
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasmon resonance dynamics and enhancement effects in tris(2,2′-bipyridine)ruthenium(II) gold nanosphere oligomers†
Umar
Yunusa
 a,
Natalie
Warren
a,
David
Schauer
ab,
Prasenjit
Srivastava
a and
Emily
Sprague-Klein
*a
a,
Natalie
Warren
a,
David
Schauer
ab,
Prasenjit
Srivastava
a and
Emily
Sprague-Klein
*a
aDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: emily_sprague-klein@brown.edu
bETH Zurich, Department of Chemistry and Applied Biosciences, LPC, Vladimir-Prelog-Weg 2, 8049 Zürich, Switzerland
First published on 22nd February 2024
Abstract
Ruthenium-based metal complexes are one of the most widely studied dyes because of their rich photochemistry and light-harvesting properties. Significant attention has been paid to the energy and charge transfer dynamics of these dyes on semiconductor substrates. However, studies on photophysical and photochemical properties of these dyes in plasmonic environments are rare. In this study, we report a plasmon-mediated resonance energy transfer in an optimized oligomer system that enhances the photoexcited population of the well known dye, tris(2,2′-bipyridine)ruthenium(II), [Ru(BPY)3]2+ adsorbed on gold nanosphere surfaces with a defluorescenced Raman signal. Structural and chemical information is collected using a range of techniques that include in situ time-resolved UV/VIS, DLS, SERS, and TA. The findings have great potential to impact nanoscience broadly with special emphasis on surface photocatalysis, redox chemistry, and solar energy harvesting.
 Emily Sprague-Klein | Emily Sprague-Klein is an Assistant Professor of Chemistry at Brown University. She worked with the late Richard Van Duyne for her PhD in Applied Physics at Northwestern University. She was then a postdoc in the Solar Energy Conversion Group at Argonne National Laboratory joint with Northwestern University advised by Lin X. Chen and David Tiede. Her research is focused on plasmon-driven chemical reactions for facilitating surface redox activity. Techniques employed in her group include the development of surface-enhanced spectroscopies both ultrafast and steady-state for elucidating light activated dynamics in nanomaterials of varying compositions and morphologies. |
Introduction
Noble metal nanoparticles possess unique properties that allow for highly localized chemical selectivity, reactivity, and stability.1–3 With the aid of localized surface plasmon resonances (LSPRs), these nanostructured materials can confine light at the nanoscale level with great tunability, and can play a critical role in creating thermodynamically favorable conditions for energy intensive reactions to proceed efficiently at high product yields.4,5 Notably, the extremely intense local fields generated on plasmonic surfaces can dramatically enhance the optical properties of proximal molecular species such as Raman scattering cross sections, light absorption, IR extinction and fluorescence processes. The relative shape, size, composition, and aggregation state of these nanostructures influences the magnitude of the plasmonic enhancement as well as the reaction dynamics of the adsorbed molecular species.6–8Ru-based polypyridyl complexes are widely studied because of their rich photochemistry.9 Upon photoexcitation, these complexes generate a long-lived excited state which enables either charge transfer or energy transfer with a substrate.10 The photophysics and interfacial charge-transfer dynamics of these complexes have been a topic of interest because they play a pivotal role in photocatalysis and light harvesting applications. In most of these applications, the dyes are typically adsorbed onto the surface of semiconductors, such as TiO2 and ZnO. The dye-substrate interactions are of fundamental importance because they strongly affect the transfer of electrons from the photoexcited molecules to the substrate, which can influence the light harvesting applications.11 For these reasons, the investigation of the adsorption modes and charge transfer mechanisms for dye/semiconductors systems has been the focus of many studies.12 At present, how adsorbing Ru-based dyes onto a plasmonic surface impacts the photophysics and charge transfer dynamics of the system has received limited attention. These plasmon–molecule systems have great potential to show intriguing photophysical and photochemical properties which can be exploited in the nascent field of plasmon-mediated chemistry. For example, a recent study exploited the plasmon–molecule interaction in the weak coupling regime to modulate the absorbance of gold nanostructures.13
Generally, isolated gold nanospheres do not offer a maximized plasmonic effect for fundamental studies and practical applications. One of the viable strategies to create highly active plasmonic platforms is to engineer the interparticle nanogap-induced “hotspots” by controlled aggregation of the nanosphere monomers. This is because when two or more particles form aggregates, the surface plasmons excited by each particle interact resulting in a dramatic enhancement of the electromagnetic field on the nanoparticle surface.14 The intense local field generated allows for ultrasensitive detection and enhanced photochemistry. Currently, one major issue in coupled molecular–plasmonic metal nanostructure systems is that there are relatively few studies which quantify surface–molecule interactions and absorption kinetics correlated with chemical properties.15–19 Elucidating the optimal nanoscale conditions for surface–molecule interactions is especially important because uncontrolled aggregation or instability of assumed active sites can severely limit applications for catalysis, spectroscopy and light-harvesting processes.15,16,20,21
Early observations of adsorbate–molecule induced aggregation of metal nanoparticles were reported by Moskovits and Vlčková where the aggregation rate constant depends nontrivially on adsorbate concentration.22 For gold and other metallic nanoparticles in general, there exists a range of competing interactions such as van der Waals, electrical double layers, hydration forces, hydrophobic forces, steric electronic interactions, and electrostatic forces that drive aggregation reactions.16,23 Recent studies have examined molecule–nanoparticle interactions while distinguishing between chemisorption energies and electrostatic aggregation pathways in characterizing electronic energy shifts in plasmon resonances resulting from molecule–plasmon orbital overlap.15,19,24–26 To the best of our knowledge, there are no reports on the impact of plasmonic field enhancements on the energy transfer or charge transfer dynamics of a Ru-based dye gold nanosphere oligomer system.
Our work provides detailed explanation of the oligomerization reaction induced by the prototypical Ru-based photoredox catalyst tris(2,2′-bipyridine)ruthenium(II), also written as [Ru(BPY)3]2+, with gold nanoparticles of varying sizes, acting as novel system for observing light-trapping properties in nanoscale confinement under plasmonic high-field conditions. We performed in situ oligomerization reactions to observe shifting absorption and plasmon resonance energies with structure–function correlations using dynamic light scattering (DLS) and transmission electron microscopy (TEM). The observed oligomer reaction deviates notably from the Langmuir isotherm with a saturation effect that depends on the size of the gold nanosphere. Implications for surface–molecule interactions include a plasmon mediated quenching of the fluorescence background in surface-enhanced Raman spectroscopy (SERS) and a significant enhancement of a photoexcited population that is stabilized near the surface following simultaneous excitation of both the molecule and gold nanosphere. The findings have far reaching implications in utilizing plasmonic oligomers as a novel approach for driving site-specific photoredox reactions and for probing excited states of molecular catalysts in plasmonic confinement.
Experimental
Nanospheres and chemicals
Aqueous suspensions of citrate capped gold nanospheres (AuNS) of two different sizes (40 and 100 nm in diameter) were purchased from nanoComposix (San Diego, CA). Tris(2,2′-bipyridine)ruthenium(II) chloride hexahydrate ([Ru(BPY)3]2+; see Fig. 1) ethanol (ACS grade), polyvinylpyrrolidone (Mw ∼ 55 kDa), nitric acid (60%, ultrapur) and hydrochloric acid (30%, ultrapur) were purchased from Sigma-Aldrich (Burlington, MA). Single element ruthenium and gold standards (1000 μg mL−1, 10% HCl) were obtained from Fisher scientific (Franklin, MA). All chemicals were used without further purification. Milli-Q water (18.2 MΩ. cm) was used for preparing samples and standards.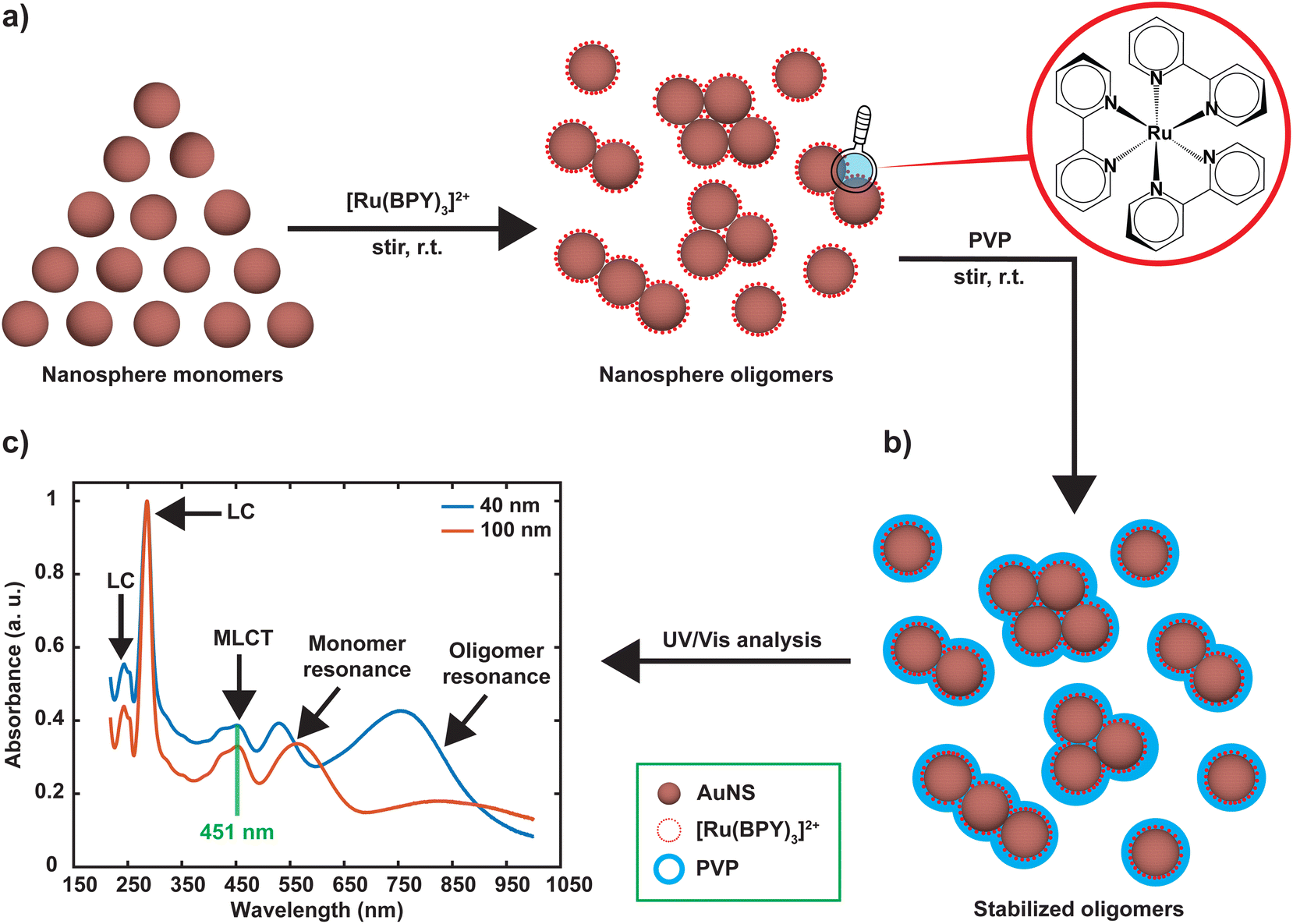 | ||
| Fig. 1 Scheme for preparation of nanosphere oligomers. (a) Oligomers are formed upon injection of 40 μM [Ru(BPY)3]2+ aqueous solution to isolated gold nanospheres suspension. (b) Oligomers formed are stabilized by injection of 200 μL of 10% aqueous PVP solution. (c) UV-vis spectra of the [Ru(BPY)3]2+/AuNS oligomers in water, electronic assignments are shown. | ||
Nanosphere aggregation with [Ru(BPY)3]2+
All preparations were done sequentially to preserve the inertness of the system which was deoxygenated under argon atmosphere using Schlenk line and glovebox techniques. Briefly, 1 mL of AuNS monomer (unaggregated) suspension in a 2 mm pathlength quartz cuvette (FUV, Spectrocell Inc.) equipped with septum was thoroughly deoxygenated with argon. Then the in situ UV–Vis spectroscopic monitoring of the aggregation was performed using a Cary 60 spectrophotometer from Agilent (Santa Clara, CA). In a typical experiment, the aggregation was induced by injecting through the septum a standard volume of deoxygenated [Ru(BPY)3]2+ ethanolic solution into the AuNS dispersion where the final dye concentration in the system was varied from nM to mM regimes. Before measurements, the AuNS-[Ru(BPY)3]2+ mixture was quickly stirred to ensure homogeneous mixing, and the spectra of the aggregated particles were continuously recorded from 200 to 1100 nm at predetermined intervals. The interval between stirring of the sample and measurement of the first spectrum was typically 8 s. Finally, the aggregation was stopped by the injection of 200 μL of deoxygenated 10% aqueous PVP solution as reported in the literature.19 For comparison, additional aggregation experiments were performed using the same conditions, but the order of injection is reversed such that AuNS dispersion was added to the [Ru(BPY)3]2+ solution.Nanosphere oligomer characterization
Samples of nanosphere oligomers which consist of aggregated 40 nm or 100 nm sphere cores and adsorbed [Ru(BPY)3]2+ were characterized as follows. Particle size distribution measurements at different aggregation times were accomplished by the dynamic light scattering using a Malvern Zetasizer Nano S90 instrument. TEM images were acquired on a JEOL 2100F microscope operating at 200 kV. The quantification of ruthenium adsorbed onto the AuNS surface was achieved based on inductively coupled plasma-optical emission spectrometry (ICPOES) using Thermo Scientific iCAP 7400 DUO instrument. The Raman and SERS spectra were obtained using a Witech Alpha 300 confocal Raman microscope with a 50×/0.6 objective equipped with a 457 nm, 532 nm, 633 nm, and 785 nm excitation laser. Transient absorption (TA) spectroscopy measurements were performed on a Spectra Physics ultrafast spectrometer using a pump–probe configuration. More details on the characterization are available in the methods section of the ESI.†Results and discussion
Formation of nanosphere oligomers
Nanosphere oligomers consisting of 40 nm or 100 nm spherical cores and [Ru(BPY)3]2+ were prepared under inert conditions of argon to exclude molecular oxygen from the system. Fig. 1a schematically illustrates the strategy for forming the oligomers, in which a standard volume of deoxygenated [Ru(BPY)3]2+ stock ethanolic solution was quickly injected to aqueous dispersion of gold monomer colloids to trigger the aggregation process which can be visually verified by a color change of the colloidal dispersion. The aggregation triggered by [Ru(BPY)3]2+ can be explained by the neutralization of the negative surface charge of the citrate-capped gold monomers leading to enhanced attractive interactions between neighboring gold nanospheres. Interestingly, the degree of aggregation can be controlled in real time by injection of polyvinylpyrrolidone (PVP), resulting in very stable oligomers (Fig. 1b).27 The ability to preserve the plasmonic properties coupled with easy tunability of the plasmonic response make these stabilized aggregates viable platforms for plasmon-mediated catalytic reactions and plasmon-enhanced spectroscopy under harsh operation conditions. The optical absorption spectra obtained after stabilizing the oligomers and assignments for the underlying transitions, are depicted in Fig. 1c. Both the 40 nm and 100 nm AuNS samples exhibit coupled plasmon resonance modes, as reflected by the broad bands at longer wavelengths (600–1000 nm) in addition to the plasmon modes of the isolated nanospheres. Absorption features centered at 451 nm, 285 nm, and 244 nm can also be seen in the spectra which correlate well with the ruthenium to bipyridine charge transfer (MLCT) transitions, as well as ligand centered (LC) π to π1* and π to π2* transitions on the bipyridine, respectively.10,28–30 We have also observed that the 453 nm MLCT band of the free dye solution is very slightly shifted to higher energies (451 nm) when the dye is in plasmonic environment, which is indicative of the interaction of the [Ru(BPY)3]2+ with the gold nanosphere surface. This finding was consistent with the blue shift (544 nm to 542 nm) that was reported when a ruthenium dye (N3) was adsorbed onto a TiO2 nanoparticle surface, which was attributed to weak electronic coupling between the adsorbed N3 and the semiconductor substrate.31In order to achieve the desired degree of oligomerization and better plasmonic characteristics, the aggregation behavior was investigated by varying the final [Ru(BPY)3]2+ concentration in the system from nM to mM regimes. We track the formation of the aggregates by an in situ UV/VIS spectroscopy with which the absorption spectra were continuously acquired for 120 min. When the final [Ru(BPY)3]2+ concentration in the colloidal dispersion is in the nM regime, neither red shifting of the monomer peak nor the appearance of secondary plasmon peak was observed for both samples even after one month, signifying the absence of any aggregation (Fig. S1†). Therefore, we adjusted to μM concentrations where secondary spectral features at longer wavelengths begin to appear for the 40 nm sample when the final dye concentration in the system reaches 2 μM. This indicates that a certain critical [Ru(BPY)3]2+ concentration is required to change the aggregation propensity of the well dispersed gold nanospheres and induce oligomer formation. Representative examples of the spectral evolution during the process of aggregation for different dye concentrations are shown in (Fig. 2 and S1†).
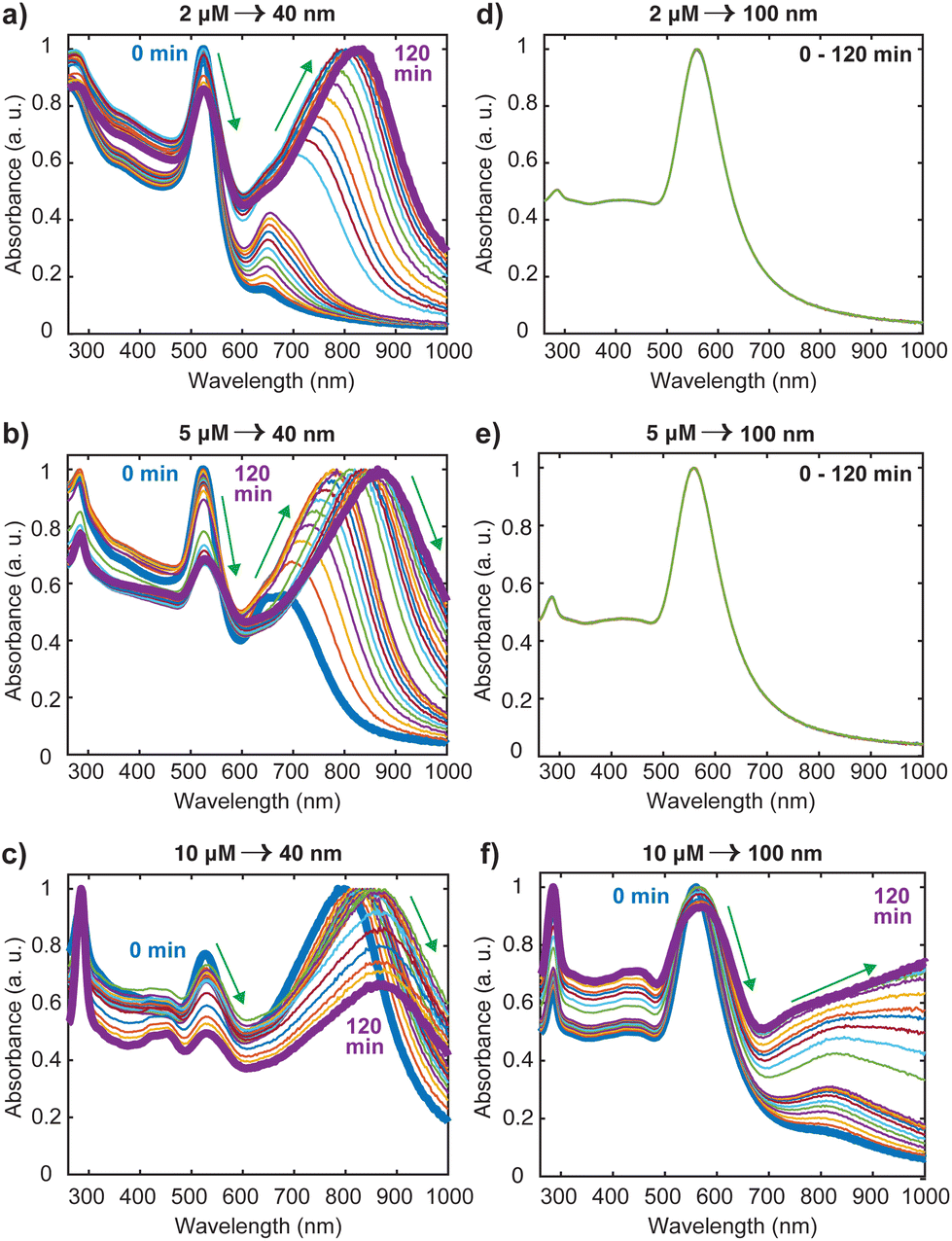 | ||
| Fig. 2 Time evolution of the absorption spectra during the aggregation of 40 nm and 100 nm gold nanospheres with different [Ru(BPY)3]2+concentrations. The systems are: (a) 2 μM, 40 nm AuNS. (b) 5 μM, 40 nm AuNS. (c) 10 μM, 40 nm AuNS. (d) 2 μM, 100 nm AuNS. (e) 5 μM, 100 nm AuNS. (f) 10 μM, 100 nm AuNS. The arrows show the decay, red shift and growth of the plasmon peaks. | ||
Using the 40 nm oligomers as a reference, the general trend is a decay of the monomer peak and concurrent growth and red-shift of the oligomer band as the time progresses (Fig. 2(a)–(c)). We observed that the aggregation rate was controlled by the dye concentration as the significantly red-shifted oligomer peaks dominate the spectrum after 60 min, 30 min, and 1 min, for the 2 μM, 5 μM, and 10 μM [Ru(BPY)3]2+ concentrations, respectively. These concentration dependent kinetics coincide with those reported recently for electrolyte-stimulated aggregation of silica nanoparticles17 and arise from the complete shielding of electrostatic repulsion at high adsorbate concentration leading to effective collision between proximal particles and a fast aggregation regime. Notably, in the case of 10 μM [Ru(BPY)3]2+ concentrations and above (Fig. 2(c) and S1†), we observed direct transition from monodisperse nanospheres to larger and polydisperse aggregates. This was followed by a simultaneous decay of both the monomer and oligomer peak, as well as a rapid decrease in the initial absorbance of the [Ru(BPY)3]2+ even though its concentration in the system was not altered. This phenomenon is interpreted as an onset of extreme aggregation when the larger nanosphere oligomers begin to irreversibly crash out of the suspension, leading to a reduced colloidal concentration of the samples. Obviously, beyond a critical size, the oligomers cannot grow further without precipitating.32 We also observed that a dye concentration on the order of mM results instantaneously in a fully aggregated system, as indicated by the disappearance of the plasmon peak in the absorption spectra (Fig. S1†).
It is worthy to note from Fig. 2 and S1† that there are obvious differences in the aggregation behavior of the 40 nm and the 100 nm nanosphere samples, which are likely due to the different number of particles per mL for the two samples. However, it was revealed previously that at high electrolyte concentration, only particles with larger sizes exhibited less aggregation susceptibility than that at high particle concentration.17 This signifies that size dependence of the aggregation behavior cannot be completely excluded in both the diffusion-controlled and reaction-controlled phase of the aggregation process.17 For example, we noticed that for the 100 nm nanospheres, there are no spectral changes when the concentration of [Ru(BPY)3]2+ in the system was below 10 μM (Fig. 2(d) and (e)), in contrast to the distinct broad spectral features that were observed for the aggregated 40 nm analogues (Fig. 2(a) and (b)). For the 10 μM concentration and above (Fig. 2(f) and S1†), aggregation was observed similar to the 40 nm nanospheres, but to a lower extent, as the monomer resonance clearly dominates over the corresponding oligomer resonance even after 120 min. The higher aggregation stability observed in the 100 nm nanospheres can be largely ascribed to surface phenomena, that become less pronounced in the large size regime with a small proportion of surface atoms,33 and that ultimately leads to a lower amount of surface adsorbed [Ru(BPY)3]2+ species compared to the 40 nm nanospheres analog.
To better understand the red shifting of the plasmon resonance for the different dye concentrations, we fitted the oligomer peak position as a function of aggregation time with an exponential function (Fig. 3a). While these fittings do not have direct interpretations on the aggregation mode, the extent of the red shift of the peak position and dependence of the decay behavior on the dye concentrations can be complementary to the data presented in Fig. 2. The red shifting observed as time progresses is an indication that a lower energy is needed to trigger the coupling of the plasmon oscillation modes of the aggregates.14 An inspection of Fig. 3a shows three regions with different decay behavior. Initially, there is a rapid decrease in oligomer resonance energy followed by a slow decrease over time. A steady state was observed after 50 minutes for the higher [Ru(BPY)3]2+ concentrations, while there is still a slow shift for the case of the 2 μM till 120 min. This verifies that the aggregation was faster with higher dye concentrations. The time dependence of monomer decay was also studied in a similar fashion. In the case of 40 nm oligomers, a decrease in monomer absorbance was observed for all dye concentrations during the course of the aggregation (Fig. 3b). However, a steadier behavior was observed for 100 nm nanospheres for concentrations below 10 μM, which stems from its aggregation stability (Fig. 3c).
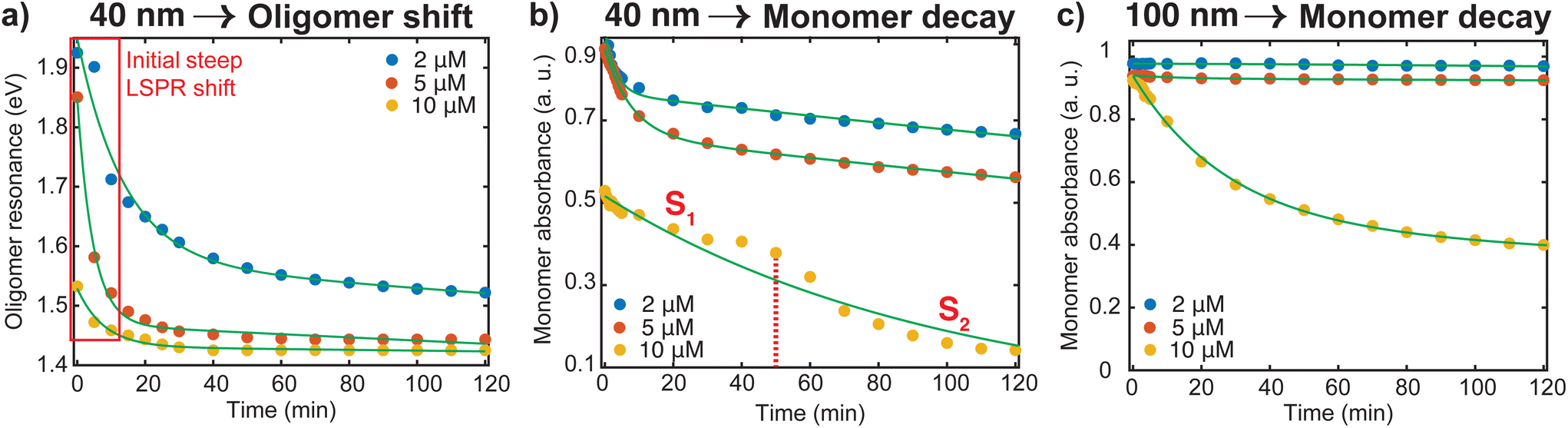 | ||
| Fig. 3 (a) Oligomer resonance energy as a function of aggregation time for different dye concentrations. The initial steep decay is highlighted. (b) Absorbance-time decay profile of 40 nm oligomers. For 10 μM, the stages are indicated as S1 and S2 in the curve where the dotted red line delineates the phase separation. (c) Absorbance-time decay profile of 100 nm oligomers. | ||
We also note from Fig. 3b that for the 10 μM dye concentration, an inverse sigmoidal shape of the monomer decay curve can be seen which contrasts with those observed for 2 μM and 5 μM concentrations. Two distinct segments highlighted as S1 and S2 can be observed which suggest the loss of two kinetically distinct monomeric species. As already mentioned, in the case of 40 nm nanosphere samples, the presence of 10 μM [Ru(BPY)3]2+ in the system results in simultaneous aggregation and precipitation of particles out of the colloidal dispersion. Therefore, we hypothesized that S1 describes the kinetics corresponding to the initial diffusion-controlled association of the monomers that leads to the fast oligomerization reaction and subsequent formation of partially condensed and precipitated system. At 50 min, which corresponds to the progression into the second stage, an equilibrium between dispersed and precipitated nanoparticles is very likely. Apparently, longer periods of time allow continued collisions between condensed particles resulting in a higher degree of oligomerization. Thus, the second stage (S2) corresponds to the pronounced precipitation of the nanoparticles, where the monomers are more readily consumed because of the relatively lower interparticle interaction energy barrier and multicollisions between nanoparticle clusters. This data provides new details on monomer transformations that are difficult to extract from the overlapping spectral data presented in Fig. 2.
Most of the studies on the aggregation behavior of nanoparticles have been unclear about the aggregation regimes the system will pass through as time elapses.34,35 However, the aggregation state of the nanostructure is a crucial characteristic that needs to be considered to simulate the plasmonic response for both fundamental and practical applications. Hence, we have chosen to study the transitions between different aggregated structures of the system, which has not received attention. The transitions between the aggregation modes are better appreciable from the spectral changes presented in Fig. 2(a) because of the slower rate of oligomer formation as compared to the higher dye concentrations. From this set of spectral evolution, the time dependence of the aggregation states can be discerned easily using the oligomer peak position and width. The deconvoluted in situ spectra reveals three distinct stages of aggregation (Fig. 4a). The first stage (0–5 min) is characterized by the growth of the oligomer band that was observed at around 652 nm. However, the color of the gold nanosphere suspension and the position of the secondary peak remains almost the same as only a 3 nm shift is observed within this period. This indicates the early stages (induction stage) of the evolution of the oligomer population as the interaction energy barrier between isolated nanospheres is gradually overwhelmed. In the second stage (5–10 min), a remarkable red shifting of the oligomer peak from 655 nm to 712 nm occurs, which is an indication of the favorable coupling of the plasmon modes of neighboring particles.14 Consequently, a dramatic red-to-dark blue color change of the suspension was observed, which probably signals the transition to intermediate aggregated states. The later stage (10–120 min) is signaled by a dark blue-to-pale blue color change and is dominated by the gradual red-shifting and broadening of the oligomer peak. This signifies a heterogeneous highly aggregated state as the oligomer peak dominates the spectrum and the particles begin to crash out of the system after 120 min. A similar behavior was reported in previous studies, where it was shown that for monodisperse particles to form highly aggregated structures, there exist pathways involving intermediate aggregated structures.18,32,36 The favored pathway was observed to be largely dependent on the degree of attractive interactions between proximal nanoparticles,18 with the intermediate pathway becoming more prevalent when the attraction is relatively milder as we observed in the low concentration regime.
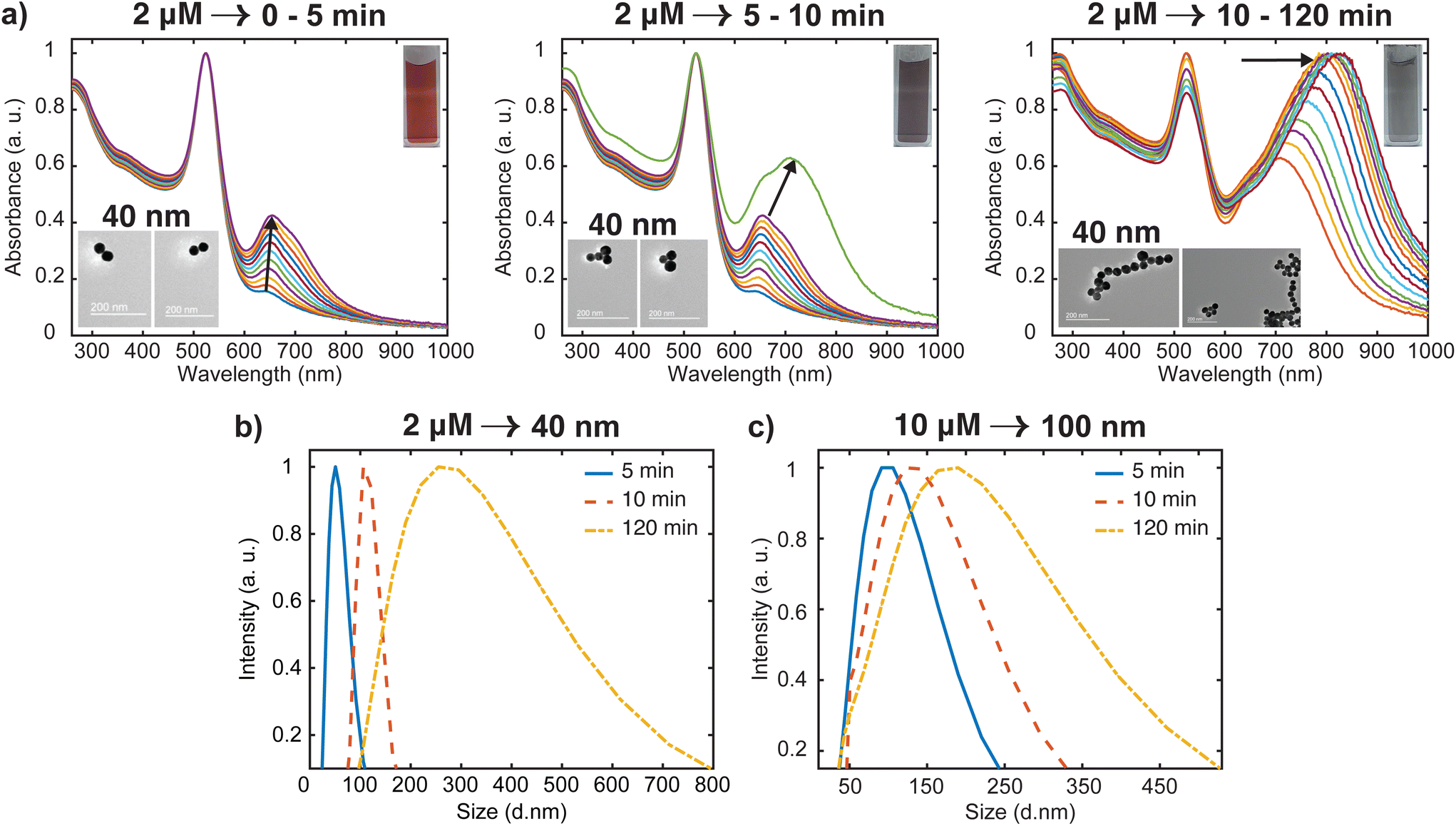 | ||
| Fig. 4 Analysis of time dependent aggregation states and particle size distributions (PSD). (a) Deconvoluted in situ spectra of data presented in Fig. 2(a), showing the spectral changes (labeled with arrows) from well dispersed state to highly aggregated state. The inset shows the representative TEM images for various stages of the aggregation (b) time-resolved evolution of PSD of 40 nm oligomers. (c) Time-resolved evolution of PSD of 100 nm oligomers. | ||
To obtain additional information on the aggregation modes of the three distinct regimes, TEM analysis was performed by taking multiple 2 μL aliquots of the sample at different times during the aggregation process. Because of the tendency of the TEM to overestimate aggregation state, we first stabilized the aggregates and diluted the samples 1000 times with Milli-Q water to obtain a single monomer or oligomer which are dropped on the grid (see ESI† for details). Representative images for the various aggregation stages are displayed in the left inset of Fig. 4(a). It appears that the majority of aggregates captured during the early stages (0–5 min) of the oligomerization are dimers. We also note that the aggregate proportion relative to the dispersed particles is very small at this stage, which corresponds to the negligible shift of the plasmon peak observed in the spectra. During the intermediate stage (5–10 min), significant trimer and tetramer configurations can be seen in addition to a dimer population. The formation of a significant population of larger and polydisperse aggregate species was seen at the later stage (10–120 min), which is consistent with the broad spectral feature seen in the spectra. Additional confirmation of the aggregation state was obtained by imaging several areas of the grid for more concentrated samples (Fig. S2†) with peak assignments of plasmon modes and Mie resonances available in the Supplementary information. These images containing many particles verified the initial aggregation configuration revealed by the dilute sample measurements.
Time-resolved dynamic light scattering (DLS) measurements were also performed to complement the aggregation data provided by the UV-Vis spectroscopy and TEM analysis. Fig. 4(b) and (c) depict the evolution of hydrodynamic diameter size distributions of oligomers during the aggregation of 40 nm and 100 nm nanosphere samples. Three distinct peaks of the size distribution can be observed in both cases, which coincides with the previous proposition of three aggregation regimes. As shown in Fig. 4b, in the early stage (5 min), the mean hydrodynamic diameter of the aggregates is around 51 nm which corresponds to a size slightly higher than that of the isolated 40 nm nanospheres. This correlates well with the previous observations of minor shifts of the plasmon peak observed in the UV spectral data and verifies the TEM data that only a small fraction of the nanospheres aggregated to largely form dimers at this stage, thus resulting in a smaller contribution to the average hydrodynamic diameter. The growth of the aggregates was reflected by the shift of the peak of the size distribution to 120 nm, which corresponds to the size of the particles having the highest intensity.37,38 This coincides with the remarkable shift of the plasmon peak position and significant trimer configurations observed in the TEM data. We also note the narrow distribution in this stage, which shows the low dispersity of the aggregates. As expected, in the later stages, the mean size of the aggregates is 300 nm, and the size distribution is much broader than the previous stages. Similar behavior was observed for the 100 nm oligomers, but the extent of the shift of the peak of the size distribution is less compared to the 40 nm analog (Fig. 4c). These findings further indicate the complementary insights on the aggregation behavior resulting from the combination of three different techniques. On the basis of our results, a schematic representation of the time-dependent aggregation regime of the AuNS-[Ru(BPY)3]2+ system is depicted in Fig. 5. We propose that the formation of the larger aggregates proceeds via interactions of the smaller aggregates leading to further oligomerization with time.
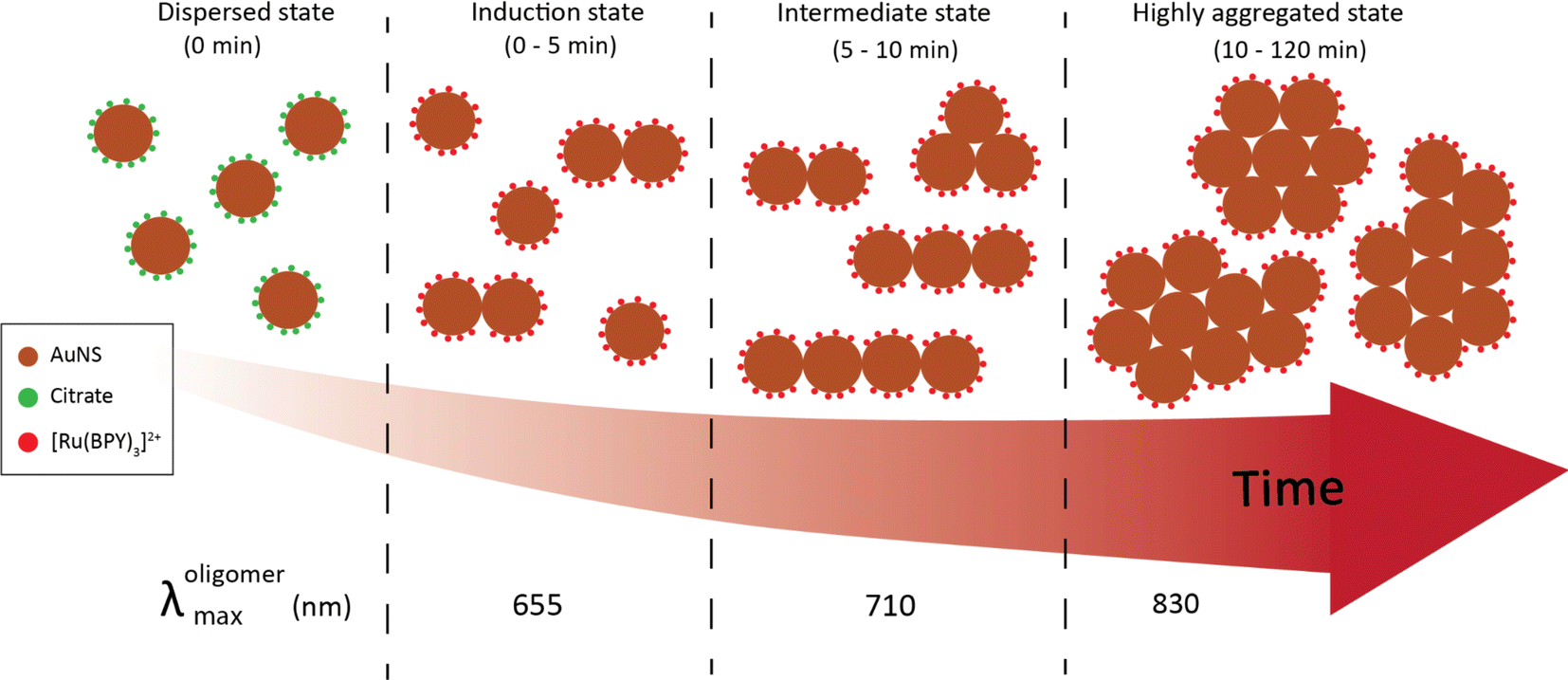 | ||
| Fig. 5 Schematic representation of the aggregation regimes of 40 nm AuNS upon injection of 2 μM [Ru(BPY)3]2+. | ||
To gain some insight into whether the order of reagent addition might affect the aggregation behavior, we altered the initial aggregation protocol such that the AuNS dispersion was injected to the [Ru(BPY)3]2+ solution. Although this produces similar spectral features and the position and width of the plasmon peaks are noticeably identical, a significant change in the aggregation rate was consistently observed (Fig. S3†). Notably, the evolution of oligomer population by this protocol is relatively slow, which in the case of the 40 nm sample, the aggregation begins only after 10 min of the AuNS injection into system. This behavior contrasts with the instantaneous aggregation that was observed using the original mixing order in which the dye solution with the same concentration was added to the gold monomer. Moreover, comparison between Fig. 4a and S3† reveals that the degree of aggregation attained after 5 min with the original procedure is equivalent to the one attained after 120 min with the reverse method. It is important to note that the observation of difference in aggregation behavior caused by inversing the order of mixing is not new. For instance, it was recently reported that the color change induced by aggregation of citrate-terminated AuNS is sensitive to the order of addition of the monomer, hydrogen peroxide, and 4-mercaptophenylboronic acid.39 However, to the best of our knowledge, the difference in aggregation kinetics due to reversing of the order addition of gold nanoparticles and other chemicals including [Ru(BPY)3]2+ has not been previously reported. We suspect that since most reports characterize the nanoparticles suspensions with low time resolution and several minutes after mixing, the difference in the aggregation rate especially in the early stages will not be captured. Our results therefore highlight the importance of the in situ study in elucidating the aggregation behavior of colloidal nanoparticle systems and provide insights into the correlation between order of reagent addition and aggregation kinetics.
To rationalize the anomalous mixing order-induced rate discrepancy mentioned above, we consider the dominant role of monomer–monomer interactions in the aggregation behaviors of colloidal particles. Previous studies have shown that nanoparticles would exhibit different aggregation kinetics when the ionic strength, pH, temperature, particle size and concentration of the system were changed.17,37 However, considering that the aggregation parameters, in particular, nanoparticle diameter and dye concentration for the two mixing orders are the same for our system, the influence of size, surface, and solution chemistry on the variations of the aggregation rate could be excluded. Then there must be enhanced effective collisions between the primary particles that can correspond to the fast aggregation rate observed with the original order of reagent addition. Based on this premise, the highest barrier to effective collisions and aggregation is likely associated with the adsorption of [Ru(BPY)3]2+ on the citrate terminated AuNS surface. Therefore, it is plausible to assume that the original mixing mode especially promotes faster binding and attainment of critical coverage of the dye molecules on the nanoparticle's surface. This was accompanied by a pronounced suppression of the electrostatic repulsion between the dispersed particles leading to effective interparticle collisions that causes aggregation in the early stages. It should be noted that the injection order can also influence the dispersion state of the nanospheres and how they coalesce towards a preferential orientation for collision and oligomerization. We expect that an in-depth theoretical simulation of the aggregation behavior under different addition modes could provide refined mechanistic understanding of these rate discrepancies, however, that is beyond the scope of this article. For the remainder of this paper, only the first mixing mode was used to trigger the oligomerization.
Evaluation of adsorption and plasmon enhancement effects
As indicated in the introduction, while a lot of adsorbate molecules have been employed to engineer plasmonic hotspots through aggregation of nanoparticle dispersion,25,27 little attention has been paid to determine the amount of molecules bound to the surface. The extent of the dye adsorption onto a surface strongly influences interfacial processes, including charge injection, surface reactions, and plasmonic enhancement. Specifically, the surface coverage of the dye molecules on a nanostructure influences the confinement of a fraction of them in the hotspots, which in turn substantially determines the overall efficiency of plasmon-mediated processes. A recent study emphasizes that precise experimental estimation and comparison of plasmonic enhancements requires accurate determination of the number of molecules adsorbed on the plasmonic platform.40 To this end, we performed three independent experiments to characterize the [Ru(BPY)3]2+ adsorption on the gold nanosphere surface at dye concentrations ranging from 2 μM to 40 μM. The amount of dye adsorbed was measured using inductively coupled plasma-optical emission spectrometry. Good recovery for the [Ru(BPY)3]2+-bound aggregates to avoid incorrect estimation of adsorbed dye concentration were achieved by repeated centrifugation until there were no traces of the plasmon mode in the spectrum of the supernatant (Fig. S4†). Before measurements, preliminary optimization of a sample digestion method41,42 and control experiments were carried out to avoid Ru losses and ensure that the quantification results were accurate (Table S1 and Fig. S4†). Further details of the sample digestion procedure can be found in the ESI.†Fig. 6a reports that the amount of dye adsorbed per mg of the nanosphere is controlled by the dye concentration in the system. Further comparison reveals that the percent adsorbed shows the opposite trend (Fig. 6b). The superior adsorptive capacity at higher concentrations can be ascribed to the large concentration gradient between the bulk solution and the nanoparticle surface. As can be observed in Fig. 6a, the adsorption behavior of the 40 nm and the 100 nm nanospheres are similar in the low concentration regime but differ significantly at higher concentrations. In the case of the 100 nm samples, maximum adsorption amount of 7.69 mg g−1 was attained when the dye concentration reaches 14.97 mg mL−1 which is then followed by a decrease in adsorption capacity (Table S1†). The decrease in adsorption can be linked to a desorption process. It can be assumed that complete coverage of the nanoparticle surface is attained leading to electrostatic repulsion between free and adsorbed [Ru(BPY)3]2+ species. Conversely, the 40 nm sample displayed a higher uptake capacity of 9.96 mg g−1 and saturation of adsorption sites was not observed within the concentration ranges studied. This signifies that a higher dye concentration is required to saturate the adsorption sites on the 40 nm nanosphere surface. Furthermore, the estimated maximum surface coverage based on the adsorption data for the 40 nm and 100 nm nanospheres was 960 molecules per nm2 and 475 molecules per nm2, respectively (see ESI† method section for details). It must be noted that saturation of adsorption sites by dye molecules was not attained in the case of the 40 nm sample. The difference in adsorptive behavior was consistent with the larger surface area and thus a high amount of adsorption sites available on the 40 nm nanosphere surface.
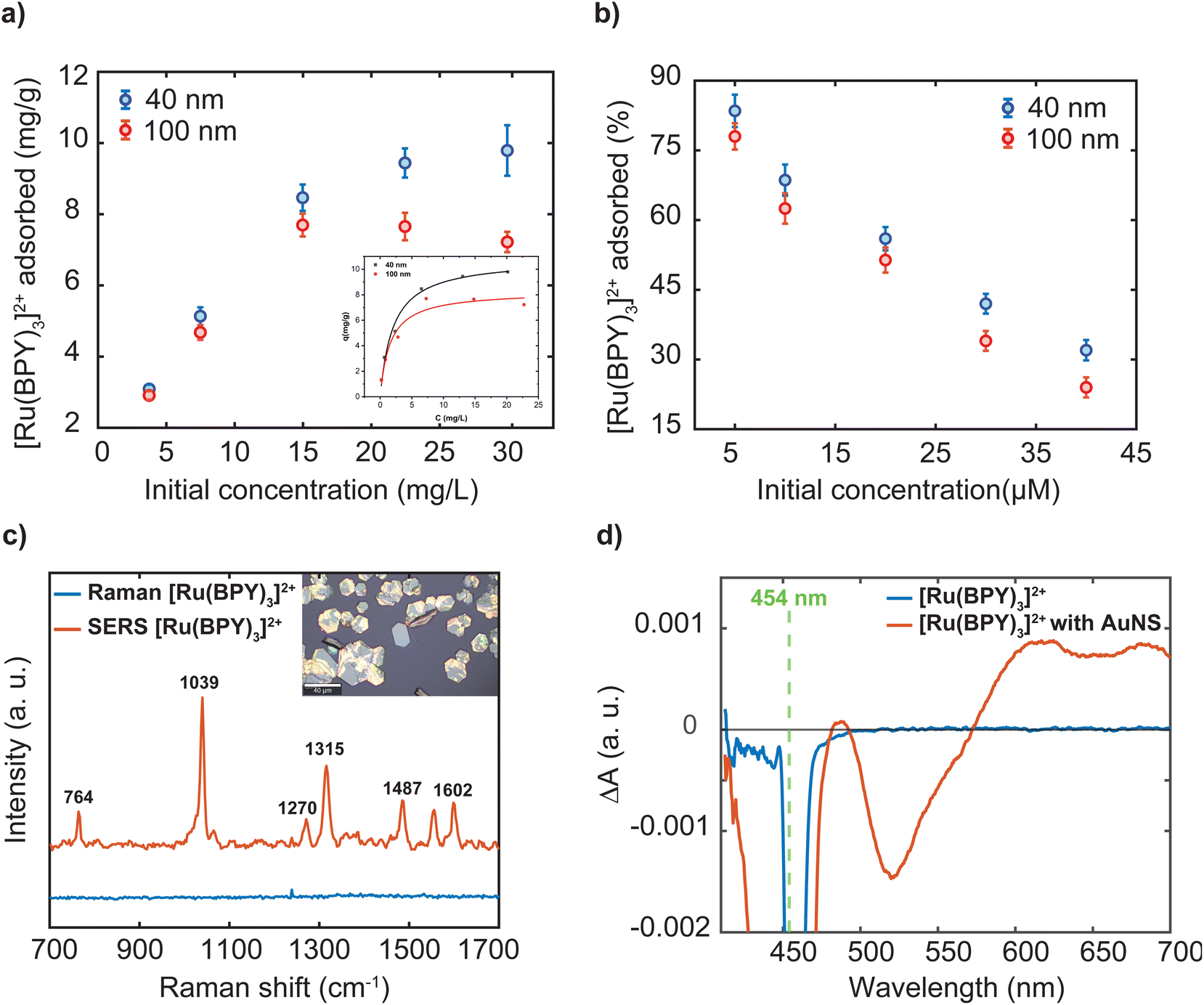 | ||
| Fig. 6 (a) Amount adsorbed as a function of [Ru(BPY)3]2+ concentration (inset: Langmuir adsorption isotherm at 25 °C). (b) Amount adsorbed expressed as percent. Error bars represent the standard deviation of three replicates. (c) Raman and SERS spectra of tris(bipyridine)ruthenium(II) [Ru(BPY)3]2+ adsorbed on the 40 nm AuNS; λex 633 nm. (d) Transient absorption spectra of unbound [Ru(BPY)3]2+ and [Ru(BPY)3]2+ adsorbed on the 40 nm nanospheres. λex 454 nm, solvent: water. | ||
Next, the adsorption data was analyzed by the nonlinear forms of the conventional Freundlich and Langmuir isotherms models. These models adopted from the ideal adsorption theory of gasses can provide some insights to the extent of adsorption and the underlying mechanism of molecular interactions at interfaces. In our case, the Langmuir model showed better fit to the experimental data and the predicted maximum adsorption capacity for both samples were found to be close to that of the experimental data (inset Fig. 6a). The Langmuir isotherm assumes monolayer adsorption to occur based on specific interactions between molecules and homogeneous surfaces.43 However, it is well-known that it is challenging to interpret the adsorption mode in colloidal systems because of the complications in determining molecular orientations.44 In addition, nanoparticles exhibit low-symmetry surface structures45 and largely sustain surface defects,46 leading to adsorption sites with different energies. Because the adsorption process is driven by reduction in surface energy, the defect sites allow the adsorption of large number of molecules as compared to a perfect crystal structure.11
Generally, chemisorption occurs in a monolayer and is deemed to be the appropriate adsorption mode for enhancement and electron transfer processes.11 However, the primary mechanism of the adsorption of many molecules on metal surfaces including our [Ru(BPY)3]2+ can be largely described as physisorption as the adsorption energy is dominated by van der Waals dispersion forces47 resulting from conjugated aromatic system, which disagrees with the Langmuir isotherm. On the basis of our data, we speculate a mixture of monolayer and multilayer adsorption modes considering the inhomogeneity of the nanoparticle surface. This implies that adsorbed [Ru(BPY)3]2+ molecules do not evenly form a monolayer but are rather concentrated at higher energy sites on the AuNS surface. Furthermore, the dye adsorption mode can switch from monolayer to multilayer mode depending on the adsorbate concentration.11 However, we cannot clarify the strength of the adsorption from this data. In other words, is the interaction between adsorbed [Ru(BPY)3]2+ and the gold nanosphere surface strong enough to maximize the photophysical and photochemical properties of the dye molecules?
Obviously, adsorption must affect the charge transfer dynamics as well Raman scattering, absorption efficiency, and fluorescence behavior of the [Ru(BPY)3]2+. For instance, some studies have shown that molecules in the vicinity of the metal surface play a dominant role for attaining the maximum effect of plasmon resonances through the local electromagnetic field enhancement and charge transfer effects.40,48 Meanwhile, it is well known that normal Raman spectrum of Ru-based dyes is difficult to obtain because of the strong fluorescence background.12 However, the intense electromagnetic field in the plasmonic environment can enhance surface-molecule energy transfer which in turn can lead to enhanced transmission and fluorescence quenching of the molecular adsorbates.24,48 The later phenomenon has made it possible to observe the Raman signal of these fluorophores. To this end, we evaluate the interaction of the [Ru(BPY)3]2+ with the colloidal gold nanosphere surface by analyzing the fluorescence quenching behavior in this system via SERS effects.
For convenience, further discussion will be centered on the surface plasmon enhancing effects in the 40 nm gold nanosphere oligomer systems following stabilization with an outer polymer shell. We start measurements with the 457 nm laser, whose energy matches the MLCT absorption band of the dye and of the oligomer system. Thus, at 457 nm, resonance effects and plasmon effects through interband transitions in gold which occur with the threshold below 516.6 nm are involved.49 The recorded Raman spectrum of the free molecules shows a strong fluorescence background which masked the dye's vibrational signals (Fig. S5†). For the [Ru(BPY)3]2+–AuNS oligomer system, significant reduction of the fluorescence background was observed which arises from the plasmon–molecule interaction effect (Fig. S5†). It is however noticed that the dye is undergoing structural transformations due to photodegradation even when the irradiation time and laser power is reduced to the operational limit of the microscope (Fig. S5†). The most obvious evidence of the photodamage is a deformation of the strong signal at 1039 cm−1 and the disappearance of some of the dye signals. For this reason, we switch to the excitation line at 633 nm, which clearly shows fluorescence quenching and improved signal to noise ratio compared to the 457 nm laser excitation (Fig. 6c). It is worth mentioning that the Raman and SERS spectra were both recorded using the 2 μM dye concentration and using the same acquisition parameters for proper assessment of the plasmon enhancing effects. Initially, we observe a small fluorescence background even when a 785 nm excitation source is used which can be associated with two-photon absorption processes. It has been previously reported that the presence of non-adsorbed molecules might contribute to the negligible fluorescence background in the spectra.12 Based on the data presented in Fig. 6b, 11.7% of the [Ru(BPY)3]2+ molecules are not adsorbed to the gold surface when the final dye concentration in the system is 2 μM. To this end, we performed repeated centrifugation until there are no traces of the absorption features of the free dye in the spectrum of the supernatant. The spectrum obtained after centrifugation for only the adsorbed species appeared with concurrent signal enhancement and fluorescence quenching (Fig. 6c). Thus, our finding has unambiguously shown that removal of free dye molecules can enhance the quenching of the fluorescence background as previously surmised from time-resolved single-photon counting measurements.50 We also note some additional changes to the spectrum of the adsorbed dye. The most pronounced difference is the stronger peak at 1039 cm−1 ascribed to the Ru–N stretch and the ring breathing of the bipyridine51 is weaker in the unadsorbed sample. The peaks at 1315 cm−1 and 1602 cm−1, assigned to the C–C stretching of the bipyridine, increase in intensity and shift to higher energies as compared to the unbound molecules. These adjustments were consistent with the [Ru(BPY)3]2+ adsorption on the plasmonic surface which changes these vibrational modes.
One possible interpretation of the overwhelming fluorescence quenching behavior is the overlap between the plasmon resonance frequency of the oligomers and the fluorescence emission frequency of [Ru(BPY)3]2+, leading to enhanced plasmon–dye interaction.52 As presented in Fig. 2a, the oligomer absorption spectrum spans from 550 nm to 950 nm with a peak centered at around 650 nm. This matches the fluorescence emission spectrum of [Ru(BPY)3]2+ which spans from 500 to 750 nm with a peak centered at 622 nm. Secondly, the adsorption of the dye on the nanoparticle surface is expected to efficiently mediate plasmon-induced resonance energy transfer and dramatically increase the number of nonradiative states.53,54 This also implies that electron transfer from the excited dye molecule to the plasmonic surface can occur. Based on our data, however, we cannot conclude whether electron transfer processes play a role in the plasmon-mediated fluorescence quenching effects.
To gain some insights into the existence of charge transfer processes, we performed transient absorption experiments. Fig. 6d shows the frequency resolved excited-state absorption spectra of the bound and unbound [Ru(BPY)3]2+ molecule with 1 μJ of a 454 nm pulse at a time delay of 1 ns. The transient spectrum of the 2 μM unbound [Ru(BPY)3]2+ molecule in water displays a stimulated emission signature at 625 nm that is consistent with previous pump–probe measurements.55 In sharp contrast, the AuNS bound [Ru(BPY)3]2+ at an equimolar concentration displays a significant enhancement of a surface-stabilized MLCT state under aqueous conditions. The [Ru(BPY)3]2+ ground-state bleach occurs at 450 nm with a delocalized spectral feature centered about 650 nm that could be indicative of a triplet10 or bipyridyl anionic species.56 Furthermore, simultaneous bleaching of the interband absorption can be seen at 520 nm leading to a depopulation of the d-band in the AuNS.57,58 Future work involves detailed studies to clarify the mechanism and kinetics of interfacial electron transfer and symmetry breaking that is surmised to give rise to a surface-stabilized MLCT state that competes with luminescence deactivation pathways.50
Conclusions
We demonstrate a controlled in situ oligomerization study of the absorption and scattering properties of plasmonic nanocomposite materials consisting of 40 nm and 100 nm gold nanospheres functionalized with the photoredox catalyst molecule [Ru(BPY)3]2+. A nonlinear aggregation time is observed with significant size dependent dynamics on the minutes timescale that correlate to structural and chemical properties at the nanoscale. Notably, 2 μM is observed to be the optimal quantity for oligomerization of 40 nm gold nanospheres as the percent of adsorbate molecule that occupies active sites on the nanoparticle decreases linearly with increasing concentration, whereby the onset of a saturation regime is observed at lower concentrations for larger nanospheres (100 nm) than for smaller nanospheres (40 nm). We identified that the order of reagent addition might introduce different aggregation mechanisms in the same mixture leading to a significant variation in the overall kinetics. Our results have significant impact on surface-enhanced spectroscopy measurements which are enabled by an optimized dye-nanoparticle coverage that brings about a localized surface plasmon resonance induced fluorescence quenching from surface mediated deactivation pathways of the molecule. The findings have broad implications for elucidating the mechanisms and transformations in the excited state properties of photoredox catalysts in plasmonic high field confinement. Knowledge and insights gained from these studies have wide ranging applications to the design of nanoscale catalytic reactors, photovoltaic devices, and light-harvesting systems in general.Author contributions
Umar Yunusa: Conceptualization, investigation, data curation & measurement, formal analysis, writing – original draft; Natalie Warren: Investigation, TA data curation and analysis, writing – editing; David Schauer: data curation; Prasenjit Srivastava: TA data curation, writing – editing; Emily Sprague-Klein: Conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the insightful discussion and research instrumentation provided by the Institute for Molecular and Nanoscale Innovation (IMNI) and Hector Garces. The authors acknowledge the facilities and technical assistance of the Brown University Environmental Geochemistry Facility for use of ICPOES. The authors also acknowledge generous research support from Brown University's start-up funds.References
- J. Langer, D. Jimenez De Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García De Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed.
- J. A. Dionne and H. A. Atwater, MRS Bull., 2012, 37, 717–724 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- C. L. Warkentin, Z. Yu, A. Sarkar and R. R. Frontiera, Acc. Chem. Res., 2021, 54, 2457–2466 CrossRef CAS PubMed.
- S. L. Kleinman, R. R. Frontiera, A.-I. Henry, J. A. Dieringer and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2013, 15, 21–36 RSC.
- Z. Yu and R. R. Frontiera, ACS Nano, 2022, 16, 847–854 CrossRef CAS PubMed.
- S. Sim, A. Beierle, P. Mantos, S. McCrory, R. P. Prasankumar and S. Chowdhury, Nanoscale, 2020, 12, 10284–10291 RSC.
- M. Liebtrau, M. Sivis, A. Feist, H. Lourenço-Martins, N. Pazos-Pérez, R. A. Alvarez-Puebla, F. J. G. de Abajo, A. Polman and C. Ropers, Light: Sci. Appl., 2021, 10, 82 CrossRef CAS PubMed.
- S. Wallin, J. Davidsson, J. Modin and L. Hammarström, J. Phys. Chem. A, 2005, 109, 4697–4704 CrossRef CAS PubMed.
- L. Ellis-Gibbings, V. Johansson, R. B. Walsh, L. Kloo, J. S. Quinton and G. G. Andersson, Langmuir, 2012, 28, 9431–9439 CrossRef CAS PubMed.
- C. Pérez León, L. Kador, B. Peng and M. Thelakkat, J. Phys. Chem. B, 2006, 110, 8723–8730 CrossRef PubMed.
- T. E. Tesema, H. Kookhaee and T. G. Habteyes, J. Phys. Chem. Lett., 2020, 11, 3507–3514 CrossRef CAS PubMed.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- B. Saini, L. Khamari and T. K. Mukherjee, J. Phys. Chem. B, 2022, 126, 2130–2141 CrossRef CAS PubMed.
- S. Shrestha, B. Wang and P. Dutta, Adv. Colloid Interface Sci., 2020, 279, 102162 CrossRef CAS PubMed.
- H. Sun, R. Jiao, G. An, H. Xu and D. Wang, J. Environ. Sci., 2021, 103, 33–42 CrossRef CAS PubMed.
- X. Yang and Z.-Y. Lu, J. Chem. Phys., 2022, 156, 214902 CrossRef CAS PubMed.
- B. Negru, M. O. McAnally, H. E. Mayhew, T. W. Ueltschi, L. Peng, E. A. Sprague-Klein, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2017, 121, 27004–27008 CrossRef CAS.
- S. Saini, J. Halldin Stenlid and F. Abild-Pedersen, npj Comput. Mater., 2022, 8, 163 CrossRef CAS.
- Z. Lu, S. Yadav and C. V. Singh, Catal. Sci. Technol., 2020, 10, 86–98 RSC.
- M. Moskovits and B. Vlčková, J. Phys. Chem. B, 2005, 109, 14755–14758 CrossRef CAS PubMed.
- T. Kim, C.-H. Lee, S.-W. Joo and K. Lee, J. Colloid Interface Sci., 2008, 318, 238–243 CrossRef CAS PubMed.
- T. E. Tesema, H. Kookhaee and T. G. Habteyes, J. Phys. Chem. Lett., 2020, 11, 3507–3514 CrossRef CAS PubMed.
- E. A. Sprague-Klein, R. Ho-Wu, D. Nguyen, S. C. Coste, Y. Wu, J. J. McMahon, T. Seideman, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2021, 125, 22142–22153 CrossRef CAS.
- C. Tang, B. Auguié and E. C. Le Ru, J. Phys. Chem. C, 2022, 126, 10129–10138 CrossRef CAS.
- R. Grumich, T. Griggs-Demmin, M. Glover and B. Negru, ACS Omega, 2021, 6, 31818–31821 CrossRef CAS PubMed.
- Y. Jiang, A. Wang, B. Ren and Z.-Q. Tian, Langmuir, 2008, 24, 12054–12061 CrossRef CAS PubMed.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1982, 21, 3967–3977 CrossRef CAS.
- P. Dongare, B. D. B. Myron, L. Wang, D. W. Thompson and T. J. Meyer, Coord. Chem. Rev., 2017, 345, 86–107 CrossRef CAS.
- L. C. T. Shoute and G. R. Loppnow, J. Am. Chem. Soc., 2003, 125, 15636–15646 CrossRef CAS PubMed.
- R. Pamies, J. G. H. Cifre, V. F. Espín, M. Collado-González, F. G. D. Baños and J. G. De La Torre, J. Nanopart. Res., 2014, 16, 2376 CrossRef.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- D. Alba-Molina, M. Martín-Romero, L. Camacho and J. Giner-Casares, Appl. Sci., 2017, 7, 916 CrossRef.
- R. De La Rica, Nanoscale, 2017, 9, 18855–18860 RSC.
- J. K. Stolarczyk, A. Deak and D. F. Brougham, Adv. Mater., 2016, 28, 5400–5424 CrossRef CAS PubMed.
- W. Zhang, J. Crittenden, K. Li and Y. Chen, Environ. Sci. Technol., 2012, 46, 7054–7062 CrossRef CAS PubMed.
- S. Martinez Legaspi and L. Segatori, Bioconjugate Chem., 2019, 30, 1986–1997 CrossRef CAS PubMed.
- R. Li, X. Gu, X. Liang, S. Hou and D. Hu, Materials, 2019, 12, 1802 CrossRef CAS PubMed.
- M. Chen, R. Solarska and M. Li, J. Phys. Chem. C, 2023, 127, 2728–2734 CrossRef CAS.
- M. Balcerzak, Talanta, 1999, 48, 39–47 CrossRef CAS PubMed.
- W. Jiang, D. B. Hibbert, G. Moran and R. Akter, J. Anal. At. Spectrom., 2012, 27, 1465 RSC.
- H. Swenson and N. P. Stadie, Langmuir, 2019, 35, 5409–5426 CrossRef CAS PubMed.
- E. C. Le Ru, S. A. Meyer, C. Artur, P. G. Etchegoin, J. Grand, P. Lang and F. Maurel, Chem. Commun., 2011, 47, 3903 RSC.
- D. Kang, S. Kim, J. Heo, D. Kim, H. Bae, S. Kang, S. Shim, H. Lee and J. Park, Nanoscale, 2023, 15, 532–539 RSC.
- C. George, D. Ricci and E. Di Zitti, Superlattices Microstruct., 2008, 44, 608–616 CrossRef CAS.
- L. Zhou, R. Johnson, T. Habteyes and H. Guo, J. Chem. Phys., 2017, 146, 164701 CrossRef PubMed.
- K. Mori, M. Kawashima, M. Che and H. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 8598–8601 CrossRef CAS PubMed.
- K. Kolwas and A. Derkachova, Nanomaterials, 2020, 10, 1411 CrossRef CAS PubMed.
- W. R. Glomm, S. J. Moses, M. K. Brennaman, J. M. Papanikolas and S. Franzen, J. Phys. Chem. B, 2005, 109, 804–810 CrossRef CAS PubMed.
- D. W. Silverstein, C. B. Milojevich, J. P. Camden and L. Jensen, J. Phys. Chem. C, 2013, 117, 20855–20866 CrossRef CAS.
- J. Huang, J. Lu, Y. Zhang, S. Xie, G. Lin, W. Lin, P. Zhuang, Z. Zhu, S. Guo, S. Zhang, Q. Ge, X. Zhang and W. Cai, J. Phys. Chem. C, 2021, 125, 14891–14896 CrossRef CAS.
- Q. Su, C. Jiang, D. Gou and Y. Long, ACS Appl. Bio Mater., 2021, 4, 4684–4705 CrossRef CAS PubMed.
- C. R. Simovski, M. S. M. Mollaei and P. M. Voroshilov, Phys. Rev. B, 2020, 101, 245421 CrossRef CAS.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2018, 9, 6670–6678 RSC.
- E. A. Sprague-Klein, B. Negru, L. R. Madison, S. C. Coste, B. K. Rugg, A. M. Felts, M. O. McAnally, M. Banik, V. A. Apkarian, M. R. Wasielewski, M. A. Ratner, T. Seideman, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2018, 140, 10583–10592 CrossRef CAS PubMed.
- X. Zhang, C. Huang, M. Wang, P. Huang, X. He and Z. Wei, Sci. Rep., 2018, 8, 10499 CrossRef PubMed.
- G. Kedawat, I. Sharma, K. Nagpal, M. Kumar, G. Gupta and B. K. Gupta, ACS Omega, 2019, 4, 12626–12631 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary results. See DOI: https://doi.org/10.1039/d3nr06129a |
| This journal is © The Royal Society of Chemistry 2024 |