
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amino acid interactions dependent on the polymerization of charged residues and surface properties of monolayers†
Akira
Nomoto
ab,
Kentaro
Shiraki
a and
Tsukuru
Minamiki
 *bc
*bc
aFaculty of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8573, Japan
bHealth and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8566, Japan. E-mail: t.minamiki@aist.go.jp
cPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 12th May 2025
Abstract
Charged amino acids are the key residues that regulate protein function and stability, and successive sequences of charged amino acids contribute significantly to protein assembly. Therefore, an in-depth understanding of the strengths and manners of amino acid–amino acid interactions (AAIs) caused by successive sequences of charged residues is required. In this study, we prepared self-assembled monolayers (SAMs) bearing charged alkanethiols as ligands to mimic protein surfaces with accumulated charged amino acids. Moreover, we used peptides as analytes to evaluate the AAIs based on chain length. The strengths of the AAIs of tri- and tetrapeptides varied per residue, depending on the properties of the SAMs, such as their densities and hydrophobicities, whereas those of mono- and dipeptides did not vary significantly. Remarkably, the strengths of the AAIs per residue decreased significantly with increasing peptide length, even when the strengths of the AAIs increased at the peptide scale. These results enabled us to quantify the microscopic changes in the AAIs, in addition to the overall interactions governed by the reaction field and alignment of the charged amino acids. Our analysis of AAIs shall be beneficial in protein engineering via genetic mutations based on charged amino acids.
Introduction
Charged amino acids regulate protein conformation via electrostatic interactions and hydrogen bonds.1,2 Such amino acids, which are highly hydrophilic, are easily exposed on protein surfaces, and thus,3,4 they are key residues in protein engineering, e.g. certain residues can be substituted with charged residues to increase protein solubility and stability.5,6 Additionally, charged amino acids are abundant in intrinsically disordered proteins (IDPs) without specific conformations.7,8 Mutations in the charged residues of IDPs drastically alter the assembly states of the proteins.9,10 Remarkably, successive sequences of charged residues often occur in IDPs and facilitate liquid–liquid phase separation.11,12 Furthermore, polymers with successive charged amino acid residues are used as additives to modulate enzyme activity and antibody stability, triggering the formation of protein assemblies.13,14 Thus, understanding the properties of the reaction field and amino acid–amino acid interactions (AAIs) induced by accumulated charged residues is critical in advancing protein engineering. Although in silico analysis is used to assess AAIs in such scenarios,15,16in situ analyses of individual interactions are challenging because of the limitations of conventional detection methods.Field-effect transistors (FETs) are semiconductor devices that detect interactions between recognition units and target molecules as electrical signals.17,18 FETs drastically improve the efficiencies of molecular big data analyses, such as genome sequencing, because of their superior levels of portability, quantitative performances, and capacities for simultaneous parallel processing.19,20 Self-assembled monolayers (SAMs) are used as molecular recognition units to enhance the responses of FET-based sensors.21,22 SAMs form spontaneously on solid surfaces via the chemical adsorption of organic molecules, providing chemically and physically stable reaction fields.23,24 Changing the SAM components produces different responses for the same target molecule.25,26 Thus, the FET-based SAM system is a powerful tool in evaluating the interactions between ligands and analytes because of its capacity to bear multiple highly sensitive detection spots.27 FET-based sensors modified with SAMs can detect not only specific intermolecular interactions, such as enzyme–substrate and antibody–antigen reactions,28,29 but also nonspecific interactions between functional groups and saccharides or biogenic amines.30,31
Based on these findings, we previously mimicked the substructures of protein surfaces using the functional groups of charged amino acids as components of SAMs.32 By modifying the side-chain-mimicking monolayer of an FET sensor (Fig. 1a), the intensities of AAIs, which depend on the ionic strengths and types of amino acids, were successfully quantified.32 In this study, we detect peptides as target molecules to gain deeper insight into the effects of successive sequences of charged amino acids on the AAIs per residue (Fig. 1b). In addition, to alter the properties of the reaction field wherein charged amino acids accumulate, we prepare SAMs using alkanethiols with different linker lengths (Fig. 1c).
 | ||
| Fig. 1 (a) Schematic of the field-effect transistor (FET) sensor functionalized with the self-assembled monolayer (SAM) mimicking the side chains of charged amino acids. Chemical structures of (b) amino acids in the analyte solutions and (c) ligand molecules of mercaptocarboxylic acids (MCAs) and aminoalkanethiols (AATs) used in preparing the SAMs. | ||
Experimental details
Materials
To evaluate AAIs, glycine (Gly), L-glutamic acid (Glu), L-lysine (Lys), and their homopeptides were used as analytes (Fig. 1b), and 3-mercaptopropionic acid (3-MPA), 4-mercaptobutanoic acid (4-MBA), 6-mercaptohexanoic acid (6-MHA), 2-aminoethanethiol (2-AET), 3-aminopropanethiol (3-APT), and 5-aminopentane-1-thiol (5-APT) were used as ligands in the formation of the SAMs (Fig. 1c). Gly, Lys, Glu, and dimethyl sulfoxide (DMSO) were obtained from FUJIFILM Wako Pure Chemical (Osaka, Japan), and 3-morpholinopropanesulfonic acid (MOPS), di-Gly, tri-Gly, tetra-Gly, di-Lys di-HCl, tri-Lys, tetra-Lys, and di-Glu were obtained from Sigma-Aldrich (St. Louis, MO, USA). Tri-Glu and tetra-Glu were obtained from GL Biochem (Shanghai, China), and isopropyl alcohol (IPA) and NaCl were obtained from Kanto Chemical (Tokyo, Japan). 3-MPA and 2-AET were obtained from Tokyo Chemical Industry (Tokyo, Japan), and 4-MBA and 6-MHA were obtained from BLD Pharmatech (Shanghai, China) and DOJINDO Laboratories (Kumamoto, Japan), respectively. 3-APT HCl and 5-APT HCl were obtained from Achem Technology (Taipei, Taiwan) and Biosynth (Staad, Switzerland), respectively. All chemicals were of reagent grade and used as received without further purification.Functionalization of FET-based sensors using the SAMs
We utilised a sensor array chip comprising 64 × 64 extended-gate-type FETs. The FET devices were equipped with Au extended-gate electrodes (BC2, BioCMOS, Nagoya, Japan). As the sensing component (extended gate) and transducing unit (FET channel) within the sensor are separated, the extended-gate configurations of the FETs ensure repeatable, stable measurements of the analytes in aqueous solutions (Fig. 1a). Before functionalization of the FET devices, the surfaces of the Au extended-gate electrodes were rinsed with IPA and Milli-Q water (resistivity: 18 MΩ cm at 25 °C). The electrodes were then cleaned via vacuum plasma treatment for 1 min (introduced gas: atmospheric air). The Au electrodes were immersed in DMSO solutions containing 1 mM of the ligands (Fig. 1c) for 1 h at room temperature. Finally, the SAM-functionalised electrodes were rinsed using IPA and Milli-Q water.Surface characterization of the SAM-modified electrodes
Elemental analyses of the SAM-modified Au surfaces were performed using X-ray photoelectron spectroscopy (XPS, Kratos AXIS Nova surface analysis spectrometer, Shimadzu, Kyoto, Japan) with an Al Kα radiation source. Quartz crystal microbalance (QCM) measurements were conducted using a QCM922A (AMETEK, Berwyn, PA, USA) to evaluate the molecular densities of the SAMs on the Au surfaces. The levels of adsorption of the SAMs were evaluated based on the changes in frequency when solutions with or without 1 mM of the ligands (Fig. 1c) in DMSO were used. The base frequency used for the QCM measurements was 8.9 MHz. The masses of the SAM monomers on the Au surfaces were calculated using the changes in frequency,33 and the sensing areas of the Au surfaces were 0.196 cm2. The changes in the work functions of the Au electrodes with and without the SAMs were evaluated using photoemission yield spectroscopy in air (PYSA, AC-2S, RIKEN KEIKI, Tokyo, Japan). The differences in the hydrophobicities of the Au surfaces modified with the SAMs were evaluated based on the water contact angles observed using contact angle goniometry (CAG, SImage AUTO 100, Excimer, Yokohama, Japan). All surface characterization studies were performed at room temperature.Electrical detection of the AAIs using the FET-based SAM system
After functionalising the extended-gate sensing electrodes, the FET sensor chip was connected to the measuring equipment (BCT-II, BioCMOS). An Ag/AgCl reference electrode with an inner solution of 3 M NaCl (RE-3VT, BAS, Tokyo, Japan) was coupled with the measurement equipment as a control gate electrode (Fig. 1a). First, a 10 mM MOPS buffer solution (pH 7.0) containing 10 mM NaCl was applied to the FET sensor chip. The change in the output potential (Vout) with time due to the fluctuation in the electrode potential in the buffer solution was measured. The device was incubated with the buffer solution for 2 h to stabilise the drift in the Vout, and the drift fraction was formulated to analyse the Vout as a sensing signal. The change in the Vout was then measured while titrating the analyte solution containing 0–10 μM peptide and 10 mM NaCl in 10 mM MOPS (pH 7.0). The calibrated Vout (ΔVout) was determined by subtracting the drift fraction from the measured Vout.32 The independent Vout was determined using 42 detection points on the SAMs, and the mean and standard error of the 42 data points were calculated. The apparent equilibrium dissociation constant (Kd) is calculated using the response curve of the electrical titration isotherm based on the Hill–Waud model:26,34 | (1) |
In this equation, [x], n, and ΔVmax respectively represent the analyte concentration, Hill coefficient of cooperativity, and maximum Vout when the analyte completely saturates the SAM surface. All electrical measurements were performed at room temperature.
Multivariate analysis of the electrical responses
For deeper insight into the interactions of each peptide with the SAMs, we performed principal component analysis (PCA) using the OriginPro software (OriginLab, Northampton, MA, USA). We used the electrical responses during the titration of each analyte, with 10 μM peptide and 10 mM NaCl in 10 mM MOPS (pH 7.0), as the dataset. This dataset is a matrix with the 12 types of analytes and 6 types of ligands in rows and columns, respectively. In the PCA, the six points with the smallest deviations were extracted from the 42 detection points and used as repeating numbers.Results
Characterization of the reaction fields with the accumulated charged residues
First, we performed elemental analysis using XPS to confirm the formation of the SAMs terminated with the ligand molecules on the Au surfaces (Fig. 2). The compositional changes in the SAMs on the Au surfaces are observed as shifts in the elemental peaks of the charged alkanethiols in the C 1s, O 1s, N 1s, and S 2p regions. The C 1s peak at approximately 285 eV in the spectrum of each SAM is attributed to the carbon attached to the sulfur atom (Fig. 2). In addition, the peaks derived from the orbital doublets of S 2p3/2 (∼162 eV) and S 2p1/2 (∼163 eV) are attributed to the sulfur of the thiol group. Based on peak separation analysis (Fig. S1†), the changes in the intensity ratios of the peaks representing S 2p3/2 and S 2p1/2 of each SAM were analysed. These changes indicate that the amounts of disulfide-like compounds are higher when using the mercaptocarboxylic acids (MCAs), which mimic the negatively charged side chains, compared to those observed when using the aminoalkanethiols (AATs), which mimic the positively charged side chains. Conversely, the amounts of thiol-derived compounds are higher when using the AATs compared to those observed when using the MCAs. In the spectra of the SAMs terminated with the MCAs, the C 1s and O 1s peaks at approximately 288 and 533 eV, respectively, are derived from the carbon and oxygen within the carboxyl groups of the MCAs (Fig. 2a). In the spectra of the SAMs terminated with the AATs, the N 1s peaks at approximately 400 eV are derived from the nitrogen within the amino groups of the AATs (Fig. 2b). Therefore, the ligands are adsorbed onto the Au surfaces, and the intermolecular distances between the thiol groups within the SAMs are shorter when using the MCAs compared to those observed when using the AATs. | ||
| Fig. 2 C 1s, S 2p, O 1s, and N 1s X-ray photoelectron spectra of the Au surfaces modified with self-assembled monolayers comprising (a) mercaptocarboxylic acids or (b) aminoalkanethiols. | ||
Second, we evaluated the densities of the ligands on the Au surfaces using QCM measurements (Table 1). These densities indicated that the charge density of the ligand molecules is comparable to, or slightly higher than, that of the oligopeptides. When using an MCA, the molecular density increases slightly with an extended alkyl chain in the order 3-MPA < 6-MHA ≈ 4-MBA. When using an AAT, the molecular density increases with an extended alkyl chain in the order 2-AET ≪ 5-APT < 3-APT. The extended alkyl chain of the ligand increases the molecular density of the SAM,35 but the interactions between the charged moieties likely result in a higher density at the intermediate alkyl length when using an MCA or AAT. In addition, the molecular density of an MCA is higher than that of an AAT with a similar alkyl chain length, which is consistent with the results of our previous studies.32 The MCAs form hydrogen bonds between their carboxyl groups,36 likely resulting in denser adsorption than that of AATs.
| Building blocks of the SAM | Density (nmol cm−2) | Work function (eV) | Contact angle (°) |
|---|---|---|---|
| Values represent the mean and standard error of three measurements. | |||
| 3-Mercaptopropionic acid | 2.15 ± 0.05 | 4.89 ± 0.02 | 44.7 ± 1.7 |
| 4-Mercaptobutanoic acid | 2.59 ± 0.12 | 4.88 ± 0.01 | 31.7 ± 1.1 |
| 6-Mercaptohexanoic acid | 2.48 ± 0.16 | 4.89 ± 0.01 | 28.1 ± 1.5 |
| 2-Aminoethanethiol | 1.17 ± 0.18 | 4.70 ± 0.01 | 52.1 ± 1.7 |
| 3-Aminopropanethiol | 2.47 ± 0.05 | 4.73 ± 0.01 | 57.5 ± 1.0 |
| 5-Aminopentanethiol | 2.30 ± 0.06 | 4.66 ± 0.01 | 50.7 ± 0.5 |
Third, we performed PYSA to confirm the donor or acceptor properties of the Au surfaces modified with the SAMs, which were determined using the shifts in the work functions (Fig. S2†). The work function of the untreated SAM is 4.81 ± 0.01 eV. After modification with an MCA, the work function shifts to approximately 4.9 eV (Table 1), suggesting the presence of electron-withdrawing moieties and thus the adsorption of carboxyl groups on the Au surfaces. In contrast, the work function shifts to approximately 4.7 eV after modification with an AAT (Table 1), suggesting the presence of electron-donating moieties and thus the adsorption of amino groups on the Au surfaces. The work function is independent of the alkyl length of the MCA, whereas it varies slightly with the alkyl length of the AAT in the order 3-APT ≈ 2-AET > 5-APT. These results suggest that the Au surfaces modified with 5-APT exhibits the strongest acceptor properties.
Finally, the water contact angles on the SAM surfaces were measured using CAG to evaluate the hydrophobicities of the reaction fields with the accumulated charged residues. When using the MCAs as the building blocks of the SAMs, the contact angle decreases as the alkyl chain length of the ligand increases, which results in an enhanced hydrophilicity (Table 1). The increased orientation of the carboxyl groups on the SAM with increasing alkyl chain length results in an enhanced hydrophilicity derived from the carboxyl groups rather than an enhanced hydrophobicity derived from the alkyl chain of the ligand. Conversely, the contact angle when using an AAT increase in the order 2-AET ≈ 5-APT < 3-APT, although the change is insignificant (Table 1). These results suggest that SAMs with 6-MHA as building blocks are the most hydrophilic and those with 3-APT are the most hydrophobic. Additionally, the surface hydrophobicities of the SAMs are higher when using the AATs compared to those observed when using the MCAs with similar alkyl chain lengths (Table 1).
The results of XPS, QCM measurement, PYSA, and CAG reveal that varying the alkyl length of the ligand from 2 to 5 results in slight changes in the density and donor/acceptor properties of the SAMs. However, varying the alkyl length of the ligand results in significant changes in the hydrophobicity of the SAMs.
Electrical detection of the interactions between the peptides and SAMs terminated with charged residues
Initially, we evaluated the interactions between the peptides and accumulated charged residues on the Au surfaces. Fig. S3† shows relationship between the peptide concentration and changes in the Vout of the FET-based SAM system. Concentration-dependent responses are observed in the titration studies with the peptide solutions (Fig. S3†). Most peptides change their electrical potentials positively, but several peptides with more than two degrees of polymerization change their electrical potentials negatively (Fig. S3†). The molecular orientations on the Au surfaces are likely influenced by the conformations of the peptides, and thus, several electrical potentials can change negatively. When using the monoamino acids as analytes, the electrical potentials change positively, independent of the type of amino acid, owing to the identical orientations of the terminal carboxyl and amino groups, which is consistent with our previous studies.32 For each electrical potential curve shown in Fig. S3,† the Kd at the peptide scale was calculated by fitting to eqn (1), where [x] is the peptide concentration (Fig. 3a). When using a SAM modified with carboxyl groups, the Kd of the Gly peptide increases slightly with increasing peptide length (Fig. 3b). It is likely that the electrostatic interaction between the terminal amino group of the Gly peptide and the carboxyl moiety on the SAM surface becomes weaker as the peptide length increases. The Kd values of the Lys peptides do not change considerably, indicating that the interactions between Lys and the carboxy groups are consistent and independent of the peptide length (Fig. 3c). The Kd values of the Glu peptides decrease slightly, indicating that the longer the Glu peptide is, the stronger the interactions between Glu and the carboxy groups are owing to the enhanced hydrogen bonds between carboxylic acids (Fig. 3d). When the SAMs are modified with amino groups, differences in Kd based on the ligand rather than the peptide length are observed, particularly when using the tri- and tetrapeptides (Fig. 3e–g). Amino groups serve as a hydrogen bond acceptor dependent on the reaction environment,37 and thus, the different properties of the amine accumulated fields should alter the hydrogen bonds between amino moiety of SAM and peptides.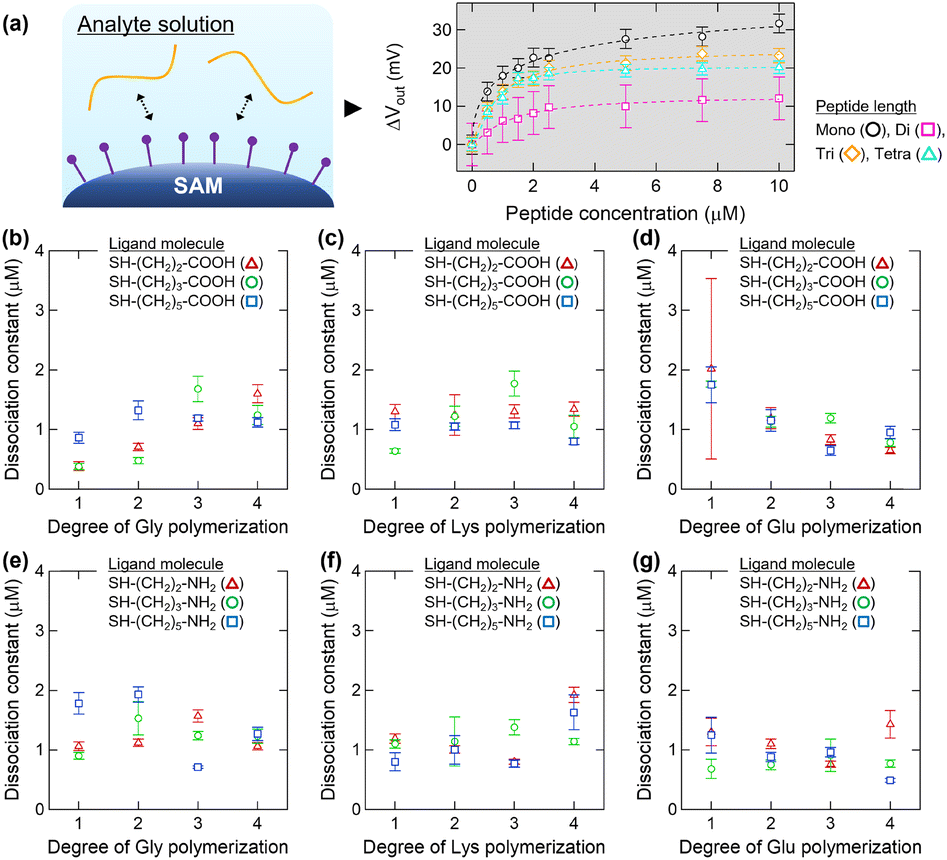 | ||
| Fig. 3 Analysis of the interactions between peptides and accumulated charged residues. (a) Schematic of AAIs and an example of an electrical response at the peptide scale. (b–g) Dissociation constants depending on the chain lengths of (b and e) Gly, (c and f) Lys, and (d and g) Glu peptides using the SAMs comprising (b–d) carboxy or (e–g) amino groups. Error bars represent the standard deviation obtained from the Hill–Waud fitting. | ||
Collectively, these results indicate that the intensities of AAIs vary with the environment provided via the accumulation of charged residues and peptide length, but the differences are smaller than those expected. In particular, a possible explanation for the lack of strong electrostatic repulsion force between similarly charged ligand and analyte molecules is the contribution of terminal amino or carboxyl groups of the peptides to the AAIs.
Analysis of converted amino acid interactions per monomer unit
We then analysed the AAIs per amino acid using the interactions between the peptides and accumulated charged residues on the Au surfaces. Fig. S4† shows the relationship between the concentration of amino acids in our measurement system and the changes in the Vout, which is obtained by converting the horizontal axis shown in Fig. S3† with respect to monomer concentration. For each electrical potential curve shown in Fig. S4,† the Kd at the monomer scale was calculated by fitting to eqn (1), where [x] is the monomer concentration (Fig. 4a). The Kd values analysed at the monomer scale for almost all SAMs increase with increasing peptide chain length, indicating that the longer the peptide is, the weaker the interactions per amino acid unit are (Fig. 4b–g). Notably, although the interactions between the peptide and accumulated charged moieties are stronger at the peptide scale (Fig. 3d), the interactions with each amino acid are weaker (Fig. 4d). Although polymers with continuously charged amino acids display stronger overall interactions than those of polymers with non-continuously charged amino acids,15 this study reveals an unintuitive result in that the relative AAIs are weaker with increasing continuity in the amino acid sequence. Consecutive sequences of identically charged amino acids could enhance electrostatic repulsion between side chains within the analyte, thereby weakening the interaction between peptides and ligand molecules. Remarkably, when using SAMs terminated with amino groups, the increase in Kd varies significantly for peptides with even and odd degrees of polymerization. This unexpected result indicates that the intensities of the interactions per amino acid unit, particularly hydrogen bonds affected by the peptide conformation, depend on the even–odd effects of the peptides in the reaction fields with accumulated amino groups.38 | ||
| Fig. 4 Analysis of the interactions per amino acid unit. (a) Schematic of AAIs and an example of the electrical response at the monoamino acid scale. (b–g) Dissociation constants depending on the chain lengths of the (b and e) Gly, (c and f) Lys, and (d and g) Glu peptides using the SAMs comprising (b–d) carboxy or (e–g) amino groups. Error bars represent the standard deviation obtained from the Hill–Waud fitting. | ||
In summary, as sequence continuity increases: i) the intensities of overall peptide interactions change slightly, ii) the individual AAIs per amino acid unit are weakened significantly, and iii) the reaction field with accumulated side chains alters not only the intensities of the overall peptide interactions but also those of the individual AAIs. Notably, the ligand effects were observed with analyte of longer chain length, indicating that the AAIs of longer peptides are more sensitive to the properties of reaction fields.
Discussion
We successfully evaluated the dependences of AAIs on peptide length using SAMs terminated with charged residues. The monomer-scale interactions are weaker, whereas the peptide-scale interactions display various changes, such as increases, decreases, and consistency with increasing peptide length (Fig. 3 and 4). The additive effects of mixed amino acids can be explained as the sum of the effects of each amino acid, whereas the effects observed when using homopeptides cannot be accounted for by simple summation.39 This trend is consistent with the findings of our study using homopeptides, indicating that future investigations using heteropeptides are necessary to evaluate the cumulative property of AAIs.PCA is a superior statistical tool for use in interpreting multidimensional datasets, and it can extract the most critical characteristics of the data.40 Hence, we performed PCA of the multidimensional electrical response pattern (6-dimensional, 6 ligands) obtained when titrating 12 types of analytes containing 10 μM peptide to consider the diverse variations in the peptide-scale interactions. Each pattern generated using the six ligands was converted to principal component (PC) scores and plotted in PC space (Fig. 5). The PCA score plot utilises the first three PCs representing approximately 75% of the variance, and thus, the interactions responsible for each peptide can be explained using three characteristics. PC1 exhibits the largest contribution (35.0%), and the large fluctuations in Lys and Glu indicate the contribution of electrostatic interactions (Fig. 5). Among the Gly peptides, only triglycine shifts negatively in the PC1 space, similar to the Lys peptides, reflecting the influence of main-chain amino groups due to peptide conformation. PC2 displays the second-largest contribution of 23.4%, with large fluctuations in Gly and Lys (Fig. 5a). Mono-Gly and tri-Gly are close to 0, and mono-Lys and tri-Lys change positively in the PC2 space, whereas di-Gly and tetra-Gly are far from 0, and di-Lys and tetra-Lys change negatively. As the even–odd effects of the peptides are represented in this space, PC2 reflects the contribution of hydrogen bonds due to the different peptide conformations.41,42 PC3 exhibits the third-largest contribution of approximately 17% and large fluctuations in all amino acids indicate the contribution of hydrophobic interactions (Fig. 5b). The hydrophobicity of Gly increases with increasing peptide chain length,43 and thus, the negative change in the PC3 space may indicate an enhanced hydrophobicity. In the PC3 space, the positive changes in tetra-Lys and tetra-Glu, with abundant hydrophilic side chains, also indicate that the hydrophilic contribution may be reflected positively in this space. These considerations are summarised as follows: in reaction fields with accumulated charged residues, the contributions of the i) hydrogen bonds and hydrophobic interactions of the Gly peptide, ii) hydrogen bonds, electrostatic and hydrophobic interactions of the Lys peptide, and iii) electrostatic and hydrophobic interactions of the Glu peptide, although hydrogen bonds should predominantly contribute to SAMs modified with carboxylic acids (Fig. 3d), depend on peptide length.
 | ||
| Fig. 5 PCA plot of peptides for SAMs modified with alkanethiols. (a) PC1 vs. PC2 and (b) PC1 vs. PC3 plot of the first three PCs describing about 75% of the total variance. PCs were created using a standardized dataset of 12 analytes × 6 ligands × 6 replicates. G, K and E mean Gly, Lys and Glu respectively, and the side numbers mean the polymerization degree of peptides. | ||
In this study, reaction fields with different properties, such as hydrophobicity and density, were fabricated using AATs and MCAs with different alkyl chain lengths (Table 1). These molecules mimic the side chains of charged amino acids, particularly the AAT with an alkyl chain length of four carbon atoms, which is identical to the side chain of Lys, and the MCA with an alkyl chain length of two carbon atoms, which is identical to the side chain of Glu. Amino acids with different alkyl side chain lengths are abundant in nature,44 but only those with specific alkyl chain lengths are used as protein building blocks. Mysteriously, hydrophobic and basic amino acids with different alkyl side chain lengths also occurred in the prebiotic world, but only approximately 20 amino acids were evolutionarily selected because of their advantages in protein folding and solubility.45 For an in-depth consideration of such evolutionary selection, the surface properties of the SAMs provide another perspective (Table 1), e.g. carboxyl groups with extended or short alkyl chains provide a hydrophilic or -phobic reaction field, respectively (Table 1). Short carboxylic acids, such as Glu, are used to produce a hydrophobic environment suitable for protein folding. Amino groups with longer alkyl lengths provide reaction fields with strong acceptor properties (Table 1). To enhance the transfer of electrons via proteins, extended amines, such as Lys, are used instead of short amines, such as diaminopropionic acid and diaminobutyric acid.45 The SAMs formation with other amino acid side chains may provide an interesting perspective in terms of explaining the existence of approximately 20 types of proteinogenic amino acids.
Conclusions
We fabricated SAMs with accumulated charged residues and treated them as pseudoprotein surfaces to understand the properties of the reaction fields generated via the assembly of charged residues. The accumulation field of the amino groups was more hydrophobic than that of the carboxyl groups, although the molecular densities of the fields were almost identical. Furthermore, to evaluate the changes in the AAIs caused by the continuity of the sequence, we detected the electrical potential during peptide titration with each FET-based SAM. Remarkably, the interactions per amino acid unit weakened with increasing peptide length, and even–odd effects of the peptides were observed in the reaction fields with accumulated amines. Multivariate analysis indicated that the contributions of hydrogen bonds, electrostatic and hydrophobic interactions varied with increasing peptide length for each amino acid. These findings provide valuable insights into the control of biological reactions in cellulo according to mutations in the charged residues and the improvement of protein stability in vitro using synthetic polymers. Furthermore, our FET-based SAM system, which can detect interactions at the single residue level, would facilitate the analysis of AAIs in hetero sequences as well as homo sequences.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. N.: conceptualization, investigation, writing – original draft. K. S.: conceptualization, supervision. T. M.: investigation, methodology, supervision. All authors: funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially conducted at the AIST Nano-Processing Facility supported by the Nanotechnology Platform Program of the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We acknowledge BioCMOS and Mitorika (Mito, Japan) for preparing and supplying the FET sensor array chips. We gratefully acknowledge the financial support provided by the JST PRESTO Program (Grant No. JPMJPR21RB), Japan Society for the Promotion of Science KAKENHI (Grant No. 23KJ0259 and 24K01970), Tanaka Kikinzoku Memorial Foundation, and Fuji Seal Foundation and a special strategic grant from AIST.Notes and references
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS PubMed.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS PubMed.
- S. Miller, J. Janin, A. M. Lesk and C. Chothia, J. Mol. Biol., 1987, 196, 641–656 CrossRef CAS PubMed.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
- S. S. Strickler, A. V. Gribenko, A. V. Gribenko, T. R. Keiffer, J. Tomlinson, T. Reihle, V. V. Loladze and G. I. Makhatadze, Biochemistry, 2006, 45, 2761–2766 CrossRef CAS PubMed.
- R. M. Kramer, V. R. Shende, N. Motl, C. N. Pace and J. M. Scholtz, Biophys. J., 2012, 102, 1907–1915 CrossRef CAS PubMed.
- F.-X. Theillet, L. Kalmar, P. Tompa, K.-H. Han, P. Selenko, A. K. Dunker, G. W. Daughdrill and V. N. Uversky, Intrinsically Disord Proteins, 2013, 1, e24360 CrossRef PubMed.
- J. C. Hansen, X. Lu, E. D. Ross and R. W. Woody, Biol. Chem., 2006, 281, 1853–1856 CrossRef CAS PubMed.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- S. Boyko, K. Surewicz and W. K. Surewicz, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 31882–31890 CrossRef CAS PubMed.
- S. K. Rai, R. Khanna, A. Avni and S. Mukhopadhyay, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216338120 CrossRef CAS PubMed.
- S. Xue, R. Gong, F. He, Y. Li, Y. Wang, T. Tan and S.-Z. Luo, Sci. Adv., 2019, 5, eaax5349 CrossRef CAS PubMed.
- T. Ura, A. Kagawa, N. Sakakibara, H. Yagi, N. Tochio, T. Kigawa, K. Shiraki and T. Mikawa, Sci. Rep., 2023, 13, 1435 CrossRef CAS PubMed.
- A. Nomoto and K. Shiraki, Int. J. Biol. Macromol., 2025, 296, 139500 CrossRef CAS PubMed.
- L.-W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1273 CrossRef PubMed.
- U. Rana, K. Xu, A. Narayanan, M. T. Walls, A. Z. Panagiotopoulos, J. L. Avalos and C. P. Brangwynne, Nat. Chem., 2024, 16, 1073–1082 CrossRef CAS PubMed.
- Y.-C. Syu, W.-E. Hsu and C.-T. Lin, ECS J. Solid State Sci. Technol., 2018, 7, Q3196 CrossRef CAS.
- P. Bergveld, Sens. Actuators, B, 2003, 88, 1–20 CrossRef CAS.
- J. M. Rothberg, W. Hinz, T. M. Rearick, J. Schultz, W. Mileski, M. Davey, J. H. Leamon, K. Johnson, M. J. Milgrew, M. Edwards, J. Hoon, J. F. Simons, D. Marran, J. W. Myers, J. F. Davidson, A. Branting, J. R. Nobile, B. P. Puc, D. Light, T. A. Clark, M. Huber, J. T. Branciforte, I. B. Stoner, S. E. Cawley, M. Lyons, Y. Fu, N. Homer, M. Sedova, X. Miao, B. Reed, J. Sabina, E. Feierstein, M. Schorn, M. Alanjary, E. Dimalanta, D. Dressman, R. Kasinskas, T. Sokolsky, J. A. Fidanza, E. Namsaraev, K. J. McKernan, A. Williams, G. T. Roth and J. Bustillo, Nature, 2011, 475, 348–352 CrossRef CAS PubMed.
- T. Sakata and Y. Miyahara, Angew. Chem., Int. Ed., 2006, 45, 2225–2228 CrossRef CAS PubMed.
- N. Crivillers, E. Orgiu, F. Reinders, M. Mayor and P. Samorì, Adv. Mater., 2011, 23, 1447–1452 CrossRef CAS PubMed.
- R. T. Weitz, U. Zschieschang, F. Effenberger, H. Klauk, M. Burghard and K. Kern, Nano Lett., 2007, 7, 22–27 CrossRef CAS PubMed.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS PubMed.
- L. J. Cristina, G. Ruano, R. Salvarezza and J. Ferrón, J. Phys. Chem. C, 2017, 121, 27894–27904 CrossRef CAS.
- F. Sun, H.-C. Hung, A. Sinclair, P. Zhang, T. Bai, D. D. Galvan, P. Jain, B. Li, S. Jiang and Q. Yu, Nat. Commun., 2016, 7, 13437 CrossRef CAS PubMed.
- T. Minamiki, Y. Ichikawa and R. Kurita, ACS Appl. Mater. Interfaces, 2020, 12, 15903–15910 CrossRef CAS PubMed.
- T. Minamiki, Y. Ichikawa and R. Kurita, Sensors, 2020, 20, 2228 CrossRef CAS PubMed.
- T. Minami, Y. Sasaki, T. Minamiki, S. Wakida, R. Kurita, O. Niwa and S. Tokito, Biosens. Bioelectron., 2016, 81, 87–91 CrossRef CAS PubMed.
- S. M. Patrie and M. Mrksich, Anal. Chem., 2007, 79, 5878–5887 CrossRef CAS PubMed.
- T. Minami, T. Minamiki, Y. Hashima, D. Yokoyama, T. Sekine, K. Fukuda, D. Kumaki and S. Tokito, Chem. Commun., 2014, 50, 15613–15615 RSC.
- T. Minamiki and R. Kurita, Anal. Methods, 2019, 11, 1155–1158 RSC.
- A. Nomoto, K. Shiraki and T. Minamiki, RSC Appl. Interfaces, 2025, 2, 243–250 RSC.
- H. Sota, H. Yoshimine, R. F. Whittier, M. Gotoh, Y. Shinohara, Y. Hasegawa and Y. Okahata, Anal. Chem., 2002, 74, 3592–3598 CrossRef CAS PubMed.
- W. I. Lencer, S. H. Chu and W. A. Walker, Infect. Immun., 1987, 55, 3126–3130 CrossRef CAS PubMed.
- D. M. Spori, N. V. Venkataraman, S. G. P. Tosatti, F. Durmaz, N. D. Spencer and S. Zürcher, Langmuir, 2007, 23, 8053–8060 CrossRef CAS PubMed.
- A. Kudelski, Surf. Sci., 2002, 502–503, 219–223 CrossRef CAS.
- T. Nagae, M. Takeda, T. Noji, K. Saito, H. Aoyama, Y. Miyanoiri, Y. Ito, M. Kainosho, Y. Hirose, H. Ishikita and M. Mishima, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2404472121 CrossRef CAS PubMed.
- A. Grabarek, Ł. Walczak and P. Cyganik, J. Phys. Chem. B, 2021, 125, 10964–10971 CrossRef CAS PubMed.
- X. Xu and F. Stellacci, J. Phys. Chem. Lett., 2024, 15, 7154–7160 CrossRef CAS PubMed.
- M. A. Palacios, Z. Wang, V. A. Montes, G. V. Zyryanov and P. Anzenbacher Jr., J. Am. Chem. Soc., 2008, 130, 10307–10314 CrossRef CAS PubMed.
- L. Sangroniz, J. L. Olmedo-Martínez, W. Hu, Y.-J. Jang, G. Liu, M. A. Hillmyer and A. J. Müller, Biomacromolecules, 2024, 25, 7500–7510 CrossRef CAS PubMed.
- H.-T. Kuo, C.-J. Fang, H.-Y. Tsai, M.-F. Yang, H.-C. Chang, S.-L. Liu, L.-H. Kuo, W.-R. Wang, P.-A. Yang, S.-J. Huang, S.-L. Huang and R. P. Cheng, Biochemistry, 2013, 52, 9212–9222 CrossRef CAS PubMed.
- J. Lu, X.-J. Wang, X. Yang and C.-B. Ching, J. Chem. Eng. Data, 2006, 51, 1593–1596 CrossRef CAS.
- V. Vranova, K. Rejsek, K. R. Skene and P. Formanek, Plant Soil, 2011, 342, 31–48 CrossRef CAS.
- M. Makarov, A. C. S. Rocha, R. Krystufek, I. Cherepashuk, V. Dzmitruk, T. Charnavets, A. M. Faustino, M. Lebl, K. Fujishima, S. D. Fried and K. Hlouchova, J. Am. Chem. Soc., 2023, 145, 5320–5329 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including analytical details of the output potentials and self-assembled monolayers. See DOI: https://doi.org/10.1039/d5lf00069f |
| This journal is © The Royal Society of Chemistry 2025 |