
Imidazolium-based zwitterionic liquid-modified PEG–PLGA nanoparticles as a potential intravenous drug delivery carrier†
Gaya S.
Dasanayake
 a,
Christine M.
Hamadani
a,
Gagandeep
Singh
a,
Sandeep
Kumar Misra
b,
Priyavrat
Vashisth
a,
Joshua S.
Sharp
ab,
Laxmi
Adhikari‡
c,
Gary A.
Baker
c and
Eden E. L.
Tanner
*a
a,
Christine M.
Hamadani
a,
Gagandeep
Singh
a,
Sandeep
Kumar Misra
b,
Priyavrat
Vashisth
a,
Joshua S.
Sharp
ab,
Laxmi
Adhikari‡
c,
Gary A.
Baker
c and
Eden E. L.
Tanner
*a
aDepartment of Chemistry and Biochemistry, University of Mississippi, University, MS 38677, USA. E-mail: eetanner@olemiss.edu; Tel: +1-662-915-1165
bDepartment of BioMolecular Sciences, University of Mississippi, University, MS 38677, USA
cDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
First published on 22nd February 2024
Abstract
Zwitterionic-based systems offer promise as next-generation drug delivery biomaterials capable of enhancing nanoparticle (NP) stimuli-responsiveness, biorecognition, and biocompatibility. Further, imidazole-functionalized amphiphilic zwitterions are able to readily bind to various biological macromolecules, enabling antifouling properties for enhanced drug delivery efficacy and bio-targeting. Herein, we describe structurally tuned zwitterionic imidazole-based ionic liquid (ZIL)-coated PEG–PLGA nanoparticles made with sonicated nanoprecipitation. Upon ZIL surface modification, the hydrodynamic radius increased by nearly 20 nm, and the surface charge significantly shifted closer to neutral. 1H NMR spectra suggests that the amount of ZIL on the nanoparticle surface is controlled by the structure of the ZIL and that the assembly occurs as a result of non-covalent interactions of ZIL-coated nanoparticle with the polymer surface. These nanoparticle-zwitterionic liquid (ZIL) constructs demonstrate selective affinity towards red blood cells in whole mouse blood and show relatively low human hemolysis at ∼5%. Additionally, we observe higher nanoparticle accumulation of ZIL-NPs compared with unmodified NP controls in human triple-negative breast cancer cells (MDA-MB-231). Furthermore, although the ZIL shows similar protein adsorption by SDS-PAGE, LC-MS/MS protein analysis data demonstrate a difference in the relative abundance and depletion of proteins in mouse and human serum. Hence, we show that ZIL-coated nanoparticles provide a new potential platform to enhance RBC-based drug delivery systems for cancer treatments.
 Eden E. L. Tanner | Dr Eden E. L. Tanner completed her undergraduate degree in Advanced Science (Chemistry) at the University of New South Wales, Sydney, Australia. She earned her doctorate in Physical and Theoretical Chemistry at the University of Oxford with Prof. Richard G. Compton, and moved to Harvard University in 2017 as a Postdoctoral Research Fellow with Samir Mitragotri. As of August 2020, Dr Tanner is an Assistant Professor of Chemistry and Biochemistry at the University of Mississippi. The Tanner Lab works at the interface of Chemistry and Bioengineering with a focus on using ionic liquids to solve problems in nanoparticle drug delivery. |
Introduction
Nanoparticle (NP) drug delivery systems have been extensively researched as a potential route to achieving selective, targeted drug delivery. However, intravenously (IV) injected nanoparticles are subjected to off-target accumulation by the liver and spleen and rapid clearance by the mononuclear phagocytic system (MPS), limiting NP dosage effectiveness and target specificity.1 Therefore, polymeric nano formulations as a platform still necessitate further development to reach the clinic. To overcome the MPS & other intravenous drug delivery barriers, red blood cells (RBCs) can serve as natural NP carriers to prolong their vascular circulation due to their ∼120-day lifespan, relative abundance, and extended surface area due to the lack of organelles.2,3 Therefore, various NP surface modification materials such as hydrophilic polymers, zwitterionic polymers, targeting moieties, choline-based ionic liquids, and natural biomacromolecules4 have recently been developed for cellular hitchhiking.5 Among these, the zwitterionic polymers and choline-based ionic liquids have been demonstrated to significantly reduce protein nanoparticle corona6 formation and engage in in situ RBC hitchhiking and enhanced tumor retention.2,5,7In nature, nitrogen-based heterocycles, particularly the imidazole ring, play a vital role in forming various essential components in the human body, leading to the formation of various proteins and enzymes, including histamine, histidine, biotin, and nucleic acid.8,9 Work by Baviskar et al. demonstrated higher anticancer potency for bicyclic N-fused aminoimidazole derivatives in kidney cancer cell lines compared to etoposide and 5-fluorouracil.10 In recent years, along with these unique properties of the imidazole ring, imidazole-derived zwitterionic amphiphiles have been used in drug delivery applications to endow nanocarriers with increased in vivo blood circulation time, demonstrating excellent biocompatibility and antifouling ability.11–14 Studies conducted on the effect of functionalization of the alkyl chains of methyl imidazolium compounds demonstrate strong interactions with lipophilic molecules such as serum proteins, leading to the formation of reversible carrier complexes.15 Imidazole-based zwitterionic liquids have been shown to enhance the selectivity of PEG–PLGA NPs towards RBCs by interacting with the exposed membrane proteins16 and functional groups present on the RBC surface, predominately driven by hydrogen bonding with the nitrogen (N) atom from the imidazole ring. Zhao et al. recently proposed that tertiary amine oxide (TAO)-containing zwitterionic polymer, poly(2-(N-oxide-N,N-diethylamino)ethyl methacrylate) (OPDEA), shows enhanced drug delivery to cancer cells by reversibly binding to the RBC cell surface, thereby improving the circulation time and thus improving the adsorption-mediated transcytosis (AMT) towards cancer cells, indicating the crucial role of zwitterionic species towards RBC selectivity.17–19
In this study, we assessed the potential of four imidazole-based zwitterionic liquids (ZILs), EO2ImBS (ZIL-1), EO2ImPS (ZIL-2), EO3ImPS (ZIL-3), and EO7ImPS (ZIL-4), to surface modify PEG–PLGA nanoparticles. ZIL-1 and ZIL-2 differ by one carbon in the alkyl chain that connects the nitrogen in the imidazolium ring to the sulfonyl group (butyl vs. propyl, respectively). ZIL-2, ZIL-3, and ZIL-4 all contain a propyl linkage but vary in the length of the ethyl ether side chain (2, 3, and 7 motifs, respectively). When designing the studied four zwitterions, we incorporated positively charged pyridinium and negatively charged sulfonate to keep the amphiphilic nature of the molecules since electrostatically balanced charged distribution could minimize interaction with charged sites on serum proteins. By changing the alkyl chain lengths and integrating the different lengths of ethyl ether side chains, we hope to see the variation in hydrophobicity leading to adsorption and dissociation of different types of proteins leading to a total reduction in protein adsorption.12,13
Moreover, the methoxy ethoxy group length has been shown to determine the efficacy in decreasing immunogenicity while maintaining normal RBC functions20,21 which can be promising for RBC affinity, eventually leading to prolonged circulation time. Despite the promise of the imidazole motif for drug delivery, the existing literature shows that imidazolium-based ionic liquids (ILs) show reasonably high toxicity, especially compared to their zwitterionic counterparts. Notably, an increase in alkyl chain lengths in imidazole-based ILs has been correlated with an escalation in toxicity due to an associated higher degree of lipid extraction from cellular membranes, disrupting cellular integrity and ultimately causing cell death.22 However, adding ester or ether functionalities to the alkyl chain has been shown to significantly improve their biocompatibility, biodegradability, and nanoparticle stability.23 Incorporation of alkyl sulfonyl groups into a pyridinium core has been shown to provide a stabilizing effect and increase triple-negative breast cancer cell affinity.24 Considering these factors, we hypothesize that the presence of the sulfonyl group and the difference in the carbon chain length will alter the polarity and flexibility of these molecules, impacting how they interact with biological targets and potentially allowing the adoption of multiple conformations.25
Furthermore, the justification for exploring zwitterionic liquids (ZILs) is manifold. (i) Firstly, ZILs mark a fundamental departure in that although these salts possess high ion density, they exhibit a unique inability to migrate along potential gradients induced in bulk, a distinctive characteristic not found in familiar molten salts.26 ZILs consequently serve as a distinctive testbed for studying charge mobility within a matrix where only the added target ions can undergo migration, such as in single-ion conductors.27 In general, zwitterions play a pivotal role in promoting lithium-ion transport within polymer electrolytes, either by serving as solvents or by augmenting ionic dissociation.28 (ii) The juxtaposition of proximal positive and negative charges within the same unit imparts remarkable features like super hydrophilicity to zwitterionic materials. However, ZILs remain largely unexplored, resulting in a restricted understanding of their overall nature and susceptibility to design. (iii) Furthermore, the cation–anion tethering in ZILs reduces the degrees of freedom compared to their untethered ionic counterparts. This characteristic makes them more compliant targets for computational techniques like ab initio molecular dynamics simulations, for instance. (iv) Most notably, zwitterionic materials exhibit non-toxic, biocompatible, and fouling-resistant capabilities. The latter is believed to arise from a dense hydration layer with a stable hydrogen-bonded structure, acting as a protective deterrent against biomolecular attachment.29 Although the translation and expansion of these appealing features hold great promise, the absence of well-defined design principles for such endeavors justifies the increased exploration of ZILs in both fundamental studies and sustainable engineering and biomedical applications.
Using the sonicated nanoprecipitation, ZIL surface modification of PEG–PLGA nanoparticles (ZIL-NP) resulted in a significant amount of nanoparticle red blood cell affinity over platelets and white blood cells compared to their unmodified PEG–PLGA control. Moreover, LC-MS/MS analysis of the protein corona determined the enrichment and depletion of different proteins adsorbed on the nanoparticle surfaces in mouse and human serum based on their surface modification. Additionally, we show that this surface modification resulted in higher nanoparticle accumulation in human breast cancer cells (MDA-MB-231) and minimal ZIL toxicity toward noncancer cells (MCF-10A; non-tumorigenic human breast epithelial cells), suggesting that zwitterionic liquids are promising candidates for cancer nanoparticle drug delivery.
Results and discussion
PEG–PLGA nanoparticles were synthesized via a sonication-assisted nanoprecipitation method by dropwise addition of 1 mL of the polymer-containing organic phase into 3 mL of the aqueous phase while sonicating. Four zwitterionic liquids (ZILs) were prepared as described in the Methods section and characterized by 1H NMR (Fig. S1†). These four ZILs were selected to explore the impact of altering the alkyl chain between the imidazolium nitrogen and the sulfonyl group (ZIL-1 vs. ZIL-2 – butyl and propyl respectively) and the role of altering the ethyl ether side chain (ZIL-2, ZIL-3, ZIL-4, with 2, 4, and 7 motifs respectively). The successful coating of zwitterionic liquid on nanoparticles was determined by 1H NMR spectroscopy and Dynamic Light Scattering (DLS). As shown in Fig. 1A, the DLS data suggested that the overall ZIL surface-modified PEG–PLGA nanoparticle average size has increased, for ZIL-1-NP, ZIL-2-NP, and ZIL-3-NP, the size increased from 65 ± 1.4 nm to 92 ± 4 nm, 88 ± 5 nm, and 89 ± 5 nm, respectively, for the nanoparticles coated with ZIL-4-NP, the size increased to 105 ± 8 nm. This size increase could be due to the bulky structure caused by the increased number of ethyl ether motifs, specifically on the ZIL-4 side chain compared to the other three ZILs (7 vs. 2 or 3). This size increase of the coated particles when compared to the bare PEG–PLGA NPs suggests the formation of a zwitterionic liquid coating on the nanoparticle surface. The polydispersity index remained below 0.2, suggesting a homogeneous particle distribution even after the ZIL coating (Fig. 1A, dots). Furthermore, the surface charge (zeta potential) after the ZIL modification for ZIL-1-NP, ZIL-3-NP, and ZIL-4-NP changed from −27 ± 2 mV to about −5 ± 3 mV, and ZIL-2-NP surface charge was observed closer to −16 mV (Fig. 1B), suggesting electrostatics are involved with ZIL reorganization on the nanoparticle. Moreover, even if the surface charge becomes closer to neutral, the remaining negative charge on the nanoparticle surface after coating suggests that the IL's anionic sulfonate groups likely resides in the outermost layer of the nanoparticle coating. | ||
| Fig. 1 Zwitterionic liquid structure affects assembly during coating of PEG–PLGA nanoparticles. NP size increase and surface charge shifts towards neutrality after coating with zwitterionic liquid. (A) DLS data of the nanoparticle average hydrodynamic diameter (nm), size distribution, polydispersity index (dots), and (B) surface charge (mV) of PEG–PLGA, ZIL-NP-1 to 4 (EO2ImBS, EO2Imps, EO3Imps, and EO7Imps). Statistical significance was performed by one-tailed Student's t-test, comparing against PEG–PLGA for change in size and zeta potential, n = 3, mean ± SD, *p < 0.0002, **p < 0.0005, ***p < 0.002. | ||
Furthermore, the ZIL coating on PEG–PLGA nanoparticles was also confirmed via1H NMR spectroscopy analysis, and ZIL quantity was measured relative to sodium trimethylsilylpropanesulfonate (DSS), a water-soluble internal chemical shift reference standard, then spectra were normalized to 9 protons ((CH3)3Si trimethylsilyl group) at 0 ppm.30 As per 1H NMR spectroscopy data, all ZIL-coated nanoparticles showed peaks corresponding to the ZIL along with the PEG–PLGA peaks. To quantify the amount of ZIL, the peak at ∼9 ppm, which represents the C-2 proton (denoted a in Fig. 2 and S2†) of the imidazolium ring, was used because of the de-shielding effect due to the adjacent N atoms (Fig. 2B and S2†). The slight downfield shift of the ZIL-NP hydrogens compared to the neat ZIL spectrum also demonstrates that there had been a change in shielding, which could be happening due to non-covalent interactions between the oxygens of ZIL sulfonate (SO3−) groups and the terminal hydrogen from the PEG–PLGA polymer (Fig. S3A–D†).9,31
 | ||
| Fig. 2 The zwitterionic liquid was successfully able to coat PEG–PLGA nanoparticles, confirmed using 1H NMR spectroscopy. 1H NMR spectrum shows the coating of ZIL on PEG–PLGA nanoparticles (1 mg NP in 0.2 mg of DSS and 600 μL of D2O) (A) ZIL-2, (B) PEG–PLGA–ZIL-2-NP. | ||
Based on ZIL quantification data, it appears that the amount of ZIL presented on the nanoparticle surface did not significantly differ for ZIL-NP 2, 3, and 4. This can also be seen explicitly for the ZIL-2 and 3 from the similar average size increase of nanoparticles based on DLS data after coating with ZIL (Fig. 1A). However, even with the similar size increase for ZIL-1-NP, the amount of ZIL was nearly two-fold less than the other three ZIL-NPs. Therefore, rather than the amount of IL, this could be happening due to the extra carbon atom on the spacer between the imidazole and the sulfonate group of the ZIL, making it arrange differently on the PEG–PLGA surface, resulting in similar hydrodynamic radius to the ZIL-2 and 3 (Fig. 3A and S3E†).
 | ||
| Fig. 3 The amount of surface-coated ionic liquid and DiD loading efficiency. (A) The amount of each ionic liquid coating per nanoparticle sample (1 mg mL−1) was calculated via1H NMR spectroscopic analysis using 0.2 mg DSS per mg polymer as an internal standard. (B) The percentage of DiD encapsulation in PEG–PLGA nanoparticles for each zwitterionic liquid was determined (n = 3) via fluorescent plate reader measurements. | ||
According to the time-dependent DLS data, the ZIL-coated nanoparticles show stability for nearly three weeks at 4 °C. After the 3rd week, the observed increase in the particle size and the PDI suggest particle instability or aggregation (Fig. S4†). The constant PDI for nearly three weeks for all the samples indicates the stable size distribution and verifies that our nanoparticle system can be effectively used in further experiments without aggregation risk within this timeframe. Despite their long-term stability for all the experiments, the nanoparticles were used within 24 hours of preparation (Fig. S4A–E†).
The encapsulation efficiency of the far-red emitting, lipophilic carbocyanine dye DiD was also calculated for the PEG–PLGA and ZIL-coated PEG–PLGA nanoparticles using fluorescence intensity measurements made by a plate reader. The percent DiD encapsulation was determined relative to the initial DiD stock solution (2% DiD wt/wt, by PEG–PLGA mass). According to the DiD encapsulation efficiency data, ZIL-1-NP demonstrates the highest percent encapsulation of DiD, the same within error as the PEG–PLGA control. As the number of ethylene oxide (E.O., –CH2CH2O–) units increased in the ZIL, a decrease in DiD encapsulation was observed. This phenomenon likely arises from an increase in the hydrophobicity of the ZIL with elongation in the E.O. chain, leading to a decrease in the observed DiD encapsulation (Fig. 3B). The in vitro release of DID load PEG–PLGA and ZIL–PEG–PLGA NPs was carried out using a dynamic dialysis technique. Briefly, DID-loaded nanoparticles were added into a dialysis membrane with a molecular cut-off of 3.5 K, and dialysis was carried out at 37 °C, pH 7.4, in 1× PBS buffer for 48 h. At each time point (3 h, 6 h, 12 h, 24 h, 48 h), 1 mL of buffer was analyzed via fluorescence plate reader (excitation 640 nm and emission 670 nm).32 Based on the DID release data (Fig. S5†), none of the nanoparticles, including the PEG–PLGA control, displayed considerable dye release (>10%) until the 48 h time point. Overall, within the monitored 48 hours, the DID release profile remained below 15%. This minimal percentage release data indicates that the observed fluorescence in nanoparticle cellular uptake after 24 h incubation can predominately be attributed to the encapsulated dye.
In vitro assessment of zwitterionic liquid-coated nanoparticles
Hemolysis refers to the breakdown of RBC membranes, which causes the release of the iron-containing protein hemoglobin into the plasma. Hemolysis can cause anemia, jaundice, and other pathological diseases in living organisms, some of which can be fatal.33 Therefore, to assess the potential hemolytic properties of our ZIL-coated nanoparticle systems in mouse and human RBCs, we have performed an in vitro hemolysis experiment following a previously described protocol. First, we separated and washed RBCs from fresh BALB/c mouse whole blood and human whole blood by centrifuging at 1000g for 10 min at 4 °C, then incubating them with ZIL-NPs for 60 min at 37 °C.34,35 Next, the supernatant was removed, and the absorbance at 405 nm was read in a plate reader, and the absorbance values were normalized against Triton X-100. The results (Fig. 4A) showed that mouse hemolysis was below 15% for all the ZIL-NP candidates, closer to the PEG–PLGA control, for human RBCs, it remained below 10% for all the candidates, including the PEG–PLGA control (Fig. 4A and C), suggesting no significant toxicity and demonstrating their potential for additional assessment. | ||
| Fig. 4 The ZIL-coated nanoparticles show minimal hemolysis and limited adsorption in mouse and human serum. (A) Mouse RBC hemolysis and (C) human RBC hemolysis does not show a significant increase compared to the control PEG–PLGA nanoparticles (n = 3). Triton-X (20% vol/vol) was used as a positive control. (B) Mouse and (D) human serum protein adsorption, using 7.5% precast SDS-gel. | ||
To qualitatively examine the level of protein adsorption following 20 min incubation in neat mouse and human serum, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out in 7.5% Mini-PROTEAN® TGX™ precast gels in 1× Tris–Gly running buffer. Among the tested group of ZIL-coated nanoparticles, it is apparent that the amount of both human and mouse serum protein adsorption did not show any observable difference compared to the PEG–PLGA control by SDS-PAGE (Fig. 4B and D). Therefore, LC-MS/MS analysis was performed to better understand how the identity of the adsorbed proteins on ZIL-coated nanoparticles differed depending on the identity of the ZIL within both human and mouse serum.
LC-MS/MS analysis of serum protein on nanoparticles
Bottom-up LC-MS/MS serum protein analysis was conducted to determine the relative adsorption or depletion onto nanoparticles using neat serum as a control. During the preparation process, the samples were subjected to centrifugation, leading to the isolation of hard protein corona (HC). The hard protein corona forms gradually and exhibits greater stability, consisting of strongly bound proteins onto nanoparticles with less possible detachment. Moreover, due to the differing levels of binding strength, the majority of the soft corona composition is not exclusive; rather, it can also be present in the hard corona.36 LC-MS/MS identified 71 common mouse serum proteins and 55 human serum proteins compared to their negative control from pure serum. These proteins were then grouped into nine categories based on their reported primary functions using the UniProt database and NIH National Library of Medicine. The full list of detected proteins, including UniProtID and the relative abundance, can be found in Fig. S6.† For human serum corona, the majority of the hard corona consists of immunoglobulins and immune-related proteins (10 proteins), lipid and lipoproteins (9 proteins), protease inhibitors and enzymes (8 proteins), and glycoproteins (7 proteins). In contrast, most of the mouse serum hard corona is comprised of immunoglobulins and immune-related proteins (20 proteins), transport and binding proteins (11 proteins), and protease inhibitors and enzymes (18 proteins). However, other significant proteins were identified for both human and mouse systems, including apolipoproteins, coagulation-related, complement, and regulatory proteins (Fig. S6†). Some identified proteins were similar in both systems (Fig. 5 and S7†). Also, most identified proteins appear abundant and agree with the previously studied nanoparticle hard protein corona compositions.37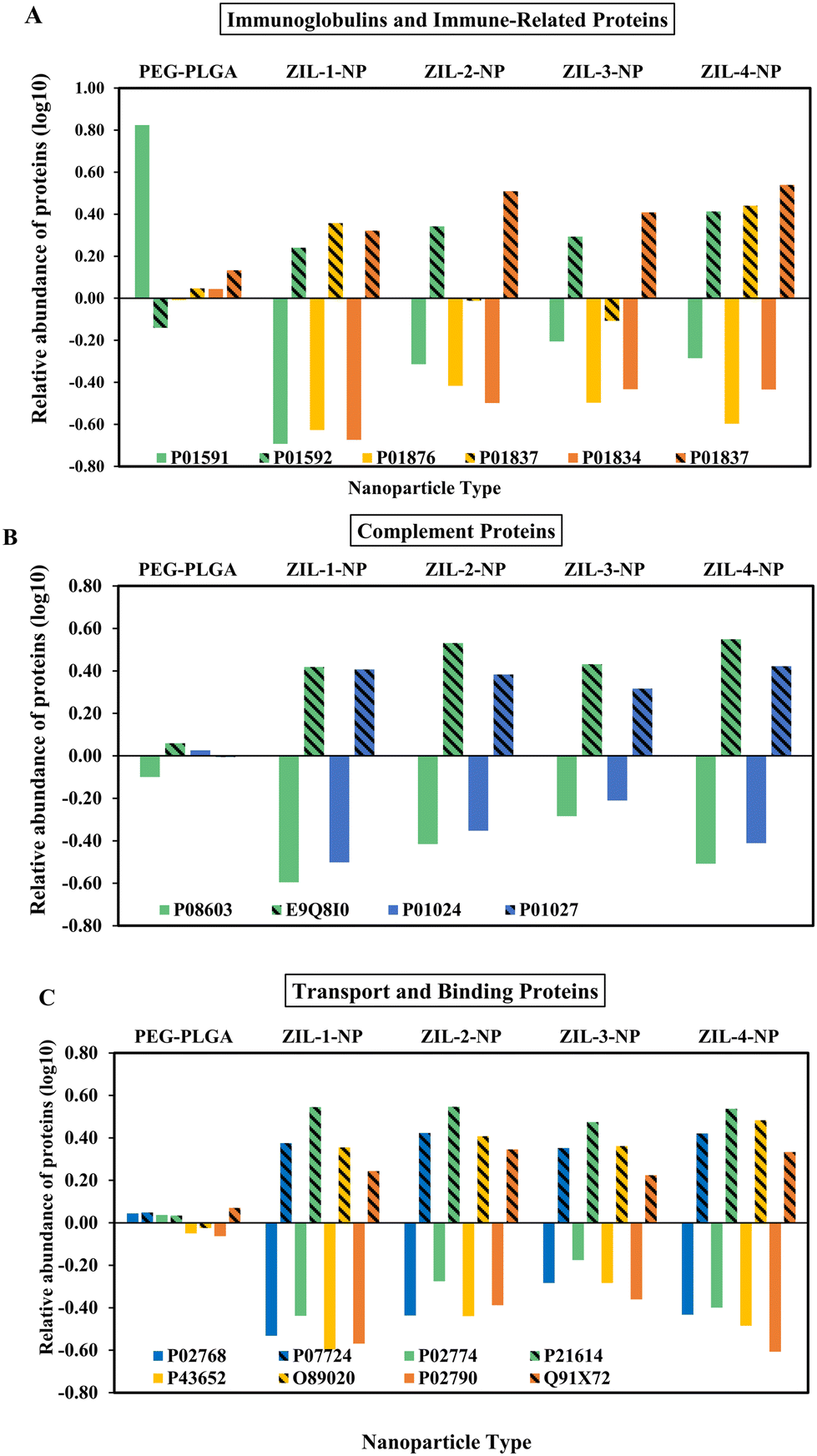 | ||
| Fig. 5 Serum protein corona composition of ZIL-NP and PEG–PLGA NPs relative to a serum control were analyzed via LC-MS/MS. The relative abundance of proteins was categorized based on their primary known functions using the UniProt database and NIH National Library of Medicine. The pattern-filled bars illustrate the abundance of mouse serum proteins, and the same solid color bar represents its corresponding human protein. | ||
Interestingly, even though within these identified similar proteins, the mouse proteins demonstrated higher absorbance relative to their human counterparts (Fig. 5 and S7†). This observed difference could be due to multiple reasons: protein concentrations, post-translation modifications, and inherent differences in protein structures,38 resulting in differences in protein localization and molecular interactions. In mouse serum samples compared to the PEG–PLGA particles, all ZIL-coated nanoparticles showed an almost two-fold depletion in alpha-1-antitrypsin 1–2 protein (P22599), a protease inhibitor that belongs to the serpin superfamily proteins and inhibits various proteases besides trypsin (Fig. S6-5.2†). In addition, it protects tissues from enzymes of inflammatory cells by inhibiting neutrophil elastase.39,40 Compared to the unmodified PEG–PLGA particles ZIL-2-NP and ZIL-3-NP nanoparticles showed particularly decreased amounts of (P01837) immunoglobulin heavy constant alpha, a protein part of serum IgA that is involved in the binding of immunoglobulin receptors and generating an immune response against bacteria and glomerular filtration41 (Fig. 5A and S6-2.2†). This suggests that the ZIL coating might imbue certain anti-inflammatory characteristics on the NPs, although this requires further investigation.
In contrast, among the 55 identified human proteins, 23 showed over two-fold depletion on ZIL-coated nanoparticles compared to the unmodified PEG–PLGA particles (Fig. 5 and S6†). Among these 23 proteins, six were identified as heterodimeric immunoglobulin-type proteins. Immunoglobulin J chain protein (P01591), also a part of serum IgA, which is involved in creating the binding site for pIgR/SC, both by determining the polymeric Ig quaternary structure and via direct interaction with the receptor protein, showed the highest depletion (over 16-fold) for ZIL coated nanoparticles compared to the control42 (Fig. 5A and S6-2.1†). Indicating up to four times depletion compared to the control, five other identified depleted proteins were apolipoproteins, a component of plasma lipoproteins responsible for transporting and metabolizing various blood lipids throughout the body.43 Additionally, among the remaining depleted proteins were glycoproteins, plasma-binding proteins, and complementary system proteins. Notably, compared to the mouse serum in human serum, the albumin protein, the most abundant protein in the blood, was depleted almost three-fold compared to the control (Fig. 5 and S6†). These variations in adsorption and depletion of proteins in both human and mouse serum on nanoparticles could partially be attributed to their different kinetic process of association and dissociation rates. It has been suggested that hydrophobic particles demonstrate faster dissociation rates and a wide span of affinities, especially for albumin and fibrinogen proteins.44 Therefore, the observed wide range of shift from −0.42 to 0.42 adsorption of albumin in human (P02768) to mouse (P07724) serum for the ZIL-4-NP respectively, may occur due to this reported kinetic and affinity difference in albumin (Fig. 5C). Interestingly, the structural variations across the four ZILs did not seem to drastically alter the hard protein corona composition compared to the large shift seen when comparing to the control NPs. This may suggest that the formation of the corona is being driven by the presence of these charged motifs (i.e., the terminal sulfonyl group) rather than small differences in the alkyl chain lengths connecting them. Broadly, the nature of protein corona composition in both systems demonstrates the importance of considering the protein composition in each human and mouse system when designing intravenous delivery systems.
Zwitterionic-coated nanoparticle affinity towards blood components
Subsequently, we investigated the competitive affinity of each zwitterionic-coated PEG–PLGA nanoparticle for different blood components in whole murine blood by following a previously described protocol.45 Briefly, whole BALB/c mouse blood was combined and treated with DiD-loaded, ZIL-coated NPs (1 mg mL−1 in 1× PBS, pH 7.4) at a treatment dose of (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) (v/v) RBCs:NPs. After the treatment, each blood component was extracted and subjected to triplicate buffer washes before analysis via fluorescence-activated cell sorting (FACS). The FACS data indicates zwitterionic coated nanoparticles exhibit over 2–3% (percentage of cells with one or more DID ZIL-NP on them) affinity towards RBCs relative to the PEG–PLGA control (Fig. 6A and S8†). This common observed affinity towards RBCs could be due to imidazolium binding to the heme protein.46 We then quantified the percent of nanoparticles associated with each blood component using a fluorescent plate reader. The data shows that ZIL-2 exhibits the highest RBC NP affinity among the tested ZIL-coated nanoparticles (Fig. 6A and B) by both FACS and plate reader. Comparing the four ZILs, the two ZIL-NPs that show highest affinity for RBCs (1 and 2) both contain two ethyl ether motifs, while ZIL-3 and ZIL-4 have longer side chains. The shorter chains may allow for slightly more compact packing in the nanoparticle coating or closer contact of the imidazole motif with the RBC membrane.
:10) (v/v) RBCs:NPs. After the treatment, each blood component was extracted and subjected to triplicate buffer washes before analysis via fluorescence-activated cell sorting (FACS). The FACS data indicates zwitterionic coated nanoparticles exhibit over 2–3% (percentage of cells with one or more DID ZIL-NP on them) affinity towards RBCs relative to the PEG–PLGA control (Fig. 6A and S8†). This common observed affinity towards RBCs could be due to imidazolium binding to the heme protein.46 We then quantified the percent of nanoparticles associated with each blood component using a fluorescent plate reader. The data shows that ZIL-2 exhibits the highest RBC NP affinity among the tested ZIL-coated nanoparticles (Fig. 6A and B) by both FACS and plate reader. Comparing the four ZILs, the two ZIL-NPs that show highest affinity for RBCs (1 and 2) both contain two ethyl ether motifs, while ZIL-3 and ZIL-4 have longer side chains. The shorter chains may allow for slightly more compact packing in the nanoparticle coating or closer contact of the imidazole motif with the RBC membrane.
 | ||
| Fig. 6 ZIL-coated PEG–PLGA nanoparticles show selective affinity for mouse RBCs. (A) FACS scatter plots showing ZIL-coated PEG–PLGA nanoparticle-treated mouse RBCs population shifts. (B) Fluorescence data showing that ZIL-coated nanoparticles have a higher affinity toward RBCs over white blood cells and platelets. Statistical significance was performed by two-tailed Student's t-test, comparing PEG–PLGA and ZIL-NP, n = 3, mean ± SD, **p < 0.01. | ||
Moreover, neither PEG–PLGA control nor zwitterion-coated nanoparticles show significant affinity towards platelets or white blood cells (Fig. 6B). Additionally, the lower quantitative serum affinity seen with zwitterionic-coated nanoparticles (ZIL-1-NP; 30 ± 1%, ZIL-2-NP; 40% ± 1%, ZIL-3 and 4 NP 35% ± 2–3%) compared to the PEG–PLGA control (60% ± 4%) could be due to the pendant zwitterionic moieties, which agrees with previously published literature based on the zwitterionic antifouling properties.47–49 Although we see no appreciable differences with SDS-PAGE after incubation with serum alone, when incubate with whole blood as we have here, we do see lower serum affinity compared with the control, suggesting that the ZIL-coating is preferentially associating with RBCs over other blood components. This is promising for later in vivo evaluation since the whole blood environment is the most clinically relevant model.
Previously published research on RBC hitchhiking materials have shown excellent in vivo profiles. Depending on the injection site used, RBC hitchhiked NPs demonstrate higher uptake in target organs at one-hour post-injection and maintain a higher level of bioavailability around ten hours, followed by gradual clearance after 24 hours.50 This phenomenon suggests that following the intravenous injection and initial interaction with the dense microvascular network, nanoparticles are mechanically sheared from RBCs into the tissue. While most hitchhiked nanoparticles accumulate in different organs (depending on injection site), the remaining RBC-bound particles may have prolonged circulation time compared to free nanoparticles due to favorable PK provided by RBC carriers.51 This suggests that the RBC hitchhiking of the ZIL-NPs may show favorable outcomes in vivo, although their exact biodistribution and pharmacokinetic profiles are the subject of future work.
The precise mechanism driving the red blood cell hitchhiking is an area of ongoing study. Due to the varying physiochemical properties of nanoparticles, the protein corona profile can influence the transformation of the identity of the ZIL-NPs. It is known that the nanoparticle size, surface charge and chemistry modulated the protein corona.52 Thus, protein corona can influence the dynamic physiological behavior of the nanoparticles.37,53 Furthermore, the dynamic nature of protein corona can facilitate a protein exchange with neighboring proteins to expose the bare fragments on the nanoparticle surface, enabling further cell association.54 Thus, the association of biomolecules with nanoparticle surface varied; techniques like TEM and cryo-EM also show that protein corona has an uneven distribution across the NP surface.55 We hypothesize possible mechanism related to immune adherence, wherein RBCs attach via complement activation. Specifically, c3b complement protein from the serum attaches to a foreign body and subsequently binds to the CR1 receptor on RBCs.56,57 Furthermore, the above-mentioned immune adherence mechanism can also function via mannose-binding lectin (MBL), which can be detected on RBCs even after performing the washing steps.58 The LCMS protein analysis on ZIL-NP protein corona composition provides us with some evidence of the presence of inactive and active complement proteins including complement factor H. Compared to the PEG–PLGA control, all four ZIL systems show elevated levels of specific mouse complement proteins (Fig. 5 and S6, S7†), possibly indicating that ZIL-coated nanoparticle hitchhiking could occur via immune adherence. Specifically, the complement factor H protein is known for binding to host cells and tissues via recognizing specific complement C3 fragment markers and protecting them from complement activation, leading to immune clearance,59,60 due to the observed presence of C3 and complement factor H in ZIL-NP after exposure to mouse serum, it is plausible that the ZIL-NP might be able to engage in the observed RBC hitchhiking though CR1 receptor docking. Certain ion channel transporters may also play a role in the observed hitchhiking, such as anion exchanger (band 3 protein) and amino acid transporter LAT 1.61,62 Since the zwitterions are electrically neutral, their transport through ion channels and pumps may not significantly disrupt the homeostasis across the RBC membrane.63 However, the complex nature of the binding occurring in whole blood as well as the dynamic protein corona on nanoparticles requires further study in order to ascertain the precise driving forces of the observed phenomena.
In vitro cell studies
The effect of neat zwitterionic liquid, zwitterion-coated nanoparticles, and its PEG–PLGA control on MCF-10A cell viability was assessed with the Promega's CellTiter-Glo® luminescent cell viability assay by adding varying concentrations of nanoparticles or neat ZIL in the cell medium and incubating for 24 h. The CellTiter-Glo assay measures the luminescence generated from the amount of ATP provided by metabolically active cells.64,65 Based on the viability data, no noticeable effect on cell viability was observed among the tested neat ZIL and ZIL-NPs concentrations (Fig. 7A and B). Hence, to investigate if there is any competitive and selective the effect of neat zwitterionic liquid, zwitterion-coated nanoparticles, and its PEG–PLGA control on MCF-10A cell viability was assessed with the Promega's CellTiter-Glo® luminescent cell viability assay by adding varying concentrations of nanoparticles or neat ZIL in the cell medium and incubating for 24 h. The CellTiter-Glo assay measures the luminescence generated from the amount of ATP provided by metabolically active cells.64,65 Based on the viability data, no noticeable effect on cell viability was observed among the tested neat ZIL and ZIL-NPs concentrations (Fig. 7A and B). Hence, to investigate if there is any competitive and selective cellular uptake for each zwitterion-coated nanoparticle, the same nanoparticle concentrations were used to assess the nanoparticle cellular uptake using MCF10A and MDA-MB-231 cells. | ||
| Fig. 7 MCF-10A cells maintain viability after 24 h of treatment with ZILs and ZIL-NPs at 37 °C, measured using CellTiter-Glo® luminescent cell viability assay. (A and B) Within the tested neat zwitterionic liquid and ZIL-NPs concentrations, cells do not show significant viability loss compared to the nontreated PEG–PLGA control (n = 3, mean ± SD). | ||
The cellular uptake studies were performed via FACS and fluorescence plate reader.24 Cells were plated on six-well plates at 3 × 105 cell density and incubated at 37 °C until they reached 80% confluency. Varying concentrations of ZIL-coated nanoparticles or a PEG–PLGA control were added and incubated for 24 h. After 24 h, the cells were washed three times with 1× PBS and analyzed for cellular uptake. Based on fluorescence quantification data for the MCF-10A cell line, both ZIL-1-NP and ZIL-2-NP demonstrated higher cellular fluorescence. This increase was more than three-fold for both of these ZIL-NPs, and the fluorescence intensity increased as the nanoparticle concentration increased. Whereas for the other ZIL-NP treated cells, no significant differences were observed compared to the control (Fig. 8A). However, the FACS data, which represents the percent of cells that contain NPs, did not correlate with the fluorescence intensity data; instead, there were no significant differences observed in cellular uptake between the PEG–PLGA control and zwitterionic liquid-coated particles, except for the ZIL-4-NP at 100 and 150 μg mL−1 concentrations where the uptake was increased nearly 15% compared to the control (Fig. 8C).
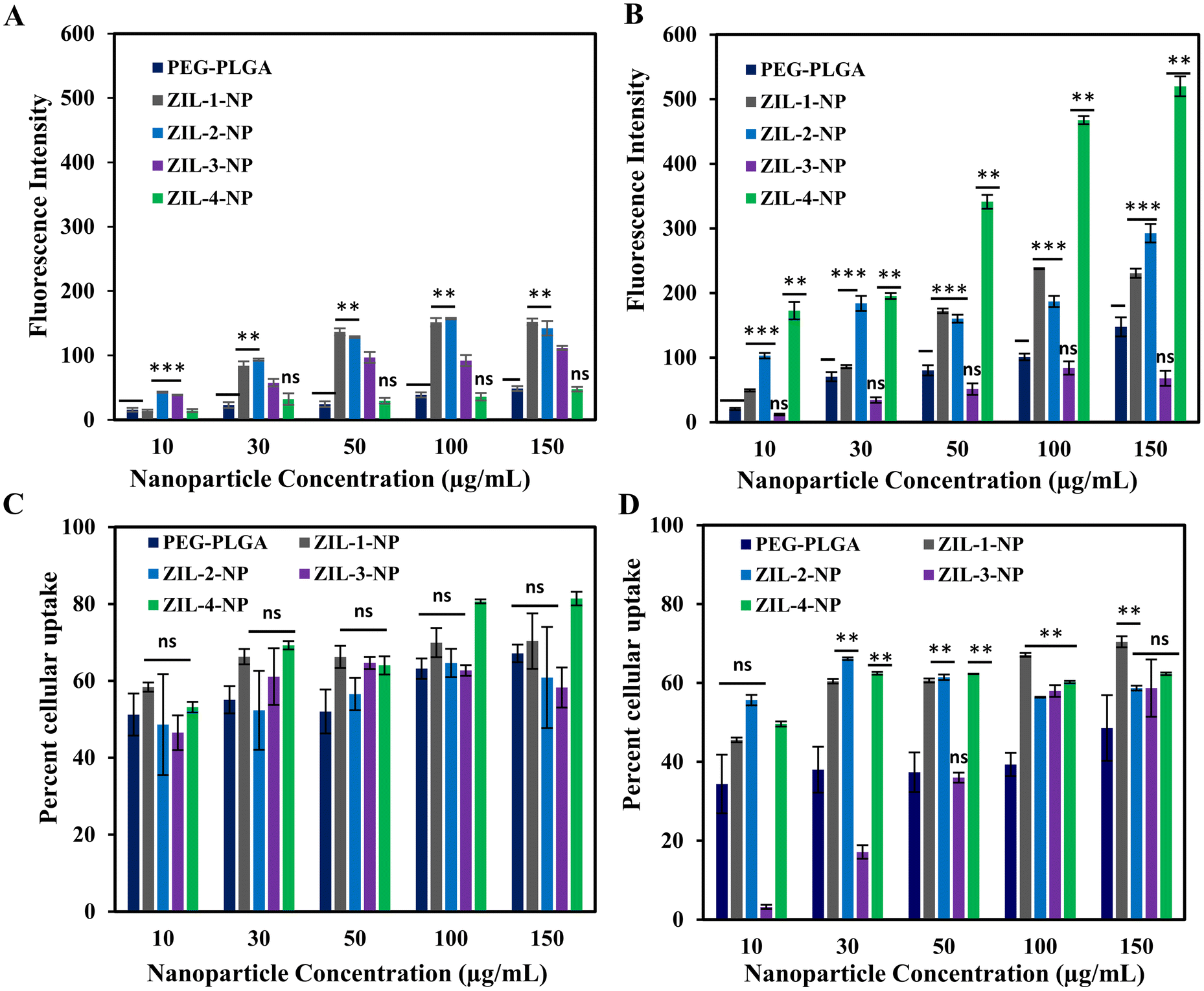 | ||
| Fig. 8 Quantitative and qualitative analysis of zwitterionic liquid-coated PEG–PLGA DiD nanoparticle cellular uptake by (A and C) MCF-10A and (B and D) MDA-MB-231 cells measured via fluorescent plate reader and FACS. (A and B) The quantification of nanoparticles uptake by MCF-10A and MDA-MB-231 cells treated with varied nanoparticle concentrations (10, 30, 50, 100, and 150 μg mL−1) at 37 °C after 24 h. (C and D) FACS analysis of nanoparticle uptake on exact conditions. Statistical significance was performed by two-tailed Student's t-test, comparing PEG–PLGA and ZIL-NP for same concentrations, n = 3, mean ± SD, **p < 0.03, ***p < 0.006, ns – nonsignificant. | ||
For the MDA-MB-231 cell line, based on quantitative fluorescence data, the ZIL-4-NP demonstrated over three-fold increase in cellular fluorescence compared to the PEG–PLGA control (Fig. 8B). Similar to MCF-10A, ZIL-1 and ZIL-2-NP also had higher cellular fluorescence. However, no increase in intensity as the nanoparticle concentration increased was observed, whereas ZIL-1-and ZIL-4-NP treated cells showed a gradual increase in fluorescence intensity with increasing nanoparticle concentration, specifically from concentrations 10 μg mL−1 to 100 μg mL−1 (Fig. 9C). Moreover, in terms of cellular fluorescence intensity, ZIL-4-NP showed the highest uptake compared to MCF-10A cells for all tested conditions (compare Fig. 8A & B). Furthermore, for MDA-MB-231 cells, all ZIL-NPs showed a higher percent cellular uptake by FACS compared to the PEG–PLGA control, and this increase in uptake did not vary as concentration increased. Except for ZIL-3-NP, the percent cellular uptake increased with the increasing nanoparticle concentration. For all ZIL-NPs at the highest nanoparticle concentration of 150 μg mL−1, no significant difference was observed to the control (Fig. 8D), indicating that although more ZIL-NPs are entering the cells, they tend to accumulate in similar numbers of cells as the control group. Compared to normal cells, cancer cells often demonstrate different membrane properties such as membrane composition, overexpression of certain receptors and greater membrane fluidity. The zwitterion coating may interact with these alternative membrane properties, resulting in an increased accumulation of nanoparticles.66 For instance, compared to non-cancer breast cells, cancer cells have elevated levels of phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol and lower levels of phosphatidylserine and sphingomyelin membrane lipids.67
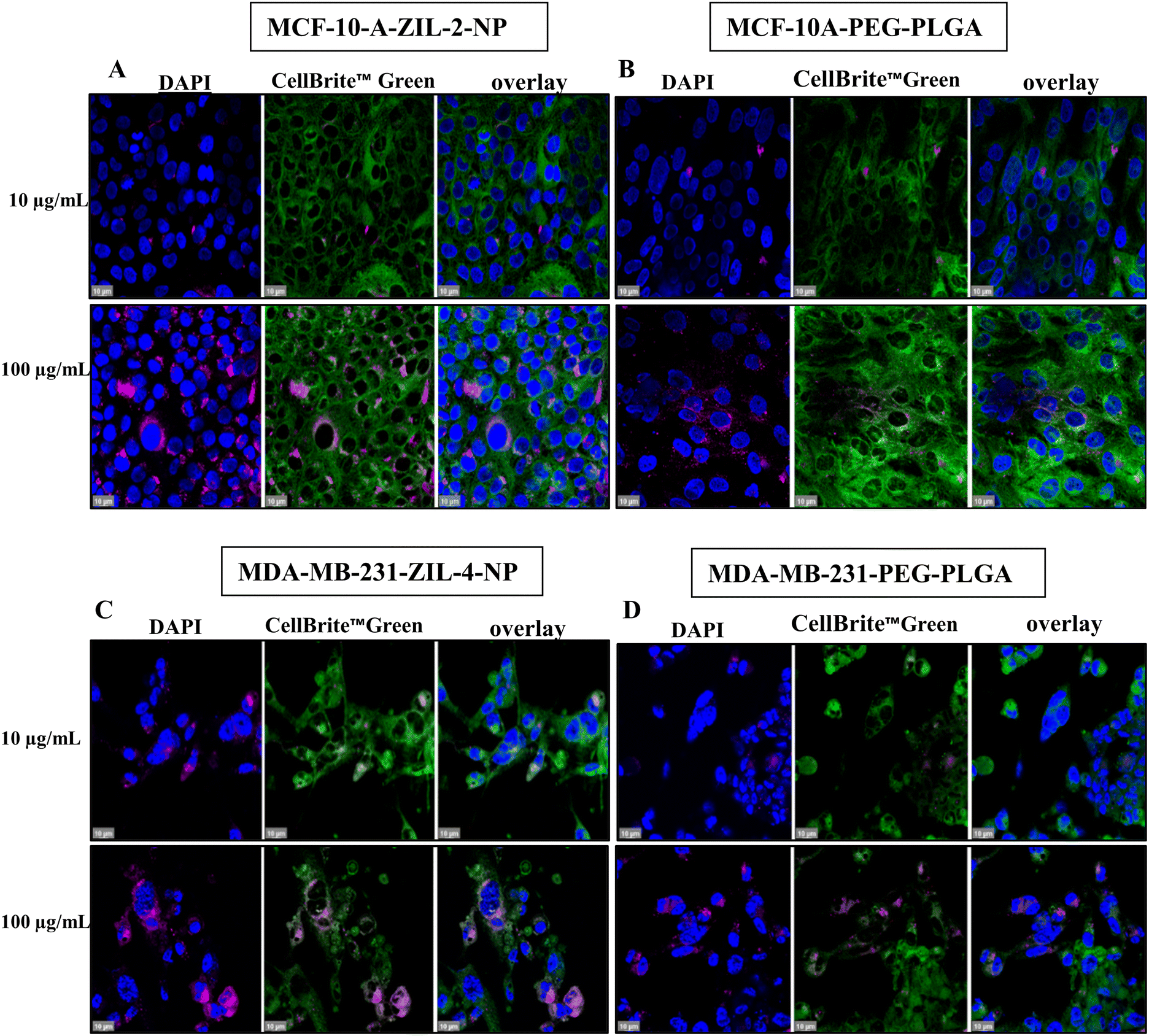 | ||
| Fig. 9 Confocal microscopy images show a higher accumulation of zwitterion-coated nanoparticle uptake per cell. (A and B) MCF-10A cells were treated with PEG–PLGA or PEG–PLGA–ZIL-2, (C and D) MDA-MB-231 cells were treated with PEG–PLGA or PEG–PLGA–ZIL-4 (10, 100 μg mL−1) at 37 °C for 24 h, fixed, stained with DAPI (nuclei) and CellBrite™ Green (cytoplasmic membrane) and observed under 63× oil immersion lens; scale bar: 10 μm. | ||
Considering this highly dynamic and complex nature of cells, higher nanoparticle internalization of specifically ZIL-4-NP in cancer cells compared to healthy cells could be due to various factors, including alterations in cancer cell membranes and function,68 along with the chemical modification of the ZIL-4 structure leading to a change in hydrophobicity and charge-specific interactions69 with the membranes. This change in lipid content results in greater cell membrane fluidity, which facilitates their entry into the bloodstream, leading to metastasis. Considering the highly metastatic character of MDA-MB231, this alteration in membrane lipidic content may have facilitated the higher internalization of the ZIL-NPs. Additionally, according to a study based on cancer cell elasticity, the F-actin percentage of MDA-MB231 cells was nearly 50% lower than the normal breast cells. This reduction of elasticity which could potentially influence the nanoparticle cellular internalization by endocytosis as well as exocytosis.70 However, when it comes to nanoparticle internalization in solid tumors, it relies on their capacity to pass through the blood vessels and the degree of nanoparticle transport endothelial cells presence within the tumor.71
In an in vitro cell culture environment as we have considered here, we can hypothesize that the nanoparticles coated with a longer ethyl ether side chain (ZIL-4-NP) can create denser hydration layers72,73 due to the presence of multiple ethyl ether units attracting more water molecules. Furthermore, the longer ethyl ether chain attached to nanoparticles could act as a steric barrier, maintaining the nanoparticles dispersion in cell media. This steric barrier along with the hydration layer could minimize the particle aggregation74 leading the nanoparticles to preferentially concentrate at the sites of endocytosis, potentially resulting in higher initial cancer cell internalization. Compared to healthy cells, cancer cell membranes have elevated levels of cholesterol and unsaturated fatty acids. This change in lipid content results in greater cell membrane fluidity, which facilitates their entry into the bloodstream, leading to metastasis.67 Considering the highly metastatic character of MDA-MB231, this alteration in membrane lipidic content may have facilitated the higher internalization of the ZIL-NPs. Due to the fluidic nature, it can create gaps within the cell membrane, increasing their permeability towards hydrophobic molecules. Therefore, after the initial higher NP internalization, the hydrophobic part of the ZIL-coated NPs may integrate into the above-mentioned fluidic regions of the cancer cell membrane more readily than into the more rigid healthy cell membrane, facilitating passive diffusion into cells. The hydrophobicity of NP is one of the key factors in nanoparticle cellular internalization.75 Several studies have demonstrated enhanced cellular uptake using nanomaterials with hydrophobic modifications, including zwitterions.6,76,77 Based on previously published molecular dynamic studies, the NPs that consist of hydrophobic moieties tend to enter the lipophilic core of the cell membrane via direct penetration of the cell membrane.78,79 A recently published study demonstrated that an amino-acid-derived zwitterionic polymer positively associated with MDA-MB 231 cells over healthy cells.80 The shared structural similarities between the studied zwitterions and the above-mentioned zwitterionic polymer also provide evidence that the hydrophobicity and the low net charge could play an important role in observed cellular specificity and interactions. Moreover, cell membrane efflux pumps also play a major role in nanoparticle cellular internalization; p-glycoprotein is one of the most well-known efflux pumps in cancer cell membranes and typically exhibits multiple hydrophobic active sites.81 The presence of multiple hydration shells and charge distribution around ZIL-coated nanoparticles could prevent binding to the active site of the efflux pump, minimizing the exocytosis and leading to further nanoparticle accumulation in the cytoplasm.
To investigate the possibility that the detected higher cellular fluorescence was caused by a higher concentration of nanoparticles in fewer cells rather than a broad distribution, we performed confocal imaging. Both MCF-10A and MDA-MB231 cells were grown on poly-D-lysine coated cover glass. When the cells reached 80% confluency, they were treated with ZIL-NPs, which had the maximum fluorescence intensity based on cellular uptake quantification data (Fig. 9A and C); i.e., ZIL-2-NP for MCF-10A cells and ZIL-4-NP for MDA-MB-231 cells, both compared alongside the PEG–PLGA control. After 24 h of incubation at 37 °C, cells were washed and stained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear visualization and CellBrite™ Green to label the cytoplasmic membrane and intracellular membrane structures. Following 4% paraformaldehyde (PFA) fixation, cells were mounted and imaged under a 63× oil immersion lens (Fig. 9). For both cell lines, the confocal images demonstrated that ZIL-NP-treated cells had higher nanoparticle accumulation per cell than PEG–PLGA controls at the same nanoparticle concentrations. When compared with the PEG–PLGA control, even though nearly a similar number of cells had nanoparticles co-localized, the density of nanoparticles seems to be higher than the control, which explains the observed higher fluorescence intensity and similar percent cellular uptake, particularly in MCF-10A cells (compare panels A and B in Fig. 9). This is consistent with the data shown in Fig. 8 whereby a similar number of cells contained NPs (panels C and D), but the amount of nanoparticles in the cancer cell line (panel B) was much higher than in the healthy cell line (panel A) in the case of ZIL-4.
Conclusions
In this work, imidazolium-based zwitterion-coated nanoparticles were synthesized and characterized. This work provides evidence that the tested imidazolium-based zwitterionic liquid can modify the PEG–PLGA surface via non-covalent, supramolecular interactions. All ZIL-NPs demonstrate minimal cellular toxicity within the tested concentrations (up to 150 μM). Additionally, this study demonstrates ZIL surface-modified nanoparticles’ competitive affinity towards RBCs over other whole blood components and increased cellular uptake as a result of high nanoparticle cellular accumulation. Moreover, after exposure to mouse serum, the LCMS analysis of the hard protein corona revealed an altered protein composition between the tested four ZIL-NPs and the control PEG–PLGA NP. ZIL-4-NP, with the longest ethyl ether side chain, showed the highest NP accumulation in MDA-MB231 over MCF-10A cells compared to PEG–PLGA control. All ZIL-NPs (ZIL-1, 2 and 4) had overall higher cellular uptake in MDA-MB231 cells, although the number of cells that contained NPs were similar to the control by FACS. Our observations in the current study of this novel zwitterion-modified nanoparticle system suggest that ZIL-NPs can potentially improve cancer treatments as a safe and effective red blood cell-targeted intravenous drug delivery system.Materials and methods
Materials
Poly(ethylene glycol) methyl ether-block-poly(L-lactide-co-glycolide), PEG average Mn 5000, PLGA average Mn 25000, lactide:glycolide 50:50, and HPLC grade acetonitrile, ≥99.9%, were purchased from Sigma Aldrich. Deuterium oxide rom Sigma Aldrich (#435767-1KG). Ultrapure Milli-Q water was obtained from a Millipore Milli-Q purifier (#Milli-Q IQ 7000) with resistivity >18 MΩ cm. CellTiter-Glo Luminescent cell viability assay was purchased from Promega Corporation. DMEM/high glucose without L-glutamine, sodium pyruvate cell culture solution, phosphate-buffered saline (PBS), and trypsin were obtained from the Cytiva Life Sciences. Fetal bovine serum (FBS), penicillin–streptomycin, and cholera toxin from Vibrio cholerae, ≥90%, and lyophilized powder were purchased from Sigma Aldrich. MEGM BulletKit (CC-3151 & CC-4136) was purchased from Lonza. The MDA-MB231 and MCF-10A cell lines were obtained from ATCC. MDA-MB 231 cells were cultured in DMEM with 10% v/v FBS and 1% penicillin–streptomycin. MCF-10A cells were cultured in MEGM complete medium supplemented with 20 ng mL−1 epidermal growth factor 10% v/v FBS, 1% penicillin–streptomycin, 100 ng mL−1 cholera toxin, 0.01 mg mL−1 human insulin, 500 ng mL−1 hydrocortisone and 5% Chelex-treated horse serum. 96-well clear plates (Corning, #353075), DiD far-red, lipophilic dye (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt) was obtained from Sigma Aldrich (Life Technologies, DiD solid, #D7757) and poly-D-lysine hydrobromide wt 70000–150000 and mounting medium with DAPI were obtained from Millipore Sigma. For cell membrane staining, CellBrite™ Green Cytoplasmic Membrane Labeling Kit was purchased from Biotium. Sodium trimethylsilylpropanesulfonate (DSS) was purchased from MilliporeSigma. For SDS-page Mini-PROTEAN® TGX™ Precast Gels, Laemmli buffer and 10× Tris/glycine/SDS electrophoresis buffer were obtained from BIO-RAD. Lyophilized normal mouse and human serum was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Phosphate buffered saline (PBS) was obtained from Sigma Aldrich (St. Louis, MO, USA), Snakeskin™ Dialysis Tubing, 3.5k MWCO, 16 mm, purchased from Thermo Fisher Scientific.
Zwitterionic liquid synthesis
The experimental details on the preparation of the zwitterionic liquids will be reported in full elsewhere and followed synthetic strategies similar to those described previously.82 Briefly, the desired monomethyl polyether (e.g., diethylene glycol methyl ether for EO2ImBS; ZIL-1) was dissolved in 1:1, v/v tetrahydrofuran (THF):water and deprotonated with sodium hydroxide on an ice bath prior to slow, dropwise addition p-toluenesulfonyl chloride. Following reaction, extraction with dichloromethane (DCM), drying over magnesium sulfate, and rotary evaporation yielded the monotosylated polyether. The monotosylated polyether was next reacted with imidazole and NaOH under reflux in THF for 24 h. After cooling and solvent removal, the dried, DCM-extracted fraction was isolated by rotary evaporation at give the corresponding imidazole-tagged polyether as a colorless liquid in ∼90% yield. The imidazole-polyether was dissolved in acetonitrile and subsequently quaternized under argon by reaction with a cyclic sulfonate ester (e.g., 1,4-butanesultone for EO2ImBS; ZIL-1) to introduce the negatively charged sulfonate terminus, producing the desired zwitterionic liquid after cleanup with diethyl ether. The NMR assignments, provided in the ESI,† are fully consistent with the targeted structures shown.
Nanoparticle synthesis and characterization
After synthesizing both coated and non-coated nanoparticles, they were centrifuge filtered in Amicon Ultra-4 centrifugal filter units, 30k MWCO (Millipore, 4 mL) at 2500 rpm for 50 min at 4 °C to remove any free unbound I.L. in the sample. The filtrate was brought up to 1 mL in cell culture grade DPBS for in vitro biological studies or D2O for in vitro characterization, and these nanoparticle solutions were stored in the dark at 4 °C.
In vitro cell studies
:50 RBC stocks were prepared in a 96-well plate; 180 μL red blood cells and 20 μL from the nanoparticle solution were added and incubated for 1 h at 37 °C. Then samples were centrifuged at 4 °C at 500g for 10 min. After the centrifugation, 100 μL of supernatant from each sample was measured at 405 nm using a plate reader (Biotek H1 Synergy Hybrid Multi-mode) to measure the hemolytic absorbance. Triton-X-100 and 1× PBS were used as the positive and negative control, respectively. After subtracting the negative control from each sample, the percentage hemolysis was calculated by normalizing it to the absorbance of the positive control Triton-X-100 (100%).
:4 ratio) and incubated for 10 min at 25 °C on a shaker (VMR orbital shaker). Then, treated samples were centrifuged with 1× PBS at 3000 rpm for 15 min and washed three times; after each centrifugation, the supernatant was carefully removed, leaving ∼100 μL liquid without disturbing the pellet. After the final centrifugation, nanoparticle samples were transferred to new 1.5 mL Eppendorf tubes, and Laemmli buffer was added at a 1:1 ratio and incubated at 100 °C for 5 min. Then samples were loaded (15 μL) to 7% Mini-PROTEAN® TGX™ precast SDS gels and ran for 40 min at 150 V in 12% Tris–Gly–SDS running buffer (Bio-Rad, California).
:1 ratio (100 μL N.P.s:100 μL serum) for 20 minutes, gently shaking at 37 °C. A negative control was also prepared by combining 100 μL serum:100 μL 1× PBS and underwent the same incubation time as the N.P.s samples. After reaching the endpoints, each sample was centrifuged at 3000 rpm for 15 min at 4 °C three times. Following each centrifugation, 100 μL of non-absorbed serum was removed and replaced with 1× PBS, thoroughly mixed before returning to the centrifuge. For all samples, final centrifugation was performed at 4500 rpm for 10 min at 4 °C.
The protein was collected and a final concentration of 50 mM Tris, 1 mM CaCl2, 0.1% rapigest was added. The samples were incubated at 60 °C for 30 min and cooled. Reduction and alkylation were performed with the 10 mM DTT and 20 mM iodoacetamide in dark. Trypsin at 1:20 ratio of protease:protein was added and incubated overnight with rotation at 37 °C.
0.1% formic acid was added, and the samples were run on Exploris 240 Orbitrap mass Spectrometer (Thermo Scientific). The samples were loaded onto an Acclaim PepMap 100 C18 nanocolumn (0.075 mm × 150 mm, 2 μm, Thermo Fisher Scientific). Peptides were separated on the chromatographic system using mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) at a flow rate of 300 μL min−1. The peptides were eluted with a gradient consisting of 2 to 32% solvent B over 23 min, ramped to 95% solvent B over 5 min, held for 4 min, and then returned to 2% solvent B over 3 min and held for 8 min. All data were acquired in positive ion mode. The spray voltage was set to 2450 V, and the ion transfer tube was set to 300 °C. The MS1 data were collected with a resolution of 60000 with a scan range of 250–2000 m/z. In CID mode, full MS scans were followed by eight subsequent MS/MS scans on the top eight most-abundant peptide ions. Collision energy was set to 30%. The data was searched by proteomics search engine Byonic V.4.4.2 (Protein Metrics, San Carlos, CA). The false discovery rate was set to 1% at the protein level. The proteins with log probability <5.0 were discarded and only proteins with at least 2 peptides were included. The intensity of the trypsin used to digest the protein samples was used for normalization of the relative abundance of the protein since the intensity of trypsin did not vary much in all the samples (Fig. S4†). To calculate the relative amount of the protein in that sample, first the intensity of a particular protein was divided by the intensity of trypsin in that particular sample. Then, the same value was obtained from the serum control for the same protein. Finally, the values for the protein from samples were divided by the values obtained from serum control and converted to log10.
Red blood cell hitchhiking: qualitative and quantitative study
Qualitative flow cytometric and quantitative plate reader studies were performed using commercially purchased BALB/c blood following a previously published protocol.5,45,83 Briefly, 1 mg mL−1 concentrated nanoparticles were combined with 500 μL of whole blood at a 1:10 ratio on ice, and samples were briefly mechanically mixed by inversion, followed by a 20 min incubation at 37 °C on a shaker (VMR orbital shaker) at 500 rpm. Subsequently, all samples were centrifuged at 1000g and 4 °C for 10 min to isolate the components in whole blood. After centrifugation, each sample was isolated and separated into serum, white blood cells, platelets, and red blood cells. Afterward, each separated component was washed three times using 1× PBS and centrifuged at 200g, 4 °C for 5 min. Following each wash, the supernatant was removed and replaced with 1× PBS pH 7.4 to its original volume. After the last wash, the volume was bought up to 1 mL with 1× PBS, then 200 μL from each isolated fraction was resuspended in 2 mL 1× PBS for fluorescent-activated cell sorting (FACS). The FACS analysis (model #AFC2) was carried out at a 12.5 μL min−1 flow rate and first gating the desired cell population side vs. forward scattering to isolate the RBCs, and then the population was gated against far-red fluorescence; this represents the percentage of cells that IL-coated nanoparticles vs. bare PEG–PLGA nanoparticles hitchhiked. For the quantification study, each separated fraction was resuspended in 1× PBS and bought up to 200 μL final volume, and the fluorescence was read by fluorescent plate reader (excitation 640 nm, emission 670 nm).
Author contributions
Gaya S. Dasanayake: investigation, methodology, visualization, writing – review and editing, formal analysis, Christine M. Hamadani: investigation, writing – review, and editing, Gagandeep Singh: investigation, writing – review and editing, Sandeep Kumar Misra: investigation, writing – review, and editing, Priyavrat Vashisth: writing – review and editing, Joshua S. Sharp: resources, supervision, writing – review & editing, Laxmi Adhikari: resources, review & editing, Gary A. Baker: resources, supervision, writing – review & editing, Eden E. L. Tanner: resources, supervision, funding, writing – original draft, conceptualization project administration.Conflicts of interest
The authors have no conflicts of interest to report.Acknowledgements
This work was supported by the College of Liberal Arts at the University of Mississippi. EELT acknowledges the PhRMA Foundation. We thank Dr Thomas Werfel for giving access to FACS and the fluorescent plate reader. We acknowledge the Glycoscience Center of Research Excellence (NIH-P20GM103460) at the University of Mississippi. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- V. V. Sheffey, E. B. Siew, E. E. L. Tanner and O. Eniola-Adefeso, PLGA's Plight and the Role of Stealth Surface Modification Strategies in Its Use for Intravenous Particulate Drug Delivery, Adv. Healthc. Mater., 2022, 11, e2101536 CrossRef PubMed.
- C. H. Villa, A. C. Anselmo, S. Mitragotri and V. Muzykantov, Red blood cells: Supercarriers for drugs, biologicals, and nanoparticles and inspiration for advanced delivery systems, Adv. Drug Delivery Rev., 2016, 106, 88 CrossRef CAS PubMed.
- J. S. Brenner, S. Mitragotri and V. R. Muzykantov, Red Blood Cell Hitchhiking: A Novel Approach for Vascular Delivery of Nanocarriers, Annu. Rev. Biomed. Eng., 2021, 23, 225–248 CrossRef CAS PubMed.
- Y. Zhang, T. Sun and C. Jiang, Biomacromolecules as carriers in drug delivery and tissue engineering, Acta Pharm. Sin. B, 2018, 8, 34–50 CrossRef PubMed.
- C. M. Hamadani, M. J. Goetz, S. Mitragotri and E. E. L. Tanner, Protein-avoidant ionic liquid (pail)-coated nanoparticles to increase bloodstream circulation and drive biodistribution, Sci. Adv., 2020, 6(48), eabd7563 CrossRef CAS PubMed.
- D. F. Moyano, et al., Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity, ACS Nano, 2014, 8(7), 6748–6755 CrossRef CAS PubMed.
- Z. Zhao, et al., Screening of Zwitterionic Liposomes with Red Blood Cell-Hitchhiking and Tumor Cell-Active Transporting Capability for Efficient Tumor Entrance, Adv. Funct. Mater., 2023, 2214369, DOI:10.1002/adfm.202214369.
- M. D. Green and T. E. Long, Designing imidazole-based ionic liquids and ionic liquid monomers for emerging technologies, Polym. Rev., 2009, 49, 291–314 CrossRef CAS.
- E. B. Anderson and T. E. Long, Imidazole- and imidazolium-containing polymers for biology and material science applications, Polymer, 2010, 51, 2447–2454 CrossRef CAS.
- A. T. Baviskar, et al., N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIα and induce apoptosis in G1/S phase, J. Med. Chem., 2011, 54, 5013–5030 CrossRef CAS PubMed.
- Z. G. Estephan, J. A. Jaber and J. B. Schlenoff, Zwitterion-Stabilized Silica Nanoparticles: Toward Nonstick Nano, Langmuir, 2010, 26, 16884–16889 CrossRef CAS PubMed.
- M. Debayle, et al., Zwitterionic polymer ligands: an ideal surface coating to totally suppress protein-nanoparticle corona formation?, Biomaterials, 2019, 219, 119357 CrossRef CAS PubMed.
- S. Chen, J. Zheng, L. Li and S. Jiang, Strong resistance of phosphorylcholine self-assembled monolayers to protein adsorption: Insights into nonfouling properties of zwitterionic materials, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS PubMed.
- W. Li, Q. Liu and L. Liu, Amino acid-based zwitterionic polymers: antifouling properties and low cytotoxicity, J. Biomater. Sci., Polym. Ed., 2014, 25, 1730–1742 CrossRef CAS PubMed.
- G. Singh, M. Kaur, T. S. Kang and V. K. Aswal, Aqueous colloidal systems of bovine serum albumin and functionalized surface active ionic liquids for material transport, RSC Adv., 2020, 10, 7073–7082 RSC.
- L. J. Bruce and G. Gyorffy, Red cell membrane proteins, HemaSphere, 2019, 3, 154–156 CrossRef PubMed.
- S. J. Nikkhah and M. Vandichel, Modeling Polyzwitterion-Based Drug Delivery Platforms: A Perspective of the Current State-of-the-Art and Beyond, ACS Eng. Au, 2022, 2, 274–294 CrossRef PubMed.
- S. Chen, et al., Enhanced tumour penetration and prolonged circulation in blood of polyzwitterion–drug conjugates with cell-membrane affinity, Nat. Biomed. Eng., 2021, 5, 1019–1037 CrossRef CAS PubMed.
- Z. Zhao, et al., Screening of Zwitterionic Liposomes with Red Blood Cell-Hitchhiking and Tumor Cell-Active Transporting Capability for Efficient Tumor Entrance, Adv. Funct. Mater., 2023, 33, 2214369 CrossRef CAS.
- A. Chen and M. Scott, Comparative analysis of polymer and linker chemistries on the efficacy of immunocamouflage of murine leukocytes, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 2006, 34, 305–322 CrossRef CAS PubMed.
- K. L. Murad, et al., Structural and Functional Consequences of Antigenic Modulation of Red Blood Cells With Methoxypoly(Ethylene Glycol), Blood, 1999, 93, 2121–2127 CrossRef CAS PubMed.
- L. R. Parisi, et al., Membrane Disruption by Very Long Chain Fatty Acids during Necroptosis, ACS Chem. Biol., 2019, 14, 2286–2294 CAS.
- C. Samori, et al., Introduction of oxygenated side chain into imidazolium ionic liquids: Evaluation of the effects at different biological organization levels, Ecotoxicol. Environ. Saf., 2010, 73, 1456–1464 CrossRef CAS PubMed.
- G. Singh, et al., Good’s buffer based highly biocompatible ionic liquid modified PLGA nanoparticles for the selective uptake in cancer cells, Mater. Chem. Front., 2023, 7(24), 6213–6228 RSC.
- F. Spyrakis, A. BidonChanal, X. Barril and F. Javier Luque, Protein Flexibility and Ligand Recognition: Challenges for Molecular Modeling, Curr. Top. Med. Chem., 2012, 11, 192–210 CrossRef PubMed.
- M. Yoshizawa, M. Hirao, K. Ito-Akita and H. Ohno, Ion conduction in zwitterionic-type moltensalts and their polymers, J. Mater. Chem., 2001, 11, 1057–1062 RSC.
- M. Yoshizawa-Fujita, T. Tamura, Y. Takeoka and M. Rikukawa, Low-melting zwitterion: effect of oxyethylene units on thermal properties and conductivity, Chem. Commun., 2011, 47, 2345–2347 RSC.
- C. Tiyapiboonchaiya, et al., The zwitterion effect in high-conductivity polyelectrolyte materials, Nat. Mater., 2003, 3, 29–32 CrossRef PubMed.
- P. Sarker, et al., Hydration behaviors of nonfouling zwitterionic materials, Chem. Sci., 2023, 14, 7500–7511 RSC.
- F. Madrid-Gambin, et al., Untargeted 1H NMR-Based Metabolomics Analysis of Urine and Serum Profiles after Consumption of Lentils, Chickpeas, and Beans: An Extended Meal Study to Discover Dietary Biomarkers of Pulses, J. Agric. Food Chem., 2018, 66, 6997–7005 CrossRef CAS PubMed.
- S. K. Lai, et al., Privileged delivery of polymer nanoparticles to the perinuclear region of live cells via a non-clathrin, non-degradative pathway, Biomaterials, 2007, 28, 2876–2884 CrossRef CAS PubMed.
- B. Gaihre, M. S. Khil, D. R. Lee and H. Y. Kim, Gelatin-coated magnetic iron oxide nanoparticles as carrier system: Drug loading and in vitro drug release study, Int. J. Pharm., 2009, 365, 180–189 CrossRef CAS PubMed.
- H. L. Gaddey and A. M. Riegel, Unexplained Lymphadenopathy: Evaluation and Differential Diagnosis, Am. Fam. Physician, 2016, 94, 896–903 Search PubMed.
- B. C. Evans, et al., Ex Vivo Red Blood Cell Hemolysis Assay for the Evaluation of pH-responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs, J. Visualized Exp., 2013, 50166, DOI:10.3791/50166.
- I. I. Slowing, C. W. Wu, J. L. Vivero-Escoto and V. S. Y. Lin, Mesoporous Silica Nanoparticles for Reducing Hemolytic Activity Towards Mammalian Red Blood Cells, Small, 2009, 5, 57–62 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, The protein corona from nanomedicine to environmental science, Nat. Rev. Mater., 2023, 8, 422–438 CrossRef PubMed.
- D. Baimanov, et al., In situ analysis of nanoparticle soft corona and dynamic evolution, Nat. Commun., 2022, 13, 1–14 Search PubMed.
- M. M. Albà and R. Guigó, Comparative Analysis of Amino Acid Repeats in Rodents and Humans, Genome Res., 2004, 14, 549 CrossRef PubMed.
- P. G. W. Gettins, Serpin Structure, Mechanism, and Function, Chem. Rev., 2002, 102(12), 4751–4804 CrossRef CAS PubMed.
- A. Abdulkarim and T. J. Craig, Alpha 1 Antitrypsin Mutation, StatPearls, 2022.
- IGHA1 immunoglobulin heavy constant alpha 1 [Homo sapiens (human)] – Gene – NCBI, https://www.ncbi.nlm.nih.gov/gene/3493.
- F. E. Johansen, R. Braathen and P. Brandtzaeg, Role of J chain in secretory immunoglobulin formation, Scand. J. Immunol., 2000, 52, 240–248 CrossRef CAS PubMed.
- T. Liu, et al., ApoPred: Identification of Apolipoproteins and Their Subfamilies With Multifarious Features, Front. Cell Dev. Biol., 2021, 8, 1583 Search PubMed.
- T. Cedervall, et al., Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- C. M. Hamadani, et al., Development of ionic liquid-coated PLGA nanoparticles for applications in intravenous drug delivery, Nat. Protoc., 2023, 18, 2509–2557 CrossRef CAS PubMed.
- J. E. Erman, D. Chinchilla, J. Studer, L. B. Vitello and J. Erman, Binding of imidazole, 1-methylimidazole and 4-nitroimidazole to yeast cytochrome c peroxidase (CcP) and the distal histidine mutant, CcP(H52L) HHS Public Access, Biochim. Biophys. Acta, 2015, 869–881, DOI:10.1016/j.bbapap.2015.04.013.
- F. Hu, K. Chen, H. Xu and H. Gu, Functional short-chain zwitterion coated silica nanoparticles with antifouling property in protein solutions, Colloids Surf., B, 2015, 126, 251–256 CrossRef CAS PubMed.
- J. Wang, et al., The effects of poly(zwitterions)s: Versus poly(ethylene glycol) surface coatings on the biodistribution of protein nanoparticles, Biomater. Sci., 2016, 4, 1351–1360 RSC.
- S. Chen, J. Zheng, L. Li and S. Jiang, Strong resistance of phosphorylcholine self-assembled monolayers to protein adsorption: Insights into nonfouling properties of zwitterionic materials, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS PubMed.
- A. C. Anselmo, et al., Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells, ACS Nano, 2013, 7, 11129–11137 CrossRef CAS PubMed.
- A. Ukidve, et al., Erythrocyte-driven immunization via biomimicry of their natural antigen-presenting function, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17727–17736 CrossRef CAS PubMed.
- M. Lundqvist, et al., Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, The protein corona from nanomedicine to environmental science, Nat. Rev. Mater., 2023, 8, 422–438 CrossRef PubMed.
- H. Mohammad-Beigi, et al., Mapping and identification of soft corona proteins at nanoparticles and their impact on cellular association, Nat. Commun., 2020, 11, 1–16 CrossRef PubMed.
- S. Sheibani, et al., Nanoscale characterization of the biomolecular corona by cryo-electron microscopy, cryo-electron tomography, and image simulation, Nat. Commun., 2021, 12(1), 573 CrossRef CAS PubMed.
- R. P. Taylor, et al., Use of heteropolymeric monoclonal antibodies to attach antigens to the C3b receptor of human erythrocytes: a potential therapeutic treatment, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3305 CrossRef CAS PubMed.
- In vivo binding and clearance of circulating antigen by bispecific heteropolymer-mediated binding to primate erythrocyte complement receptor – PubMed, https://pubmed.ncbi.nlm.nih.gov/1373171/.
- J. S. Brenner, S. Mitragotri and V. R. Muzykantov, Red Blood Cell Hitchhiking: A Novel Approach for Vascular Delivery of Nanocarriers, Annu. Rev. Biomed. Eng., 2021, 23, 225 CrossRef CAS PubMed.
- V. P. Ferreira, M. K. Pangburn and C. Cortés, Complement control protein factor H: the good, the bad, and the inadequate, Mol. Immunol., 2010, 47, 2187 CrossRef CAS PubMed.
- S. R. Moore, S. S. Menon, C. Cortes and V. P. Ferreira, Hijacking Factor H for Complement Immune Evasion, Front. Immunol., 2021, 12, 602277 CrossRef CAS PubMed.
- M. L. Jennings, Landmark Review: Cell physiology and molecular mechanism of anion transport by erythrocyte band 3/AE1, Am. J. Physiol.: Cell Physiol., 2021, 321, C1028 CrossRef CAS PubMed.
- A. Bouthelier, et al., Erythroid SLC7A5/SLC3A2 amino acid carrier controls red blood cell size and maturation, iScience, 2022, 26(1), 105739 CrossRef PubMed.
- M. von Lindern, S. Egée, P. Bianchi and L. Kaestner, The Function of Ion Channels and Membrane Potential in Red Blood Cells: Toward a Systematic Analysis of the Erythroid Channelome, Front. Physiol., 2022, 13, 824478 CrossRef PubMed.
- C. M. Chism, et al., Antimicrobial Effects of Anion Manipulation with Biocompatible Choline Carboxylic Acid-Based Ionic Liquids, ACS Appl. Eng. Mater., 2023, 1, 23–31 CrossRef CAS.
- P. Wang, S. M. Henning and D. Heber, Limitations of MTT and MTS-Based Assays for Measurement of Antiproliferative Activity of Green Tea Polyphenols, PLoS One, 2010, 5, e10202 CrossRef PubMed.
- A. B. Hendrich and K. Michalak, Lipids as a Target for Drugs Modulating Multidrug Resistance of Cancer Cells, Curr. Drug Targets, 2003, 4(1), 23–30 CrossRef CAS PubMed.
- G. Preta, New Insights Into Targeting Membrane Lipids for Cancer Therapy, Front. Cell Dev. Biol., 2020, 8, 571237 CrossRef PubMed.
- K. Kettler, K. Veltman, D. van de Meent, A. van Wezel and A. J. Hendriks, Cellular uptake of nanoparticles as determined by particle properties, experimental conditions, and cell type, Environ. Toxicol. Chem., 2014, 33, 481–492 CrossRef CAS PubMed.
- C. Cortez, et al., Influence of size, surface, cell line, and kinetic properties on the specific binding of A33 antigen-targeted multilayered particles and capsules to colorectal cancer cells, ACS Nano, 2007, 1, 93–102 CrossRef CAS PubMed.
- S. Kwon, W. Yang, D. Moon and K. S. Kim, Comparison of cancer cell elasticity by cell type, J. Cancer, 2020, 11, 5403–5412 CrossRef CAS PubMed.
- B. R. Kingston, et al., Specific Endothelial Cells Govern Nanoparticle Entry into Solid Tumors, ACS Nano, 2021, 15, 14080–14094 CrossRef CAS PubMed.
- J. Zhao, et al., Long circulating micelles of an amphiphilic random copolymer bearing cell outer membrane phosphorylcholine zwitterions, Acta Biomater., 2015, 16, 94–102 CrossRef CAS PubMed.
- H. Ou, et al., Surface-adaptive zwitterionic nanoparticles for prolonged blood circulation time and enhanced cellular uptake in tumor cells, Acta Biomater., 2018, 65, 339–348 CrossRef CAS PubMed.
- F. Gambinossi, S. E. Mylon and J. K. Ferri, Aggregation kinetics and colloidal stability of functionalized nanoparticles, Adv. Colloid Interface Sci., 2015, 222, 332–349 CrossRef CAS PubMed.
- D. Chen, et al., Promoting Inter-/Intra-Cellular Process of Nanomedicine through its Physicochemical Properties Optimization, Curr. Drug Metab., 2017, 19, 75–82 CrossRef PubMed.
- T. Bewersdorff, et al., Amphiphilic nanogels: influence of surface hydrophobicity on protein corona, biocompatibility and cellular uptake, Int. J. Nanomed., 2019, 14, 7861 CrossRef CAS PubMed.
- Z. Zhao, A. Ukidve, V. Krishnan and S. Mitragotri, Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers, Adv. Drug Delivery Rev., 2019, 143, 3–21 CrossRef CAS PubMed.
- E. M. Curtis, A. H. Bahrami, T. R. Weikl and C. K. Hall, Modeling nanoparticle wrapping or translocation in bilayer membranes, Nanoscale, 2015, 7, 14505–14514 RSC.
- P. Sabourian, et al., Effect of Physico-Chemical Properties of Nanoparticles on Their Intracellular Uptake, Int. J. Mol. Sci., 2020, 21, 1–20 Search PubMed.
- M. N. Leiske, B. G. De Geest and R. Hoogenboom, Impact of the polymer backbone chemistry on interactions of amino-acid-derived zwitterionic polymers with cells, Bioact. Mater., 2023, 24, 524–534 CAS.
- K. Robinson and V. Tiriveedhi, Perplexing Role of P-Glycoprotein in Tumor Microenvironment, Front. Oncol., 2020, 10, 515011 Search PubMed.
- S. Tang, G. A. Baker and H. Zhao, Ether- and alcohol-functionalized task-specific ionic liquids: attractive properties and applications, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC.
- C. M. Hamadani, et al., Improved nanoformulation and bio-functionalization of linear-dendritic block copolymers with biocompatible ionic liquids, Nanoscale, 2022, 14, 6021–6036 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06349f |
| ‡ Current address: U.S. Food and Drug Administration, Bedford Park, IL 60501, USA. |
| This journal is © The Royal Society of Chemistry 2024 |