
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando insights into correlating CO coverage and Cu–Au alloying with the selectivity of Au NP-decorated Cu2O nanocubes during the electrocatalytic CO2 reduction†
Clara
Rettenmaier
 a,
Antonia
Herzog
a,
Daniele
Casari
b,
Martina
Rüscher
a,
Hyo Sang
Jeon
a,
David
Kordus
a,
Mauricio Lopez
Luna
a,
Stefanie
Kühl
a,
Uta
Hejral
a,
Earl M.
Davis
a,
See Wee
Chee
a,
Janis
Timoshenko
a,
Duncan T.L.
Alexander
b,
Arno
Bergmann
a and
Beatriz Roldan
Cuenya
*a
a,
Antonia
Herzog
a,
Daniele
Casari
b,
Martina
Rüscher
a,
Hyo Sang
Jeon
a,
David
Kordus
a,
Mauricio Lopez
Luna
a,
Stefanie
Kühl
a,
Uta
Hejral
a,
Earl M.
Davis
a,
See Wee
Chee
a,
Janis
Timoshenko
a,
Duncan T.L.
Alexander
b,
Arno
Bergmann
a and
Beatriz Roldan
Cuenya
*a
aDepartment of Interface Science, Fritz-Haber-Institute of the Max-Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: roldan@fhi-berlin.mpg.de
bElectron Spectrometry and Microscopy Laboratory (LSME), Institute of Physics (IPHYS), École Polytechnique Fédérale de Lausanne (EPFL), Lausanne CH-1015, Switzerland
First published on 25th October 2023
Abstract
Electrochemical reduction of CO2 (CO2RR) is an attractive technology to reintegrate the anthropogenic CO2 back into the carbon cycle driven by a suitable catalyst. This study employs highly efficient multi-carbon (C2+) producing Cu2O nanocubes (NCs) decorated with CO-selective Au nanoparticles (NPs) to investigate the correlation between a high CO surface concentration microenvironment and the catalytic performance. Structure, morphology and near-surface composition are studied via operando X-ray absorption spectroscopy and surface-enhanced Raman spectroscopy, operando high-energy X-ray diffraction as well as quasi in situ X-ray photoelectron spectroscopy. These operando studies show the continuous evolution of the local structure and chemical environment of our catalysts during reaction conditions. Along with its alloy formation, a CO-rich microenvironment as well as weakened average CO binding on the catalyst surface during CO2RR is detected. Linking these findings to the catalytic function, a complex compositional interplay between Au and Cu is revealed in which higher Au loadings primarily facilitate CO formation. Nonetheless, the strongest improvement in C2+ formation appears for the lowest Au loadings, suggesting a beneficial role of the Au–Cu atomic interaction for the catalytic function in CO2RR. This study highlights the importance of site engineering and operando investigations to unveil the electrocatalyst's adaptations to the reaction conditions, which is a prerequisite to understand its catalytic behavior.
Broader contextEnvironmentally friendly technologies for storage and reutilization of energy are important to maintain a sustainable carbon cycle. Here, the electrocatalytic reduction of CO2 attracts by recycling CO2 with renewable energy to form highly valued chemicals. By using Cu-based materials as catalysts, the performance of the CO2 reduction reaction can be tuned towards C2+ products, such as ethanol or ethylene. However, major challenges are the in-depth understanding of reaction mechanism, including the performance towards selective products, as well as creating stable catalysts. Thus, careful characterization of the catalysts is necessary to understand how the catalysts evolve during the applied reaction. Herein, we studied a bimetallic Cu-based material by adding increasing amounts of CO-forming Au nanoparticles to understand the structural, compositional and morphological changes under reaction conditions, correlating them with the obtained selectivities and thus extracting structure–selectivity relationships. The results from this work highlight the essential changes and requirements for Cu–Au based catalysts that are relevant to create high-performing catalysts for the electrocatalytic CO2 reduction reaction. |
Introduction
The electrochemical reduction of CO2 (CO2RR) is an attractive technology for closing the anthropogenic carbon cycle by using renewable energy such as solar- or wind-power to convert the greenhouse gas CO2 into energy-dense feedstock chemicals or liquid fuels.1,2The ideal electrocatalysts for this reaction require low overpotentials, high stability and excellent selectivity for C2+ products while minimizing the parasitic hydrogen evolution reaction (HER). Whereas various materials may reduce CO2 into C1 products (CO, CH4, HCOOH), copper-based materials are unique in producing C2+ hydrocarbons (e.g., C2H4), alcohols (e.g., C2H5OH) or carbonyls, due to their ability to enable the C–C coupling. Cu2O nanocubes (NCs) are reported as one of best catalysts for C2+ products.3 The Cu-based catalysts owe their good selectivity to an optimal binding and stabilization of the CO intermediates4e.g., *CO and *CO2−). However, these catalysts still suffer from low energy efficiency and lack of selectivity toward a specific reaction product.5
Among the multitude of approaches to improve the selectivity towards C2+ products, sequential catalysis has been proposed for optimizing the selectivity by combining both, beneficial electronic effects via alloying and an increased CO coverage.6–10 By adjusting the local electronic structure of the catalyst though the use of bimetallic systems and alloys, the binding energies of the reaction intermediates such as *CO might be altered and thus, the reaction pathways might be modified.11–14 Furthermore, a higher local concentration and a subsequently higher coverage of adsorbed CO molecules on Cu can be induced through CO-selective co-catalysts, such as Au, Ag or Zn.4 This may lead to subsequent C–C coupling and thus, enhanced selectivity for C2+ products.15–17 Despite their similar abilities to efficiently reduce CO2 to CO, Au, Ag and Zn differ from each other in the onset potential for CO production with −0.25 V (vs. RHE) for Au, −0.52 V for Ag and −0.6 V for Zn.4 Thus, Au has the lowest overpotential and shows the highest CO partial current density, which can be explained by its weak *CO binding strength.4,18,19 Furthermore, it has been proposed that the thus produced CO might follow a CO spillover mechanism, which has also been reported for Ag/Cu catalysts.20,21
Moreover, Au is an attractive co-catalyst for CO2RR due to its particular alloying tendency with Cu, which allows tuning the electronic structure better than the less miscible Ag–Cu catalysts.18,22 In particular, CuAu alloys can form three ordered phases with different compositions (Cu3Au, CuAu, CuAu3),23 and CuAu superstructures24 as well as unordered phases, which present a wide variety of possible active species for CO2RR. CuAu alloyed systems have been reported to lead to enhanced CO2RR performance, which was attributed to different factors, including a possible protection from the formation of Cu oxide;22,25 an increased formation of CO, paralleled by the suppression of HER and CH4 formation;26–28 a shift in the onset potential for CO2RR towards lower overpotentials;22,29 *COOH stabilization12,30,31 and to synergistic geometric and electronic effects that boost C2+ production toward alcohols.32–36 However, the type and the influence of the alloyed structure under CO2RR conditions as well as the high CO coverage on Cu through CO producing co-catalysts remains an open question. Additionally, a systematic study of CuAu NP catalysts with a complex mixing pattern of the constituent active elements, alloy formation and the interfaces of its multiple phases under operando conditions has not yet been reported. These insights are critically needed to enable true catalyst design based on comprehensive scientific understanding of catalyst adaptations under reaction conditions and ensuring sustainable utilization such as noble metals co-catalysts.
Hence, in this work we use Cu2O nanocubes (NC) decorated with varying amounts of Au NPs as tandem catalysts to reveal the role of alloying and of the CO coverage on the product selectivity by analyzing the alloy formation and the influence of CO-rich micro-environments under CO2RR conditions. The compositional restructuring was investigated with scanning transmission electron microscopy and X-ray photoemission spectroscopy, while the active phases under reaction conditions were deconvoluted with operando high energy X-ray diffraction, operando X-ray absorption fine structure spectroscopy, and quasi in situ X-ray photoelectron spectroscopy. The CO coverage on Cu was followed using operando surface-enhanced Raman spectroscopy. Correlations between the alloy type and its evolution during CO2RR as well as the CO coverage and the product distributions were drawn, unveiling that optimized alloyed phases and CO coverages result in increased C2+ product selectivity.
Results and discussion
We prepared Cu2O NCs decorated with different Au NP loadings and applied scanning transmission electron microscopy (STEM) imaging using the high angle annular dark field (HAADF) detector to verify the cubic shape of the as-prepared Cu2O NCs. With Au loading, the cubic shape of the as-prepared catalysts remains recognizable, but the corners become progressively rounded as the Au loading increases, Fig. S1 (ESI†). The Au NPs are observed to be evenly dispersed on the surfaces of the NCs, for all loadings from lowest (Au0.4/Cu2O NCs) to highest (Au2.7/Cu2O NCs). The lower limit of Au loading was set by the decrease in the homogeneity of the Au NP decoration on the surface of the Cu2O NCs. Interestingly, using atomic resolution aberration-corrected HAADF STEM, the Au NPs are observed to exhibit both single grain and multigrain structures, Fig. 1b.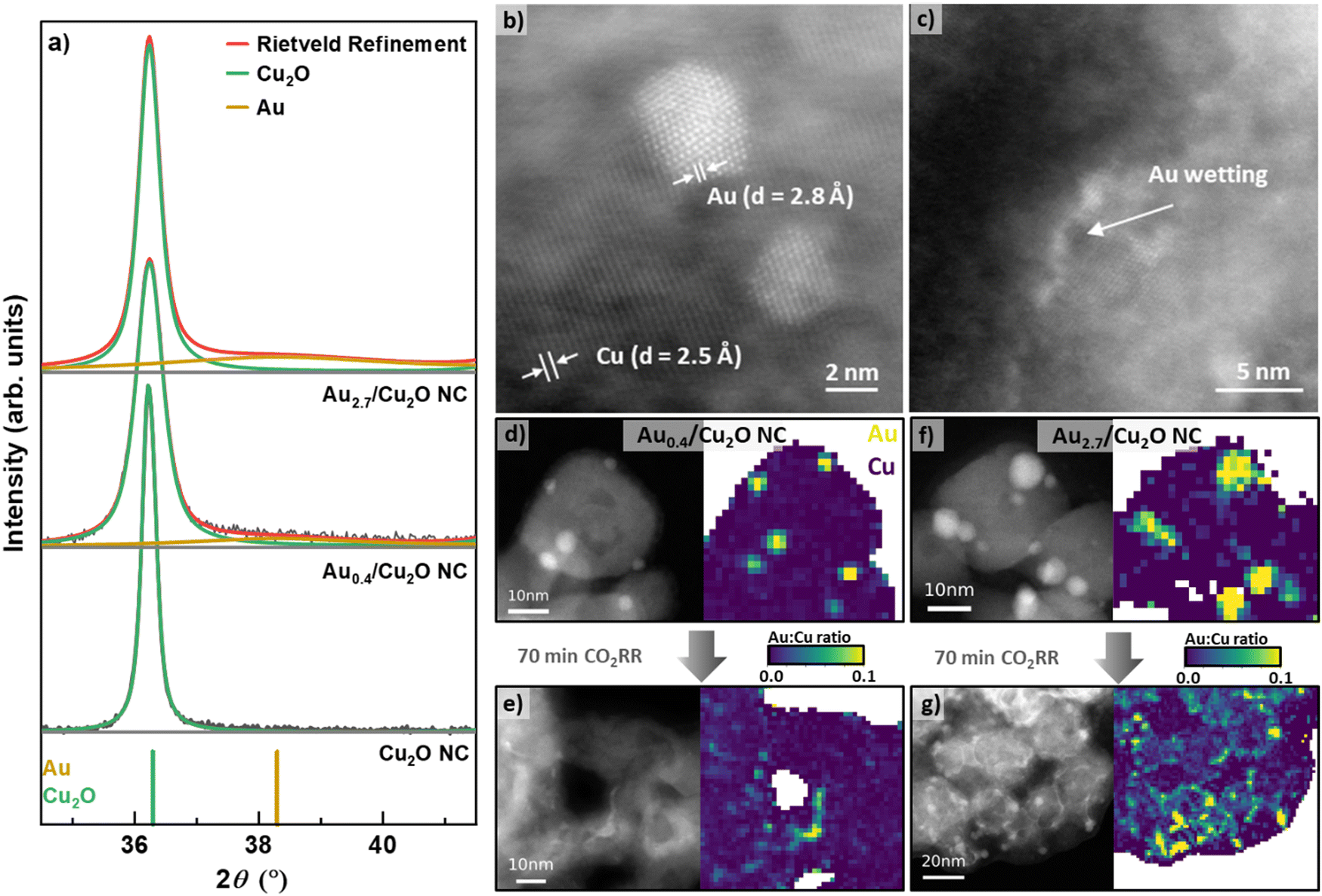 | ||
| Fig. 1 (a) Ex situ XRD patterns of the Cu2O NCs, Au0.4/Cu2O NCs and Au2.7/Cu2O NCs and their corresponding Rietveld refinement fits. STEM-HAADF images of (b) Au monocrystalline and polycrystalline NPs on the surface of the Au0.4/Cu2O NCs in their as-prepared state and (c) an example of Au wetting on the surface of the same Au0.4/Cu2O NCs after 70 min CO2RR. STEM-HAADF images with corresponding EDXS mappings of (d) and (e) Au0.4/Cu2O NCs and (f) and (g) Au2.7/Cu2O NCs in the as-prepared state (d) and (f) and after 70 min CO2RR (e) and (g). The reaction was carried out in CO2-saturated 0.1 M KHCO3 at −1.05 VRHE. | ||
Using Inductively Coupled Plasma–Mass Spectrometry (ICP–MS), we determined the bulk compositions of Au with varying loadings on the Cu2O NCs to be 0.43 at%, 0.77 at%, 1.08 at%, and 2.69 at% for Au0.4/Cu2O NC, Au0.8/Cu2O NC, Au1.1/Cu2O NC and Au2.7/Cu2O NC, respectively, Table S1 (ESI†). X-ray diffraction (XRD) analysis of the as-prepared NCs demonstrates the presence of Cu2O, with its most intense reflections (111) at 36.4° and (200) at 42.3°, Fig. 1a and Fig. S2 (ESI†). The addition of the Au NPs induces broad and weak Au(111) reflections at 37.34°, which are present with increasing intensity for all catalysts with increasing Au loading. The coherence lengths obtained by Rietveld refinement for the Cu2O NCs of all catalysts vary between 21 and 42 nm, Table S2 (ESI†). These findings are in agreement with the Cu2O NC edge length and Au NP size distributions displayed in Fig. 1d, f and Fig. S3 and Table S3 (ESI†), revealing cube edge lengths of ca. 20 nm and Au NPs sizes of 3–4 nm. We furthermore characterized the oxidation behavior of our catalysts by cyclic voltammetry (CV), Supplementary Note 2, Fig. S13 and Table S6 (ESI†).
The evolution of the catalysts under CO2RR conditions was studied to understand the catalyst function and to correlate the findings with the product selectivity. Therefore, we carefully analyzed STEM images, together with STEM energy dispersive X-ray spectroscopy (EDXS) elemental maps, of samples before and after CO2RR to identify any irreversible morphological restructuring and Au wetting effects on the Cu surface. Fig. 1 and Fig. S4–S7 (ESI†) depict the HAADF-STEM images (including aberration-corrected) and the EDXS elemental and ratio maps of the catalysts before and after CO2RR. Note that the EDXS maps are spatially binned in order to improve signal to noise ratio for their quantification. Two selected catalysts show well-dispersed Au NPs on the surface of the Cu2O NCs in the as-prepared state, Fig. 1d, f and Fig. S7 (ESI†). After 70 min of CO2RR at −1.05 V, strong morphological changes are observed for the Au–Cu2O NCs, losing their cubic shape and appearing porous, Fig. S5–S7 (ESI†). Note here that the catalysts in the present STEM study have undergone exposure to air before the microscopic analysis, but the results are in line with recent in situ TEM studies in which the shape of the pure Cu2O NCs was investigated during CO2RR.37 The NC edge lengths and the Au NP sizes did not change upon CO2RR as compared to the as-prepared catalysts within the experimental uncertainty. It is noteworthy that the morphological changes of the Au0.4/Cu2O NCs after CO2RR resemble the hollow CuOx frames that have been previously observed for pure Cu2O NCs.9,38 The other catalysts exhibit morphological changes dominated by the surface Au coverage such as the density of Au NPs decreased and Au was incorporated into Cu as fine stripes in between the retained Au NPs, Fig. 1e, g and Fig. S7 (ESI†). This Au wetting leads to a skeleton-like structure in which Cu reshapes into a new frame. A more detailed analysis on the wetting effect of the Au2.7/Cu2O NCs was performed via aberration-corrected STEM, Fig. S4 and S5 (ESI†). The images clearly show separate phases in the as-prepared state and an Au-wetted Cu surface after CO2RR framing around the nanostructures, Fig. 1b and c. This Au wetting phenomenon is more pronounced for the higher Au loadings, rearranging the catalyst structure towards an Au-rich frame, Fig. 1e, g and Fig. S7 (ESI†). Indeed, significant changes in the Cu morphology were found for the low Au loading catalysts, partially with coalescence and dissolved Cu. On the other hand, when higher Au loadings on the Cu2O catalysts are considered, a framed-like shape is more easily preserved, Fig. S7 (ESI†). However, for the Au2.7/Cu2O NCs, even after 1 h of CO2RR there are still Au NPs preserved, suggesting that the Au is not completely alloyed in this sample, which increases the compositional complexity of the catalysts, with different Au and Cu-rich phases being present during catalysis.
We also followed the changes in the composition by quantifying spatially integrated EDX spectra from the EDXS maps, Table S4 (ESI†), and found the expected increased Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu ratio trend with increasing nominal Au loading, though with a slight offset as compared to the results obtained by ICP–MS, Table S5 and Fig. S8 (ESI†). Note that EDXS is a semiquantitative method without external standards; while Au:Cu ratios show a definitive trend, the obtained values should not be treated as absolutes. The Au:Cu ratio was also locally mapped with the spatially-binned EDXS data, Fig. 1d–g and Fig. S7 (ESI†). Thus, for the as-prepared catalysts, defined NPs are observed, while after CO2RR, both distorted Au NPs and Au wetting of the Cu surfaces were observed. The Au wetting and skeleton-like structures are clearly visible.
:Cu ratio trend with increasing nominal Au loading, though with a slight offset as compared to the results obtained by ICP–MS, Table S5 and Fig. S8 (ESI†). Note that EDXS is a semiquantitative method without external standards; while Au:Cu ratios show a definitive trend, the obtained values should not be treated as absolutes. The Au:Cu ratio was also locally mapped with the spatially-binned EDXS data, Fig. 1d–g and Fig. S7 (ESI†). Thus, for the as-prepared catalysts, defined NPs are observed, while after CO2RR, both distorted Au NPs and Au wetting of the Cu surfaces were observed. The Au wetting and skeleton-like structures are clearly visible.
The electrocatalytic performance of the catalysts was evaluated by chronoamperometric measurements at −1.07 V for 1 h in CO2-saturated 0.1 M KHCO3 for the different Au loadings. Fig. 2a–c shows the selectivity trends in form of faradaic efficiencies (FE) as a function of the Au loading and the corresponding current densities (d). The potential-dependent FEs and corresponding currents for each catalyst are given in Fig. S9 (ESI†). In Fig. 2b, the increased formation of CO with increasing Au loading up to a FE of 56% for Au2.7/Cu2O NC is evident.
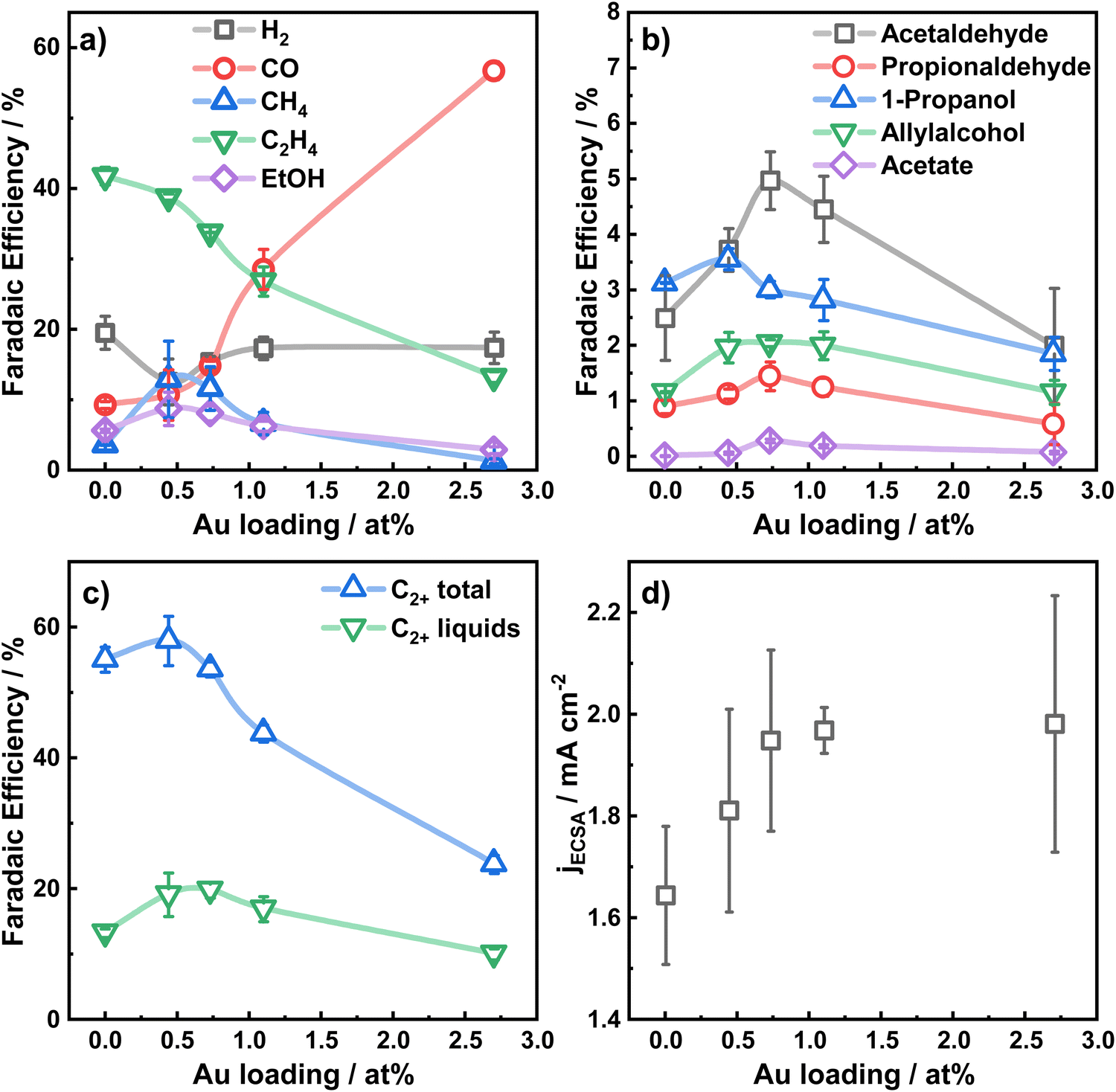 | ||
| Fig. 2 Faradaic efficiencies at −1.07 V in 0.1 M KHCO3 as a function of the Au NP loading for (a) H2, CO, CH4, C2H4, (b) minor liquid products, (c) C2+ total products, C2+ liquid products; (d) Au-loading dependency of the current density normalized by the electrochemical surface area. | ||
This high production of CO on Au suggests a high local CO concentration around the Au NP perimeter in the proximity of Cu. In return, the C2H4 production is indirectly correlated to the CO production, sacrificing the FE of C2H4 for CO for large Au coverages on the Cu2O NCs. Hydrogen production decreases for the lower Au loadings (<1%), while a slightly higher CH4 production is found. The FE of ethanol increases slightly by 2 percentage points for Au0.4/Cu2O NCs and Au0.8/Cu2O NCs. Remarkably, the FEs of the minor liquid products such as propionaldehyde, acetaldehyde and allylalcohol are also boosted for the lowest Au loadings. Acetaldehyde, which is considered as a possible precursor of ethanol,39 is improved for all Au loadings investigated and is highest for Au0.8/Cu2O NC, with 4.8% FE, 2.6 times higher than for the pure Cu2O NCs. For this catalyst, the highest FE is also observed for propionaldehyde, while allylalcohol has its peak FE for the three low Au loading catalysts and 1-propanol is the highest for Au0.4/Cu2O NC.
For the bimetallic catalysts, the increased ethanol production is typically linked to a weaker binding strength of the *CO intermediates to the secondary catalysts and to a CO spillover mechanism, which is described well for Ag–Cu systems.5,9,40,41 Although Au–Cu systems tend to alloy under reaction conditions, the CO spillover mechanism for these systems is not clearly understood. In our case, and in contrast to the similarly prepared Ag–Cu2O NCs,9 the addition of Au as co-catalyst does not improve the ethanol production significantly, despite the increased production of acetaldehyde. Ethanol is understood to be produced either directly from the *CH2CHO intermediate through protonation, or in small parts also through re-adsorption of acetaldehyde (CH3CHO) with subsequent protonation.39 We explain the preferred production of acetaldehyde and the impeded production of ethanol by the CO-richer environment that hinders the protonation of the *CH3CHO intermediate and the re-adsorption of acetaldehyde on the catalyst.
The lowered hydrogen production, observed for our Aux/Cu2O NC catalysts, is in agreement with impeded *H coverage on the surface and OH− formation.
Fig. 2c displays the total C2+ product FE, which is the highest for Au0.4/Cu2O NC and exceeds slightly the ones of Cu2O NC and Au0.8/Cu2O NCs. With increasing Au loading, the C2+ product formation reduces drastically, which results mainly from the decreased FE of ethylene and ethanol. The C2+ liquid product formation for the two lowest Au loading catalysts exceeds that of the pure Cu2O NCs by up to 5% FE. The C2+ carbonyl formation, however, increases for the low Au loadings and decreases drastically for the high Au loadings, demonstrating the beneficial effect of low amounts of Au on Cu2O NCs for CO2RR. In a similar way, the FE of the liquid C2+ products for the low Au loadings increases to a maximum of 21% FE and decreases for the Au2.7/Cu2O NCs. The combined liquid products follow the same trend, Fig. 2c. Thus, a switch in the selectivity for the gaseous products from C2H4 to CO takes place, and for the Au2.7/Cu2O NCs the CO selectivity is so high that fewer liquid products are formed.
The total and partial current densities at −1.07 V vs. RHE, normalized by the electrochemical surface area (ECSA) are shown as a function of the Au loading in Fig. 2d and Fig. S10 (ESI†) and represent the intrinsic catalytic turnover. The total current density, Fig. 2d, increases slightly with increasing Au loading, indicating a higher activity of Au compared to Cu, and consequently higher CO production rate, in accordance with literature.31 Furthermore, the partial current densities for C2+ products, Fig. S10 (ESI†), are the highest for the low Au loadings, with decreasing activities for increasing Au loadings. Notably, the lowest Au loading of 0.4 at% leads to a 2-fold increase in C2+ current density and a 4-fold increase for the C2+ liquid product current density compared to pure Cu2O NCs. Thus, the turnover of CO to C2+ products appears to be sensitive to the presence of small amounts of Au (low loadings), leading to a slightly CO-richer environment, while higher Au loadings and thus, increased CO formation, are detrimental.
Stability tests over 20 h on all catalysts confirm stable product distributions after the initial alloy formation during the first hour and are further described in Supplementary Note 1 and Fig. S11 and S12 (ESI†).
To understand the irreversible changes of the surface composition, chemical state and alloy formation of the bimetallic catalysts upon CO2RR, we employed quasi in situ and X-ray photoemission spectroscopy (XPS). In comparison to ex situ measurements, this avoids post-reaction air exposure of the catalysts that may induce surface re-oxidation and possible restructuring. Fig. 3 shows the Cu L3M4,5M4,5 Auger electron spectra (a) and the Au 4f and Cu 3p (b) core level XPS regions for the Au2.7/Cu2O NCs before and after 1 h CO2RR at −1.05 V, as well as the surface composition (c) and the binding energy of the Au 4f7/2 peak (d) as a function of the nominal Au loading. The Cu 2p, Au 4f, Cu38 3p XPS and Cu LMM XAES for all catalysts are shown in Fig. S14–S16 (ESI†). The Cu LMM Auger spectra in Fig. 3a show a combination of Cu0, Cul and Cull in the as-prepared state and a reduction of Cull/Cul to Cu0 after CO2RR. In the as-prepared states, the catalysts present similar ratios of Cu2O (80–90%) and CuO (10–20%), whereas, after 1 h under CO2RR at −1.05 V, all catalysts are reduced to metallic Cu within the error margins, Fig. S14, S15 and Table S7 (ESI†). The Au 4f spectra in Fig. 3b show peaks at EBE,7/2 = 84.2 eV and EBE,5/2 = 87.9 eV. This increased binding energy of +0.2 eV compared to metallic Au (84.0 eV) can be assigned to alloy formation or to reduced interfacial charge transfer between Cu2O and Au.42
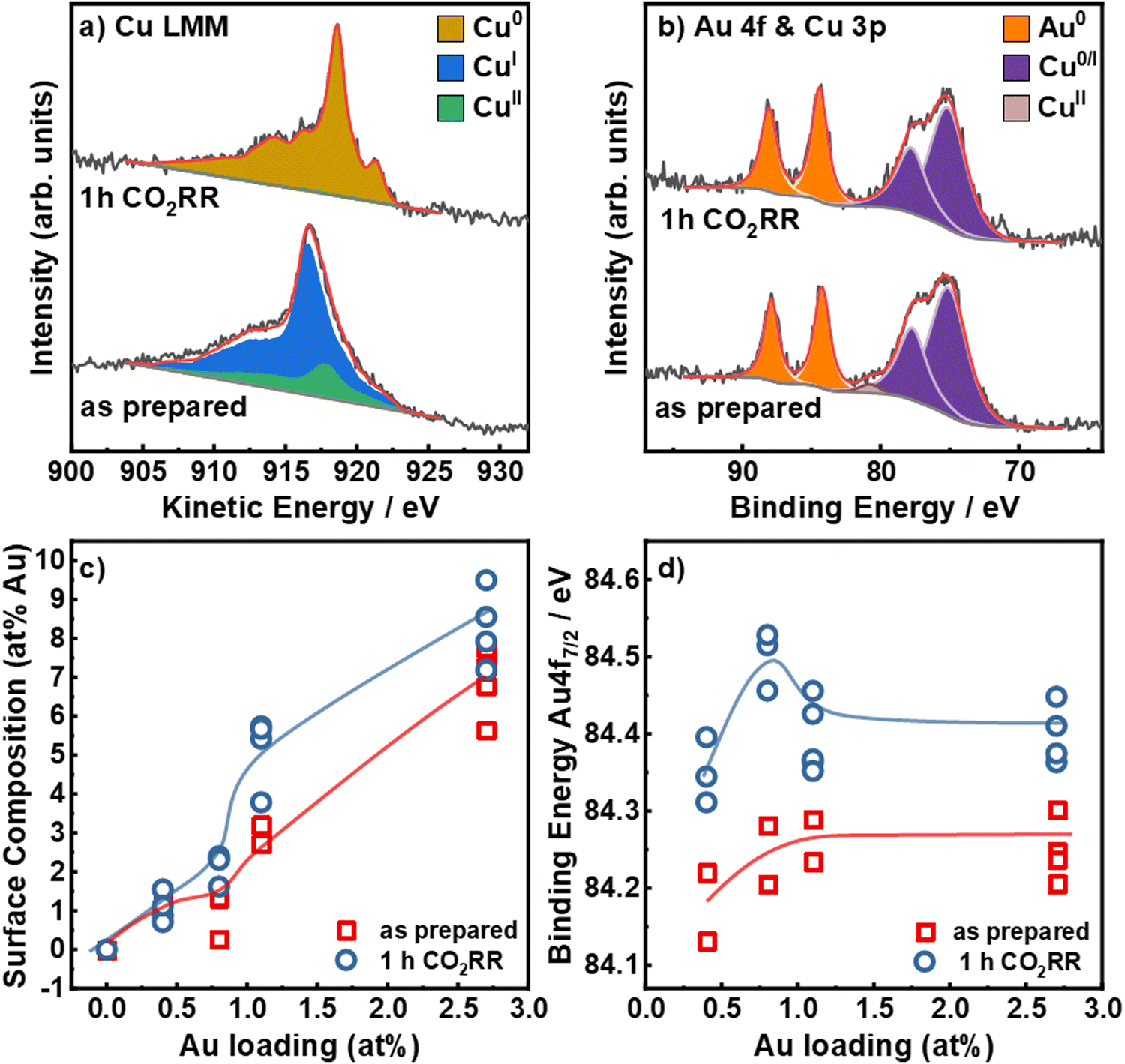 | ||
| Fig. 3 Quasi in situ XPS spectra for the Au2.7/Cu2O NCs: (a) Cu LMM Auger and (b) Au 4f and Cu 3p in the as-prepared state and after 1 h CO2RR at −1.05 V vs. RHE in 0.1 M KHCO3. (c) Au:Cu ratio (at%) at the catalysts surface in the as prepared state and after CO2RR as a function of the nominal Au loading (determined by ICP–MS). (d) Binding Energies of the Au 4f7/2 core level region in the as-prepared state and after 1 h CO2RR. | ||
The surface composition of the catalysts was determined by comparing the Cu 3p and Au 4f areas, Fig. 3b, c and Table S8 (ESI†). In the as-prepared state, the Au surface loadings are with ∼1 to ∼6% notably higher than the average bulk composition. The Au surface composition increased after 1 h CO2RR for the higher Au loadings. This effect can be assigned to a combination of dynamic redistribution, segregation and wetting of Au on the Cu surface. These findings agree well with the STEM-EDXS and ICP–MS data presented in the supporting information, Tables S4 and S5 (ESI†).
Fig. 3d demonstrates that the Au 4f binding energy does not vary strongly with Au loadings in the as-prepared state. However, the Au 4f binding energy increased upon CO2RR by 0.2 eV, suggesting a change of the electronic structure around the Au NPs and the possible formation of an alloy between Au and Cu.43 However, this change could also include the contribution of a charge transfer effect, which would similarly lead to higher binding energy shifts, as detected for the catalysts in the as-prepared state. Furthermore, depending on the Au loading, the binding energy after CO2RR varies from 84.3 eV to 84.4 eV. Remarkably, the Au0.8/Cu2O NCs show relatively high binding energies of the Au 4f7/2, indicating higher alloy formation and also possibly the presence of sample regions containing pure Au clusters or small NPs which are also characterized by large positive binding energy shifts. This observation coincides with a lower CO, C2+ liquid products and H2 specific activities, Fig. S10 (ESI†), as compared to the Au0.4/Cu2O NCs. Our findings suggest that a large content of CuAu alloy formation is detrimental for C2+ production from CO2RR. As we expected to have a large fraction of CuAu alloy regions with increasing Au loading after CO2RR, we conclude that the shift in binding energy detected must be the convolution of the regions with CuAu alloys and those with very small pure Au clusters of NPs. It is plausible that in some of our samples, like Au0.8/Cu2O NCs there exists a larger compositional heterogeneity combining CuAu alloyed regions and low-coordinated Au regions. Overall, our data also reveal a significant decrease in the density of pristine Cu sites during CO2RR.
To extract comprehensive information on the catalyst's bulk structure during CO2RR, we employed operando high energy XRD (HE-XRD) to understand the formation of crystalline Cu1−xAux alloy phases during CO2RR. Note that highly disordered metallic or cationic phases cannot be detected using XRD. We have previously shown that bare Cu2O NCs mostly reduce to metallic Cu using operando HE-XRD.44 Fig. S17 (ESI†) presents the HE-XRD pattern of the Cu2O NCs recorded at 67 keV at open circuit potential (OCP) and after 1 h at −1.05 V. For the Aux/Cu2O NCs, at OCP, the diffraction pattern agrees well with the Cu2O phase, in addition to the strong background caused by the electrolyte. During CO2RR, the Cu2O Bragg peaks disappear almost completely, and a diffraction pattern of metallic Cu develops, in which the broad feature at ∼4.65° could be caused by Cu1−xAux phase(s). Compared to the as-prepared Au2.7/Cu2O NCs at OCP, the Bragg peaks of metallic Cu of Au2.7/Cu2O NCs during CO2RR are significantly broader, suggesting a shorter structural coherence length. Rietveld refinement reveals a coherence length of ∼7 nm of the metallic Cu domains compared to ∼17 nm for the Cu2O domain at OCP. The structural properties of the potential Cu1−xAux minority phase were not reliably resolvable using Rietveld refinement. The Cu lattice parameter of ∼3.6499 Å did not show any evidence for Au incorporation into the main Cu phase during CO2RR. Thus, operando HE-XRD shows the reduction of the Au2.7/Cu2O NCs during CO2RR, which consists mainly of a bulk Cu phase with a potential highly disordered Cu1−xAux minority phase on the surface. Further details are given in Table S9 (ESI†).
To understand the chemical composition, interatomic interactions and alloy formation during CO2RR were extracted from operando X-ray absorption spectroscopy (XAS). This technique is highly complementary to the XRD data presented above, since it unveils the disordered phases present in these samples under the different environments and reaction conditions. The analysis of the Cu K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data is shown in Fig. 4a–c and Fig. S18, S19 and Table S10 (ESI†).
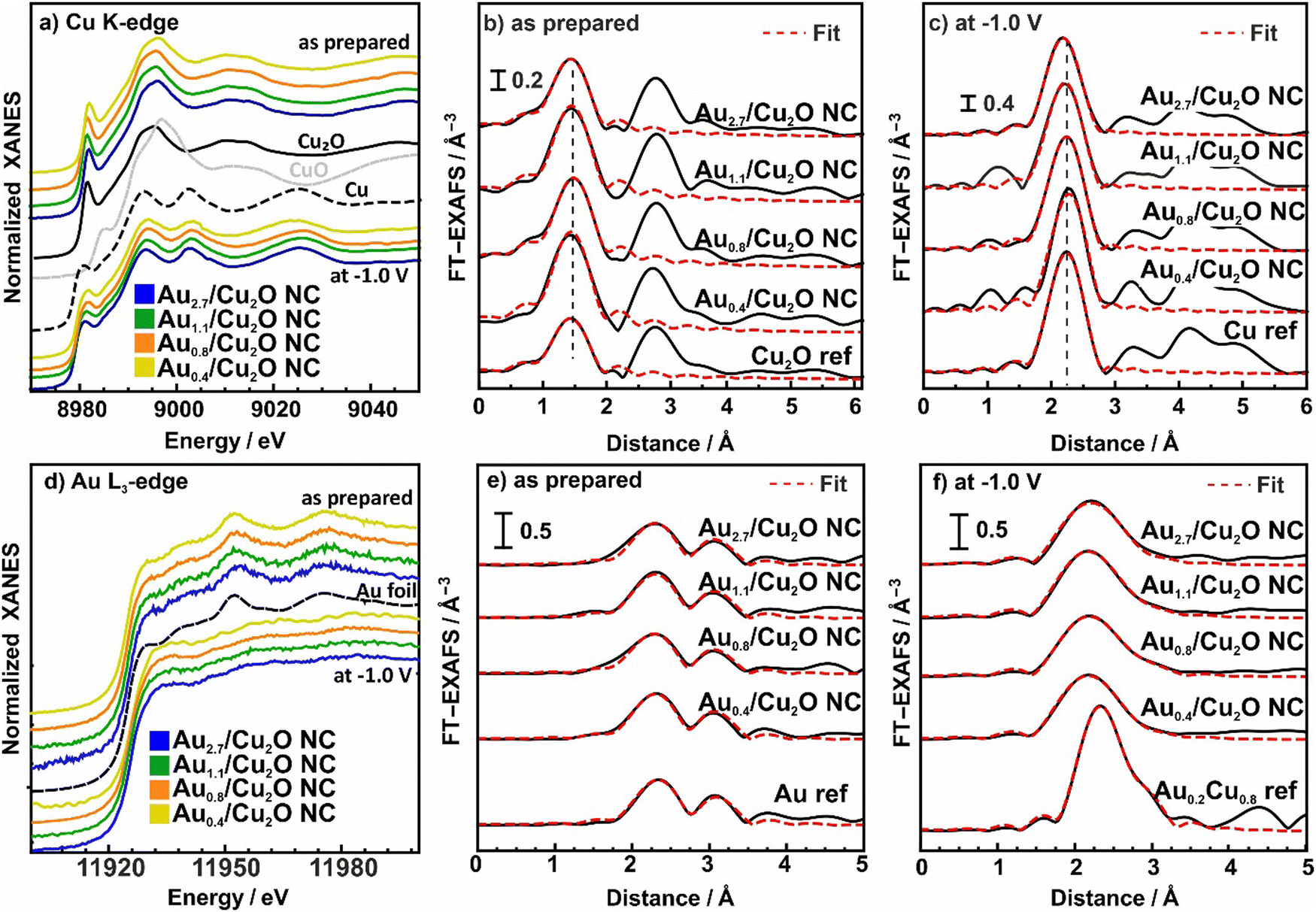 | ||
| Fig. 4 Normalized XANES spectra from (a) the Cu K-edge and (d) Au-L3 edge of bimetallic AuCu NCs with different Au:Cu ratios measured as-prepared and under CO2RR at −1 VRHE, plotted together with bulk Cu, Cu2O and CuO references. FT- EXAFS data from the Cu K-edge (b) and (c) and Au-L3 edge (e) and (f) are also shown together with the corresponding fits for the as-prepared samples (b) and (e) and those during 4 h of CO2RR (c) and (f). Reference EXAFS spectra of an Au foil and a Cu0.2Au0.8 reference alloy are shown for comparison. Cu0.2Au0.8 reference was redrawn from ref. 46. | ||
Fig. 4a shows the XANES of the Cu K-edge for the samples in the as-prepared state and during CO2RR in steady state at 4 h CO2RR at −1.0 V. The XANES spectra for as-prepared catalysts demonstrate the presence of oxidized Cu species, mostly in the Cul state, as suggested by the characteristic pre-edge feature. Nonetheless, significant amounts of Cu are also oxidized to Cull. After 4 h of CO2RR, the Cu of all catalysts is mostly reduced to Cu0. These results are in agreement with the previously discussed XPS data.
The evolution of the local atomic structure around Cu under CO2RR was tracked by studying the Fourier transformed (FT-EXAFS) spectra of the Cu K-edge, Fig. 4b, c and Table S10 (ESI†). In the as-prepared state, Cu K-edge FT-EXAFS exhibits prominent peaks at 1.85 Å and 2.8 Å (phase-uncorrected) corresponding to Cu–O and Cu–Cu bonds in bulk Cu2O-like structure. Data fitting revealed that the coordination number of the Cu–O bonds are 3.0–3.6, which is higher than the one for Cu2O (2), indicating significant amounts of CuO in our catalysts. Under CO2RR, the FT-EXAFS features corresponding to Cu oxide decrease while a new peak corresponding to the Cu–Cu distances in metallic Cu appears at 2.5 Å. Data fitting suggest that the corresponding Cu–Cu coordination number after reaching the equilibrium state after 4 h is close to 12, which fits well to the fcc structure of metallic Cu.45 Note here that no significant contribution of Cu–Au bonds to Cu K-edge FT-EXAFS data are observed, due to the low Au-to-Cu ratio of the catalysts.
The corresponding Au L3-edge XANES data in Fig. 4d demonstrate that the Au NPs in the as-prepared catalysts are in a metallic state, with a local environment similar to that in the Au foil reference material. However, during CO2RR, a white line feature at 11930 eV appears, reflecting a more cationic character of the Au atoms, and thus, changes in the electronic structure due to alloying with Cu and an accompanying charge transfer to the Cu.43,47 Moreover, we observe significant changes in the post-edge features, suggesting strong differences in the interatomic distances and/or changes in the types of nearest neighbors of the absorbing Au atoms as compared to bulk Au. We note that the evolution of the Au L3-edge XANES spectra proceeds similarly for all our catalysts, regardless of the Au loading.
We obtained further insights into the local atomic structure of the Au atoms from the fitting of the FT-EXAFS spectra, Fig. 4e, f and Table S11 (ESI†). The as-prepared catalysts exhibit a prominent peak at 2.3 Å (phase uncorrected), resembling Au–Au bonds with a distance RAu−Au of 2.8 Å and a coordination number (CN) of around 12(2), matching the results obtained for the Au foil (RAu−Au 2.9 Å, CN = 12). This is analogous to the conclusions extracted from the XANES spectra as well as with the TEM and XPS data showing Au NPs that are well dispersed and attached to the Cu2O NC surfaces. During CO2RR, the Au–Au coordination number decreases, while the Au–Cu contribution evolves with coordination numbers between 8 and 10. Interestingly, we observe a significant mismatch between Au–Au and Au–Cu bond lengths for all catalysts, where the former remains similar to that in the Au foil (2.86 Å), while the latter is ca. 2.62 Å, which is clearly larger than the Cu–Cu bond lengths in bulk metallic Cu (2.54 Å).45 These results suggest a heterogeneous structural evolution in which Au-rich regions coexist with regions of a Cu-rich CuAu alloy. The average interatomic distance for Au–Cu of 2.62 Å agrees best with a 1:1 AuCu-like phase, following Vegard's rule. Furthermore, we note that the CN ratio of the Au–Au and Au–Cu distances during CO2RR seems to decrease with increasing Au content for loadings above 0.4 at%. Thus, our operando analysis shows that for all Au loadings a CuAu alloy of low crystallinity and/or domain size forms during CO2RR. This conclusion is compatible with the Au wetting behavior observed with STEM and EDXS analysis.
Furthermore, we investigated the time evolution of the species from both the Cu and Au perspective over 60 min by collecting XAS spectra every 1 s for Au2.7/Cu2O NC (in QXAS mode) and every 12 min for Au0.4/Cu2O NC and Au0.8/Cu2O NC, Fig. 5 and Fig. S19 and S20 (ESI†). The chemical state components were quantified via linear combination analysis (LCA) using reference spectra. For the Cu-K edge, the initial states in the electrolyte show varying contributions of Cu2O and CuO, which might be attributed to an aging effect of the catalysts and/or a beam damage effect, Fig. 5a, b and Fig. S19 (ESI†). Upon application of −1.0 V, while the majority of the Cu2O reduces within 20 s to metallic Cu, after 4 h of CO2RR fractions of 10% Cu2O are nevertheless observed. Given that quasi in situ XPS showed a purely metallic catalyst surface after 1 h CO2RR, this suggests that a robust Cu2O phase remains in the core of the catalysts. These remnant Cu oxide species even after prolonged exposure to CO2RR conditions were also observed for analogous pure Cu2O and Ag-decorated Cu2O nanocubes.9,44 Meanwhile, the Cu–Cu coordination numbers during CO2RR suggest a bulk-like metallic Cu as the dominant phase, which is in agreement with the insights from operando HE-XRD. Interestingly, the CuO of the Au2.7/Cu2O catalysts first reduces to Cu2O as the Cu2O LCA weight first increases by about 5–10 percentage points before the catalysts reduces further, which is also in accord with previous observations.44
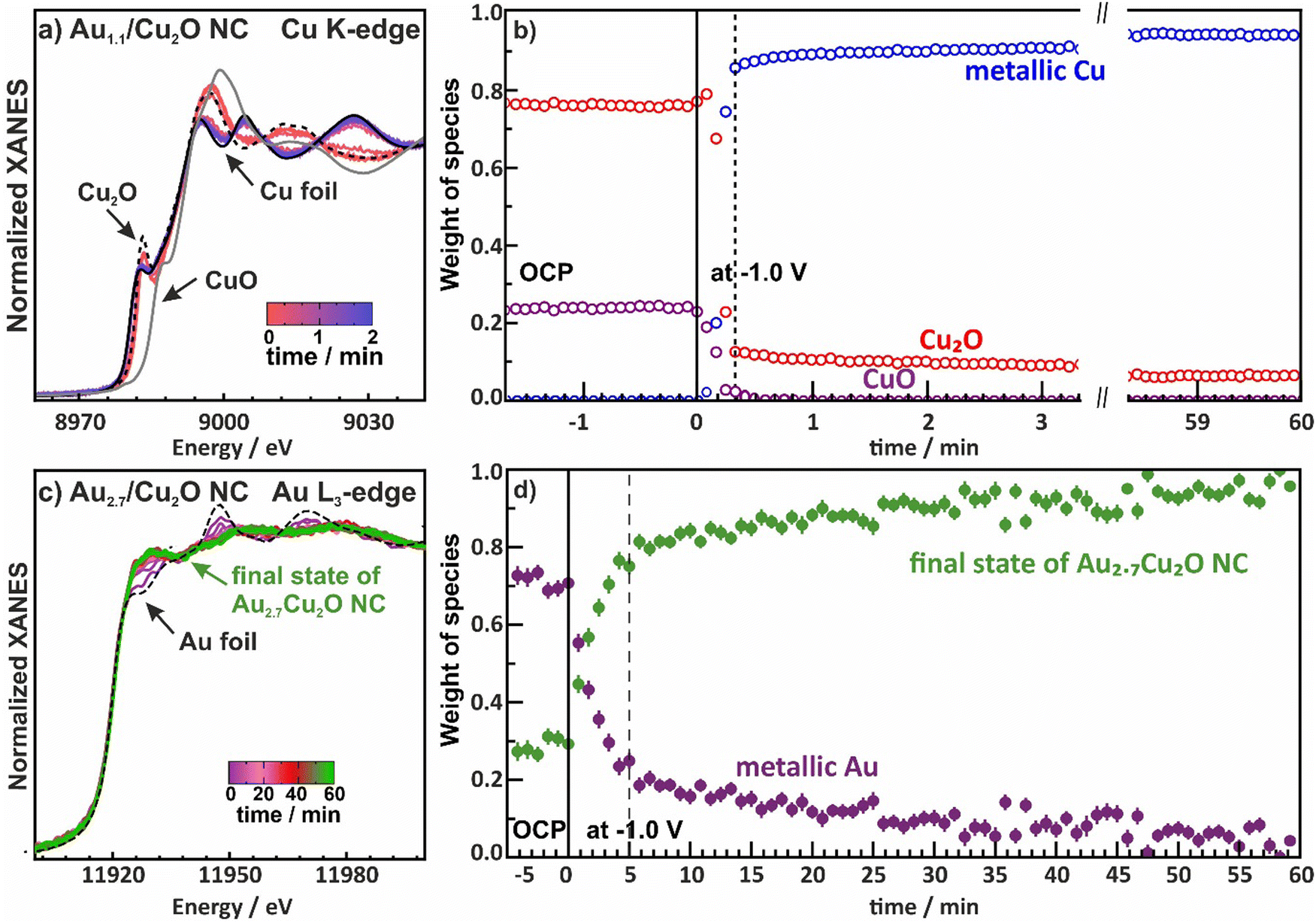 | ||
| Fig. 5 (a) Normalized Cu K-edge XANES spectra and (b) weight of the observed species as a function of time during CO2RR at −1.0 V for the Au1.1/Cu2O NCs as an example. (c) Normalized Au L3-edge XANES spectra and (d) weight of the species as a function of time during CO2RR at −1.0 V unveiling the formation of an CuAu alloy for the Au2.7/Cu2O NCs as example. The final state of Au2.7/Cu2O NCs is a mixed state with Cu-rich, Au-rich and CuAu alloy phases. | ||
We then evaluated the time evolution of the Au L3-edge and show the weight of the different species as a function of time with a final state of the Au2.7/Cu2O NC as reference for the alloy phase, Fig. 5c, d and Fig. S20 (ESI†). It should be noted that the chosen final state of the Au2.7/Cu2O NC is a mixture of Cu-rich, Au-rich and CuAu-alloy phases. The local structure around Au evolves much slower than the reduction of Cu, with about one hour needed to reach a stable alloy phase, where the alloy formation is not evolving significantly anymore. Thus, these results might explain the slow product adaptation of the Au-containing catalysts in the long-term CO2RR measurements over 20 h. Restructuring of the catalyst during alloy formation represents significant atom mobility and continuous altering of the active sites which in turn continuously affects the product distribution.
We furthermore study the role of the potential-dependent CO-related adsorbates with varying Au loading by using operando surface-enhanced Raman spectroscopy (SERS), Fig. 6 and Fig. S21, S22 (ESI†). Fig. 6a shows the SERS data recorded at potentials between OCP and −1.1 V vs. RHE for the Au2.7/Cu2O NCs, as an example for all investigated catalysts (Fig. S22, ESI†). At OCP, the Raman scattering of the Cu2O exhibits the typical bands at 415 cm−1 (multiphonon process), 527 cm−1 (Raman active F2g vibrational mode), 623 cm−1 (IR active F1u mode) and 220 cm−1 (overtone 2Eu). These bands were found to disappear at 0.2 V for all catalysts, which reflects the electrochemical reduction of Cu2O to Cu at the surface.48 Between 0.2 V and 0.1 V, bands at 360 cm−1 appear with corresponding bands at 706 cm−1, 1050 cm−1 and 1074 cm−1, which can been assigned to surface copper carbonate or bidentate carbonate species, Fig. S22 (ESI†).38,49 Remarkably, a CO stretching band around 2090 cm−1 was observed at 0.2 V for the three higher Au loadings and may be linked to carbonates or Had. These CO stretching bands around 2090 cm−1 are significantly stronger for the Au2.7/Cu2O NCs compared to the Au0.8/Cu2O NCs and Au1.1/Cu2O NCs. The two peaks at 2039 cm−1 and 2090 cm−1 overlap for the Au2.7/Cu2O NCs, while the former band decreases in intensity until −0.4 V.
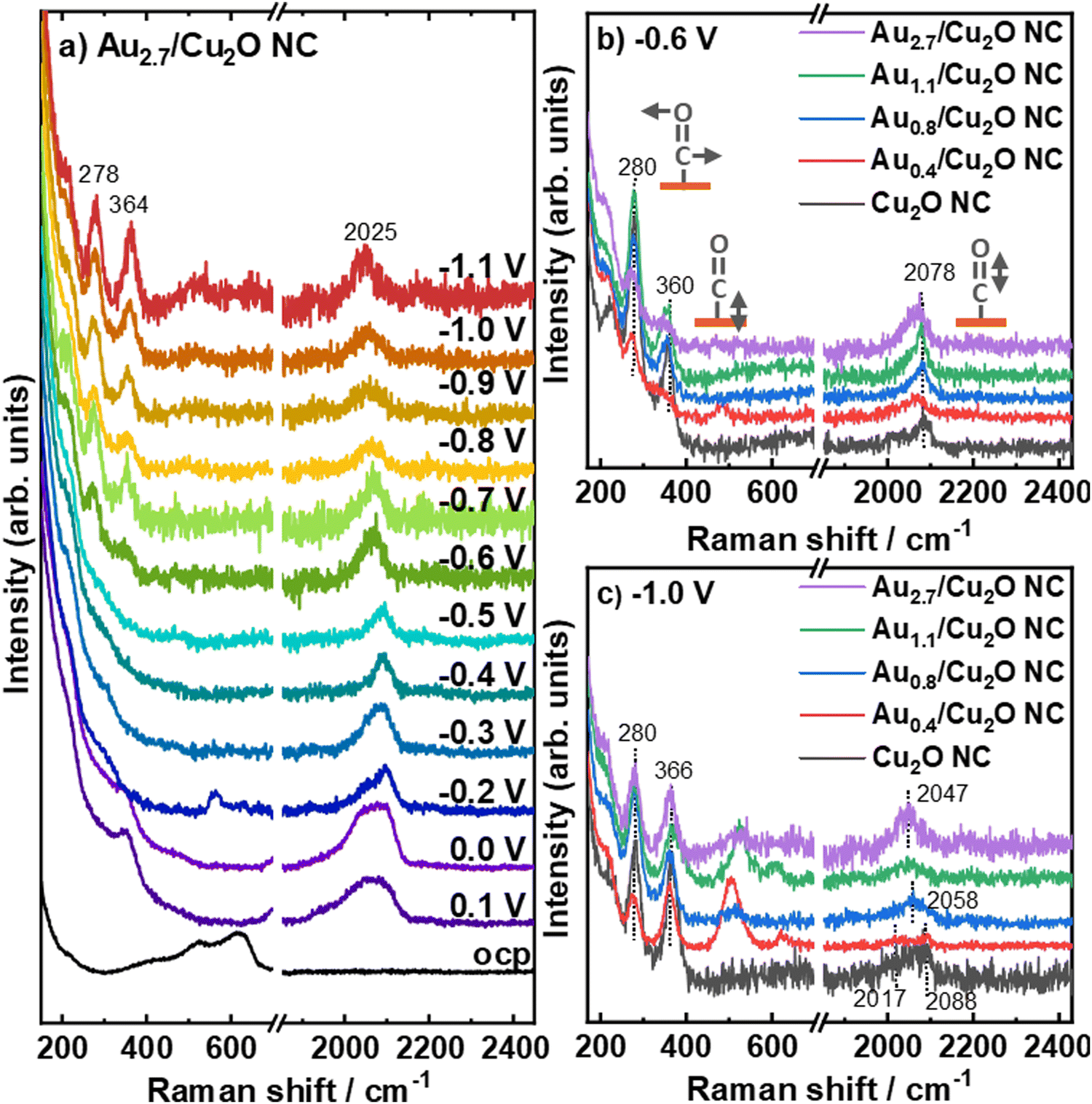 | ||
| Fig. 6 Operando SERS of (a) Au2.7/Cu2O NC under potential dependence and of all Au–Cu2O catalysts at (b) −0.6 V and (c) −1.1 V in CO2-saturated 0.1 M KHCO3. Data for the Cu2O NC are redrawn from ref. 9. | ||
At more cathodic potentials, new bands appear at 280 cm−1 and 366 cm−1, which correspond to the restricted rotation of *CO on Cu (COrot) and Cu–CO stretching (COstretch), respectively. Following our previously established link of their intensity ratio (COstretch/COrot) to the formation of C2+ products,38 we determined COstretch/COrot ratios for the Au-decorated Cu2O NC catalysts, which are lower than those found previously for the pure Cu2O NCs38 as well as those for the Ag/Cu2O NCs during CO2RR.9 We also did not detect a potential-dependent correlation between the COstretch/COrot ratios and the C2+ product FE for the Aux/Cu2O NCs as was the case for the bare Cu2O NCs or Ag/Cu2O NCs, Fig. S23 (ESI†). This result suggests a lower CO coverage on Cu for the Au-decorated Cu2O NC catalysts as compared to the pure Cu2O NCs38 and Ag/Cu2O NCs,9 and indicates a more complicated mechanism. Thus, the enhanced C2+ (liquid) product FE identified for low Au loadings on the Cu2O does not appear to stem from differences in the CO coverage on the Cu surface, as it was the case for the Ag-decorated Cu2O NCs. Instead, our new findings for the Au–Cu system suggest a more complex mechanism involving the presence of the more easily formed CuAu alloy as compared to the Cu/Ag system. Notably, bands between 500 cm−1 and 700 cm−1 develop during CO2RR, which has not yet been unambiguously assigned and might evolve from Cu/Au–OH species.41,50 These bands display a shift towards higher Raman frequencies with increasing Au loading without a specific trend in the intensity, and suggest a decreasing OH binding energy with increasing Au loading.
Fig. 6b shows the Raman spectra for the different Au loadings at −0.6 V and verifies that the C–O stretching bands have similar Raman shifts of ∼2078 cm−1. At −1.0 V, the C–O stretching bands of the Au2.7/Cu2O NC and the Au1.1/Cu2O NC blue-shift towards 2047 cm−1, Fig. 6c. For these high Au loadings we can also identify two additional bands at ∼1900 and 2200 cm−1,51 which stem from the stretching vibrations of CO on Au sites and can be linked to the significant increase of the FE of CO. For the Au0.8/Cu2O NC, the C–O stretching band shifts only to 2058 cm−1, while no peak shift is observed for Au0.4/Cu2O NC and the pure Cu2O NC with 2088 cm−1.9,41 Interestingly, the Au0.4/Cu2O NC shows an additional weak Raman band at ∼2017 cm−1, suggesting the presence of multiple CO adsorption sites. The position of the C–O stretching band is directly linked to the average CO binding energy to the surface50 and thus, the observed variation in its position during CO2RR towards lower Raman shifts reflects a weaker binding of the CO with increasing Au loading, which appears to stabilize for Au loadings higher than 1.1 at%. This effect agrees with the increasing fraction of CuAu alloy with increasing Au loading which exhibits a weaker CO binding energy and can be explained with a downshift of the d-band center from the Fermi level with an increasing Au fraction.52,53 It has also been discussed that adjacent OH adsorption to the COads sites may influence the C–O stretching band, but we did not detect a link between the C–O band position and the intensity of the Cu/Au–OH band.54 Thus, we attribute the variations in the C–O band position to variations in the CO binding primarily induced by CuAu alloy formation, leading to a weaker bound CO on the catalyst surface for the higher Au loadings. For low Au loadings, multiple e.g. Cu- and Au-like adsorption sites with clearly different binding energies are present.
Lastly, we have to note that the intensity of the C–O stretching bands at high overpotentials, thus, highest C2+ product yield, increases with Au loading compared to the pure Cu2O NCs, while the Cu–CO related bands do not vary strongly in intensity. This suggests that a CO-richer surface and/or microenvironment forms during CO2RR in the presence of Au-rich regions, while any increase in CO surface coverage would be linked to Au-related sites as the Cu surface coverage does not increase following the COstretch/COrot analysis. These results suggest a complex mechanism, where the CO coverage on Cu as well as on the Au-containing adjacent regions will play a role, with the optimum surface composition that should be desirable to stabilize under reaction conditions involving Au–Cu alloyed regions in close proximity to Cu regions that would benefit from CO spillover.
Overall, our comprehensive study of the active catalysts state and the catalytic function of Au-decorated Cu2O NC suggest that there is a two-stage catalytic role of the Au decoration within the complex mixed phase between the Cu host, CuAu alloy and Au-rich NPs. Clearly, our Au/Cu2O catalysts easily form CuAu alloys in situ under CO2RR conditions, which can be detected within minutes during CO2RR and even after CO2RR in the near-surface. A higher Au loading leads to stronger alloy formation and results in a weaker average CO binding to the catalyst surface during CO2RR. The weaker (average) binding energy agrees with the d-band theory52 and enhances CO production.
This interpretation is in agreement with the current state of knowledge for the formation of C2+ products over AuCu,31,34–36 which has also been discussed in the literature for CuAg systems,9,20,40,45,55 see also Table S12 (ESI†). Homogeneous Cu-rich CuAu alloy generates only a very small fraction of C2+ products, while Au-rich alloys produce mostly CO.31 Interestingly, the high CO production obtained in the presence of an increasing amount of CO-forming Au and Cu–Au areas has associated the observation of a low CO coverage, as determined from operando Raman data. This result suggests a likely weaker binding of CO to the mixed Au–Cu surface obtained for the larger Au loadings, which leads to preferential direct CO desorption versus subsequent protonation. Thus, the Au–Cu alloy itself is not considered to improve the CO2RR catalytic function for C2+ formation, since this occurs on Cu–Cu sites. While CO may be produced in the Au–Cu and Au regions of our catalysts, the actual CO dimerization occurs on the Cu surface in a sequential fashion and an optimal ratio between all regions (Au, the CuAu interface and the Cu–Cu surface) is therefore crucial. The higher fraction of near-surface AuCu alloy likely decreases the density of Cu–Cu sites which are better for C2+ product formation and, thus, the formed CO cannot be utilized for dimerization at high Au loadings. Therefore, as depicted in Fig. 7, the beneficial effect of Au with respect to the formation of CO is concurrent with the detrimental effect of the Au–Cu alloy in the subsequent C–C coupling, for which Cu–Cu sites are sacrificed. These results are in agreement with literature indicating a lower formation energy for Au as skin-layer, which would increase the surface area of Au compared to Cu.56
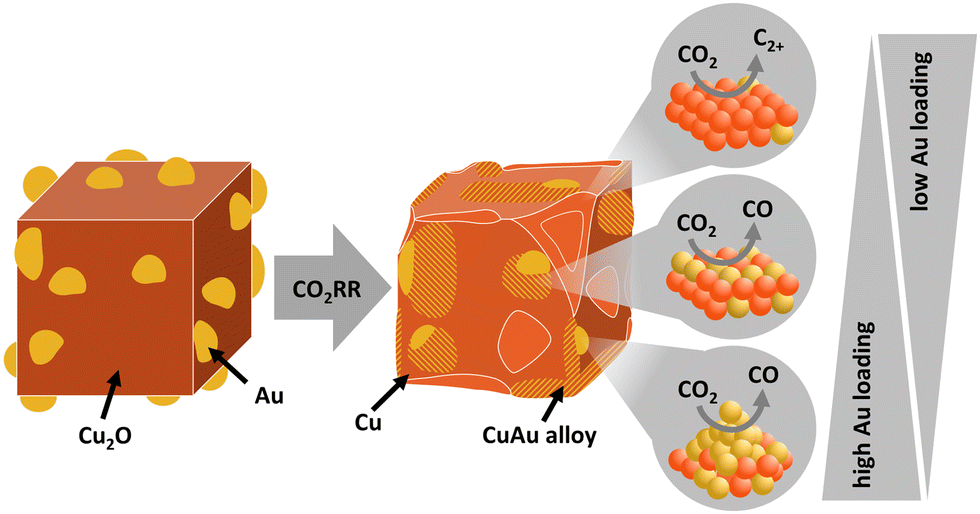 | ||
| Fig. 7 Schematic illustration of the catalyst structure in the as-prepared state and during/after the CO2RR, with their corresponding main products. The triangles indicate the amount of Au loading favoring the different Au–Cu atomic structures/redistributions. | ||
Nevertheless, the Au-decorated Cu2O NCs did not improve the ethanol performance as much with respect to prior literature reports.35,36 However, comparing CuAu and CuAg bimetallic systems for improved C2+ selectivity to pure Cu, we observe substantial differences between both systems in the onset potential of CO, the CO production rate as well as their ability to form alloyed structures. Table S12 (ESI†) displays the overall better performances towards C2+ products of CuAg systems, which are mainly attributed to CO spillover and/or to a good Ag/Cu miscibility without pronounced alloy formation.9,20 In contrast, CuAu systems, with its pronounced alloy formation during reaction conditions, may benefit from synergistic effects between the electronic structure and the morphology of the catalytic system.33
Unlike other studies in literature, we present here an Au-loading dependent study and found optimal Au loadings <1 at% for improving the C2+ products. Only highly Au-diluted CuAu alloys lead to an enhanced C2+ FE. Remarkably, the specific activity for C2+ (liquid) product formation increased by up to 400%. In this optimal case, a significant fraction of the Cu domains remains unalloyed and the catalysts exhibit a stronger average or even improved CO binding sites during CO2RR. The latter ones can act as active sites for CO dimerization, while the minority CuAu alloyed regions still reduce CO2 to CO and the tandem catalytic function boosts the intrinsic C2+ formation significantly. Our findings suggest that low Au loadings, i.e. decoration on the atomic level, can be a promising approach to further enhance the C2+ product formation. Here, we see a striking similarity to our findings on Ag-decorated Cu2O NCs, which form disperse Ag sites on the Cu surface even at higher Ag loadings.9
Our findings strengthen the hypothesis that (i) optimal amounts of additionally formed CO are beneficial for the formation of C2+ liquid products on Cu, (ii) CuAu alloys, formed under CO2RR operation mainly contribute to the CO formation but not to C2 product formation and (iii) ethylene formation is maintained without addition of Au. We therefore conclude that the CuAu systems are worth exploring further for C2+ product promotion, in particular liquid products. When low amounts of Au are available in the vicinity of Cu–Cu sites, the indispensable alloy formation during CO2RR is low compared to the pure Cu regions. Sufficiently low amounts of Au allow improved C2+ selectivity with CuAu bimetallic systems.
Conclusion
In summary, we demonstrated that highly-diluted Au-decorated Cu2O NCs yield notably enhanced C2+ production in the presence of a CO-rich environment around copper, which results from the highly CO producing Au NPs, and due to significant restructuring towards CuAu alloy formation. Under CO2RR relevant conditions, Cu2O NCs with large amounts of Au NPs demonstrate significant restructuring and redistribution by forming CuAu alloyed frames with increased Au-to-Cu ratios on their surface, which appears to stabilize their initial cubic shape. High loadings of Au on Cu2O NCs produce predominantly CO, while a small Au loading leads to an enhancement of C2+ products. This is assigned to the favourable coexistence in the latter samples of small superficial areas covered with CO-producing CuAu alloy and Au NPs and large areas in their vicinity consisting of pure Cu sites which make the C2+ products.Operando XAS enabled following the alloy formation and observing structural changes between Cu and Au that occur within one hour, which led to a variation of the selectivity trends, in agreement to the long-term reactivity measurements. Increasing Au loadings result in a proportional increase of CO, while low Au loadings lead to a notable increase (4-fold) in C2+ liquid products such as ethanol, acetaldehyde, 1-propanol, allylalcohol and propionaldehyde.
With operando Raman spectroscopy, we could link the catalytic function, in particular the strong CO formation, to a weaker average CO binding to the catalyst surface in a CO-richer microenvironment and/or higher coverage. The preferred C2+ product formation is linked to minority CuAu alloy species being formed in close proximity with the stronger CO binding Cu regions. In contrast to our findings for pure Cu2O NCs, we did not detect a clear correlation between the Cu surface coverage with CO and the C2+ product formation. Linking to the Ag–Cu2O system, we emphasize the importance of dispersed CO-producing sites on a Cu host in order to facilitate the CO2RR faradaic efficiency. Thus, it is essential to achieve an in-depth understanding of the atomic scale interaction of Au clusters or single sites with oxide-derived Cu using model systems under CO2RR to verify its importance over the CO spill-over mechanism. Understanding the selectivity dependencies on the restructuring of a Cu–Au system, which undergoes continuous transformation under CO2RR relevant conditions, provides opportunities for a rational design of highly active and selective bimetallic catalysts. Thus, our work provides crucial input to enable knowledge driven catalyst design of bimetallic CO2RR electrocatalysts and in particular to explore the potential of ultralow Au decoration of Cu nanocatalysts towards facilitating specific product distribution in CO2RR.
Author contributions
C. R. conceptualization, data curation, investigation, writing – original draft. A. H. data curation, investigation, writing – review and editing. D. C. data curation, investigation, writing – review and editing. M. R. data curation, investigation, writing – review and editing. H. S. J. investigation, writing – review and editing. D. K. investigation, writing – review and editing. M. L. L investigation, writing – review and editing. S. K. investigation, writing – review and editing. U. H. investigation, writing – review and editing. E. M. D investigation, writing – review and editing. S. W. C. data curation, writing – review and editing. J. T. data curation, investigation, writing – review and editing. D. T. L. A. data curation, writing – review and editing. A. B. conceptualization, investigation, writing – original draft. B. R. C. conceptualization, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council under grant ERC-OPERANDOCAT (ERC-725915) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat and the SPP 2080 – project no. 406944504. C. R, A. H, D. K. acknowledge support by the IMPRS for Elementary Processes in Physical Chemistry. The Max Planck-EPFL Center for Molecular Nanoscience and Technology is acknowledged for supporting the collaborative electron microscope measurements performed at EPFL, which were made using the instruments at the Interdisciplinary Centre for Electron Microscopy (CIME) XAS experiments were performed at CLAESS beamline at ALBA synchrotron with the collaboration of ALBA staff and we would like to thank Dr Carlo Marini for assistance. The Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76F00515 and we would like to thank Dr Adam Hoffman for assistance. Further, we acknowledge SOLEIL for provision of synchrotron radiation facilities and we would like to thank Dr Andrea Zitolo for assistance in using beamline SAMBA. We also acknowledge the Paul Scherrer Institut, Villingen, Switzerland for provision of synchrotron radiation beamtime at beamline SuperXAS of the SLS and we would like to thank Dr Adam H. Clark for assistance. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA lll and we would like to thank Dr Zoltan Hegedues for assistance in using the P21.2 facilities. Finally, we acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal number CH-5914 and we would like to thank Dr Jakub Drnec for assistance and support in using beamline ID31. Open Access funding provided by the Max Planck Society.References
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef.
- T. Möller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya, A. S. Varela and P. Strasser, Angew. Chem., Int. Ed., 2020, 59, 17974–17983 CrossRef PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- Y. Hori, Modern Aspects of Electrochemistry, 2008, pp. 89–189 Search PubMed.
- H. Hu, Y. Tang, Q. Hu, P. Wan, L. Dai and X. J. Yang, Appl. Surf. Sci., 2018, 445, 281–286 CrossRef CAS.
- H. S. Jeon, J. Timosnenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. R. Cuenya, J. Am. Chem. Soc., 2019, 141, 19879–19887 CrossRef CAS PubMed.
- Y. Feng, Z. Li, H. Liu, C. Dong, J. Wang, S. A. Kulinich and X. Du, Langmuir, 2018, 34, 13544–13549 CrossRef CAS PubMed.
- A. Herzog, A. Bergmann, H. S. Jeon, J. Timoshenko, S. Kühl, C. Rettenmaier, M. Lopez Luna, F. T. Haase and B. R. Cuenya, Angew. Chem., Int. Ed., 2021, 60, 7426–7435 CrossRef CAS.
- D. Karapinar, C. E. Creissen, J. G. Rivera De La Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- K. Liu, M. Ma, L. Wu, M. Valenti, D. Cardenas-Morcoso, J. P. Hofmann, J. Bisquert, S. Gimenez and W. A. Smith, ACS Appl. Mater. Interfaces, 2019, 11, 16546–16555 CrossRef CAS.
- H. A. Hansen, C. Shi, A. C. Lausche, A. A. Peterson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2016, 18, 9194–9201 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- J. W. Vickers, D. Alfonso and D. R. Kauffman, Energy Technol., 2017, 5, 775–795 CrossRef CAS.
- H. Mistry, R. Reske, P. Strasser and B. R. Cuenya, Catal. Today, 2017, 288, 30–36 CrossRef CAS.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, B. Xu, M. J. Cheng and Q. Lu, Nat. Commun., 2019, 10, 1–9 CrossRef.
- Z. Xu, E. Lai, Y. Shao-Horn and K. Hamad-Schifferli, Chem. Commun., 2012, 48, 5626–5628 RSC.
- S.-H. Wei, A. A. Mbaye, L. G. Ferreira and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 4163–4185 CrossRef CAS.
- P. P. Fedorov and S. N. Volkov, Russ. J. Inorg. Chem., 2016, 61, 772–775 CrossRef CAS.
- D. Friebel, F. Mbuga, S. Rajasekaran, D. J. Miller, H. Ogasawara, R. Alonso-Mori, D. Sokaras, D. Nordlund, T. C. Weng and A. Nilsson, J. Phys. Chem. C, 2014, 118, 7954–7961 CrossRef CAS.
- J. Christophe, T. Doneux and C. Buess-Herman, Electrocatalysis, 2012, 3, 139–146 CrossRef CAS.
- P. Hirunsit, J. Phys. Chem. C, 2013, 117, 8262–8268 CrossRef CAS.
- Z. Liu, M. N. Hossain, J. Wen and A. Chen, Nanoscale, 2021, 13, 1155–1163 RSC.
- E. Andrews, Y. Fang and J. Flake, J. Appl. Electrochem., 2018, 48, 435–441 CrossRef CAS.
- S. Back, J. H. Kim, Y. T. Kim and Y. Jung, ACS Appl. Mater. Interfaces, 2016, 8, 23022–23027 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- F. Jia, X. Yu and L. Zhang, J. Power Sources, 2014, 252, 85–89 CrossRef CAS.
- W. Zhu, K. Zhao, S. Liu, M. Liu, F. Peng, P. An, B. Qin, H. Zhou, H. Li and Z. He, J. Energy Chem., 2019, 37, 176–182 CrossRef.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- S. Shen, X. Peng, L. Song, Y. Qiu, C. Li, L. Zhuo, J. He, J. Ren, X. Liu and J. Luo, Small, 2019, 15, 1902229 CrossRef.
- J. Gao, D. Ren, X. Guo, S. M. Zakeeruddin and M. Grätzel, Faraday Discuss., 2019, 215, 282–296 RSC.
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. R. Cuenya, Nat. Commun., 2021, 12, 6736 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- Z. Jiang, W. Zhang, L. Jin, X. Yang, F. Xu, J. Zhu and W. Huang, J. Phys. Chem. C, 2007, 111, 12434–12439 CrossRef CAS.
- M. Kuhn and T. K. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 1647–1661 CrossRef CAS.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. R. Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- D. Higgins, A. T. Landers, Y. Ji, S. Nitopi, C. G. Morales-Guio, L. Wang, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 2947–2955 CrossRef CAS.
- A. I. Frenkel, V. S. Machavariani, A. Rubshtein, Y. Rosenberg, Y. Rosenberg, A. Voronel and E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 9364–9371 CrossRef CAS.
- T. K. Sham, Y. M. Yiu, M. Kuhn and K. H. Tan, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 11881–11886 CrossRef CAS PubMed.
- A. Singhal, M. R. Pai, R. Rao, K. T. Pillai, I. Lieberwirth and A. K. Tyagi, Eur. J. Inorg. Chem., 2013, 2640–2651 CrossRef CAS.
- S. Jiang, K. Klingan, C. Pasquini and H. Dau, J. Chem. Phys., 2019, 150, 041718 CrossRef.
- C. M. Gunathunge, J. Li, X. Li, J. J. Hong and M. M. Waegele, ACS Catal., 2020, 10, 6908–6923 CrossRef CAS.
- G. L. Beltramo, T. E. Shubina and M. T. Koper, ChemPhysChem., 2005, 6, 2597–2606 CrossRef CAS.
- S. Jiao, X. Fu and H. Huang, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- T. K. Sham, A. Bzowski, M. Kuhn and C. C. Tyson, Solid State Commun., 1991, 80, 29–32 CrossRef CAS.
- G. Iijima, T. Inomata, H. Yamaguchi, M. Ito and H. Masuda, ACS Catal., 2019, 9, 6305–6319 CrossRef CAS.
- Y. C. Li, Z. Wang, T. Yuan, D. H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. De Arquer, Y. Wang, C. T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS.
- M. Bernal, A. Bagger, F. Scholten, I. Sinev, A. Bergmann, M. Ahmadi, J. Rossmeisl and B. R. Cuenya, Nano Energy, 2018, 53, 27–36 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary notes, complementary XRD, TEM, XAS, XPS, Raman and ICP-MS Figures, analysis results. See DOI: https://doi.org/10.1039/d3ey00162h |
| This journal is © The Royal Society of Chemistry 2024 |