role in enhancing the electrochemical stability while maintaining the OER activity. Its degree of crystallinity can be modified by thermal treatments.37–40 Thermally treated supported IrO2 nanoparticles demonstrate high mass activity retention after a potentiostatic stability test.41 However, a decrease in the OER activity is expected due to the drop in intrinsic OER activity for Ir(IV) species formed upon thermal treatment over Ir(III) species.12,42 A trade-off needs to be found for iridium oxide, allowing sufficient crystallinity and stabilisation and electroactivity.
This work aims at tackling the main challenge of PEMWE anodes of keeping high activity and stability with minimum PGM loading and reports a low-loaded anode of homogenously dispersed IrO2 nanoparticles supported on doped tin oxide nanofibres. Antimony and tantalum doped tin oxide (ATO and TTO, respectively) fibres are promising supports for electrodes experiencing high potentials (anodes in water electrolysis and cathodes in fuel cell) with high resistance to corrosion combined with decent electronic conductivity (maxima for 10 at% Sb and 1 at% Ta) and surface area, and high electronic interaction with the catalyst.24,27,28,33 In particular, 10 at% ATO presents a conductivity similar to that of carbon (1 S cm−1), but suffers from Sb leaching at potentials higher than 1.9 V/RHE.24 1 at% TTO, with a ten-time lower conductivity was still adapted for the application, evidenced by the lack of leaching of the doping agent up to 2.5 V, even though it has a slightly lower surface area than ATO (27 vs. 35 m2 g−1).28 The objective of this work is to compare their use as supports in OER in the rotating disk electrode (RDE) and a single electrolysis cell and their interaction with the iridium-based catalyst as a function of the treatment temperature and the crystallisation of the latter.
The results of the physico-chemical and electrochemical characterisation in three-electrode configuration and the PEMWE single cell are used to rationalise the enhanced electrochemical performance and resistance to degradation compared to unsupported commercial IrO2. Furthermore, the role of catalyst crystallinity and of the tin oxide support on the electrocatalytic properties is discussed.
2. Results and discussion
Tantalum and antimony doped tin oxides (TTO and ATO) prepared by single-needle electrospinning and subsequent calcination27,28,33,34 were used as electrocatalyst supports for iridium oxide (IrOx) nanoparticles synthesised by a microwave-assisted polyol method.24 The corresponding TEM micrographs are depicted in Fig. 1 (more micrographs and the corresponding diameter distribution histograms are displayed in Fig. S1†). Unsupported IrOx nanoparticles were also prepared using the same procedure for comparative purposes (Fig. S1l†). | ||
| Fig. 1 TEM micrographs of 16/TTO at different magnifications (b is the magnified area in red in a). | ||
IrOx nanoparticles deposited on TTO nanofibres at several loadings ranging from 5 to 25 wt% (Fig. S1†) show a progressively complete nanofibre coverage, resulting in agglomerate-free homogeneous distribution. The loading on ATO was set at 23 wt% according to a previous optimisation.28,33 The surface saturation of TTO seemed to be reached at lower loading than that for 23/ATO, which may be related to its lower surface area (–23% compared to ATO) as reported elsewhere.28,33 Comparable narrow size distributions of IrOx nanoparticles, with an average diameter of 1.2 nm, homogeneously dispersed onto the different supports were observed in each case (Fig. S1†). The iridium loading for supported materials was determined by XRF and SEM-EDX analysis (Table 1), and in agreement with the nominal value. The O/Ir atomic ratio for unsupported IrOx nanoparticles determined by XRF and EDX was 2.3 and 2.4, respectively, indicating that a proportion of oxygen species (OH and/or H2O groups) were higher than expected from stoichiometry, which will be further discussed after TG-QMS-DSC and XPS analyses. For this reason, the as-prepared, non-thermally treated catalysts are referred to as IrOx in the manuscript.
| Electrocatalyst | IrOx wt% | ||
|---|---|---|---|
| Nominal | XRF | SEM-EDX | |
| 6/TTO | 5 | 5.7 ± 0.2 | 5.9 ± 0.2 |
| 12/TTO | 10 | 11.9 ± 0.3 | 12.2 ± 0.4 |
| 16/TTO | 15 | 16.1 ± 0.4 | 16.3 ± 0.7 |
| 19/TTO | 20 | 19.0 ± 0.6 | 19.3 ± 0.9 |
| 24/TTO | 25 | 24.2 ± 0.7 | 24.0 ± 0.9 |
| 23/ATO | 25 | 22.8 ± 0.8 | 23.2 ± 1.0 |
XRD patterns analysis of all the catalysed materials as well as on pristine doped-tin oxide supports and unsupported IrOx nanoparticles are displayed in Fig. 2. IrOx nanoparticles appear amorphous, and only diffraction peaks corresponding to SnO2 rutile structure (JCPDS 41-1445) are observed after their deposition on both electrocatalyst supports. Meanwhile, no diffraction peaks corresponding to iridium oxide were detected. Similar results have been previously reported,11,35,43 indicating that the catalyst phase is formed by amorphous clusters of hydrated IrOx and/or consists of very small crystals lacking sufficient long-range ordering to be detected by XRD, which is in agreement with the average nanoparticle size obtained by TEM analysis (ca. 1.2 nm). As the amount of amorphous IrOx deposited on SnO2 increased, the intensity of the peaks of SnO2 decreased (Fig. 2 and S2†).
 | ||
| Fig. 2 XRD diffraction patterns for doped tin oxide supports and with different IrOx loadings (6–24 for TTO (a) and 23 wt% ATO (b)) as well as unsupported IrOx. | ||
After the synthesis, thermal treatments in the range 300–550 °C were carried out on the prepared electrocatalysts to increase the crystallinity of iridium oxide and assess the evolution of its OER activity and resistance to degradation.
Thermogravimetry quadrupole mass spectrometry differential scanning calorimetry (TG-QMS-DSC), X-ray thermodiffractometry (TDX) and TEM analysis were carried out on supported and unsupported IrOx nanoparticles (16/TTO, 24/TTO and 23/ATO) to investigate the effect of post-synthesis thermal treatments on the thermal stability, crystallinity and size of the nanocatalysts.
The results obtained by TG-QMS-DSC are reproduced in Fig. S3.† In the TG curve of unsupported IrOx nanoparticles, a continuous weight loss (5.6 wt%) is observed up to 193 °C related to the initial H2O loss, and there is a weight loss of 11.4 wt% up to 600 °C (Fig. S3b†), which is attributed to decomposition of OH groups on the IrOx surface. In agreement with this conclusion, mass spectrometry analysis was carried out simultaneously with TG-DSC analysis (Fig. S3a†), revealing a constant removal of water and OH groups (m = 17 and 18 a.u.),44 the corresponding signals reaching their maximum at 185 and 300 °C. These results are consistent with DSC curves displayed in Fig. S3b,† indicating three exothermic peaks at 193, 297 and 460 °C assigned to the removal of physisorbed water, crystallisation of IrOx nanoparticles44,45 and oxidation of Ir(III) to Ir(IV).46 This might suggest that unsupported IrOx nanoparticles prepared by the microwave-assisted polyol method exhibit an elevated percentage of hydroxyl groups and can be considered as an Ir–oxyhydroxide, IrOx(OH)y. This could explain the high amount of oxygen detected by XRF compared with the expected IrO2. Hydrous IrOx, highly covered by electrophilic oxygen ligands, and covalently linked to the metal possess high OER catalytic activity.47,48
Comparable TG curves were obtained for the catalysed materials although they exhibited lower mass losses (Fig. S3c and d†), despite performing the analysis with similar IrOx mass as with unsupported material. This may be attributed to the high dispersion and improved interaction of IrOx nanoparticles on the oxide supports. Higher mass loss was observed for the catalyst supported on ATO than on TTO, in agreement with its higher catalyst loading. From the DSC curves of the catalysed materials, only two exothermic peaks were observed, the former at slightly lower temperature (180 °C) than for unsupported IrOx, and the latter around 297–310 °C, attributed to the phase transition from amorphous to crystalline iridium oxide, referred as IrO2.
The crystallisation of IrOx to IrO2 with temperature was monitored by X-ray thermodiffractometry (Fig. 3). Peaks of the rutile structure of IrO2 appeared by increasing the treatment temperature, confirming the presence of crystalline particles. The analysis of supported and unsupported IrOx nanoparticles demonstrated that no new diffraction peaks were observed at temperatures lower than 300 °C, unless those corresponding to SnO2 rutile structure.
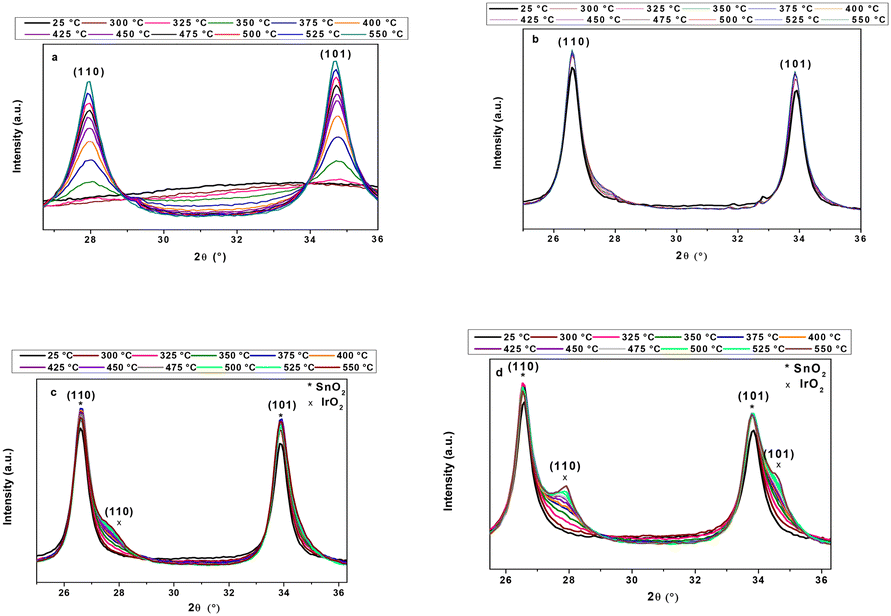 | ||
| Fig. 3 X-ray thermodiffraction patterns of a) unsupported IrOx nanoparticles, b) 16/TTO, c) 24/TTO and d) 23/ATO. | ||
Starting at 300 °C, diffraction peaks at 28° and 34.7° appeared corresponding to iridium oxide (JCPDS 15-0870). Such peaks became progressively more defined as the treatment temperature increased, which may be related to the progressive crystallisation of IrOx nanoparticles, as well as to their coalescence and growth. It is interesting to notice the effect of the support on the crystallisation and growth of IrO2 particles. A reduced IrO2 peak formation is detected for TTO compared to ATO (Fig. 3c and d). At the same conditions of catalyst loading and treatment temperature, the crystallisation of IrOx is favoured on ATO, suggesting a different catalyst/support interaction. The average crystallite sizes at 350 °C (when IrO2 peaks are more prominent, allowing peak deconvolution) were 5.8 and 4.2 nm (Scherrer equation applied to the (110) peak) for unsupported and supported (24/TTO and 23/ATO) IrO2, respectively. At lower loading (16 wt%), the diffraction peaks of iridium oxide are barely observed in TTO-supported IrO2 due to the low size and high dispersion of the nanoparticles. Raman spectroscopy was performed to assess top surface crystallisation. Comparison of the Raman spectra of 16/TTO/300 and TTO reveals the appearance of two lines at 542 and 713 cm−1 (Fig. S6†), confirming the presence of crystallised iridium oxide at the support surface. The former line corresponds to the Eg mode, while the latter is attributed to the overlapping vibrational modes B2g and A1g. Regarding major vibration modes Eg and B2g for IrO2 at 561 and 728 cm−1, respectively,49–51 these bands are red-shifted and assigned to the Ir–O stretching vibrations coupled with O–H bending vibrations in the Ir–(OH) structure,51,52 in agreement with the coexistence of amorphous–crystalline IrO2. All these results correlate with the DSC analysis and lead us to conclude that a post-synthesis treatment at around 300 °C is needed for the transition from amorphous to crystalline structure, which is already demonstrated to be more stable to degradation.53–55 However, a loss of electrochemical activity is expected due to the reduced amount of Ir(III) and iridium hydroxide groups.55–57
The evolution of supported and unsupported IrOx nanoparticles upon thermal treatment was also monitored by TEM and electron diffraction analyses (Fig. S4 and S5†). Although as-synthesised unsupported IrOx nanoparticles exhibited no diffraction pattern after treatment at 300 °C (Fig. S4b†), the SAED displayed an initial transition to a crystalline material, which was finalised at 325 °C (Fig. S4d and f†), in agreement with thermal and structural analysis results. In the absence of support, no isolated nanoparticles were detected by HRTEM; instead there is a self-organised array (Fig. S4a†), which might be related to nanoparticle self-interactions. Such particles sinter and increase in size after thermal treatment at 300 °C. There is an initial coalescence with an average nanoparticle size of 2.4 nm (Fig. S4c†), which increases at 325 °C, leading to the formation of agglomerates (Fig. S4e†).
Significant changes in size and distribution of supported iridium oxide nanoparticles after treatment at 300 °C were detected by TEM (Fig. S5†). The nanoparticles supported on ATO nanofibres exhibited an increase in size to ca. 2.0 nm (Fig. S5a and b†). The lower average nanoparticle size compared to unsupported catalysts suggests that their interaction with the support stabilise their growth, avoiding agglomeration and favouring homogeneous dispersion. 16 wt% IrOx loaded TTO treated at 300 °C exhibited homogeneous nanocatalyst dispersion (Fig. S5c†), while at higher loadings, for instance 24/TTO/300, only IrO2 nanoparticles agglomerates were observed (Fig. S5e†). This might indicate that nanofibre over-coverage was reached. For the same catalyst loading, the ATO based catalyst did not present over-coverage, which may be related to the slightly higher developed surface area of this support. These results suggest that a low catalyst loading is desired to achieve high dispersion without agglomeration on tantalum doped SnO2 fibres. The optimal IrO2 loading on the TTO nanofibres support (16 wt% sample or 16/TTO) was further investigated by electrochemical methods.
X-ray photoelectron spectroscopy (XPS) was carried out to investigate the surface chemical state of unsupported IrOx nanoparticles and of IrOx/TTO with several catalyst loadings ≥16 wt% compared with 23/ATO and the bare support (Fig. 4). High resolution spectra were recorded for Ir, O and Sn. Owing to the small size of the IrOx particles, it was assumed that both bulk and surface species contribute to the XPS signals. Ir 4f core level spectra for all catalysed materials and unsupported IrOx nanoparticles presented the conventional asymmetrical shape,58 indicating two oxidation states related to Ir(IV) (A and A′ corresponding to Ir 4f7/2 (61.9 eV) and Ir 4f7/2 satellite (63.1 eV), respectively) and Ir(III) (B and B′ corresponding to Ir 4f7/2 (62.4 eV) and Ir 4f7/2 satellite (63.5 eV), respectively) (Fig. 4a).10,30,44,59 A Ir(III)/Ir(III) + Ir(IV) proportion of 26.7% was found for the IrOx particles synthesised by the microwave-assisted polyol method, in agreement with the amorphous nature of the catalyst.60 The XPS core level spectra of Ir 4f for all supported-IrOx electrocatalysts exhibits a negative shift compared to unsupported IrOx nanoparticles (62.1 eV) (Fig. 4b), suggesting an increase of highly oxidised iridium. This is in contrast to previous observations of lower average Ir and Pt oxidation states in the oxide-supported IrOx nanoparticles compared to the carbon-supported ones.27,30 This peak shift is more evident for lower catalyst loadings onto TTO (exacerbating IrOx/TTO interactions vs. IrOx/IrOx interactions). The XPS high-resolution spectra of the O 1s and Sn 3d regions for bare and catalysed TTO are reported in Fig. 4c and d, respectively. Core level spectra of Sn 3d shows a doublet at 486.9 and 495.3 eV, attributed to Sn(IV) considering the peak position and their gap. Sn(II) may also be present but it is difficult to analyse by XPS.61,62 For O 1s core level spectra, a broad peak ranging from 528 to 535 eV can be deconvoluted in two contributions. The peak at lower binding energy (band A′) was assigned to O2− in an oxide lattice structure and that at higher binding energy (band B′) to H2O, OH or CO species adsorbed on the surface, in agreement with the presence of a hydrous IrOx phase. No third contribution at 533 eV (band C) was detected for catalysed materials in contrast with unsupported IrOx nanoparticles due to higher proportion of electrocatalyst support. As for Ir 4f peaks, a shift of Sn 3d and O 1s peaks is observed compared to bare supports. These results suggest an electronic coupling effect corresponding to interfacial electron transfer between the support and the iridium oxyhydroxide centres of the catalyst nanoparticles.27,28,30,63,64 The catalyst with the mower IrOx loading on TTO shows a higher downshift for the Ir 4f band binding energy (Fig. 4b), suggesting a stronger interaction and charge transfer with the support. For 16/TTO, the shift was of 0.3 eV. For 24% IrOx loading, this effect disappeared, and the binding energy was almost the same as that of unsupported nanoparticles, suggesting that nanofiber over-coverage leads to prevailing interparticle interactions over nanoparticle/support ones. After thermal treatment, 16/TTO/300 exhibited a lower shift to higher binding energy (0.1 eV), which suggests a reduced electronic coupling effect compared to the as-synthesised material. For IrOx deposited onto ATO a slight shift of the Ir 4f peak (61.8 eV) was observed compared to TTO. The shift in binding energy for Sn 3d and O 1s core level spectra compared to the bare support (Fig. 4c and d) also depends on the amount of catalyst deposited on TTO. The shift is positive for the catalysed ATO, and negative in the case of TTO. As already observed for the IrOx crystallisation (TDX analysis), this result demonstrates the role of the nature of the doping agent for the support of the interaction with the electrocatalyst and its overall properties. Treatment of 16/TTO at 300 °C shows a lower downshift in the Sn 3d and O 1s core level spectra compared to the non-treated one (0.2 eV and 0.3 eV, respectively), suggesting lower interactions and a smaller proportion of the hydrous IrOx phase, in agreement with XRD and TG-QMS-DSC analysis.
 | ||
| Fig. 4 Deconvoluted XPS core level spectra of Ir 4f for the unsupported IrOx nanoparticles (a) and XPS core level spectra of Ir 4f (b), O 1s (c) and Sn 3d (d) for catalysed materials after thermal treatment in comparison with unsupported IrOx and the bare supports. | ||
The OER activity of the catalysed supports before and after the thermal treatment at 300 °C was determined in N2-saturated 0.5 M H2SO4 at 20 °C using a rotating speed of 1600 rpm. OER polarisation curves normalised to iridium mass are reproduced in Fig. 5 after iR correction and the extracted electroactivity data listed in Table 2. OER polarisation curves normalised to the geometric disk area before and after iR correction are presented for comparison in Fig. S7a and b.† The corresponding Tafel plots are presented in Fig. S8.† As prepared 16/TTO and 23/ATO exhibited the highest OER activities (137 and 144 A gIr−1 at 1.5 V/RHE, respectively). Despite the lower electrical conductivity of TTO and the lower IrOx loading on this support, the electrocatalytic activity of 16/TTO is the same as that of 23/ATO, which may be ascribed to the synergy between the catalyst and the support (similar XPS peak shifts observed for those samples). Samples with IrOx loading lower than 16 wt% show a reduced OER activity, probably due to a more pronounced resistive effect of the support. On the other hand, lower performance was observed with IrOx loading higher than 16 wt% on TTO, which can be ascribed to the overcoverage of the support, leading to a reduced catalyst–support interaction effect as observed by XPS.57 Analysis of the electrochemical surface area (ECSA) of the catalysed materials58 indicate higher ECSA values for 16/TTO and 23/ATO (Table S1†). These results confirm enhanced catalyst utilisation and align with the higher electrocatalytic activity observed for these electrocatalysts.
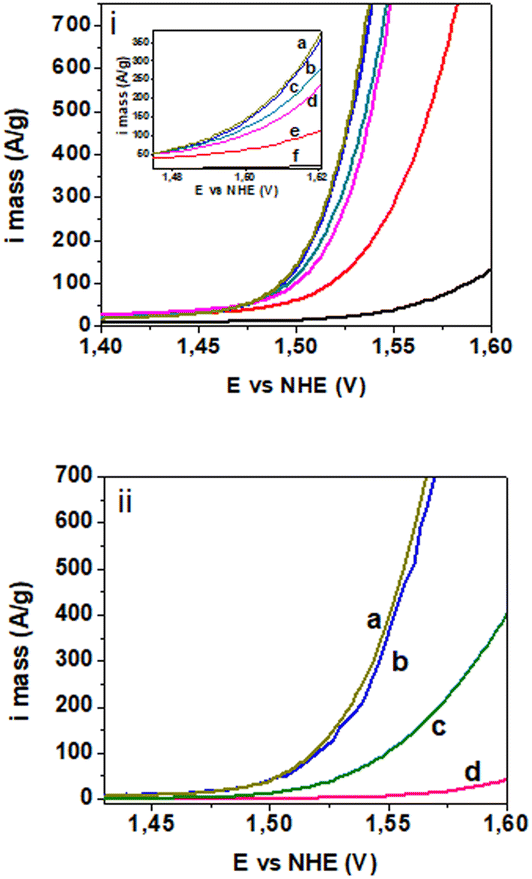 | ||
| Fig. 5 OER polarisation curves of Ir-normalised mass current density for i) non-treated electrocatalyst (inset: enlarged graphic) and ii) thermally treated electrocatalysts: a) 23/ATO/300, b) 16/TTO/300, c) 19/TTO/300, d) 24/TTO/300, e) 12/TTO and f) 6/TTO. | ||
| Electrocatalyst | Mass activity (A g−1) at 1.5 V/RHE | Overpotential (mV) at 10 mA cm−2 |
|---|---|---|
| 6/TTO | 14 ± 4 | 449 |
| 12/TTO | 62 ± 7 | 342 |
| 16/TTO(300) | 137 ± 11 (48 ± 8) | 302 (332) |
| 19/TTO(300) | 122 ± 9 (14 ± 4) | 309 (389) |
| 24/TTO(300) | 103 ± 8 (2 ± 1) | 314 (505) |
| 23/ATO(300) | 144 ± 13 (51 ± 10) | 301 (326) |
Electrochemical characterisation was also performed on the electrocatalysts treated at 300 °C presenting the beginning of crystallisation of the iridium oxide phase. As expected, the activity dropped and the overpotential increased. 16/TTO/300 demonstrated the highest OER activity (48 A g−1 at 1.5 V) among the treated IrO2/TTO (Table 2). As the catalyst loading increased, the electroactivity dropped until 2 A g−1 at 1.5 V for 24/TTO/300, demonstrating the effect of overcoverage combined with particle growth and aggregation due to the thermal treatment. Thermally treated 23/ATO/300 presented similar activity (51 A gIr−1 at 1.5 V) as that of 16/TTO/300.
The 16/TTO electrocatalyst presented the highest activity. To further optimise and verify the superiority of this sample of the thermal treatment at 300 °C, its electrochemical activity and stability was assessed after thermal treatments between 200 °C and 325 °C. OER polarisation curves normalised to the geometric disk area before and after iR correction are displayed in Fig. S7c.† The electrochemical degradation of the different treated TTO based catalyst was monitored by evaluating the OER activity before and after performing chronoamperometry measurements at a current density of 10 mA cm−2 for 20 h (Fig. 6). As expected, an increase in treatment temperature led to a reduced OER activity while enhancing the resistance to degradation. All Tafel slopes increased under similar accelerated stress test conditions, indicating electrocatalyst degradation ascribed to increased nanoparticle sizes and/or Ir-dissolution.40,65 This study highlighted that the 16/TTO catalyst treated at 300 °C gave rise to the best activity–stability trade-off (Fig. 6c).
 | ||
| Fig. 6 Ir-mass normalised OER polarisation curves obtained with the catalysts indicated in the legend before (a) and after (b) chronopotentiometry measurements at 10 mA cm−2 for 20 h (c). | ||
These results agree with a) those obtained by TG-QMS and XPS, indicating that the decrease in the activity is related to the loss of OH groups and b) those of TG-DSC, TDX and HRTEM, confirming the formation of the IrO2 crystalline phase at 300 °C with homogeneous distribution. Despite the similarity highlighted by the range of characterisation techniques for 16/TTO/300 and 23/ATO/300 (Fig. 6b), 23/ATO/300 displays a lower resistance to degradation, which could be related to the instability of antimony as dopant as reported elsewhere.24,66 These results shed light on the double role of tantalum doping agent on the catalysed material activity (48 ± 8 A g−1 at 1.5 V/RHE) and the electrocatalyst support stability after 20 h (32 ± 4 A g−1 at 1.5 V) keeping a mass activity retention of 65% and a slight increase on the overpotential of 22 mV. Overall, 16/TTO/300 demonstrated higher OER activity and stability compared to similar supported catalysts.16,36,41,67–69
Finally, the electrocatalyst 16/TTO treated at different temperatures around 300 °C was characterised at the anode side of a water electrolysis cell to find the best compromise between performance and durability in the membrane electrode assembly (MEA) operating conditions. The charge of the anode was kept ultra-low (<0.2 mgIr cm−2), and the MEA results were compared with those obtained with the commercial benchmark with conventional high charges.
The MEAs investigated included 0.18 mgIr cm−2 anodes based on 16/TTO treated at 275–300–325 °C, a Nafion 115 membrane and a 0.5 mgPt cm−2 Pt/C commercial GDE cathode. Their corresponding polarisation curves in the electrolysis cell and electrochemical impedance spectra at 80 °C and a range of current densities are displayed in Fig. 7 and S9, Tables 3 and S2.† The average voltage at 1.0 and 1.5 A cm−2 increased with higher treatment temperatures of the anode catalyst according to the i/V curves, which agrees with the ex situ electrochemical measurements. EIS analysis showed an almost identical ohmic resistance for all the MEAs, and enhanced charge transfer resistance as the treatment temperature of the supported anode catalyst increased. This is in agreement with a lower electrocatalytic activity and results reported in the literature.70 To assess the effect of electrocatalyst thermal treatment on its stability, the i/V curves were recorded before and after an accelerated stress test in the polarising the cell at 2.0 V (Fig. 7a and Tables 3 and SI2†).
 | ||
| Fig. 7 I/V curves at 80 °C for MEAs based on a) 16/TTO treated at different indicated temperatures at the anode side before and after the stability test at 2.0 V and on b) 16/TTO/300 and 23/ATO/300 at the anode side before and after the stability test at 2.0 V. | ||
| OER catalyst | Cell voltage (V) | Mass activity (A gIr−1) | Tafel slope (mV dec−1) | Overpotential at 10 mA cm−2 (mV) | |
|---|---|---|---|---|---|
| @1 A cm−2 | @1.5 A cm−2 | @1.5 V | |||
| 16/TTO/275 | 1.70 (1.89) | 1.81 (>2) | 901 (508) | 61 (72) | 184 (188) |
| 16/TTO/300 | 1.73 (1.78) | 1.83 (1.89) | 498 (429) | 56 (58) | 210 (213) |
| 16/TTO/325 | 1.76 (1.86) | 1.88 (>2) | 408 (310) | 59 (66) | 214 (217) |
| 23/ATO/300 | 1.74 (1.80) | 1.84 (1.92) | 617 (490) | 60 (64) | 200 (202) |
All MEAs lost performance after polarisation at high potential. The drop was higher for the MEA containing the anode electrocatalyst treated at 275 °C (1.89 V at 1 A cm−2) and lower for that containing the anode electrocatalyst treated at 300 °C (1.77 V at 1 A cm−2), as expected with the increasing degree of crystallisation assessed by TDX and TG-DSC analysis. However, the stability was lower for the MEA comprising the most crystalline electrocatalyst, 16/TTO/325 °C, (1.85 V at 1 A cm−2), probably due to the agglomeration state of the catalyst induced by the higher treatment temperature. Such results were further interpreted after carrying out end of test (EoT) analysis on the different MEAs by SEM-EDX and ICP-MS of anode exhaust water (Table S3 and Fig. S10†). The latter indicated no significant leaching of elements from the oxide support from the electrocatalysts treated at 300 and 325 °C. TEM and EDX analysis of both MEAs after the test (Fig. S11 and Table S3†) demonstrate that iridium oxide particles coalescence was more prominent for the sample treated at 325 °C, in agreement with its higher performance drop.
In conclusion, MEA characterisation confirmed that 16/TTO/300 presents the best trade-off between OER activity and stability. A further comparison was made with an MEA with antimony doped-tin oxide as OER electrocatalyst support. Fig. 7b and Tables 3 and S2† present the comparison of results between MEAs with a charge of 0.18 mgIr cm−2 at the anode side based on 23 wt% IrO2/ATO and 16 wt% IrO2/TTO treated at 300 °C. The slight improvement in the performance and ohmic and charge transfer resistance in the activation region for the 23/ATO/300 might be attributed to the 10-time higher electrical conductivity of the support, while similar resistance is observed for the ohmic and mass transport region (the used membrane was the same). However, greater performance loss and ohmic and charge transfer resistances were evident after the degradation protocol up to 2.0 V. This result was ascribed to the instability of antimony in SnO2 detected by EDX and ICP-MS analysis (Table S3 and Fig. S10†) and reported elsewhere,24,66 possibly leading to a drop in conductivity of the support material and to nanocatalyst deactivation.
In general, similar performances are reported for MEAs including Ir-based catalysts supported on other types of metal oxide, but using a higher noble metal loading at the anode than that used in this work (0.18 mgIr cm−2 of 16/TTO/300).15,68,69,71–74
The promising results for 16/TTO/300 as a PEMWE anode with a low loading (0.18 mgIr cm−2) confirm that the catalysed material exhibits a high activity (498 A gIr−1 at 1.5 V) despite the lower conductivity and relatively low surface area of the tantalum doped tin oxide nanofibrous support, and a remarkable resistance to corrosion (83% of mass activity retention after AST at 1 A cm−2), which can be rationalised by the strong metal oxide–support interaction and the stability of TTO. MEAs bearing such anode catalyst with low iridium loading (0.18 mg cm−2) with an otherwise identical MEA based on a commercial unsupported IrO2 catalyst with conventional 7 times higher PGM loading (1.28 mg cm−2) have similar performance (Fig. 8).
 | ||
| Fig. 8 Polarisation curves of the MEAs including 16/TTO/300 or commercial IrO2 with seven times higher Ir loading at the anode. | ||
In conclusion, the 16 wt% IrOx/TTO catalyst treated at 300 °C demonstrated a high stability and activity in RDE as well as in the PEMWE single cell, reaching the performance of an MEA including an unsupported commercial OER catalyst with seven time more iridium, with great perspectives towards the reduction of this PGM metal for decarbonated hydrogen production.
3. Experimental
3.1 Preparation of TTO and ATO nanofibres
1 at% tantalum doped SnO2 fibres (TTO) and 10 at% antimony doped SnO2 fibres (ATO) were prepared by electrospinning and thermally treated according to a previously reported synthesis.27,28,33,343.2 Preparation of IrOx supported onto TTO and ATO
A microwave-assisted polyol method was used to prepare nanoparticles of iridium oxide.24,75 In a typical experiment, a 0.78 g L−1 solution of hydrogen hexachloroiridate(IV) hydrate (H2IrCl6·xH2O, Sigma Aldrich 99.98%) in nitrogen-saturated ethylene glycol (40 mL, Sigma Aldrich 99.8%) was prepared and its pH was adjusted to 11 with NaOH (1 M solution, 98%, Sigma Aldrich). The solution was transferred to a microwave reactor (MiniFlow 200XX, Sairem) and the reaction carried out at 180 °C for 20 minutes at 160 W under an oxygen atmosphere. The resulting IrOx nanoparticle suspension was cooled down to room temperature, and then sonicated for 20 min (Branson Digital Sonifier®) after addition of the corresponding TTO or ATO doped-SnO2 support materials to reach the targeted loading. Afterwards, the suspension was diluted by the addition of 50 mL of water, and the pH was adjusted to 1 by adding HNO3 (65%, Sigma Aldrich). After five minutes at constant pH, the IrOx/doped-SnO2 materials were recovered by filtration, washed with Milli-Q® grade water, and dried at 80 °C for 24 h. The samples were labelled x/TTO and x/ATO, where x indicates the targeted catalyst loading on TTO and ATO: namely 5, 10, 15, 20 and 25 wt% IrOx on TTO nanofibres and 25 wt% IrOx on ATO nanofibres since this loading provides the highest performance, as we previously reported.24Further thermal treatment of the supported IrOx materials using a ramp rate of 5 °C min−1 for 2 h at temperatures ranging from 300 °C to 550 °C was carried out to study its effect on catalyst nanoparticle size and crystallinity degree, and the resulting impact on the electrochemical activity and stability of the supported catalysts. Thermally treated samples were labelled x/TTO/y and x/ATO/y, where y indicates the temperature of the applied thermal treatment.
3.3 Physicochemical characterisation of IrOx/TTO
The morphology of IrOx/TTO was analysed using a scanning electron microscope (SEM) FEI Quanta FEG 200 equipped with energy-dispersive X-ray spectroscopy (EDS) analysis and using a JEOL 1200 EXII transmission electron microscope (TEM) operating at 120 kV, equipped with a SIS Olympus Quemesa CCD camera (11 million pixels). For TEM analysis, the samples were suspended and sonicated in ethanol, then deposited onto carbon-coated copper grids, while a microtome was used to cut resin-encapsulated electrocatalyst powders for cross-sectional analysis in SEM, and the corresponding slices were deposited onto copper grids. The average size of iridium oxide particles and tin oxide fibres was determined by measuring 200 selected objects using ImageJ software. A FEI Talos F200X microscope operating at 200 kV (Schottky-FEG emitter) and fitted with an in-column omega-filter and a GATAN Ultrascan CCD 2048 × 2048 px2 camera was used to obtain high-resolution TEM (HRTEM) micrographs and the corresponding selected area electron diffraction (SAED) (0.23 nm point resolution).Powder X-ray diffraction (XRD) patterns were recorded at 20 °C in Bragg–Brentano configuration using a PANalytical X'pert diffractometer, equipped with a hybrid monochromator, operating with CuKα radiation (λ = 1.541 Å). A step size of 0.1° 2θ was employed within the 2θ domain from 20° to 80°.
X-ray thermodiffractometry (TDX) was performed to study crystal structure changes and its characterisation was monitored using an in situ high temperature XRD with a Malvern-Panalytical Empyrean diffractometer series 2, equipped with an Anton Paar HTK 16 high-temperature camera (CoKα-radiation). For each sample, a sequence of XRD measurements was performed at room temperature employing a step size of 0.1° and 2θ range from 25° to 36°, and again after each heating step of 25 °C (ramp rate of 5 °C min−1) between 300 °C to 550 °C, with the sample cell under vacuum of 10−3 Torr, and a recording time of 2 h for every diffractogram. After recording the XRD pattern at 550 °C, the samples were cooled to 25 °C (5 °C min−1) under vacuum.
X-ray fluorescence (XRF) was used to determine the iridium loading onto the metal oxide supports. Samples were prepared by grinding 50 mg of IrOx/doped-SnO2 with 25 mg of cellulose to form a powder that was placed in a cavity in a boric acid matrix and subsequently pressed to obtain a pellet of 32 mm diameter with scanned surface of ca. 12 mm. The same protocol was used to prepare six standards using 5, 10, 15, 20, 25 and 30 wt% of IrO2 (Sigma Aldrich, 99.9%) and doped-SnO2 to obtain a calibration line. The XRF analyses were performed with a PANalytical Axios Max spectrometer fitted with a Rh (4 kW) tube and equipped with a LiF200 crystal and Omnian software.
Thermogravimetry/differential scanning calorimetry coupled with mass spectrometry (TG/DSC/MS) analysis was carried out using a simultaneous thermal analyser model STA449F1 Jupiter® (Netzsch) (TGA/DSC) coupled to a Aeolos Quadro QMS 403 mass spectrometer between 25 °C to 600 °C with a heating rate of 5 °C min−1 under air.
Raman analysis was performed on a Renishaw inVia spectrometer with a wavelength of 532 nm, a power of 200 μW and a resolution of 1 cm−1.
The surface chemical state of the materials was investigated by X-ray photoelectron spectroscopy (XPS) on an ESCALAB 250 (Thermo Fisher Scientific) spectrometer. The X-ray excitation was provided by a monochromatic Al Kα (1486.6 eV) source, and the analysed surface area was 400 μm2. A constant analyser energy mode was used for the electron detection (20 eV pass energy), which was performed perpendicularly to the sample surface. The data were analysed in the Avantage and CASA XPS software, removing the background signal using the Shirley method. The surface atomic concentrations were determined from photoelectron peak areas using the atomic sensitivity factors reported by Scofield.76 Binding energies of all core levels refer to the C–C bond of C 1s at 284.8 eV.
3.4 Electrochemical characterisation of TTO and IrO2/TTO
Electrochemical analyses were carried out at 25 °C using a Pine bipotentiostat model AFCBP1 in a conventional three-electrode cell composed of a reversible hydrogen electrode (RHE, reference electrode), a gold rotating disk electrode (RDE) (working electrode, geometric area 0.196 cm2), and a platinum wire (counter electrode). The reference and counter electrodes were separated from the working electrode compartment by a glass frit. Prior to evaluation of the OER activity, 10 cyclic voltammograms (CVs) were recorded from 0.05 to 1.4 V vs. RHE in N2 saturated 0.5 M H2SO4 at 50 mV s−1. Linear sweep voltammetry (LSV) was performed between 1.2 and 1.6 V vs. RHE at 10 mV s−1 at 25 °C using a rotation speed of 1600 rpm. All the potential values were referred to the RHE and were corrected for the ohmic drop in solution. The current densities are reported as current per gram of iridium oxide for mass activity evaluation at 1.5 V/RHE. For instance, the catalyst ink of 24/TTO was prepared by dispersing 3 mg in 297.8 μL of Milli-Q® grade water, 893.4 μL of isopropanol (Aldrich) and 17.1 μL 5 wt% Nafion® EW1100 solution in alcohols (Aldrich) and sonicating for 15 min using a VWR ultrasonic cleaner. A micropipette was used to deposit 7 μL aliquots onto the RDE surface and dried in air. The Ir loading on the electrode was 17.7 μg cm−2 for all the electrocatalysts. To estimate the electrochemically active surface area (ECSA), the double-layer capacitance (Cdl) was calculated using CV scans at different scan rates (10, 20, 30, 40 and 50 mV s−1) in the non-faradaic region (0.5–0.6 VRHE). After plotting the average between the maximum anodic current density and the minimum cathodic current density against the scan rate, the corresponding slope (Cdl) was obtained for every catalysed material. Then, Cdl was converted to ECSA by diving it by the specific capacitance in acidic conditions (0.035 mF cm−2)77 and the mass loading deposited on the electrode.58Chronopotentiometry measurements were performed to investigate the resistance of IrOx/TTO and IrOx/ATO to electrochemical degradation by holding the working electrode (gold, geometric area of 0.196 cm2) at a constant current density of 10 mA cm−2 for 20 h in 0.5 M H2SO4 at 25 °C and assessing the evolution of the potential up to a cut-off potential of 2.0 V vs. RHE. LSV was performed to determine the decay of OER activity after each 20 hour potential hold.
3.5 Preparation of membrane electrode assemblies
Membrane-electrode assemblies (MEAs) with an active area of 6.75 cm2 were prepared using the decal method for the anode and a commercial Pt/C cathode. For the anode, 0.18 mgIr cm−2 were deposited from an ink composed of the catalyst (22.7 mg of 16/TTO, 15.8 mg 23/ATO and 22.2 mg commercial IrO2 99.9 wt% Surepure® Chemetals) and a mass ratio of 20 wt% of Nafion (from a 5 wt% Nafion EW1100 solution in alcohols) with respect to the catalysed material dispersed in isopropanol/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (V/V) homogenised in ultrasonic bath. This ink was sprayed onto a Teflon sheet using a Nadetech Innovations ND-SP Spray Coater placed over a heating pad at 80 °C. A Nafion 115 membrane (Ion Power) was pre-treated to re-establish the acidic sites,78 and then employed for the decal transfer of the above catalyst layer to produce a catalyst coated membrane (CCM). For this, the membrane was placed over the sprayed catalyst layer, covered with a Teflon-glass-fibre fabric, and hot-pressed using the following steps: pre-heating at 80 °C, applying a pressure of 8.1 MPa, increasing the temperature to 140 °C and then the pressure to 15.7 MPa for 15 min. Similar hot-pressing conditions were used for the assembly of the cathode side (0.5 mg Pt cm−2 supported on a Sigracet 22BB GDL from Baltic Fuel Cells) to the prepared CCM, when 90 s were needed for its complete transfer to obtain the final MEA. A similar procedure was carried out for the preparation of a reference MEA for which commercial unsupported IrO2 (Surepure® Chemetals) was used at the anode, with a loading of 1.28 mgIr cm−2.
:1) (V/V) homogenised in ultrasonic bath. This ink was sprayed onto a Teflon sheet using a Nadetech Innovations ND-SP Spray Coater placed over a heating pad at 80 °C. A Nafion 115 membrane (Ion Power) was pre-treated to re-establish the acidic sites,78 and then employed for the decal transfer of the above catalyst layer to produce a catalyst coated membrane (CCM). For this, the membrane was placed over the sprayed catalyst layer, covered with a Teflon-glass-fibre fabric, and hot-pressed using the following steps: pre-heating at 80 °C, applying a pressure of 8.1 MPa, increasing the temperature to 140 °C and then the pressure to 15.7 MPa for 15 min. Similar hot-pressing conditions were used for the assembly of the cathode side (0.5 mg Pt cm−2 supported on a Sigracet 22BB GDL from Baltic Fuel Cells) to the prepared CCM, when 90 s were needed for its complete transfer to obtain the final MEA. A similar procedure was carried out for the preparation of a reference MEA for which commercial unsupported IrO2 (Surepure® Chemetals) was used at the anode, with a loading of 1.28 mgIr cm−2.
3.6 Single-cell electrochemical characterisation of the prepared MEAs
The MEA was integrated into the cell set-up employing fluorinated ethylene propylene gaskets and reaching a final compression of 50–55%. After flooding the anode side with deionized water (Milli-Q quality, 18 MW), a 200 mL h−1 flow rate of deionized water was passed through the anode chamber and the MEA was conditioned at 80 °C for 12 h at 0.2 A cm−2 to ensure complete membrane hydration. The characterisation of the cell was carried out at a cell temperature of 80 °C using a Bio-logic SP-150 potentiostat with a 20 A booster. Polarisation curves were recorded between 0 to 2 A cm−2 current density. A fixed current density was maintained until a variation of the steady-state potential was lower than 1 mV min−1. Thereafter, a potentiostatic accelerated stress test (AST) was performed by holding at a potential from 1.5 to 2.0 V (with 0.1 V steps) for 4 h to evaluate the stability of the catalysed materials. A polarisation curve was recorded after every potentiostatic step. Furthermore, ca. 1 L of exhaust water was recovered from the anode side and 20 mL of water was withdrawn. One hundred μL of aqua regia was added to dissolve any solid and analysed by ICP-MS. Electrochemical impedance spectroscopy (EIS) measurements were performed between 30 kHz to 100 mHz at 0.025, 0.05, 0.125 and 0.2 A cm−2 to measure the MEA resistance before and after the AST.4. Conclusions
This work investigated the OER electrocatalytic activity and stability trade-off of iridium oxide nanoparticles supported onto doped-SnO2 fibres (ATO and TTO) upon thermal treatments at different temperatures. The treatment at 300 °C led to crystallised, well dispersed IrO2 particles with high OER electrochemical activity and resistance to degradation. In particular, 16 wt% IrO2 catalysts supported on TTO (16/TTO/300) demonstrated higher stability in the RDE and in the single-cell electrolysis test, with a mass activity retention after AST of 83% at 1 A cm−2. The performance of the MEAs comprising an anode based on 16/TTO/300 overcome that of MEAs based on commercial unsupported IrO2 but with seven times lower iridium loading (0.18 mgIr cm−2vs. 1.28 mgIr cm−2). These results confirm that 16/TTO/300 is a promising anode electrocatalyst for PEMWE and that the optimisation of crystallinity of IrOx and the use of conducting and stable supports paves the way for the design of OER catalysts with ultra-low iridium loading. Further optimisation of the support porosity and catalyst surface area and composition can further reduce this loading, while keeping high mass activity and durability, tackling one of the greatest challenges for PEMWE development.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
I. J.-M. carried out the experimental work and wrote the initial draft. D. J., J. R. and S. C. provided supervision and revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the H2020 European Union's Programme/ERC-2019-POC in the frame of the HYDROGEN project (Grant Agreement no. 875573) and the French National Research Agency in the frame of the MOISE (Grant Agreement no. ANR-17-CE05-0033) and MATHYLDE projects (Grant Agreement no. ANR-22-PEHY-0001).References
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 Search PubMed.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukauloo and C. Etiévant, Int. J. Hydrogen Energy, 2011, 36, 4134–4142 CrossRef CAS.
- A. Marshall, B. Børresen, G. Hagen, M. Tsypkin and R. Tunold, Energy, 2007, 32, 431–436 CrossRef CAS.
- O. Kasian, S. Geiger, P. Stock, G. Polymeros, B. Breitbach, A. Savan, A. Ludwig, S. Cherevko and K. J. J. Mayrhofer, J. Electrochem. Soc., 2016, 163, F3099–F3104 CrossRef CAS.
- T. Audichon, E. Mayousse, S. Morisset, C. Morais, C. Comminges, T. W. Napporn and K. B. Kokoh, Int. J. Hydrogen Energy, 2014, 39, 16785–16796 CrossRef CAS.
- T. Audichon, T. W. Napporn, C. Canaff, C. Morais, C. Comminges and K. B. Kokoh, J. Phys. Chem. C, 2016, 120, 2562–2573 CrossRef CAS.
- S. Siracusano, N. Van Dijk, E. Payne-johnson, V. Baglio and A. S. Aricò, Appl. Catal., B, 2015, 164, 488–495 CrossRef CAS.
- D. F. Abbott, D. Lebedev, K. Waltar, M. Povia, M. Nachtegaal, E. Fabbri, C. Copéret and T. J. Schmidt, Chem. Mater., 2016, 28, 6591–6604 CrossRef CAS.
- C. Felix, B. J. Bladergroen, V. Linkov, B. G. Pollet and S. Pasupathi, Catalysts, 2019, 9, 1–16 CrossRef.
- M. Faustini, M. Giraud, D. Jones, J. Rozière, M. Dupont, T. R. Porter, S. Nowak, M. Bahri, O. Ersen, C. Sanchez, C. Boissière, C. Tard and J. Peron, Adv. Energy Mater., 2019, 9, 1–11 Search PubMed.
- S. Siracusano, N. Hodnik, P. Jovanovic, F. Ruiz-Zepeda, M. Šala, V. Baglio and A. S. Aricò, Nano Energy, 2017, 40, 618–632 CrossRef CAS.
- H. Yu, N. Danilovic, Y. Wang, W. Willis, A. Poozhikunnath, L. Bonville, C. Capuano, K. Ayers and R. Maric, Appl. Catal., B, 2018, 239, 133–146 CrossRef CAS.
- C. Van Pham, M. Bühler, J. Knöppel, M. Bierling, D. Seeberger, D. Escalera-López, K. J. J. Mayrhofer, S. Cherevko and S. Thiele, Appl. Catal., B, 2020, 269, 118762 CrossRef.
- H. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC.
- C. Van Pham, D. Escalera-López, K. Mayrhofer, S. Cherevko and S. Thiele, Adv. Energy Mater., 2021, 11, 2101998 CrossRef.
- L. Moriau, M. Smiljanić, A. Lončar and N. Hodnik, ChemCatChem, 2022, 14, e202200586 CrossRef CAS PubMed.
- J. Y. Choi, J. G. Kim, H. J. Lee and C. Pak, Int. J. Hydrogen Energy, 2025, 97, 57–65 CrossRef CAS.
- C. Baik, J. Cho, J. In, Y. Cho, S. Soon and C. Pak, J. Power Sources, 2023, 575, 233174 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179–F3189 CrossRef CAS.
- I. Jiménez-Morales, S. Cavaliere, J. Rozière, M. Dupont and D. Jones, Sustainable Energy Fuels, 2019, 3, 1526–1535 RSC.
- H. P. Tran, H. N. Nong, M. Zlatar, A. Yoon, U. Hejral, M. Rüscher, J. Timoshenko, S. Selve, D. Berger, M. Kroschel, M. Klingenhof, B. Paul, S. Möhle, K. N. Nagi Nasralla, D. Escalera-López, A. Bergmann, S. Cherevko, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2024, 146, 31444–31455 CrossRef CAS PubMed.
- P. K. Mohanta, C. Glökler, A. O. Arenas and L. Jörissen, Int. J. Hydrogen Energy, 2017, 2, 1–12 Search PubMed.
- I. Jiménez-Morales, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2018, 20, 8765–8772 RSC.
- I. Jiménez-Morales, F. Haidar, S. Cavaliere, D. Jones and J. Rozière, ACS Catal., 2020, 10, 10399–10411 CrossRef.
- J. Ma, A. Habrioux, Y. Luo, G. Ramos-Sanchez, L. Calvillo, G. Granozzi, P. B. Balbuena and N. Alonso-Vante, J. Mater. Chem. A, 2015, 3, 11891–11904 RSC.
- H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- Y. Wang, T. Brezesinski, M. Antonietti and B. Smarsly, ACS Nano, 2009, 3, 1373–1378 CrossRef CAS PubMed.
- H. Ohno, S. Nohara, K. Kakinuma, M. Uchida and H. Uchida, Catalysts, 2019, 9, 74 CrossRef.
- S. Cavaliere, I. Jiménez-Morales, G. Ercolano, I. Savych, D. Jones and J. Rozière, ChemElectroChem, 2015, 2, 1966–1973 CrossRef CAS.
- S. Cavaliere, S. Subianto, I. Savych, M. Tillard, D. J. Jones and J. Rozière, J. Phys. Chem. C, 2013, 117, 18298–18307 CrossRef CAS.
- A. T. Marshall and R. G. Haverkamp, Electrochim. Acta, 2010, 55, 1978–1984 CrossRef CAS.
- D. Böhm, M. Beetz, M. Schuster, K. Peters, A. G. Hufnagel, M. Döblinger, B. Böller, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2020, 30, 1906670 CrossRef.
- T. Reier, D. Teschner, T. Lunkenbein, A. Bergmann, S. Selve, R. Kraehnert, R. Schlögl and P. Strasser, J. Electrochem. Soc., 2014, 161, F876–F882 CrossRef CAS.
- M. Möckl, M. F. Ernst, M. Kornherr, F. Allebrod, M. Bernt, J. Byrknes, C. Eickes, C. Gebauer, A. Moskovtseva and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 064505 CrossRef.
- M. Malinovic, P. Paciok, E. S. Koh, M. Geuß, J. Choi, P. Pfeifer, J. P. Hofmann, D. Göhl, M. Heggen, S. Cherevko and M. Ledendecker, Adv. Energy Mater., 2023, 13, 2301450 CrossRef CAS.
- M. Povia, D. F. Abbott, J. Herranz, A. Heinritz, D. Lebedev, B. J. Kim, E. Fabbri, A. Patru, J. Kohlbrecher, R. Schäublin, M. Nachtegaal, C. Copéret and T. J. Schmidt, Energy Environ. Sci., 2019, 12, 3038–3052 RSC.
- Z. S. H. S. Rajan, T. Binninger, P. J. Kooyman, D. Susac and R. Mohamed, Catal. Sci. Technol., 2020, 10, 3938–3948 RSC.
- C. Daiane Ferreira Da Silva, F. Claudel, V. Martin, R. Chattot, S. Abbou, K. Kumar, I. Jiménez-Morales, S. Cavaliere, D. Jones, J. Rozière, L. Solà-Hernandez, C. Beauger, M. Faustini, J. Peron, B. Gilles, T. Encinas, L. Piccolo, F. H. Barros De Lima, L. Dubau and F. Maillard, ACS Catal., 2021, 11, 4107–4116 CrossRef CAS.
- V. K. Puthiyapura, M. Mamlouk, S. Pasupathi, B. G. Pollet and K. Scott, J. Power Sources, 2014, 269, 451–460 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- F. Karimi, B. A. Peppley and A. Bazylak, ECS Trans., 2015, 69, 87–98 CrossRef CAS.
- C. Massué, X. Huang, A. Tarasov, C. Ranjan, S. Cap and R. Schlögl, ChemSusChem, 2017, 10, 1958–1968 CrossRef PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- Y. S. Huang, S. S. Lin, C. R. Huang, M. C. Lee, T. E. Dann and F. Z. Chien, Solid State Commun., 1989, 70, 517–522 CrossRef CAS.
- W. Sun, L. Cao and J. Yang, J. Mater. Chem. A, 2016, 4, 12561–12570 RSC.
- C. Ma, C. Hu, X. Liu, Y. Li, J. Cui, Y. Wu and J. Huang, ACS Omega, 2022, 7, 28985–28993 CrossRef CAS PubMed.
- Z. Pavlovic, C. Ranjan, Q. Gao and M. Van Gastel, ACS Catal., 2016, 6, 8098–8105 CrossRef CAS.
- S. Geiger, O. Kasian, B. R. Shrestha, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2016, 163, F3132–F3138 CrossRef CAS.
- S. Cherevko, T. Reier, A. R. Zeradjanin, Z. Pawolek, P. Strasser and K. J. J. Mayrhofer, Electrochem. Commun., 2014, 48, 81–85 CrossRef CAS.
- G. C. da Silva, N. Perini and E. A. Ticianelli, Appl. Catal., B, 2017, 218, 287–297 CrossRef CAS.
- F. Karimi, A. Bazylak and B. A. Peppley, J. Electrochem. Soc., 2017, 164, F464–F474 CrossRef CAS.
- J. Cheng, J. Yang, S. Kitano, G. Juhasz, M. Higashi, M. Sadakiyo, K. Kato, S. Yoshioka, T. Sugiyama, M. Yamauchi and N. Nakashima, ACS Catal., 2019, 9, 6974–6986 CrossRef CAS.
- J. Ruiz Esquius, D. J. Morgan, I. Spanos, D. G. Hewes, S. J. Freakley and G. J. Hutchings, ACS Appl. Energy Mater., 2020, 3, 800–809 CrossRef CAS.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- C. Roiron, C. Wang, I. V. Zenyuk and P. Atanassov, J. Phys. Chem. Lett., 2024, 15, 11217–11223 CrossRef CAS PubMed.
- V. Senthilkumar, P. Vickraman, M. Jayachandran and C. Sanjeeviraja, J. Mater. Sci.: Mater. Electron., 2010, 21, 343–348 CrossRef CAS.
- Q. Liu, L. Zhang, J. F. Chen and Y. Le, Mater. Lett., 2014, 137, 339–342 CrossRef CAS.
- L. P. Yuan, W. J. Jiang, X. L. Liu, Y. H. He, C. He, T. Tang, J. Zhang and J. S. Hu, ACS Catal., 2020, 10, 13227–13235 CrossRef CAS.
- S.-C. Sun, H. Jiang, Z.-Y. Chen, Q. Chen, M.-Y. Ma, L. Zhen, B. Song and C.-Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202519 CrossRef CAS PubMed.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed.
- L. Dubau, F. Maillard, M. Chatenet, S. Cavaliere, I. Jiménez-Morales, A. Mosdale and R. Mosdale, Energies, 2020, 13, 403 CrossRef CAS.
- S. S. Karade, R. Sharma, S. Gyergyek, P. Morgen and S. M. Andersen, ChemCatChem, 2023, 15, e202201470 CrossRef CAS.
- G. Li, H. Jia, H. Liu, X. Yang and M.-C. Lin, RSC Adv., 2022, 12, 28929–28936 RSC.
- J. I. Cha, C. Baik, S. W. Lee and C. Pak, Catal. Today, 2022, 403, 19–27 CrossRef CAS.
- H. Guan, Q. Ke, C. Lv, N. Zeng, C. Hu, S. Wang, X. Ge and J. Cai, Chem. Lett., 2020, 49, 705–708 CrossRef CAS.
- S. W. Lee, C. Baik, D. H. Kim and C. Pak, J. Power Sources, 2021, 493, 229689 CrossRef CAS.
- Y. Liu, M. Zhang, C. Zhang, H. Zhang and H. Wang, Nanoscale, 2024, 16, 9382–9391 RSC.
- J. Islam, S.-K. Kim, M. M. Rahman, P. T. Thien, M.-J. Kim, H.-S. Cho, C. Lee, J. H. Lee and S. Lee, Mater. Today Energy, 2023, 32, 101237 CrossRef CAS.
- G. H. J. Changsoo Lee, K. Shin, Y. Park, Y. H. Yun, H. Y. K. Gisu Doo, M. J. Kim, W.-C. Cho, C.-H. Kim, H. M. Lee, H.-S. C. Sechan Lee and G. Henkelman, Adv. Funct. Mater., 2023, 33, 2301557 CrossRef.
- T. Audichon, B. Guenot, S. Baranton, M. Cretin, C. Lamy and C. Coutanceau, Appl. Catal., B, 2017, 200, 493–502 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- C. C. L. Mccrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135(45), 16977–16987 CrossRef CAS PubMed.
- I. Savych, S. Subianto, Y. Nabil, S. Cavaliere, D. Jones and J. Rozière, Phys. Chem. Chem. Phys., 2015, 17, 16970–16976 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lf00008d |
| ‡ Current address: Department of Inorganic Chemistry, University of Salamanca, GIR-QUESCAT Group, Pl. Caídos, 37008 Salamanca, Spain. |
| This journal is © The Royal Society of Chemistry 2025 |