
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of metal ions in the electron-transfer mechanism on the photovoltaic performance of SALPHEN-based DSSC: experimental and theoretical studies†
José Guadalupe
Hernández
a,
Carlos Alberto Huerta
Aguilar
b,
Jayanthi
Narayanan
 *c,
Eduardo Daniel Tecuapa
Flores
c,
Pandiyan
Thangarasu
*d,
Aldo Hernández
Ramírez
c,
Karthika
Shanmugam
e and
Mayra Margarita Luna
Martinez
c
*c,
Eduardo Daniel Tecuapa
Flores
c,
Pandiyan
Thangarasu
*d,
Aldo Hernández
Ramírez
c,
Karthika
Shanmugam
e and
Mayra Margarita Luna
Martinez
c
aCentro Tecnológico, Facultad de Estudios Superiores (FES-Aragón), Universidad Nacional Autónoma de México (UNAM), Estado de México, CP 57130, Mexico
bInstituto Tecnológico de Estudios Superiores de Monterrey (ITESM), Puebla, CP 72453, Mexico
cDivisión de Ingeniería en Nanotecnología, Universidad Politécnica del Valle de México, Av. Mexiquense s/n esquina Av. Universidad Politécnica, Estado de México, CP 54910, Mexico. E-mail: jnarayanan@upvm.edu.mx
dFacultad de Química, Universidad Nacional Autónoma de México (UNAM), Ciudad Universitaria, CDMX, CP 04510, Mexico. E-mail: pandiyan@unam.mx
eSchool of Chemical Engineering, Vellore Institute of Technology, Vellore 632014, Tamil Nadu, India
First published on 22nd February 2024
Abstract
Herein, we report the synthesis and characterization of metal complexes of Fe(III), Co(II), and Cu(II) with SALPHEN (N,N-bis(salicylimine)-o-phenyldiammine) and their potential application as sensitizers in dye-sensitized solar cells (DSSCs). The high thermal stability up to 550 °C observed for these compounds suggests that they may be possible candidates for the fabrication of solar cell devices without pyrolysis or thermo-oxidative degradation during the solar cell operation. The photovoltaic parameters of SALPHEN and its metal complexes were investigated experimentally, and it was observed that the metal ion coordination generally enhanced the power conversion efficiency (η) where the maximum η of 46.2% was obtained for the Co(II) complex due to its high molar extinction coefficient and enhanced charge transfer by improved π-conjugation and electron-withdrawing capability. In the theoretical calculations, the B3LYP functional was employed to compute the ground-state geometries and the frontier molecular orbitals for all the complexes. The ground-state and excited-state energy transfer, which specifically contribute to the light harvesting properties of SALPHEN and its metal complexes, were analyzed using the functionals with corrected dispersion CAM-B3LYP/dgdzvp basis set (double zeta) and functional (B3PW91, B3P86/6-311++G(d,p) basis set (triple-zeta)). It was observed that the HOMOs of the metal complexes were localized on the d orbitals with π(SALPHEN nitrogen and O− atoms) orbital character, whereas their LUMOs have π*(O−) orbital character. The light-harvesting efficiency (LHE) was analyzed using the theoretically obtained band-gap values for SALPHEN and its metal complexes. In general, metal ion chelation showed increased charge transfer transitions in both the ground and excited states. Moreover, the charge density difference in each compound was observed through three-dimensional (3D) visualization of its electron density isosurface (contour 0.05 e Å−3), which supports understanding the mechanism involved in the energy transfer.
1. Introduction
Transition metal complexes based on Schiff base ligands are widely used as potential photosensitizers in dye-sensitized solar cells (DSSCs) because of their strong visible light absorption ability, facilitating the transfer of electrons to the conduction band via metal-to-ligand charge transfer (MLCT).1–4 Especially, aromatic azo derivatives containing a conjugated azo (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) group act as D–π–A charge-transfer chromophores, showing excellent photo-harvesting properties, which is attributed to the wide-ranging delocalized π-systems between the acceptor (A) and donor (D) units through the azo group.5–7 Furthermore, their photophysical activity can often be enhanced by metal ion coordination, which influences their solar light-harvesting behavior. In particular, metal ions with incomplete valence shell configurations show intrinsic chemical characteristics when there are multi-dentate ligands in the metal complex, which has attracted considerable attention in the development of renewable energy technologies.8–11 The incorporation of suitable metal ions with a suitable ligand usually increases the HOMO–LUMO energy gap, which allows the excited-state electron to have a longer lifetime even at high temperatures, increasing the overall luminescence quantum yield of DSSCs.12 It is known that the efficiency of a DSSC is dependent on the photosensitizers used between the electron donor and acceptor electrodes, which should have a broad and strong light absorption capable of covering the entire visible and near-infrared regions (NIR) and be stable to a certain temperature. In this case, the understanding of the power conversion efficiency (PCE), spectral response range, and stability of the DSSC performance are dependent on the molecular-level study of the photosensitizers.13–16 Specifically, dynamic photo-induced intramolecular charge transfer (ICT) in the excited state reflects the change in both the electronic and structural properties of photosensitizers, which involve π-conjugation extending from the donor to the acceptor and often differ significantly from the ground-state molecular structure.17–20 Hence, it is important to explore the electronic and optoelectronic characteristics of sensitizer molecules to reveal their electron-donating and electron-withdrawing behavior, given that they play a vital role in photo-induced ICT after photoexcitation. Density functional theory (DFT) calculations can be employed to elucidate the role of LMCT (ligand-to-metal charge transfer), MLCT, and MC (metal-centered) in the excited states of photosensitizers present in DSSCs and to corroborate experimental interpretation.21–23 Time-dependent density functional theory (TD-DFT) studies have been carried out to understand the various multifaceted processes in DSSCs and metal–organic complexes.24–26 For instance, the DFT and TD-DFT methods were used to explore the light-harvesting efficiency (LHE), open circuit voltage (Voc), and driving force (ΔGinject) for Ru-based and Fe-based dye molecules, which depend on the relative energy levels of the HOMOs of Ru and Fe with respect to the π (dye) orbital and the LUMOs with π*(dye) orbital.27 A similar study was performed for diamine-dithiolate-based Pt complexes, together with an NBO (natural bond orbital) analysis, helping to identify suitable anchoring groups that enhance the overall electronic and optical properties of sensitizers.27–29 In another study, the enhancement in solar cell efficiency was explored by determining the excited-state energies by DFT for porphyrin analogues over Zn-meso-tetraphenylporphyrin complexes having different β-substituents given that an increase in π-conjugation on porphyrin impacted the electron-withdrawing capability of the substituents.30 SALPHEN-based metal complexes have interesting properties because their conjugation and rigidity are important structural features that their dictate molecular properties. Recently, these complexes have been applied in various fields of research, including metal–organic frameworks, fluorescent sensors, and potential catalysts, and even in biological applications.31 The adaptability of SALPHEN complexes has also been extended to the field of new materials, such as solar cells and scaffold materials.32 SALPHEN ligands can be easily fabricated and tuned and are alternatives to synthetically more challenging ligands, such as porphyrins, while conserving similar functionality.33 However, despite their benefits reported to date, there is limited information available on the effect of the conjugation and structural rigidity of these complexes on their optical performance.34–37 Therefore, SALPHEN-based metal complexes continue to attract attention for exploring new mechanistic insights to understand the process of photo-induced charge transfer during their DSSC performance, which may increase their applicability.35–37 Herein, we report the fabrication of a DSSC using metal complexes of an azo-Schiff base, i.e., N,N′-bis(salicylimine)-o-phenyldiammine [SALPHEN], with Fe, Co, and Cu and its photovoltaic parameters including short circuit current (Isc), open circuit voltage (Voc), and fill factor (FF) were explored. The proposed DSSC design approach is based on the use of earth-abundant metals, which can replace ruthenium; also, the multivalent nature of these metal ions can provide metal-to-ligand charge transfer in the presence of conjugated π-organic ligands, resulting in a high molar extinction coefficient. Furthermore, DFT and TD-DFT studies were performed for the metal complexes to determine their molecular and optical properties. With the use of natural transition orbitals (NTO), the nature of charge transfer in their electronic absorption and emissions was determined in detail. The obtained photovoltaic parameters for the DSSC with the metal complexes were compared with that from theoretical calculations.
N–) group act as D–π–A charge-transfer chromophores, showing excellent photo-harvesting properties, which is attributed to the wide-ranging delocalized π-systems between the acceptor (A) and donor (D) units through the azo group.5–7 Furthermore, their photophysical activity can often be enhanced by metal ion coordination, which influences their solar light-harvesting behavior. In particular, metal ions with incomplete valence shell configurations show intrinsic chemical characteristics when there are multi-dentate ligands in the metal complex, which has attracted considerable attention in the development of renewable energy technologies.8–11 The incorporation of suitable metal ions with a suitable ligand usually increases the HOMO–LUMO energy gap, which allows the excited-state electron to have a longer lifetime even at high temperatures, increasing the overall luminescence quantum yield of DSSCs.12 It is known that the efficiency of a DSSC is dependent on the photosensitizers used between the electron donor and acceptor electrodes, which should have a broad and strong light absorption capable of covering the entire visible and near-infrared regions (NIR) and be stable to a certain temperature. In this case, the understanding of the power conversion efficiency (PCE), spectral response range, and stability of the DSSC performance are dependent on the molecular-level study of the photosensitizers.13–16 Specifically, dynamic photo-induced intramolecular charge transfer (ICT) in the excited state reflects the change in both the electronic and structural properties of photosensitizers, which involve π-conjugation extending from the donor to the acceptor and often differ significantly from the ground-state molecular structure.17–20 Hence, it is important to explore the electronic and optoelectronic characteristics of sensitizer molecules to reveal their electron-donating and electron-withdrawing behavior, given that they play a vital role in photo-induced ICT after photoexcitation. Density functional theory (DFT) calculations can be employed to elucidate the role of LMCT (ligand-to-metal charge transfer), MLCT, and MC (metal-centered) in the excited states of photosensitizers present in DSSCs and to corroborate experimental interpretation.21–23 Time-dependent density functional theory (TD-DFT) studies have been carried out to understand the various multifaceted processes in DSSCs and metal–organic complexes.24–26 For instance, the DFT and TD-DFT methods were used to explore the light-harvesting efficiency (LHE), open circuit voltage (Voc), and driving force (ΔGinject) for Ru-based and Fe-based dye molecules, which depend on the relative energy levels of the HOMOs of Ru and Fe with respect to the π (dye) orbital and the LUMOs with π*(dye) orbital.27 A similar study was performed for diamine-dithiolate-based Pt complexes, together with an NBO (natural bond orbital) analysis, helping to identify suitable anchoring groups that enhance the overall electronic and optical properties of sensitizers.27–29 In another study, the enhancement in solar cell efficiency was explored by determining the excited-state energies by DFT for porphyrin analogues over Zn-meso-tetraphenylporphyrin complexes having different β-substituents given that an increase in π-conjugation on porphyrin impacted the electron-withdrawing capability of the substituents.30 SALPHEN-based metal complexes have interesting properties because their conjugation and rigidity are important structural features that their dictate molecular properties. Recently, these complexes have been applied in various fields of research, including metal–organic frameworks, fluorescent sensors, and potential catalysts, and even in biological applications.31 The adaptability of SALPHEN complexes has also been extended to the field of new materials, such as solar cells and scaffold materials.32 SALPHEN ligands can be easily fabricated and tuned and are alternatives to synthetically more challenging ligands, such as porphyrins, while conserving similar functionality.33 However, despite their benefits reported to date, there is limited information available on the effect of the conjugation and structural rigidity of these complexes on their optical performance.34–37 Therefore, SALPHEN-based metal complexes continue to attract attention for exploring new mechanistic insights to understand the process of photo-induced charge transfer during their DSSC performance, which may increase their applicability.35–37 Herein, we report the fabrication of a DSSC using metal complexes of an azo-Schiff base, i.e., N,N′-bis(salicylimine)-o-phenyldiammine [SALPHEN], with Fe, Co, and Cu and its photovoltaic parameters including short circuit current (Isc), open circuit voltage (Voc), and fill factor (FF) were explored. The proposed DSSC design approach is based on the use of earth-abundant metals, which can replace ruthenium; also, the multivalent nature of these metal ions can provide metal-to-ligand charge transfer in the presence of conjugated π-organic ligands, resulting in a high molar extinction coefficient. Furthermore, DFT and TD-DFT studies were performed for the metal complexes to determine their molecular and optical properties. With the use of natural transition orbitals (NTO), the nature of charge transfer in their electronic absorption and emissions was determined in detail. The obtained photovoltaic parameters for the DSSC with the metal complexes were compared with that from theoretical calculations.
2. Experimental
2.1. Material and methods
All reagents used in the experiments were purchased from Sigma-Aldrich-Mexico. Elemental analyses (percentage of C, H, and N) were carried out principally on a Fisons analyzer (Model EA 1108 CHNSO) for the compounds. Fourier-transformed infrared spectra (FTIR) were recorded in the range of 4000–400 cm−1 on a PerkinElmer Frontier instrument. Powder X-ray diffraction (XRD) analysis was performed on a D8 Advance Davinci equipped with a Bruker AXS theta–theta configuration with CuKα radiation (l = 1.54 Å). A UV-Vis spectrophotometer (PerkinElmer Lambda 25) was employed to record the electronic spectra of the metal complexes and analyze their electronic behaviors. Magnetic susceptibility measurements were performed using a Gouy balance at 300 K, which was calibrated with Hg[Co(SCN)4] as the standard, and the effective magnetic moments were calculated using the expression: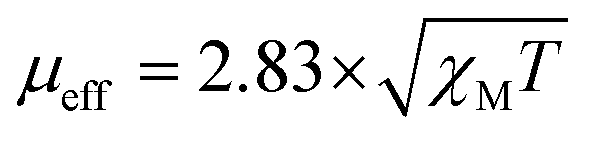 B.M.38,39 A thermogravimetric study was conducted for all the compounds using a thermal analysis system consisting of a simultaneous thermogravimetric analysis (TGA)-differential scanning calorimeter (DSC) (NETZSCH model STA 449 F1 Jupiter coupled to a Bruker FT-IR spectrometer and a nitrogen-generating instrument by Parker Balston). The thermal behavior of all the compounds was analyzed using two techniques, i.e., TGA and DSC, which were carried out on a PerkinElmer TGA-4000 and DSC1 (Mettler Toledo) instrument, respectively. Experiments were carried out from 30 °C to 800 °C for TGA and 30 °C to 550 °C for DSC at a heating rate of 10 °C min−1 under a flowing nitrogen atmosphere (20 mL min−1). The molar conductance of the metal complexes in CH3CN, DMF, and DMSO (0.001 M) solutions at 25 °C was measured using a YK-43CD LUTRON digital conductivity meter. The current–voltage measurement of the solar cell was carried out using a SANWA RD701 digital multimeter.
B.M.38,39 A thermogravimetric study was conducted for all the compounds using a thermal analysis system consisting of a simultaneous thermogravimetric analysis (TGA)-differential scanning calorimeter (DSC) (NETZSCH model STA 449 F1 Jupiter coupled to a Bruker FT-IR spectrometer and a nitrogen-generating instrument by Parker Balston). The thermal behavior of all the compounds was analyzed using two techniques, i.e., TGA and DSC, which were carried out on a PerkinElmer TGA-4000 and DSC1 (Mettler Toledo) instrument, respectively. Experiments were carried out from 30 °C to 800 °C for TGA and 30 °C to 550 °C for DSC at a heating rate of 10 °C min−1 under a flowing nitrogen atmosphere (20 mL min−1). The molar conductance of the metal complexes in CH3CN, DMF, and DMSO (0.001 M) solutions at 25 °C was measured using a YK-43CD LUTRON digital conductivity meter. The current–voltage measurement of the solar cell was carried out using a SANWA RD701 digital multimeter.
2.2. Synthesis of SALPHEN (C20H16O2N2)
SALPHEN was synthesized similarly to the method reported in the literature.40 To a solution of 2-hydroxybenzaldehyde (2.0 mmol) dissolved in ethanol (30 mL), o-phenylenediamine (1.0 mmol) was added, and then the resulting solution was stirred for 30 min at 40 °C to yield a deep-yellow product, which was filtered and dried under vacuum (Scheme 1). Yield, 99.15%. Elemental analysis: calcd (%) for C20H16O2N2: C, 75.93; H, 5.10; N, 8.86; found (%): C, 76.03; H, 4.99; N, 8.71. FT-IR (νmax/cm−1) (Fig. 1): stretching vibrations at 1610m [ν(CN)] and 1560m [ν(CC)]; benzene ring in aromatic compounds (ring-stretch, sharp bands) at 1478m [ν(C–C)] and 1278 [ν(C–N)]; O–H bending (in-plane) at 1330–1430s; α-CH2 bending at 1400–1450s; C–N in aromatic amines at 1280–1180s; C–O–H in alcohols (C–O stretch) at 1200–1015vs; ring vibrations in-plane (C–H) deformation at 1330–1300s; stretching vibrations at 1191m [ν(C–O)]. In addition, CH2CH– as (–CH2 out-of-plane wag) at 950–900vs; pyridine ring (in-plane ring formation) at 757m [ν(CN)] and 501s [ν(CC)]; C–N–C in amines (bend) at 510–400s; ring in benzene derivatives (in-plane and out-of-plane and ring-deformations as two bands) at 580–420m-s.
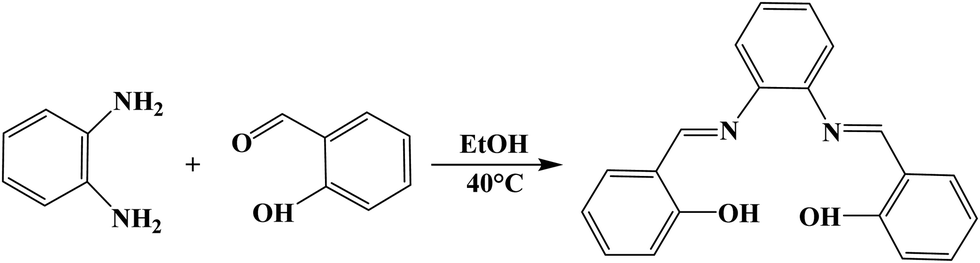 | ||
| Scheme 1 Synthesis of SALPHEN. | ||
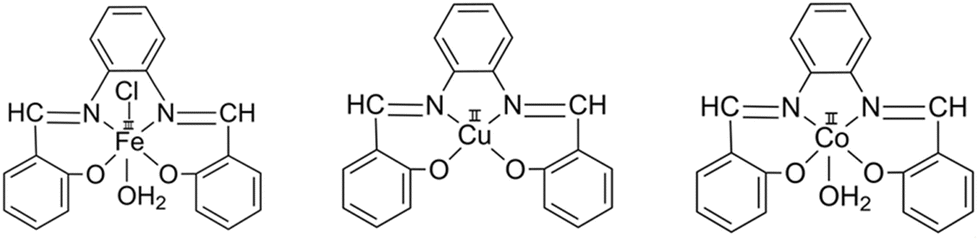 | ||
| Scheme 2 Coordination of SALPHEN with metal ions. | ||
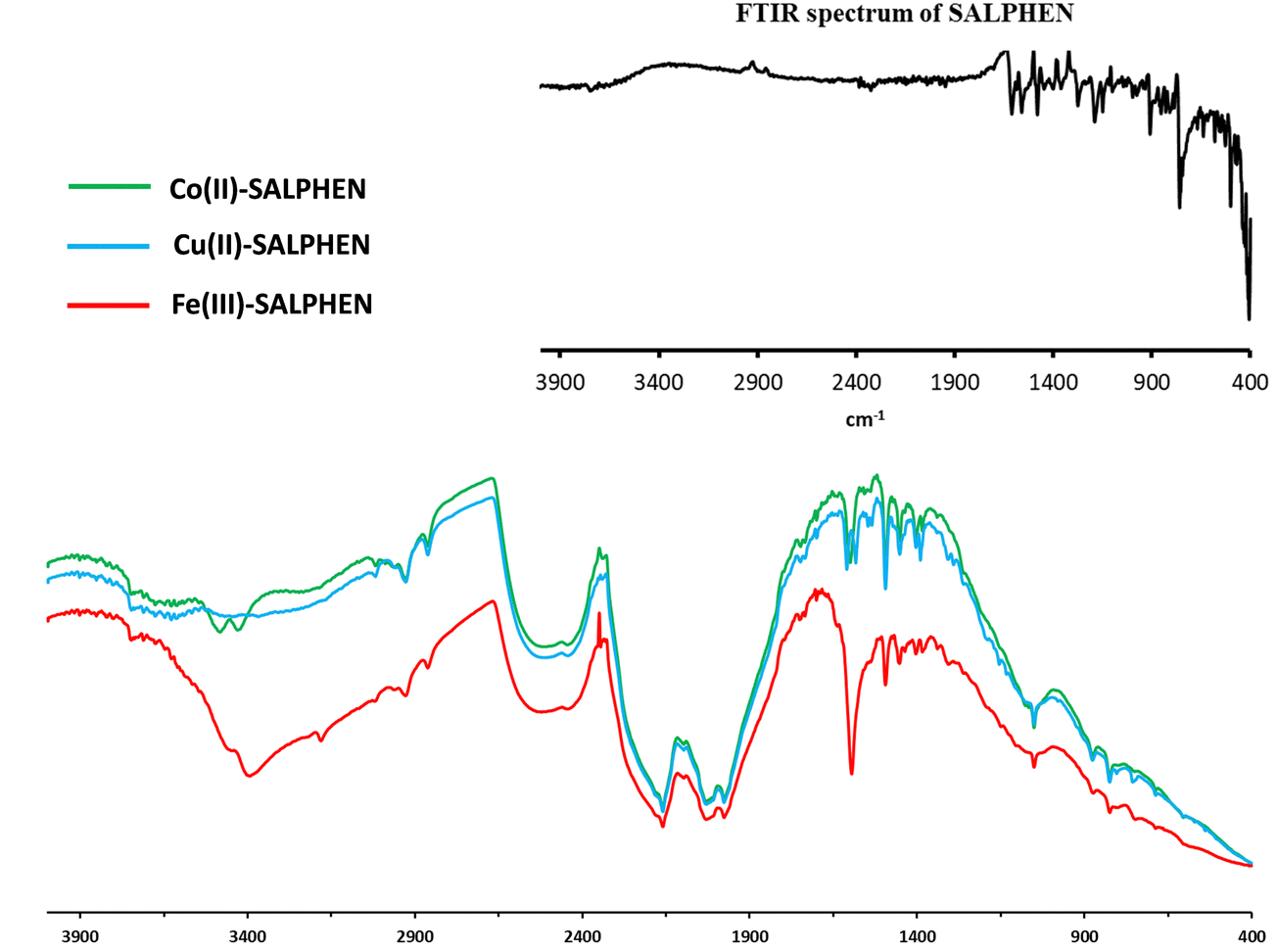 | ||
| Fig. 1 FTIR spectra of SALPHEN (inset) and its metal complexes at 25 °C. | ||
2.3. Synthesis of metal complexes (Scheme 2)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Elemental analysis: calcd (%) for C20H16O3N2ClFe: C, 56.70; H, 3.81; N, 6.61; found (%): C, 56.31; H, 3.59; N, 6.59. FT-IR (νmax/cm−1) (Fig. 1): medium-sized stretching vibration in the range of 3458–3392 [ν(O–H)] due to the crystal waters in the Fe(III) complex. Stretching frequency in the range of 2160s, 2034s, and 1978s [ν(Fe–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Fe)]; bending vibration at 822m [δ(Fe–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ (CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.82 B.M.
:1, v/v). Elemental analysis: calcd (%) for C20H16O3N2Co: C, 61.39; H, 4.12; N, 7.16; found (%): C, 61.01; H, 4.33; N, 7.03. FT-IR (νmax/cm−1) (Fig. 1): small bands of two stretching vibrations at 3480 and 3429 [ν(O–H)] due to the crystal waters in the Co(II) complex; stretching frequency in the range of 2160s, 2034s, and 1978s [ν(Co–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Co)]; bending vibration at 822m [δ(Co–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ (CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.78 B.M.
:1, v/v). Elemental analysis: calcd (%) for C20H14O2N2Cu: C, 63.57; H, 3.73; N, 7.41; found (%): C, 63.17; H, 3.53; N, 7.01. FT-IR (νmax/cm−1) (Fig. 1): stretching frequencies at 2160s, 2034s, and 1978s [ν(Cu–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Cu)]; bending vibration at 822m [δ(Cu–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ(CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.91 B.M.
:1, v/v). Elemental analysis: calcd (%) for C20H16O3N2ClFe: C, 56.70; H, 3.81; N, 6.61; found (%): C, 56.31; H, 3.59; N, 6.59. FT-IR (νmax/cm−1) (Fig. 1): medium-sized stretching vibration in the range of 3458–3392 [ν(O–H)] due to the crystal waters in the Fe(III) complex. Stretching frequency in the range of 2160s, 2034s, and 1978s [ν(Fe–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Fe)]; bending vibration at 822m [δ(Fe–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ (CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.82 B.M.
:1, v/v). Elemental analysis: calcd (%) for C20H16O3N2Co: C, 61.39; H, 4.12; N, 7.16; found (%): C, 61.01; H, 4.33; N, 7.03. FT-IR (νmax/cm−1) (Fig. 1): small bands of two stretching vibrations at 3480 and 3429 [ν(O–H)] due to the crystal waters in the Co(II) complex; stretching frequency in the range of 2160s, 2034s, and 1978s [ν(Co–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Co)]; bending vibration at 822m [δ(Co–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ (CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.78 B.M.
:1, v/v). Elemental analysis: calcd (%) for C20H14O2N2Cu: C, 63.57; H, 3.73; N, 7.41; found (%): C, 63.17; H, 3.53; N, 7.01. FT-IR (νmax/cm−1) (Fig. 1): stretching frequencies at 2160s, 2034s, and 1978s [ν(Cu–O)]; stretching frequency at 907s [ν(C–O), ν(C–O–Cu)]; bending vibration at 822m [δ(Cu–O)]; stretching frequency at 1496m [ν(CN)]; bending vibration at 590 [δ(CN)]. Molar conductance/DMF: 3.93–5.44 Ω−1 cm2 mol−1. Magnetic moment (μeff): 1.91 B.M.
2.4. Fabrication of DSSC using SALPHEN and its metal complexes
Fluorine-doped tin oxide (FTO) glass (from Sigma-Aldrich) was employed as the substrate (thickness 2.2 mm), which was sequentially cleaned by ultra-sonicating it in surfactant, deionized water, acetone, and ethanol. The photoanodic layer was prepared on the above-mentioned glass by depositing nanocrystalline TiO2 nanopowder over each metal complex (substrate) using the doctor blading method. For this, a solid paste of TiO2 nanopowder (1.0 g), Triton X-100 surfactant (0.05 mL), acetic acid (1.0 mL, 0.1 M), and polyethylene glycol (0.1 mL) were mixed and pasted on the glass surface to produce a uniform layer. The porous TiO2 films had an average thickness of ∼10 μm, and the doctor blading method was repeated to vary their thickness. The TiO2-coated electrodes were sintered at 500 °C for 40 min and cooled to room temperature. The activated TiO2 film was dipped in each metal complex of [Fe(SALPHEN)(OH2)Cl], [Co(SALPHEN)(OH2)], and [Cu(SALPHEN)] (0.3 mM in ethanol), followed by drying. The active area of 5 mm by 5 mm squares of thin films was used for the solar conductivity measurements. A graphite counter electrode was fabricated by applying a thin layer of graphite on the FTO substrate, followed by heat treatment in the air for 30 min at 400 °C. I−/I3− solution as the electrolyte was used to drop-cast the TiO2 photoanode sensitized by each of the metal complexes. The concentration of the adsorbed quantity of dye was calculated by dipping the DSSC photoanode in 0.1 M NaOH in an ethanol:water (1:1) solution until complete desorption of the dye, and the desorbed dye solution was measured using a UV-visible spectrophotometer.
2.5. Computational methods
DFT was performed using the Gaussian 09 software and the B3LYP/DGDZVP basis set was employed to optimize the ground-state geometry of the Fe(III), Co(II), and Cu(II) complexes of SALPHEN.41,42 Employing density functional theory (DFT), the ground-state geometry of the compounds was fully optimized after considering different hybrid functionals such as B3LYP, CAM-B3LYP43 (Takeshi Yanai, 2004), with DGDZVP (double-zeta), and another 6-311++G(d,p) (triple-zeta) basis set with different exchange–correlation hybrid functionals B3LYP44–51 (A. D. Becke, 1993, Becke, 1988, C. Lee, 1988, B. Mielich, 1989), B3PW91 (J. P. Perdew, 1992), and B3P86 (J. P. Perdew, 1986, 1987. A. D. Becke, 1993). The obtained results were analyzed. The electronic absorption transitions and optical properties were calculated using time-dependent density functional theory (TD-DFT) using the B3LYP, CAM-B3LYP/DGDZVP basis set (double-zeta)52 and B3PW91, B3P86/6-311++G(d,p) triple-zeta basis sets.53 The obtained results for the metal complexes of SALPHEN in the gaseous state were compared with the experimental data. It was expected that the combination of dgdzvp (double-zeta) with the (B3LYP and CAM-B3LYP) functionals and 6-311++G(d,p) (triple-zeta) with the (B3LYP, B3PW91, B3P86) functionals can yield the most precise optical spectral measurements, as suggested in the literature.54The vertical excitation energy and oscillation strengths were also calculated for the complex geometry in the ground state. The natural transition orbitals (NTOs) were derived for [Fe(SALPHEN)Cl(H2O)], [Co(SALPHEN)(H2O)], and [Cu(SALPHEN)] to determine the particle-hole position in the excited-state using B3LYP/DGDZVP.55,56 The charge difference density was also calculated by three-dimensional (3D) visualization of the electron density isosurface (contour 0.05 e Å−3). The HOMO and LUMO were derived for the complexes using B3LYP/DGDZVP to understand the influence of their structure on the performance of the DSSC.57 The visualization of the molecular system in the excited state can support the mechanism involved in the photo-induced charge transfer and energy transfer.58
3. Results and discussion
3.1. Infrared spectra
The FTIR frequency (νmax/cm−1) in the range of 400–4000 cm−1 was recorded for the free ligand (SALPHEN) (Fig. 1, inset), where the absence of an O–H stretching frequency around 3100 to 2900 cm−1 was observed due to the internal hydrogen bridge between both hydroxyl groups [–O–H⋯O–H] present in the ligand.59 The benzene ring stretching vibrations were observed at 1610 cm−1 [ν(CN)] and 1560 cm−1 [ν(CC)] and the bending vibrations at 757 cm−1 and 501 cm−1, which are characteristic of the Schiff base ligands.60 In addition, the stretching vibrations corresponding to [ν(C–C)] for the benzene ring, ν[C–O] for the phenolic, and [ν(C–N)] for the azomethine groups were observed at 1478, 1278, and 1191 cm−1, respectively.61
All the IR frequencies (νmax/cm−1) were consistent with the theoretical DFT-IR spectra (Fig. S1–S5, ESI†). Similarly, the experimental FTIR spectra of the metal complexes were compared with that of the ligand (SALPHEN), and it was observed that there was a remarkable shift in the frequency for the metal complexes compared to the ligand, especially the peaks for the azomethine ν[CN] and phenolic ν[O–H] groups shifted to a lower frequency for the complexes, which suggests the involvement of the nitrogen and oxygen atoms in the coordination bond formed with the metal(II) ions. For example, for the Fe(III) complex (Fig. 1), two peaks appeared at 3458 and 3392 cm−1 due to the ν[–OH] stretching vibration, which correspond to the crystal water molecules, suggesting the presence of crystal water in the iron complex and confirmed by the thermal and elemental analyses.62 The coordination of water molecules with Fe(III) was confirmed by the FTIR signal at 600 cm−1.63 The appearance of a broad peak at around 2160–1978 cm−1 originated from the ν[M–O] and ν[M–N] vibrations as the characteristic of the metal coordination with the Schiff base ligand and confirms the involvement of the nitrogen and oxygen atoms of azomethine and phenolic (–OH) groups. Also, there was a red shift in the CN stretching vibration, which was detected for the ligand at 1610 cm−1 and shifted to 1592 cm−1 for the complex due to a shift of the azomethine nitrogen lone pair density toward the metal ion during the coordination of the metal ion. This was further supported by the signals at 822, 717, and 671 cm−1, corresponding to M–N and M–O originating from the azomethine and phenolic groups, respectively. Furthermore, the peak for ν(C− ≪ O) observed at 1278 cm−1 for the free ligand shifted to 907 cm−1 for the complex due to the formation of the C–O–M bond.64 The same observation was found for the Co(II) complex (Fig. 1), where the ν[O–H] stretching vibration observed at around 3480–3429 cm−1 corresponds to the crystal water molecules in the coordination sphere. This was further confirmed by the bands corresponding to ν[O–H] (645 cm−1), which indicate the presence of coordinated water molecules.65 The coordination of azomethine and phenolic groups (from the ligand) with the Co(II) ion generated the IR peaks at 2156, 2031, and 1977 cm−1 assigned to the ν[M–O] and ν[M–N] bonds, which were further supported by the bands at 876, 822 and 670 cm−1.66 Alternatively, for the Cu(II) complex (Fig. 1), the vibrations corresponding to the azomethine nitrogen ν[CN] and phenolic oxygen ν[O–H] to shifted to lower frequencies for the complex, which were observed at 1632, 1664 cm−1, and 3216 cm−1, while these peaks for the ligand were observed at 1680 cm−1 and 3208 cm−1, confirming the coordination of nitrogen and oxygen atoms with the Cu(II) ion.67 As observed for the other complexes, no ν[–OH] stretching vibration was observed in the range of 3480–3429 cm−1, suggesting the absence of water molecule coordination with Cu(II) in the complex.
In addition, the number of vibrational modes were compared with the theoretically obtained values. The number of vibrational modes for the Co complex is 120; for the Fe complex, it is 123; and for Cu, it is 111. However, according to the theoretical calculations by DFT, the vibrational mode was 129 for the Co complex, 132 for the Fe complex, and 120 for the Cu complex. There is was slight increase in the vibration frequency in the optimized geometry due to the gaseous state used for the theoretical calculations.
3.2. Electronic properties
In the UV-visible spectrum, electronic transitions were observed at 247, 319, and 387 nm for SALPHEN (Fig. 2 and Table 1), which originated from the π → π* and n → π* transitions corresponding to azomethine (CN) and phenolic oxygen (OH) of SALPHEN.68 In the UV spectra of the metal complexes, the transition at around 250 nm remained unchanged during the formation of the complex due to the intra-molecular π → π* transition within the ligand molecule. The transition at around 331–393 nm is associated with the n → π* transition originating from the phenolic groups involving a bathochromic shift during the complexation, indicating the ligand coordination with the metal ion. For example, the Fe(III) complex exhibited transitions at 299 and 288 nm originating from the LMCT and n → π*, which correspond to phenolic oxygen and coordinated water molecule present in the coordination sphere, respectively. In addition, bands were observed at 329 and 334 nm, corresponding to the 6A1g → 6T2g and 6A1g → 5T1g spin and multiplicity forbidden transitions, respectively, confirming the presence of a low-spin octahedral configuration for Fe(III) (d5).69 The low-spin nature of Fe(III)-SALPHEN was further supported by measuring the magnetic moment (μeff) of 1.82 B.M. at room temperature, showing its paramagnetic nature with one unpaired electron.
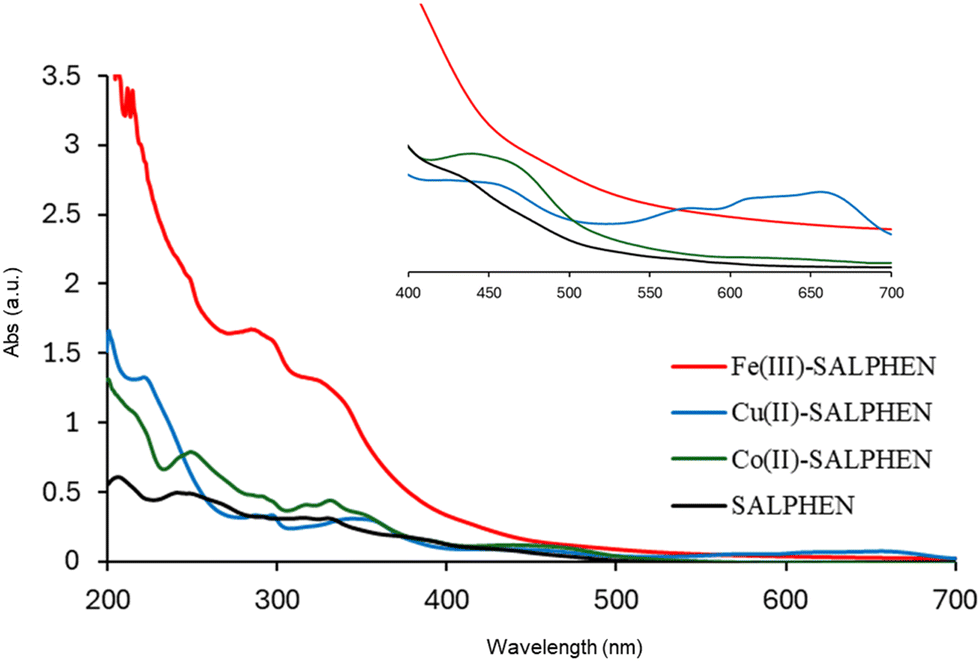 | ||
| Fig. 2 UV-Vis absorption spectra of SALPHEN and its metal complexes in DMF (1 × 10−3 M) at 25 °C. | ||
| Compound | (λ) nm | Electronic transitions | Molar extinction coefficient (ε) (M−1 cm−1) |
|---|---|---|---|
| SALPHEN | 247 | π → π*; n → π* | 4.91 × 103 |
| 319 | n → π* | 3.12 × 103 | |
| 387 | n → π* | 3.06 × 103 | |
| Fe(III)-SALPHEN | 249 | π → π*; n → π* | 2.04 × 105 |
| Low spin d5 | 288 | n → π* | 1.66 × 105 |
| Octahedral55 | 299 | LMCT | 1.57 × 105 |
| 335 | 6A1g → 6T2g | 1.21 × 105 | |
| 378 | 6A1g → 5T1g | 5.08 × 103 | |
| Co(II)-SALPHEN- | 250 | π → π*; n → π* | 7.83 × 104 |
| Low spin d7 | 294 | MLCT | 4.54 × 104 |
| Square pyramidal56 | 331 | 4A2 + 4E → 4A2(P) | 4.37 × 104 |
| 393 | 4A2 + 4E → 4E(P) | 1.41 × 104 | |
| 458 | 4A2 + 4E → 4B1 | 1.15 × 104 | |
| Cu(II)-SALPHEN | 287 | n → π* | 1.32 × 105 |
| Low spin d9 | 350 | LMCT | 3.28 × 104 |
| Square planar57 | 457 | 2B1g → 2B2g (dx2−y2 → dzy) | 3.02 × 104 |
| 657 | 2B1g → 2A1g (dx2−y2 → dz2) | 7.71 × 103 |
In the case of Co(II)-SALPHEN, a typical d–d transition at 458 nm corresponding to 4A2 + 4E → 4B1 was observed. The bands (319 and 387 nm) observed for the ligand shifted to 331 and 393 nm for Co(II)-SALPHEN, where the latter peaks are assigned to MLCT, 4A2 + 4E → 4E(P) and 4A2 + 4E → 4A2(P), respectively, which originated from the square pyramidal geometry of the Co(II) complex.70 Similar to the iron complex, the band at 294 nm corresponding to the n → π* transition originated from the coordinated water molecule present in the coordination sphere. The magnetic moment (μeff, 1.78 B.M.) determined for the Co(II) complex corresponds to one unpaired electron, suggesting its low spin and paramagnetic character.
The Cu(II) complex exhibited a n → π* transition at 287 nm and LMCT transition at 350 nm. The typical d–d absorption bands for Cu(II) in the square planar coordination were observed at 657 and 618 nm, which are generated from the 2B1g → 2B2g (dx2−y2 → dzy) 2B1g → 2A1g (dx2−y2 → dz2) transition, confirming its geometry with SALPHEN.57 Also, it was found to be paramagnetic with a magnetic moment, μeff, of 1.91 B.M., which is in good agreement with the spin-only value of the square planar complex.71,72 In addition, the corresponding molar extinction coefficients (ε) were determined and shown in Table 1. In general, the metal complexes exhibit higher molar extinction coefficients compared to SALPHEN. Among the studied metal complexes, Fe(III)-SALPHEN showed the highest molecular extinction coefficient at its maximum absorption values (2.04 × 105, 1.66 × 105, 1.57 × 105, and 1.21 × 105 M−1 cm−1), as well as broader and stronger UV-Vis absorption bands compared to other metal ions. However, Cu(II)-SALPHEN presented the maximum molar extinction coefficient of 1.32 × 105 M−1 cm−1 and the longest wavelength absorption band, reaching up to 657 nm. Furthermore, a very low molar conductance was calculated for all the complexes (Fe(III)-SALPHEN, 3.93; Co(II)-SALPHEN, 2.15; Cu(II)-SALPHEN, 5.44 Ω−1 cm2 mol−1 in DMF), which indicates the existence of neutral and non-electrolytic natures.
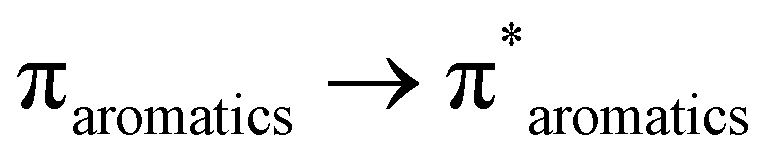 ) (250–350 nm), and above 250 nm is normally from the oxide ion (O−)
) (250–350 nm), and above 250 nm is normally from the oxide ion (O−) 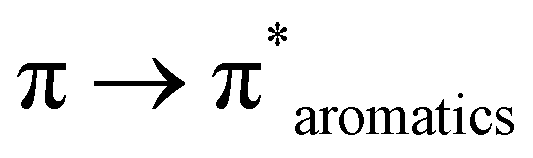 ;
; 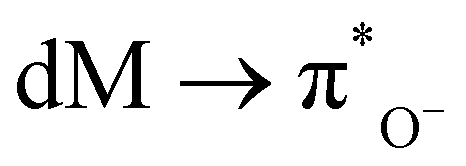 ;
; 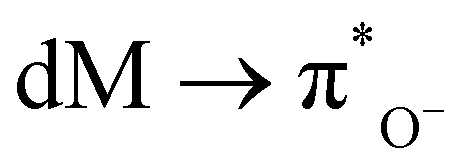 (MLCT); O− (π) → Md (LMCT); and also from some weak d–d transitions.73
(MLCT); O− (π) → Md (LMCT); and also from some weak d–d transitions.73
| Compounds | Ground state (GS) | Excited state (ES) |
|---|---|---|
| [Fe(SALPHEN)Cl(H2O)] | S = 1/2 | S = 3/2 |
| Doublet-zeta basis set | Wavelength (λ, nm) | |
| B3LYP/DGDZVP | 369, 420, 499, 1024 | 424, 556, 615, 766 |
| CAM-B3LYP/DGDZVP | 460, 530, 610, 746, 1090 | 468, 514, 645 |
| Triple-zeta basis set | ||
| B3LYP/6-311++g(d,p) | 495, 570, 698 | 537, 607, 738 |
| B3PW91/6-311++g(d,p) | 548, 608, 1056 | 541, 604, 746 |
| B3P86/6-311++g(d,p) | 494, 570, 709 | 520, 599, 761 |
| [Co(SALPHEN)(H2O)] | S = 1/2 | S = 3/2 |
| Doublet-zeta basis set | Wavelength (λ, nm) | |
| B3LYP/DGDZVP | 340, 385, 451 | 319, 342, 408, 493 |
| CAM-B3LYP/DGDZVP | 270, 340, 349 | 270, 291, 354 |
| Triple-zeta basis set | ||
| B3LYP/6-311++g(d,p) | 327, 386, 495 | 292, 339, 397 |
| B3PW91/6-311++g(d,p) | 318, 374, 518 | 304, 328, 377 |
| B3P86/6-311++g(d,p) | 326, 385, 504 | 300, 330, 387 |
| [Cu(SALPHEN)] | S = 1/2 | S = 3/2 |
| Doublet-zeta basis set | Wavelength (λ, nm) | |
| B3LYP/DGDZVP | 241, 289, 327, 439 | 404, 525 |
| CAM-B3LYP/DGDZVP | 268, 288, 377, 537 | 267, 294, 316, 366, 460, 691 |
| Triple-zeta basis set | ||
| B3LYP/6-311++g(d,p) | 297, 352, 408 | 377, 437, 527, 1013 |
| B3PW91/6-311++g(d,p) | 291, 352, 412, 439 | 340, 391, 402, 477, 802, 1312 |
| B3P86/6-311++g(d,p) | 291, 352, 406 | 378, 388, 473, 717, 1241 |
Nevertheless, there was an alteration in the band position for the different metal ions in [M-SALPHEN]n+ (n = 3, M = Fe; n = 2, M = Co and Cu), where the greater intensity of the higher-energy peaks originated from the metal–ligand charge transfer (MLCT). The following orbital electronic transitions were obtained in the ground state and in the excited state by employing the B3LYP/DGDZVP double-zeta basis set: for [Fe(SALPHEN)Cl(H2O)] in the ground state, its orbital transitions were HOMO−X to LUMO+Y (X = 0–6, 10–13; Y = 0 + 4); while in the excited state, they were (X = 0–12, Y = 0 + 5); similarly, for [Co(SALPHEN)(H2O)] in the ground state, its transitions of HOMO−X to LUMO+Y were (X = 0–7, 11; Y = 0 + 2, 4) and its excited-state transitions were (X = 0–7, 11; Y = 0 + 2); and for [Cu(SALPHEN)], its ground-state transitions were (X = 0–4, 6; 9, 14, 15, 21, Y = 0 + 2), its excited-state orbital transitions were (X = 0–4, 9–10, 13–14, 18, 21–22). After analyzing the above-mentioned transitions, the high energy band (413 nm) was assigned to the mixing of the metal orbital LUMO or LUMO+Y (Y = 0, 7) with the HOMOs of the donor (Nazo) in the excited state, and the peaks (997, 507, and 423 nm) for [Fe(SALPHEN)Cl(H2O)]; the signal (488 nm and 416 nm) for [Co(SALPHEN)(H2O)]; and (346 nm and 463 nm) for [Cu(SALPHEN)] (Fig. 3) confirm the coordination of nitrogen atoms with the metal ion.74
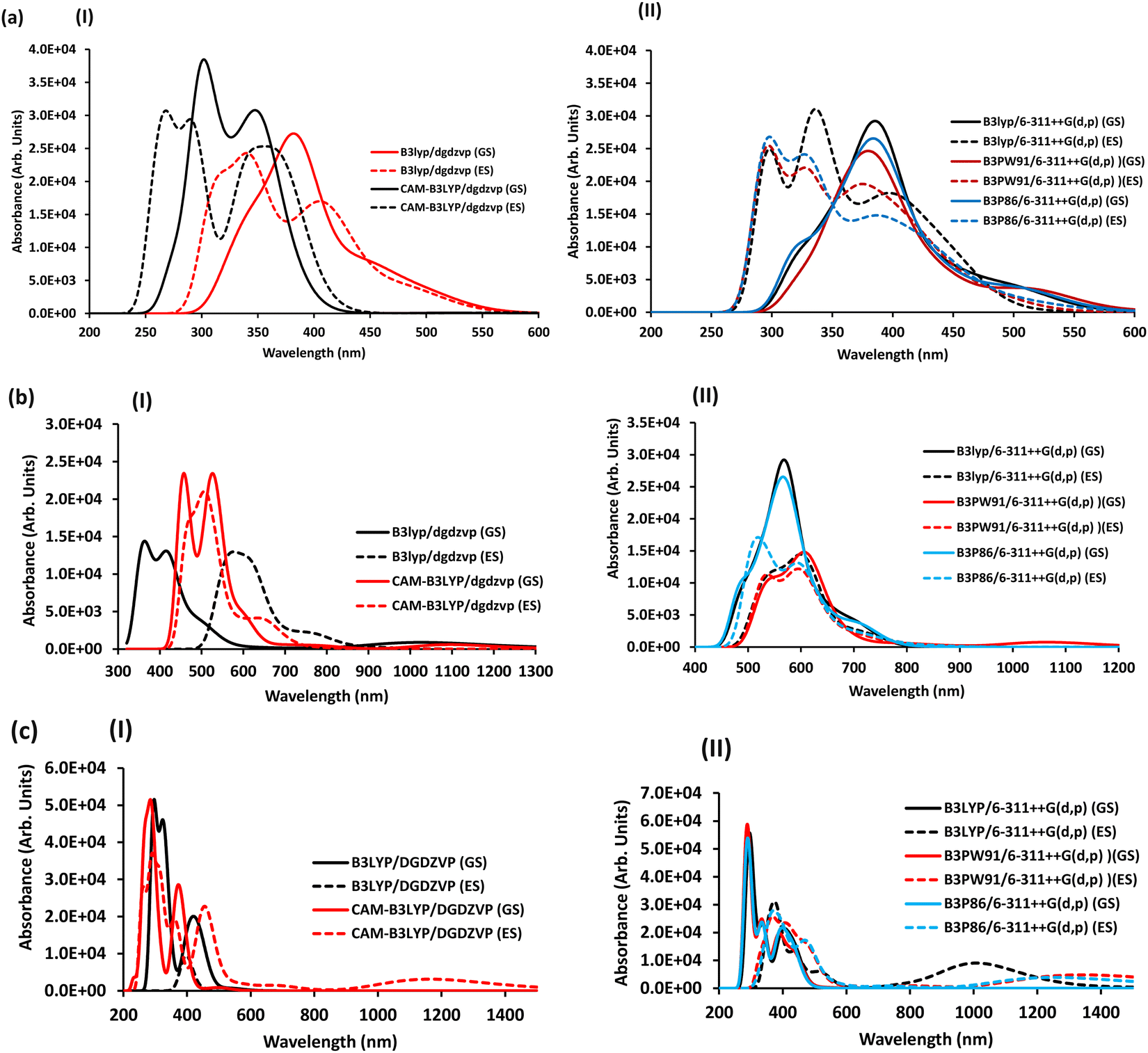 | ||
| Fig. 3 TD-DFT spectra in the gaseous state: (I) DGDZVP double-zeta basis set and (II) 6-311++G(d,p) triple-zeta basis set of [CoL1(H2O)] in the ground state (GS) and excited state (ES). (a) [Co(SALPHEN)(H2O)], (b) [Fe(SALPHEN)Cl(H2O)] and (c) [Cu(SALPHEN)]. | ||
Similarly, for the complexes, the electronic orbital transitions such as HOMO−X to LUMO+Y using the B3PW91/6-311++G(d,p) triple-zeta basis set were obtained as follows: for [Fe(SALPHEN)Cl(H2O)] in the ground state, the transitions were (X = 0–7, 11–12; Y = 0 + 4), and in the excited state, they were (X = 0–7, 12; Y = 0 + 5, 11); for [Co(SALPHEN)(H2O)], its ground-state transitions were (X = 0–6, 11; Y = 0 + 2, 4) and its excited-state transitions were (X = 0–6, 8; Y = 0 + 3, 5, 11); and finally, for [Cu(SALPHEN)], its transitions in the ground state were (X = 0–8, 10-11, 13, 20, Y = 0 + 4, 6 + 7) and its transitions in the excited state were (X = 0–5, 7–10, 12–13); Y = 0 + 3, 6, 8 + 13.
As observed in Fig. 3(a)-I, in the double zeta basis set DGDZVP with B3LYP and CAM-B3LYP without diffuse and polarized functions, there is a slight shift in the absorption peak in the spectra. For example, with B3LYP/DGDZVP in the ground state, for [Co(SALPHEN)(H2O)], three peaks (340, 385, and 451 nm) appeared in the ground state spectra are converted to four peaks (319, 342, 408, and 493 nm) in the excited state. The accurate displacement of peaks was observed using CAM-B3LYP/DGDZVP with a dispersion of correlated data. This indicates that better displacement of the bands (270, 340, and 349 nm) was obtained for [Co(SALPHEN)(H2O)] in the ground-state spectrum compared to its excited-state signals (270, 291, and 354 nm). However, Table 2 and Fig. 3(a)-II show that with the use of a triple-zeta basis set 6-311++G(d,p) with B3LYP, B3PW91, and B3P86 with diffuse and polarized functions, a clear shift in the excited-state electronic transitions was obtained compared to its ground-state peaks. For example, for [Co(SALPHEN)(H2O)], with B3PW91/6-311++G(d,p), the best displacement of its electronic transitions was seen in the ground-state peak (318 nm), which appears at 304 nm in the excited state; similarly, the other ground state peaks (374 nm and 518 nm) shifted to 328 nm and 377 nm, respectively, in the excited state (see Table 2 and Fig. 3(a)-II).
The above-mentioned methods were employed to optimize the geometry of [Fe(SALPHEN)Cl(H2O)] and [Cu(SALPHEN)]. After analyzing the results, in contrast to the above-mentioned [Co(SALPHEN)(H2O)] complex, a mixture of peaks was obtained for [Fe(SALPHEN)Cl(H2O)] in the red region of the spectrum, showing a small displacement with a low absorption intensity both in the ground state and in the excited state for both the double-zeta basis set and triple-zeta basis set. For example, for [Fe(SALPHEN)Cl(H2O)], with the triple zeta B3PW91/6-311++G(g,p) basis set, a slight spectral shift to the blue region was found, namely, the ground state peak (548 nm) shifted to 541 nm (towards the blue region) in its spectrum in the excited state. Similarly, the other GS peaks (608 nm and 1056 nm) shifted to 604 nm and 746 nm, respectively, in the ES. In the case of the double zeta basis set, the absorption bands (364 nm, 420 nm, and 499 nm in the ground state) shifted to 420 nm, 556 nm, and 615 nm in the excited state, respectively (in the red region of the electronic spectrum; see Fig. 3(b)(I) and (II).
Similar results were obtained for [Cu(SALPHEN)], where its GS peaks (291 nm, 352 nm, and 406 nm) shifted to 378 nm, 388 nm, and 473 nm in the excited state, respectively. Additionally, two new bands (717 and 1241 nm) were also observed in the excited state in the red region of its spectrum, as shown in Fig. 3(c)(I) and (II).
3.3 DFT optical properties
To understand the accuracy of the computational methods, especially TD-DFT, we employed four different functionals and analyzed the results comparatively for the complexes in the gaseous state. In particular, by combining the DGDZVP basis set (double zeta) with B3LYP and CAM-B3LYP or the 6-311++G(d,p) basis set (triple zeta) with B3PW91 and B3P86, reliable spectra can be obtained for the metal complexes,75,76 and the results are presented in Table 3. In the case of [Co(SALPHEN)(H2O)], [Fe(SALPHEN)Cl(H2O)], and [Cu(SALPHEN)], four different functionals (B3LYP, CAM-B3LYP, B3PW91 and B3P86) in combination with the DGDZVP (double-zeta) and 6-311++G(d,p) (triple-zeta) basis sets were applied in the calculations after considering the polarizability and diffuse functions. With the B3LYP and CAM-B3LYP having the DGDZVP (double-zeta) basis set, the results were almost constant, although there was a tendency for the energy to decrease from the ground state to the excited state. For instance, the root-mean square of deviation (RMSD) decreased from the GS to the ES for both functionals (3.92 × 10−9 (GS) to 2.91 × 10−9 (ES) for B3LYP/DGDZVP) and from 4.70 × 10−9 (GS) to 2.81 × 10−9 (ES) in the CAM-B3LYP/DGDZVP dispersion correction for [Co(SALPHEN)Cl(H2O)], although in the latter, the calculation was performed in a larger interval and the average energy increased from (6.0 to 69.0 kcal mol−1) from the GS to the ES for both B3LYP/DGDZVP and CAM-B3LYP/DGDZVP (dispersion correction) (see Fig. 4(a) and (b)), respectively. Similarly, the data were obtained by the combination of the triple-zeta basis set, 6-311++G(d,p), with B3PW9, B3LYP, and B3P86, showing that the root-mean square of deviation (RMSD) increased from the GS to the ES in the B3PW91 and B3P86/6-311++G(d,p) triple-zeta basis set (see Fig. 4(a)). Also, the relative energy in the range of 194 to 238 kcal mol−1 was observed in the ES (see Table 3).| Double-zeta basis set | RMSD (Debye) | Dipole moment (μ, Debye) | Polarizability anisotropy (Δα, Debye) | Relative energy values (kcal mol−1) | ||||
|---|---|---|---|---|---|---|---|---|
| [Co(SALPHEN)(H2O)] | (GS) | (ES) | (GS) | (ES) | (GS) | (ES) | (GS) | (ES) |
| B3LYP/dgdzvp | 3.92 × 10−9 | 2.91 × 10−9 | 3.318 × 10−18 | 4.967 × 10−18 | 2.44 × 10−18 | 3.51 × 10−18 | 0 | 6.3 a |
| 3.270 × 10−18 | 0.93 × 10−18 | 62.7 | ||||||
| CAM-B3LYP/dgdzvp | 4.70 × 10−9 | 4.80 × 10−9 | 7.388 × 10−18 | 3.563 × 10−18 | 5.11 × 10−18 | 2.61 × 10−18 | 0 | 6.3 a |
| 1.359 × 10−18 | 1.17 × 10−18 | 69.0 | ||||||
| Triple zeta basis set | ||||||||
| B3LYP/6-311++G(d,p) | 9.0 × 10−9 | 5.45 × 10−9 | 3.342 × 10−18 | 4.141 × 10−18 | 2.24 × 10−18 | 2.22 × 10−18 | 0 | 6.3 a |
| 56.5 | ||||||||
| B3PW91/6-311++G(d,p) | 6.66 × 10−9 | 5.48 × 10−9 | 3.241 × 10−18 | 2.804 × 10−18 | 2.72 × 10−18 | 1.67 × 10−18 | 0 | 3.3 |
| B3P86/6-311++G(d,p) | 8.48 × 10−9 | 3.14 × 10−9 | 3.229 × 10−18 | 3.385 × 10−18 | 2.40 × 10−18 | 1.95 × 10−18 | 0 | 194.5 a |
| 2.499 × 10−18 | 1.17 × 10−18 | 238.5 | ||||||
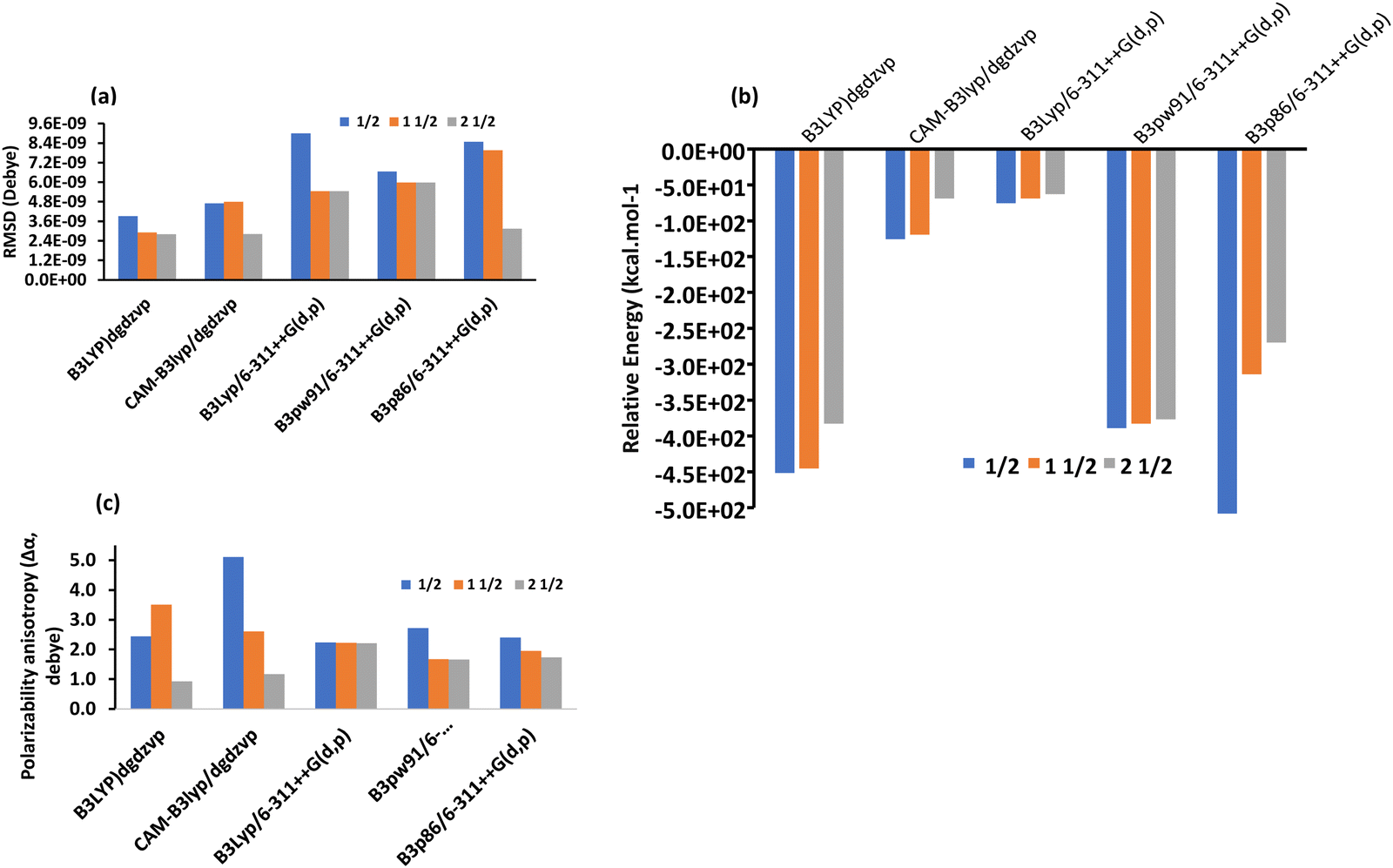 | ||
| Fig. 4 (a) Root-mean square of deviation (RMSD) value; (b) relative energy; and (c) mean polarizability (Δα) of [Co(SALPHEN)(H2O)] in the gaseous state. | ||
3.4. XRD analysis of the complexes
The powder XRD patterns of [Fe(SALPHEN)(OH2)Cl], [Co(SALPHEN)(OH2)], and [Cu(SALPHEN)] were recorded in the 2θ range of 10–80° using an X-ray diffractometer with Cu as the anode material, K-alpha [nm] = 0.154060, and the generator settings of 30 mA and 40 kV (Fig. 5). The results show well-defined crystalline peaks, suggesting that the metal-SALPHEN complexes exist in a crystalline phase. The Miller indices h, k, and l were calculated and refined using the Match software, and the lattice parameters including a, b, c, α, β, γ and V (volume) are presented in Table 4. The d values were obtained by Bragg's equation (nλ = 2dsinθ) using the values of sin2θ considering the h, k, and l data, and the correction of the d-values was carried out by comparing the observed density with that calculated using the X-ray powder diffractogram.77
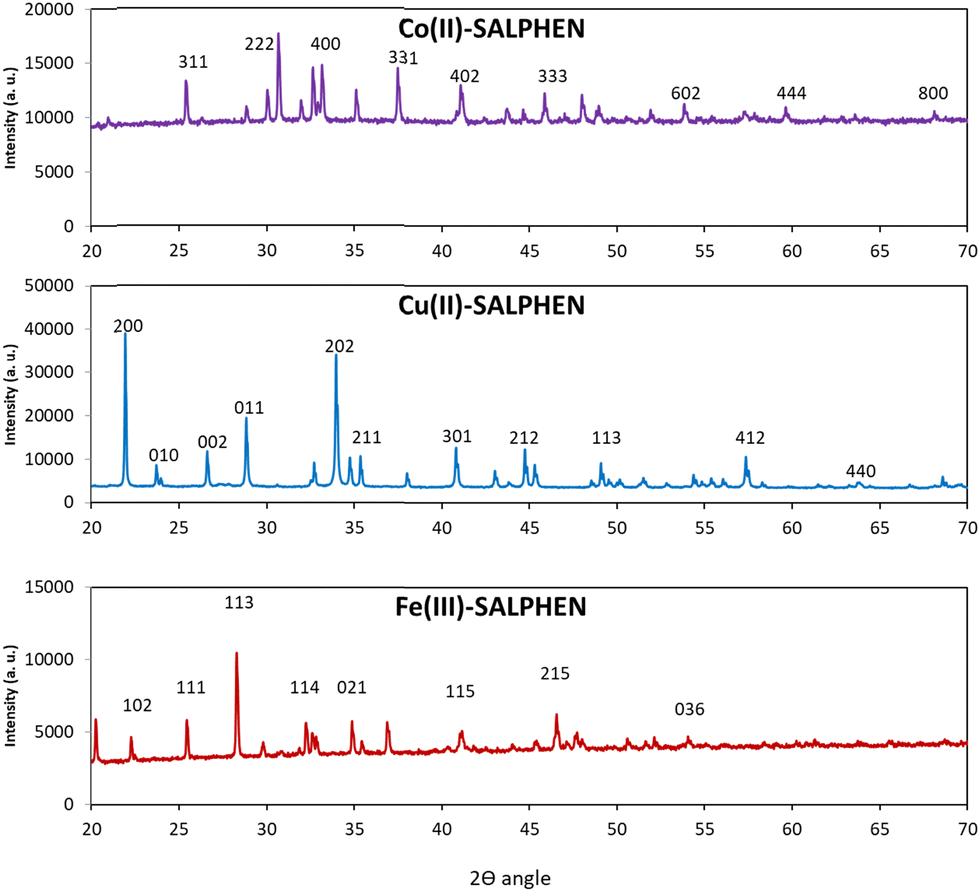 | ||
| Fig. 5 Powder XRD patterns of Fe3+, Co2+, and Cu2+ complexes with SALPHEN. | ||
| Lattice constants (Å) | Unit cell volume (Å3) | Crystal system | Space group | Angle (2θ) | d value (Å) | Density (g cm−3) | Axial angle (°) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | |||||||
| Co(II)-SALPHEN | 10.78 | 10.78 | 10.78 | 1252.73 | Cubic |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
41.0815 | 2.1945 | 2.500 | 90 | 90 | 90 |
| Cu(II)-SALPHEN | 8.104 | 3.757 | 7.433 | 226.311 | Orthorhombic | Pmna | 21.9183 | 4.0503 | 2.500 | 90 | 90 | 90 |
| Fe(III)-SALPHEN | 5.34 | 5.20 | 15.02 | 417.71 | Monoclinic | P1c1 | 34.87 | 2.57 | 2.028 | 90 | 90 | |
In the case of the Fe(III) complex, 23 reflections with the maximum at 2θ = 34.87° were observed, with the following unit cell parameters: a = 5.34 Å, b = 5.20 Å, c = 15.02 Å and α = 90°, β = 90°, γ = 90°. Given that the complex satisfies the conditions of a ≠ b ≠ c and α = γ = 90° β ≠ 90°, it considered to be monoclinic. Alternatively, for the Co(II) complex, 27 reflections with the maximum at 2θ = 41.08° were detected, satisfying the conditions of a = b = c and α = β = γ = 90° with unit cell parameters of a = b = c = 10.78 Å and α = β = γ = 90° for the cubic system. Similarly, for the Cu(II) complex, it showed 22 reflections with the maximum at 2θ = 21.91° and it obeyed the crystallographic rule of a ≠ b ≠ c and α = β = γ = 90° for an orthorhombic structure, with the unit cell parameters of a = 8.104 Å, b = 3.757 Å, c = 7.433 Å and α = 90°, β = 90°, γ = 90°.78–80 The increase in the number of reflections for all the metal complexes indicates that these structures possess a large specific surface area and provide good surface permeability, and thus the irradiation of light on their planes is expected to be greater compared to other complexes.81
According to the results, it was observed that for the Co(II) complex, there are several crystal planes (311, 222, 400, and 331, appearing in the 2θ range of 25° to 40°) with significant intensities; in contrast, for the Cu or Fe complexes in the same 2θ range, they had a lower number of crystal planes (002, 011, and 202 for the Cu(II) complex and 111 113, and 114 for the Fe(II) complex). In addition, the inter-plane distance (d value) was determined to be 2.19, 4.05, and 2.57 for the Co, Cu and Fe compounds, respectively, showing that the Co(II) complex possesses a comparatively lower value than the other complexes, which indicates that the array of crystal planes is near to each other, facilitating the greater absorption of light.82
3.5. Thermal analysis
Thermogravimetric studies were performed at a heating rate of 10 °C min−1 under an N2 atmosphere from 30 °C to 800 °C to determine the thermal stability and the change in mass with an increase in temperature. The TGA curves in Fig. 6(a) show the temperatures associated with thermal events occurring in SALPHEN and its metal complexes. According to the TGA curve of SALPHEN, it is observed that there is a clean and sharp decomposition with a weight loss of 65% from 248 °C to 409 °C, which can be attributed to the possible vaporisation of part of the SALPHEN molecule in this temperature range, while it also displayed 80% mass loss at 712 °C.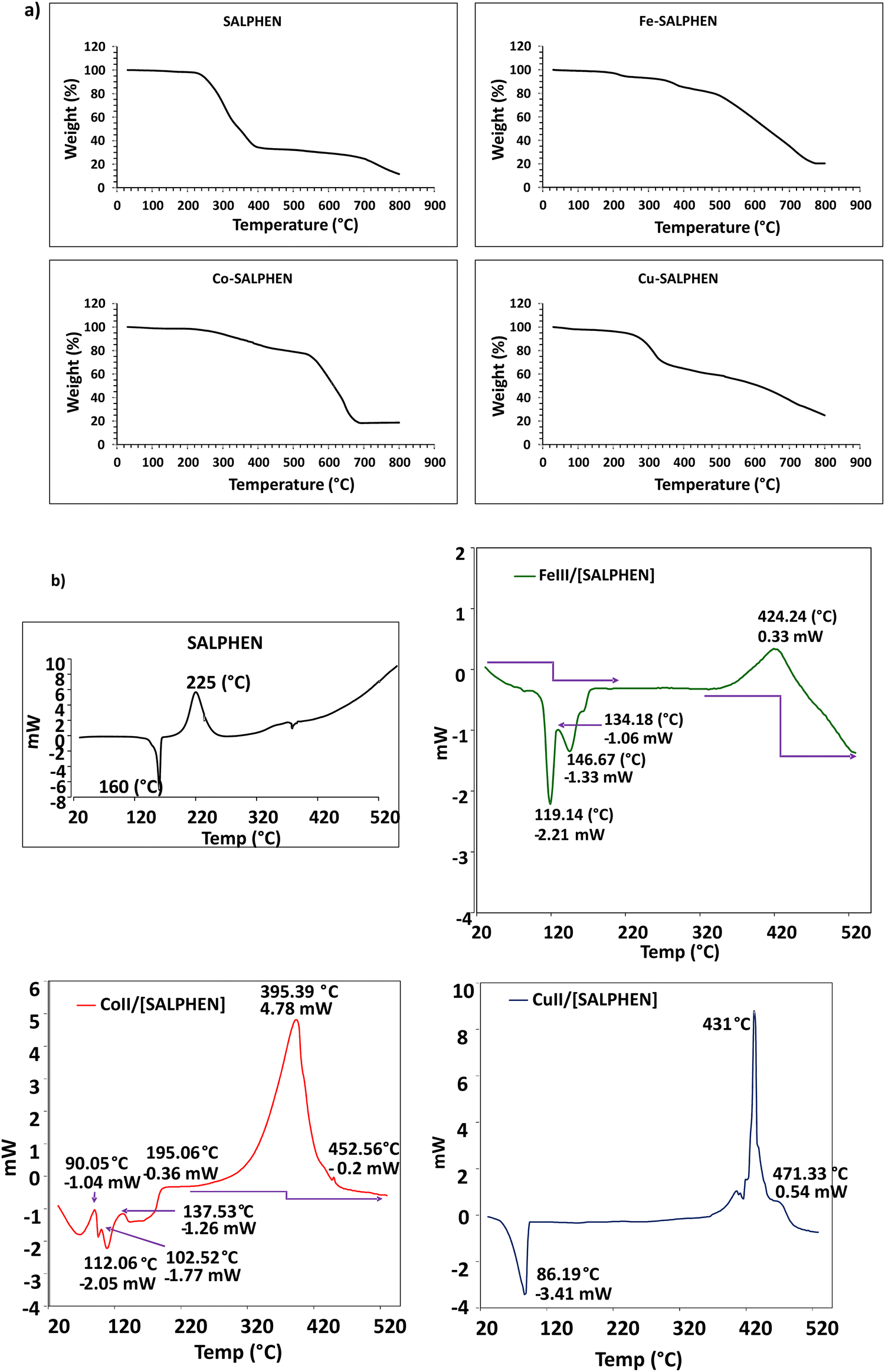 | ||
| Fig. 6 (a) TGA curves and (b) DSC curves of SALPHEN and its metal complexes. | ||
In general, the metal coordination increased the thermal stability of SALPHEN, suggesting that these metal complexes can be possible candidates for application in DSSCs. For example, the Co and Fe complexes remained stable up to 555 °C. However, a very small weight loss of approximately 3% was observed at around 250 °C for both complexes, which can be attributed to the evaporation of coordinated water molecules present in both complexes.83 The further mass loss of 23% (Fe-SALPHEN) and 30% (Co-SALPHEN) was observed at around 520 °C, which may be related to the decomposition of some of the fragments resulting from the complex at this temperature.72 In addition, Fe-SALPHEN showed a gradual decomposition between 520 °C and 730 °C, with the weight loss of 23% and 80%, respectively. In contrast, Co-SALPHEN presented a deep decomposition with 30% to 82% weight loss between 550 °C and 700 °C, respectively.
In contrast, Cu-SALPHEN only remained stable up to 285 °C; after that, it started to decompose with a rapid weight loss from 15% to 35% at 335 °C. This is attributed to the thermal degradation of Cu(II) complexes at higher temperatures,84,85 which also showed 75% weight loss at 798 °C.
According to the differential scanning calorimetry (DSC) results for SALPHEN and its metal complexes, as shown in Fig. 6(b), SALPHEN showed a glass transition peak at 133 °C and sharp endothermic peak at 160 °C; however, the presence of metal ions generally decreased the glass transition temperature, which was observed at 106 °C, 67 °C, and 53 °C for the Fe, Co, and Cu complexes, respectively. This is attributed to the decrease in the structural rigidity of the SALPHEN molecule in the presence of metal ions. Similarly, the metal ions expand the thermal-related phase changes in their crystal systems in comparison to the SALPHEN molecule, which can be observed as a decrease in the thermal hysteresis temperature peaks in the metal compounds, i.e., 119 °C, 112 °C, and 86 °C for Fe, Co, and Cu, respectively, whereas that for SALPHEN was at 160 °C, suggesting that these metal compounds can be used as phase change materials (PCM) for energy renewable applications.86 Alternatively, SALPHEN showed an exothermic melting temperature of 225 °C, whereas that of the Fe, Co, and Cu complexes was 424 °C, 395 °C, and 431 °C, respectively. This increased exothermic peak temperature is due to the increased crystalline nature of the metal complexes.87
3.6. Experimentally obtained photovoltaic parameters of the DSSC assembled using the metal complexes
The power conversion efficiency (η) of each DSSC was determined through their photovoltaic parameters such as short circuit current (Isc), open circuit voltage (Voc), and fill factor (FF). For this, the current–voltage capacities (I–V) obtained from the newly fabricated DSSCs were applied to explore their photovoltaic parameters, illustrating the I–V characteristics of the DSSC samples under 100 mW cm−2 light intensity. Fig. 7 and Table 5 show the photovoltaic parameters derived from the I–V curves to explore the performance of the solar cells.88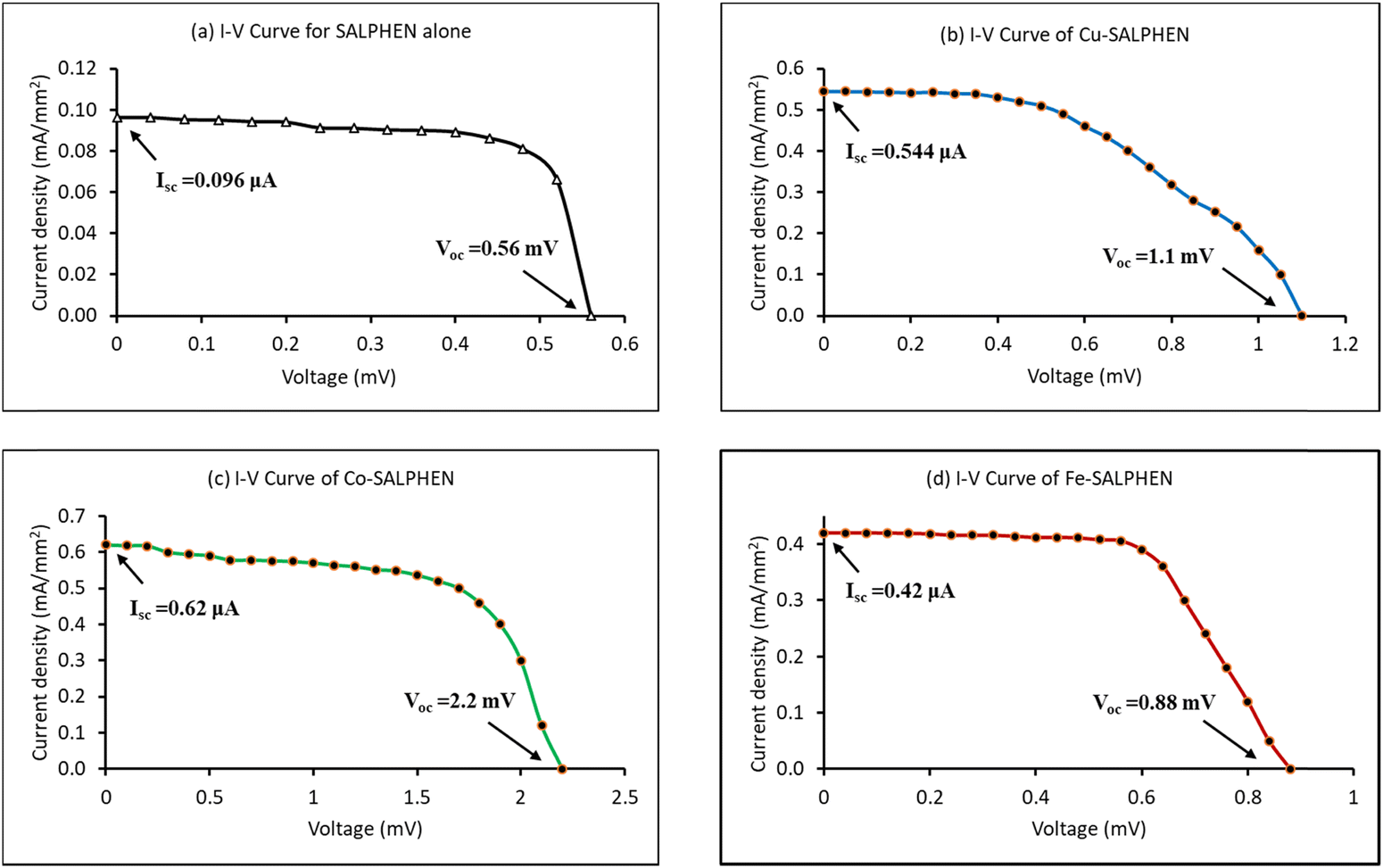 | ||
| Fig. 7 I–V curves of SALHEN and its metal complexes: (a) SALPHEN alone; (b) Cu-SALPHEN; (c) Co-SALPHEN; (d) Fe-SALPHEN. | ||
| V oc (mV) | I sc (μA) | FF (%) | η (%) | |
|---|---|---|---|---|
| where: Voc = open circuit voltage, Isc = short circuit current, FF = fill factor, η = efficiency of power conversion of DSSC. | ||||
| SALPHEN | 0.56 | 0.096 | 18.08 | 14.7 |
| [Cu(SALPHEN)(OH2)] | 1.1 | 0.544 | 11.78 | 13.3 |
| [Co(SALPHEN)(OH2)] | 2.2 | 0.620 | 15.58 | 46.2 |
| [Fe(SALPHEN)(OH2)Cl] | 0.88 | 0.420 | 15.82 | 15.0 |
The power conversion efficiency of the DSSCs was calculated using eqn (1), as follows:
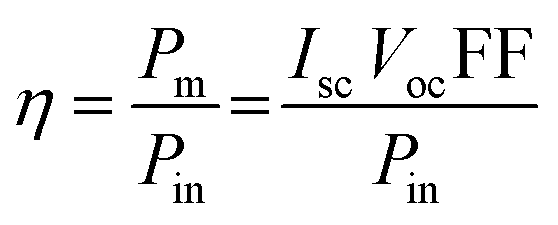 | (1) |
In addition, the fill factor, FF, is defined as the ratio of the maximum power output product (Pm) to the product of short circuit photocurrent and open circuit voltage, which can be derived from eqn (2), as follows:
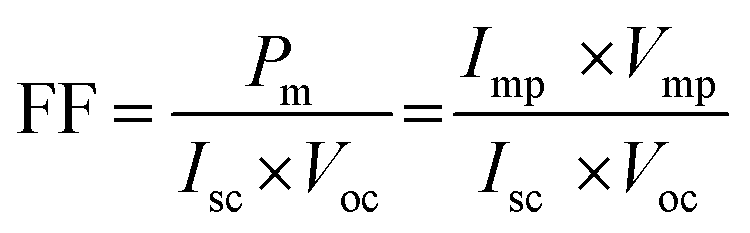 | (2) |
The data show that the I–V properties of M(SALPHEN) exhibited a significant change with a change in the metal ion of the complex, given that the nature of the metal ion coordination with SALPHEN differs, impacting the electron-transfer mechanism of the DSSC. Similarly, the metal coordination enhanced the power conversion efficiency (η) of SALPHEN, where the maximum (η) was obtained for the DSSC fabricated using the Co(II) complex (46.2%). In addition, to understand the power conversion efficiency and its corresponding electron transfer mechanism for the newly fabricated metal complex-based DSSCs, theoretical energy calculations of the various excited states of each metal complex were systematically performed by DFT.
3.7. DFT calculations
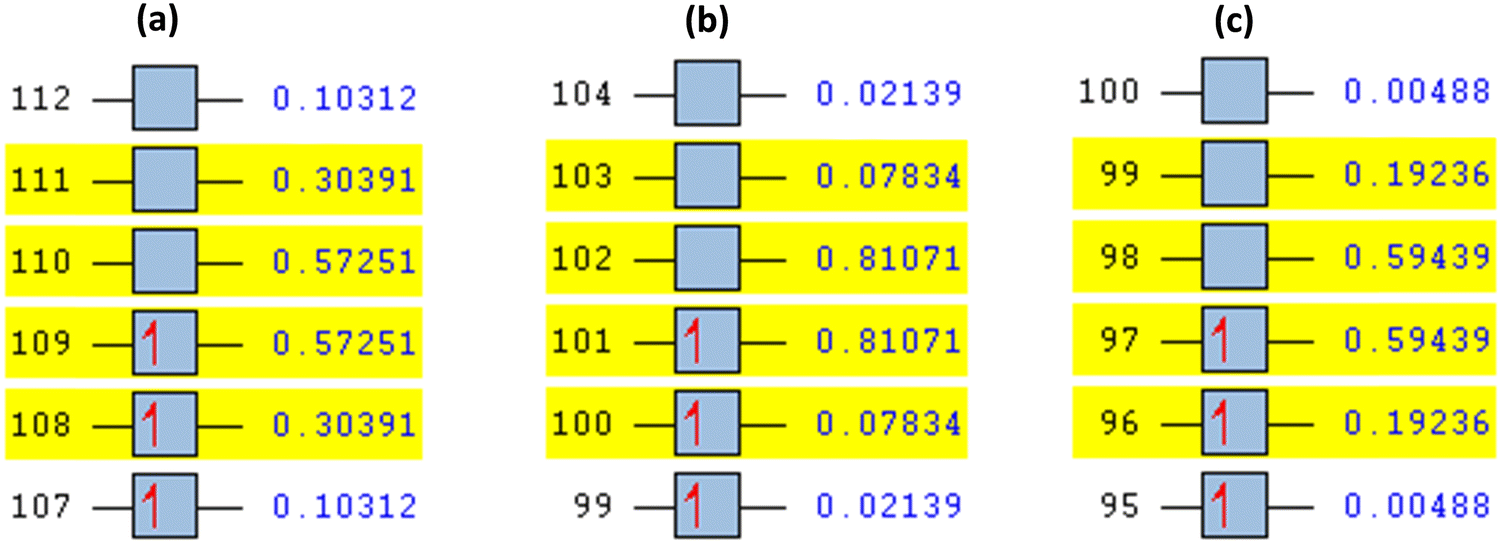 | ||
| Fig. 8 Natural transition orbitals (NTOs) derived from the B3LYP/DGDZVP basis set at S = 1/2, orbital 75 (particle), and orbital 76 (hole) for (a) [Fe(SALPHEN)Cl(H2O)], excited-state number 25, orbital 109 (particle), and orbital 110 (hole); (b) [Co(SALPHEN)(H2O)], excited-state number 20, orbital 101 (particle), and orbital 102 (hole); and (c) [Cu(SALPHEN)], excited-state number 35, orbital 97 (particle), and orbital 98 (hole). | ||
This was further explained by the NTOs of the canonical base (Fig. 9) of each complex, which illustrate the hole contribution with the delocalization from the LUMO over HOMO, showing that the hole NTO localization on the metal center and the electron NTO delocalization over the σ* orbital for Cl, π* for the H2O moiety, and σ and π orbitals for the SALPHEN. For example, the absorption maximum (413.17 nm) obtained for [Fe(SALPHEN)Cl(H2O)] gives NTO pairs related to the MLCT states (charge-transfer), suggesting that the excitation electron moves from the occupied orbitals (hole) to the unoccupied orbitals. Thus, the transition would be 109 (HOMO) and 110 (LUMO) for [Fe(SALPHEN)Cl(H2O)] at 0.57251 eV, and its orbital energy is 13.17 kcal mol−1 in the excited state. Similarly, [Co(SALPHEN)(H2O)] yields 101 (HOMO) and 102 (LUMO) at 0.81071 eV with an orbital energy of 18.65 kcal mol−1 in the excited state. [Cu(SALPHEN)], in the excited state (f = 0.6320, λ = 294.64 nm), the transition gives 97 (HOMO) and 98 (LUMO) at 0.59439 eV with an orbital energy of 13.67 kcal mol−1. The decreased HOMO–LUMO energy gap observed for the copper complex suggests its increased charge-transfer (CT) excitations and that it can be used in photovoltaic applications to achieve high solar power generation.
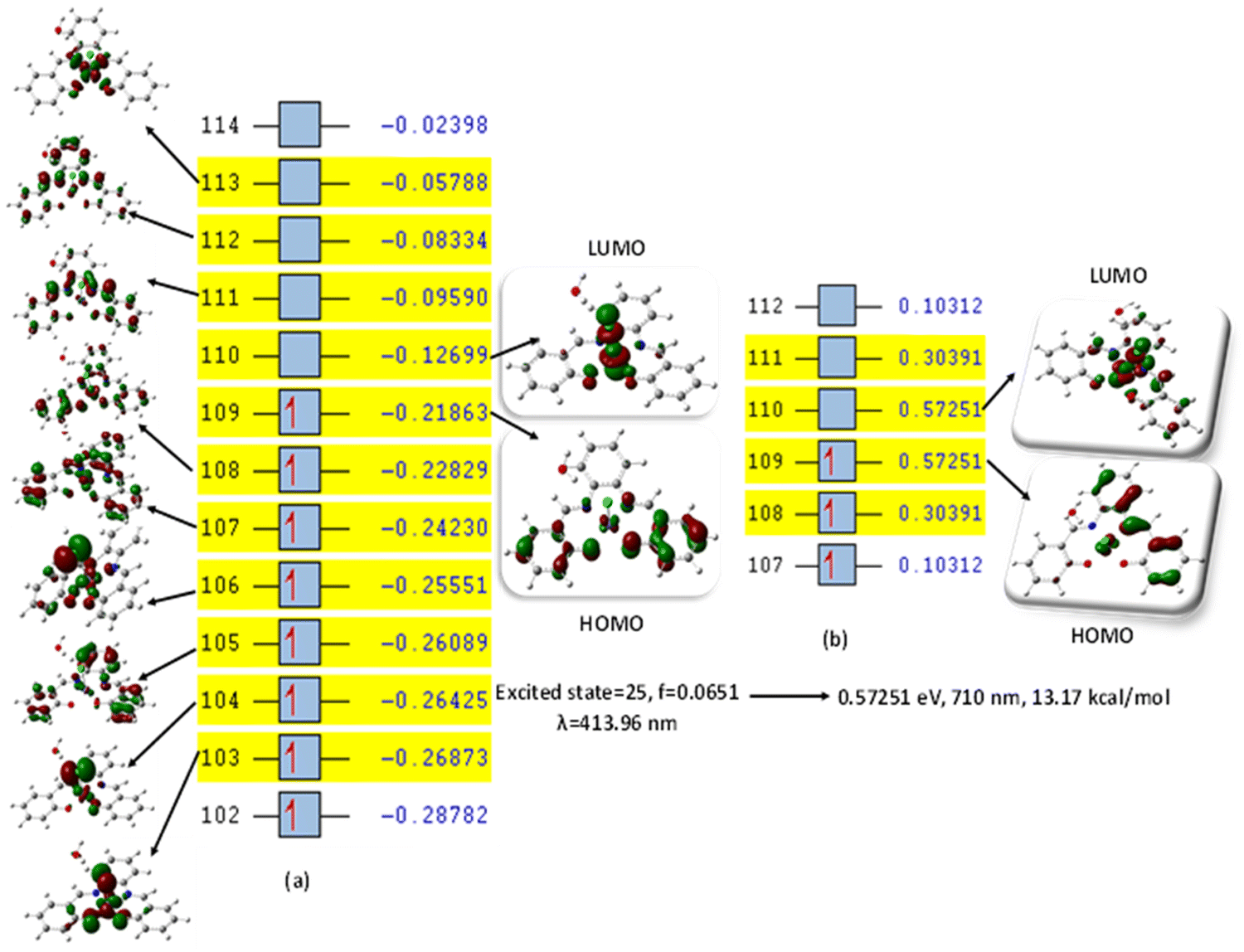 | ||
| Fig. 9 (a) Canonical orbitals involved in the 16th excited state according to TD-DFT. (b) NTOs for [Fe(SALPHEN)Cl(H2O)]. Orbital 109 (particle) and orbital 110 (hole) exhibit the largest occupations for excited state no. 25. [Fe(SALPHEN)Cl(H2O)] at a multiplicity of S = 1/2, determined with B3LYP/DGDZVP. | ||
Alternatively, to understand the light harvesting properties of the synthesized metal complexes, different excited-state levels were considered. After analyzing approximately 40 excited-state levels for [Fe(SALPHEN)Cl(H2O)], excited energy level 25 was chosen as the excited state given that it describes a relatively greater oscillator strength (f = 0.0651 and λ = 413.17 nm). Similarly, for [Co(SALPHEN)(H2O)] and [Cu(SALPHEN)], the NTOs resulted in the particle and hole at excited state level 20 for the former and level 35 for the latter complex (Fig. 10 and 11), respectively.
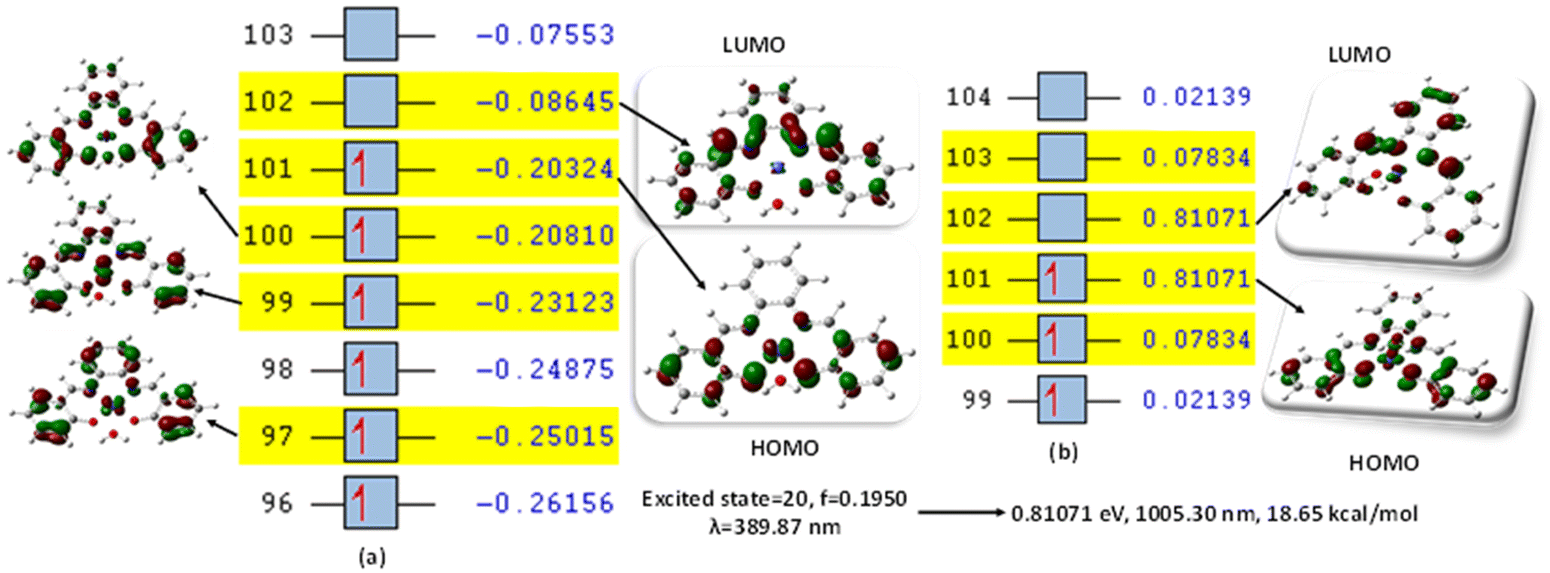 | ||
| Fig. 10 (a) Canonical orbitals involved in the 16th excited state according to TD-DFT. (b) NTO for [Co(SALPHEN)(H2O)]. Orbital 101 (particle) and orbital 102 (hole) exhibit the largest occupations for excited state no. 20. [Co(SALPHEN)(H2O)] at a multiplicity of S = 1/2, determined with B3LYP/DGDZVP. | ||
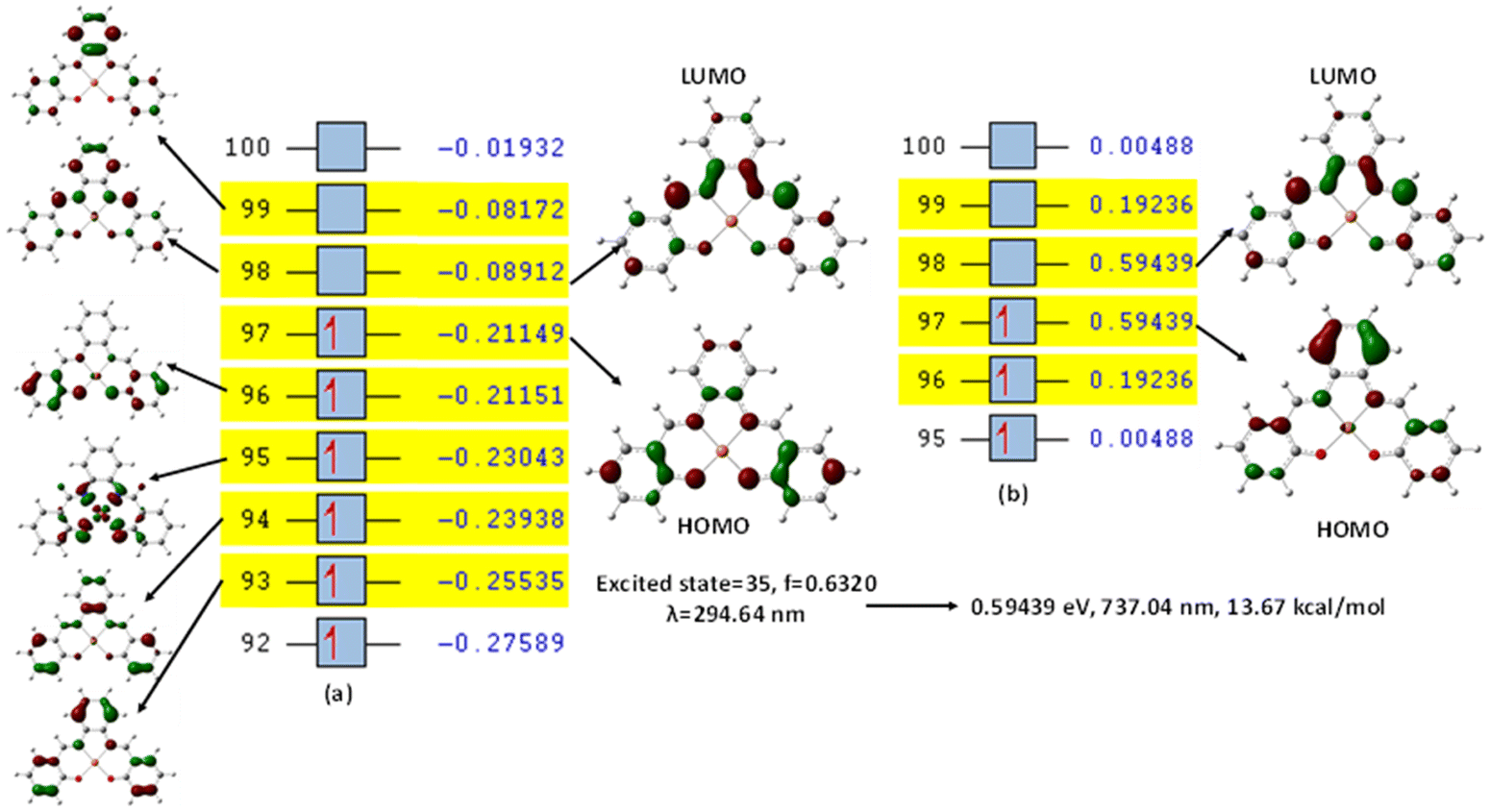 | ||
| Fig. 11 (a) Canonical orbitals involved in the 16th excited state according to TD-DFT. (b) NTO for [Cu(SALPHEN)]. Orbital 97 (particle) and orbital 98 (hole) exhibit the largest occupations for excited state no. 35. [Cu(SALPHEN)] at a multiplicity of S = 1/2, determined with B3LYP/DGDZVP. | ||
The hole contribution with the delocalization from LUMO over HOMO was identified for each complex, and it shows the hole NTO localization on the metal center and the electron NTO delocalization over the σ* orbital for Cl and π* for the H2O moiety and the ligand σ and π orbitals. The absorption maximum (413.17 nm) obtained for [Fe(SALPHEN)Cl(H2O)] gives NTO pairs related to the MLCT states (charge-transfer), indicating that the excitation electron moves from the occupied orbitals (hole) to the unoccupied orbitals. Thus, the transition would be 109 (HOMO) and 110 (LUMO) for [Fe(SALPHEN)Cl(H2O)] at 0.57251 eV, and its orbital energy is 13.17 kcal mol−1 in the excited state. Similarly, [Co(SALPHEN)(H2O)] yields 101 (HOMO) and 102 (LUMO) at 0.81071 eV with an orbital energy of 18.65 kcal mol−1 in the excited state. [Cu(SALPHEN)], in the excited state (f = 0.6320 and λ = 294.64 nm), the transition gives 97 (HOMO) and 98 (LUMO) at 0.59439 eV with an orbital energy of 13.67 kcal mol−1.
According to the CT excitation in the gas phase, the Co(II)-SALPHEN chromophore can be used in photovoltaic applications to achieve high solar power generation, which determines the efficiency of the photovoltaic device in the open-circuit voltage (Voc) related to the charge transfer excitation energy.
3.8 Molecular orbitals (MOs)
For the metal complexes, the binding nature of SALPHEN with the metal centers was analyzed by the MOs, where the frontier molecular orbitals (Fig. 12 and 13) show clear overlapping of the metal d orbital, i.e., [M(II), dx2−y2], with that of the p orbital from the ligands to result HOMOs in the ground and excited state compared to the trivalent metal ion. For example, in the case of [Co(II)(SALPHEN)(H2O)] in the ground state, overlapping of the MOs exists between HOMO to HOMO−X (X = 0–7, 11), while that for the excited state is HOMO−X (X = 0–8); for [Cu(II)(SALPHEN)] in the ground state, overlapping of the Mos was observed from HOMO to HOMO−X (X = 0–4, 9, 14, 15, and 21), while for the excited state, it was HOMO−X (X = 0–4, 9–10, 13–14, 18, 21–22). These interactions result in increased partial charge transfer the through metal complexes. Alternatively, for [Fe(SALPHEN)Cl(H2O)], less significant interaction of the d orbital (M3+) with π/p of the ligands as HOMO to HOMO−X, (X = 0–6, 10–13) was observed in the ground state, while that for the excited state was HOMO−X (X = 0–12). This is because the axial bond in M(III)–OH2 was observed in the Oh geometry (see Fig. 12 and 13; ESI,† Fig. S1–S6 and Tables S1–S16), confirming the involvement of a singly occupied orbital (SOMO) of the metal ion with the π* orbital (O−) in the intermediate-spin states, increasing the covalent nature of the bond through the distribution of electron density over Fe–O.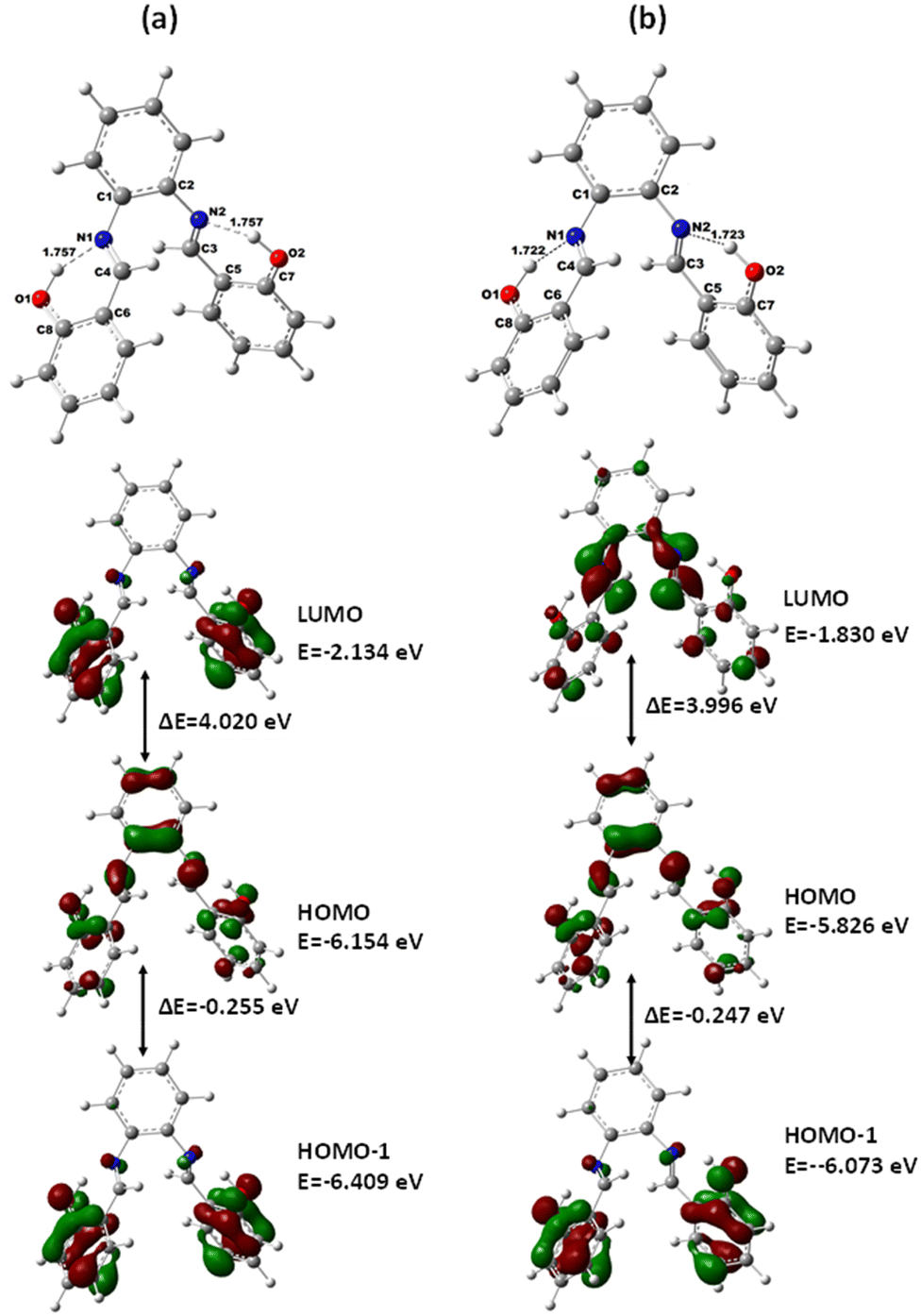 | ||
| Fig. 12 Optimized molecular structures by B3LYP functional: SALPHEN and its frontier molecular orbitals (HOMO and LUMOs) in the gaseous state (a) B3LYP/DGDZVP and (b) B3LYP/6-31G**. | ||
In addition, the observed lower energy unoccupied orbitals (LUMOs) show the overlapping of π-type [SALPHEN nitrogen and O− atoms] with d (metal ion) orbital, explaining the overlapping of dx2−y2 (metal ion) with the orbitals from O−. Therefore the excitations can be from HOMO−X to the unoccupied antibonding orbitals, i.e., π* (LUMO and LUMO+X).
According to the MOs, the decrease in the dx2−y2 energy was observed to follow the order of [Fe(SALPHEN)Cl(H2O)] (−2.63 eV) > [Cu(SALPHEN)] (−2.44 eV) > [Co(SALPHEN)(H2O)] (−2.37 eV) in the ground state (Fig. 13). Similarly, the dz2 energy decreases in the order of [Fe(SALPHEN)Cl(H2O)]2+ (−2.76 eV) > [Cu(SALPHEN)]2+ (−2.22 eV) > [Co(SALPHEN)(H2O)]2+ (−2.06 eV) (Fig. 13). Alternatively, in the excited state, the dx2−y2 dz2 energy decreased considerably for all the metal complexes, which was observed to follow the order of [Cu(SALPHEN)] (−4.50 eV) > [Fe(SALPHEN)Cl(H2O)] (−3.37 eV) > [Co(SALPHEN)(H2O)] (−2.11 eV) and [Cu(SALPHEN)] (−3.06 eV) > [Fe(SALPHEN)Cl(H2O)] (−2.64 eV) > [Co(SALPHEN)(H2O)] (−1.79 eV). In both the ground and the excited state, a decrease in the d orbital energy was observed in the cobalt complexes, suggesting that there is increased repulsion on the d orbitals, especially with the dx2−y2 energy. This allows the easier charge transfer (CT), which is consistent with the Jahn–Teller distortion because of the electrostatic axial interaction (O−) with the Co(II) ions (see ESI,† Fig. S7–S9).
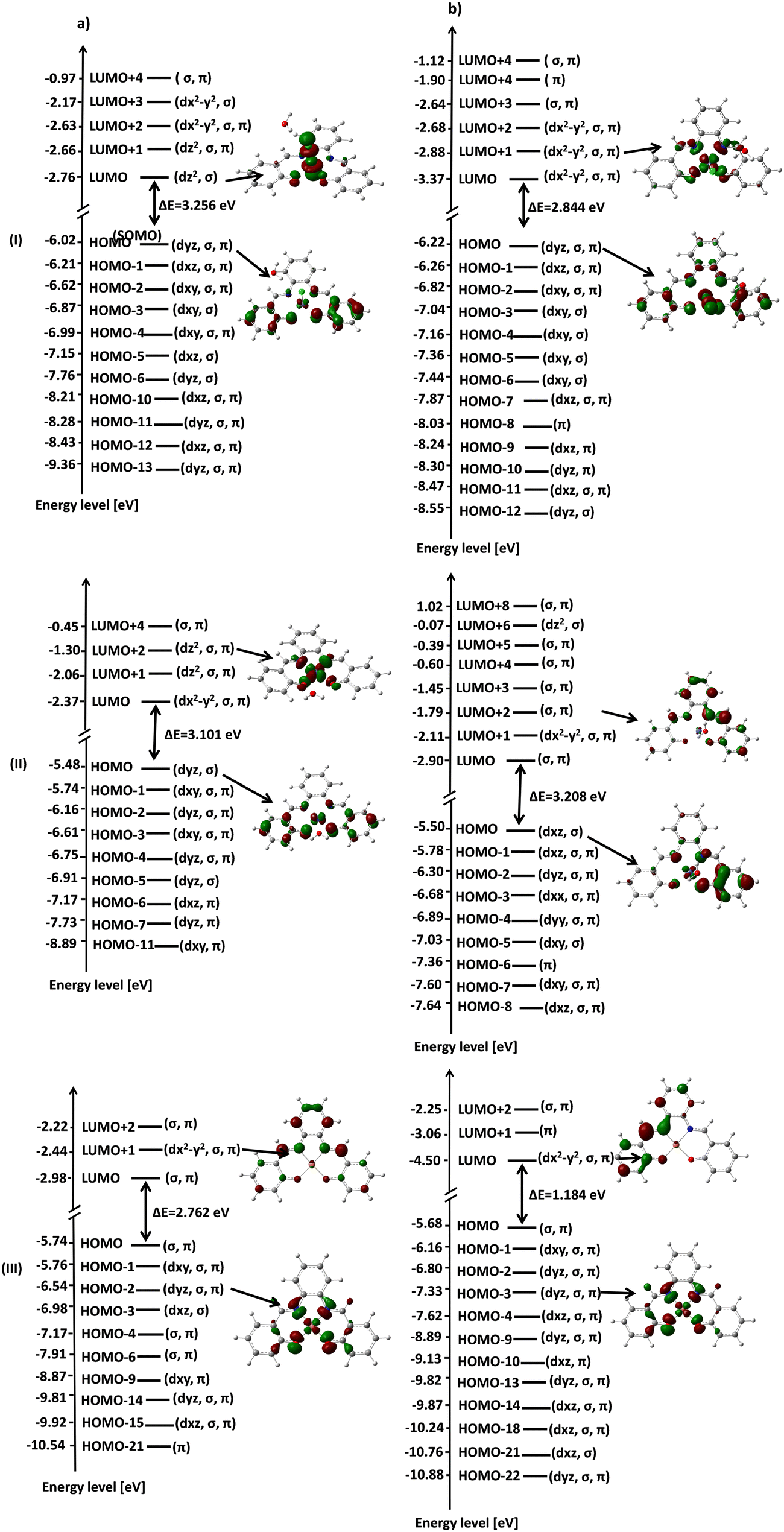 | ||
| Fig. 13 Frontier molecular orbital energy level diagram: (a) in the ground state and (b) in the excited state, low spin. (I) [Fe(SALPHEN)Cl(H2O)]; (II) [Co(SALPHEN)(H2O)]; and (III) [Cu(SALPHEN)]. | ||
In addition, the electron density isosurface of the HOMO and LUMO for all the complexes was analyzed at the B3LYP/DGDZVP level to understand the polarization and charge transfer contributions with the connectivity of atoms through the electron density movements, as shown in Fig. 14. According to the molecular graph for [Fe(III)(SALPHEN)Cl(H2O)], the contour electronic density over the molecular orbital surface shows the connectivity of the atoms (N1, N2, O1, O2, and axial Cl) with the metal ions forming a square pyramid base. A negative electron density for oxygen (−0.501), nitrogen (−0.345), and chloride (−0.282), and a positive electron density (0.623) for Fe(III) in Fe(III)–O in the excited state were detected. Similarly, for [Co(II)(SALPHEN)(H2O)], the electron density was (−0.818) for oxygen, (−0.400) for nitrogen, and (0.720) for Co(II) in Co(II)–O. In the case of [Cu(II)(SALPHEN)], the electron density of (−0.488) for oxygen, (−0.352) for nitrogen and (0.583) Cu(II) was observed for Cu(II)–O. The bonding of oxygen with the metal ions follows the order of Co–O > Cu–O > Fe–O and the negative electron density observed for Co–O suggests that the Co–O bond is so strong that it forms a coordination bond with the ligand. The maximum and minimum of the electron density indicate that there is an interaction between the metal (acceptor) and O or N (donor). According to the results, it is observed that the iron and copper complexes can participate in the higher charge transfer in the gaseous state.
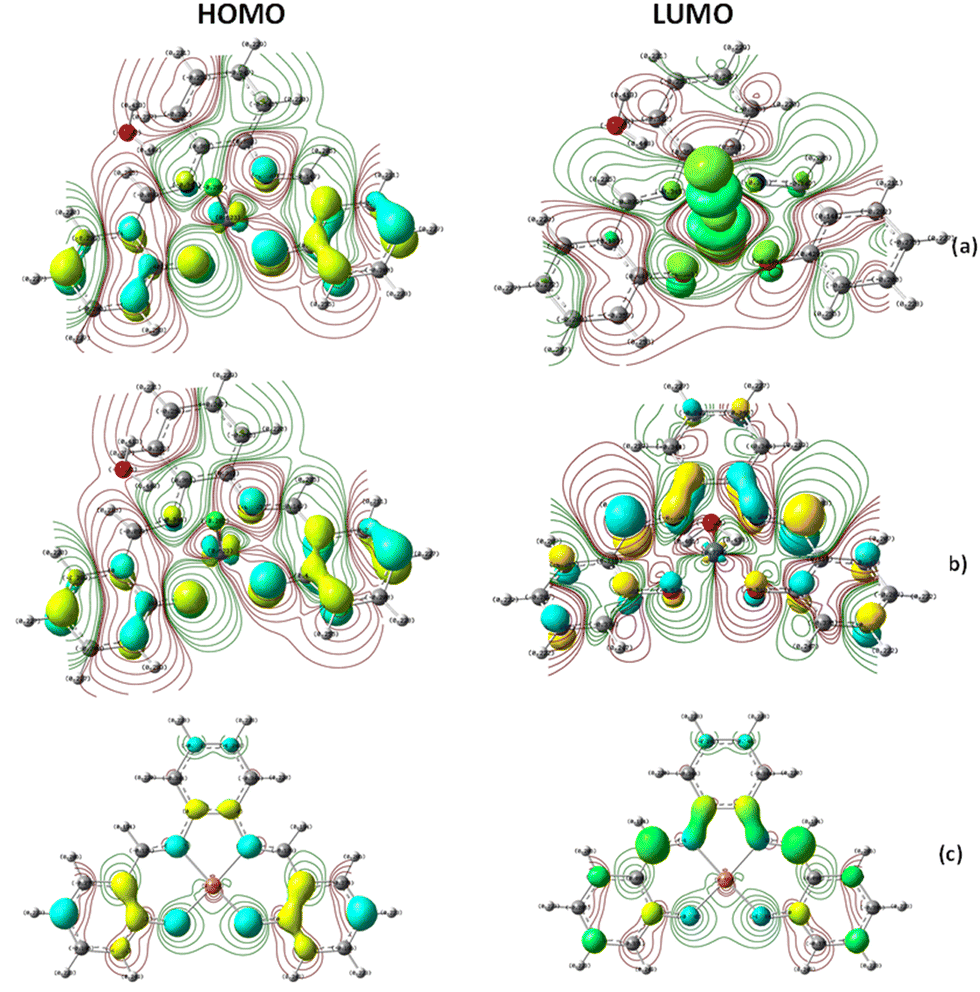 | ||
| Fig. 14 Electron density isosurface (contour 0.05 e Å−3), HOMO, and LUMO derived at the B3LYP/DGDZVP level. HOMO and LUMO contour plots (isosurface value = 0.05 a.u.) of the complexes: (a) [Fe(SALPHEN)Cl(H2O)], (b) [Co(SALPHEN)(H2O)], and (c) [Cu(SALPHEN)] in the gaseous state. Color code used for the structure of molecules: C (grey), H (white), N (blue), O (red) and metal ions (turquoise/blue for Fe(III) and Co(II); turquoise/red for Cu(II), respectively). | ||
3.9 Theoretical photovoltaic parameters
To explain both the ground-state and the excited-state behaviour in the electron transfer process during the solar cell performance, the energy level of the molecular orbitals of each complex was calculated using different functionals and basis sets, including CAM-B3LYP, B3PW91, B3P86 with DGDZVP, and 6-311++G(d,p), (Tables S7 and S8, ESI†), and used to determine the photovoltaic parameters theoretically, which were further compared with the experimentally obtained power conversion efficiency of each metal complex-based DSSC. The efficiency of a DSSC depends mainly on the electron transition between the excited state (LUMO) and the ground state (HOMO) orbitals of the photosensitizer. The obtained results show that the molecular orbitals of the metal complexes are involved in the electron transition; for example, the HOMO, HOMO−1, LUMO, and LUMO+1 energy levels are greatly involved in the process given that the position of the LUMO level of the sensitizer must be above the conduction band (CB) of the semiconductor (TiO2, −4.0 eV) and its HOMO level should be below the redox potential of the electrolyte used (I−/I3−, −4.8 eV). The above-mentioned levels allow charge injection from the sensitizer to conduction band of TiO2 and the regeneration of the sensitizer occurs by electrons from the electrolyte.81 According to the results obtained from the excited energy level calculations of these metal complexes, it was observed that these complexes are suitable candidates for application as the photosensitizer in the fabrication of DSSCs.Therefore, to determine the energy conversion efficiency of each solar cell device fabricated using SALPHEN and its metal complex sensitizers, the following equations were applied from the literature.89 For example, to determine theoretical open circuit voltage, the energy of each sensitizer molecule (ELUMO), which requires electron injection from the LUMO to the conduction band energy of TiO2, was used, as shown in the following equation:
| Voc = ELUMO − ECB | (3) |
Similarly, the short circuit current (Isc) for the DSSCs was determined theoretically using the following equation:90
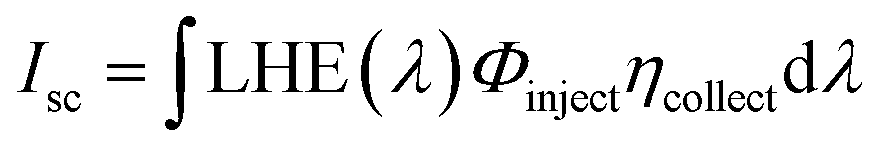 | (4) |
| LHE = 1 − 10−f | (5) |
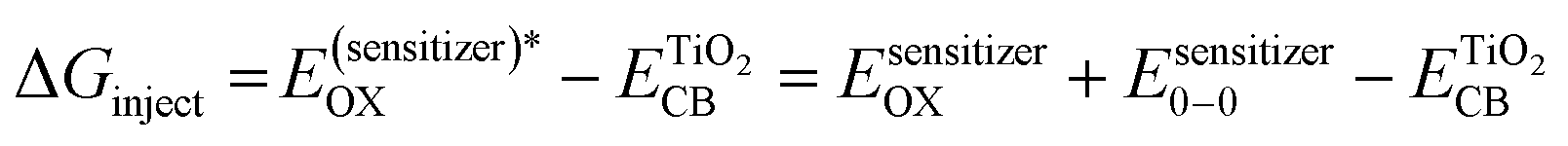 | (6) |
Thus, the theoretical power conversion efficiency of each of the synthesized complex-based DSSCs was calculated using the eqn (3)–(6) and summarized in Table 6.
| Compound | Spin | E HOMO (eV) | E LUMO (eV) | E 00 | E Sensitizer (eV) |
|
ΔGinject (eV) | ΔGreg (eV) | LHE | V oc (eV) | Oscillator strength (f) | Half-life (ns) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Functional/double-zeta basis set | |||||||||||||
| SALPHEN | B3LYP/DGDZVP | GS | −6.154 | −2.134 | 4.02 | 6.154 | 2.134 | 1.866 | 1.354 | 0.95984 | 1.866 | 0.4016 | 0.09855824 |
| B3LYP/6-31G** | GS | −5.826 | −1.83 | 3.996 | 5.826 | 1.83 | 2.17 | 1.026 | 0.97155 | 2.17 | 0.2845 | 0.15523098 | |
| Fe-SALPHEN | B3LYP/DGDZVP | GS | −6.02 | −2.764 | 3.256 | 6.02 | 2.764 | 1.236 | 1.22 | 0.99349 | 1.236 | 0.0651 | 0.6353714 |
| ES | −6.217 | −3.372 | 2.844 | 6.217 | 3.373 | 0.627 | 1.417 | 0.99435 | 0.628 | 0.0565 | 0.6864224 | ||
| Co-SALPHEN | GS | −5.476 | −2.375 | 3.101 | 5.476 | 2.375 | 1.625 | 0.676 | 0.9805 | 1.625 | 0.195 | 0.25635403 | |
| ES | −5.498 | −2.29 | 3.208 | 5.498 | 2.29 | 1.71 | 0.698 | 0.98259 | 1.71 | 0.1741 | 0.28483502 | ||
| Cu-SALPHEN | GS | −5.742 | −2.98 | 2.762 | 5.742 | 2.98 | 1.02 | 0.942 | 0.9368 | 1.02 | 0.632 | 0.07193797 | |
| ES | −5.683 | −4.499 | 1.184 | 5.683 | 4.499 | −0.499 | 0.883 | 0.97066 | −0.499 | 0.2934 | 0.15819262 | ||
| Functional/triple-zeta basis set | |||||||||||||
| SALPHEN | B3PW91/6-311++G(d,p) | GS | −6.305 | −2.307 | 3.998 | 6.305 | 2.307 | 1.693 | 1.505 | 0.97059 | 1.693 | 0.2941 | 0.12821429 |
| Fe-SALPHEN | GS | −6.066 | −2.739 | 3.327 | 6.066 | 2.739 | 1.261 | 1.266 | 0.99078 | 1.261 | 0.0922 | 0.44184088 | |
| ES | −6.314 | −3.198 | 3.116 | 6.314 | 3.198 | 0.802 | 1.514 | 0.99393 | 0.802 | 0.0607 | 0.61944648 | ||
| Co-SALPHEN | GS | −5.482 | −2.441 | 3.041 | 5.482 | 2.441 | 1.559 | 0.682 | 0.98548 | 1.559 | 0.1452 | 0.3435239 | |
| ES | −5.606 | −2.339 | 3.267 | 5.606 | 2.339 | 1.661 | 0.806 | 0.98635 | 1.661 | 0.1365 | 0.3494321 | ||
| [Cu(SALPHEN)]2+ | GS | −5.787 | −2.529 | 3.258 | 5.787 | 2.529 | 1.471 | 0.987 | 0.96657 | 1.471 | 0.3343 | 0.13389313 | |
| ES | −5.558 | −4.872 | 0.686 | 5.558 | 4.872 | −0.872 | 0.758 | 0.96887 | −0.872 | 0.3113 | 0.15587822 | ||
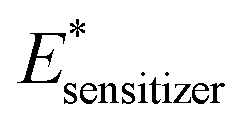
The calculated photovoltaic parameters of the metal complex photosensitizers show that their metal ions and corresponding electronic properties have a significant influence on the power conversion efficiency of the DSSCs. For example, ΔGinject, ΔGreg, LHE, and the excited-state lifetime (τ), which were used to determine the open circuit voltage (Voc) and short circuit current (Isc), have a direct influence on the effectiveness of the DSSC, showing a significant change upon metal coordination. As shown in Table 6, the metal ion generally increased the LHE and half-life time compared to SALPHEN alone, revealing the higher absorption ability of the photons by the metal complexes in the UV-visible region, which can assist in the higher injection of photo-excited electrons into the CB of TiO2. This observation is consistent with the experimental results (Table 5), where the metal complexes showed a higher power conversion efficiency than SALPHEN alone. Furthermore, the decrease in the ΔGinject value after the metal ion coordination suggests the easier injection of electrons into the CB of TiO2. Among the studied complexes, Cu(II)-SALPHEN showed the lowest ΔGinject due to its multi-valence response behavior, which supports its strongest metal to ligand electron transfer. In addition, the greater value of ΔGreg for Co(II)-SALPHEN and Fe(III)-SALPHEN indicates the longer life time of excited electrons in their CB, which can enhance the overall luminescence quantum yield of the DSSC.
In addition, the reactivity parameters such as chemical potential (μ), chemical hardness (η), softness (S), and electronegativity (χ) of the sensitizers (metal complexes) were calculated using EHOMO and ELUMO, which were used to explain the influence of the physicochemical properties on the electronic structure and PCE. The following equations (eqn (7)–(9)) were used and the data are shown in Table 7.
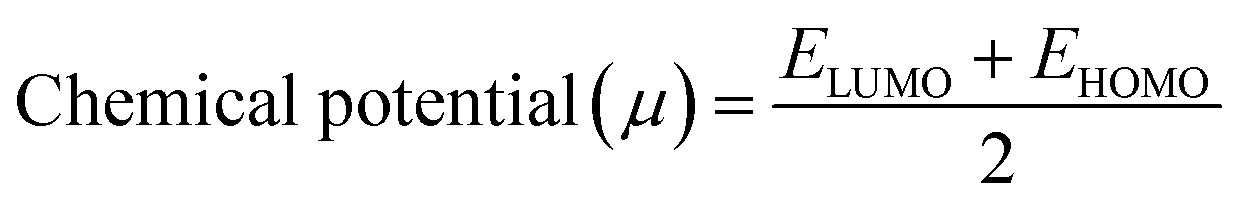 | (7) |
| Electronegativity (χ) = −μ | (8) |
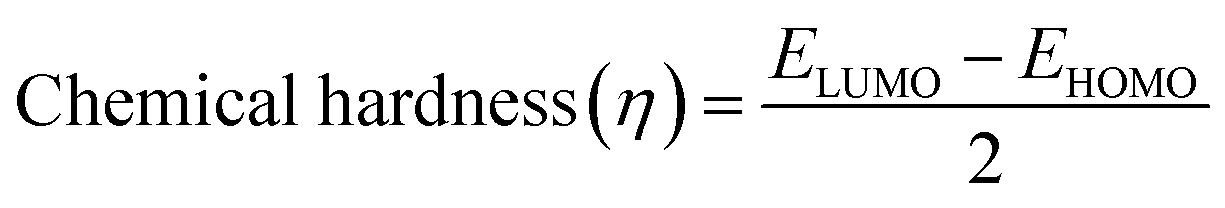 | (9) |
| Compounds | E HF | HOMO | LUMO | ΔE | Hardness (η) | Softness (σ) | Chemical potential (μ) | Electronegativity (χ) |
|---|---|---|---|---|---|---|---|---|
| B3LYP/dgdzvp (double-zeta basis set) | ||||||||
| SALPHEN | ||||||||
| −1031.81 | −6.154 | −2.134 | 4.020 | 2.010 | 0.467 | 4.14 | ||
| [Fe(SALPHEN)Cl(H2O)] | ||||||||
| GS | −2830.85 | −6.020 | −3.256 | 3.256 | 1.628 | 0.614 | −4.390 | 4.390 |
| ES | −2830.87 | −6.217 | −3.372 | 2.844 | 1.422 | 0.703 | −4.795 | 4.795 |
| [Co(SALPHEN)(H2O)] | ||||||||
| GS | −2489.72 | −5.476 | −2.375 | 3.101 | 1.551 | 0.645 | −4.360 | 4.360 |
| ES | −2489.61 | −5.498 | −2.290 | 3.208 | 1.604 | 0.624 | −3.894 | 3.894 |
| [Cu(SALPHEN)] | ||||||||
| GS | −2670.90 | −5.742 | −2.890 | 2.762 | 1.381 | 0.724 | −3.930 | 3.930 |
| ES | −2670.54 | −5.683 | −4.499 | 1.184 | 0.592 | 1.689 | −5.091 | 5.091 |
| B3LYP/6-311++G(d,p) (triple-zeta basis set) | ||||||||
| SALPHEN | ||||||||
| −1031.28 | −6.305 | −2.307 | 3.998 | 1.999 | 0.500 | −4.306 | 4.306 | |
| [Fe(SALPHEN)Cl(H2O)] | ||||||||
| GS | −2830.44 | −6.066 | −2.739 | 3.327 | 1.664 | 0.601 | −4.403 | 4.403 |
| ES | −2830.46 | −6.314 | −3.198 | 3.116 | 1.558 | 0.642 | −4.756 | 4.756 |
| [Co(SALPHEN)(H2O)] | ||||||||
| GS | −2489.62 | −5.482 | −2.441 | 3.041 | 1.521 | 0.658 | −3.962 | 3.962 |
| ES | −2489.61 | −5.606 | −2.339 | 3.267 | 1.633 | 0.612 | −3.973 | 3.973 |
| [CU(SALPHEN)] | ||||||||
| GS | −2670.62 | −5.787 | −2.529 | 3.258 | 1.629 | 0.614 | −4.158 | 4.158 |
| ES | −2670.54 | −5.558 | −4.872 | 0.686 | 0.343 | 2.914 | −5.215 | 5.215 |
The chemical reactivity parameters (Table 7) such as the chemical potential (μ) of the metal complex sensitizers have higher values than that of the TiO2 semiconductor (−4.66 eV),93 suggesting that the electron transfer from the sensitizer to the CB of TiO2 would be relatively straightforward. The chemical hardness (η) of all the compounds was lower than that of TiO2 (3.78 eV),94 indicating that all the sensitizers can release electrons compared to TiO2. Also, the electronegativity (χ) of the metal complexes is lower than that of TiO2 (−4.66 eV), illustrating that TiO2 may easily accept electrons from these sensitizers.
The influence of the excited-state electron lifetime for each sensitizer was determined using eqn (10), as follows:95
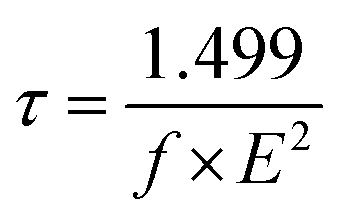 | (10) |
4. Conclusion
SALPHEN-based Schiff base metal complexes with Co(II), Cu(II), and Fe(III) ions were synthesized and characterized and the structural and electronic properties of the synthesized complexes were analyzed through experimental results and theoretical calculations using DFT. All the metal complexes presented high thermal stability, indicating their possible application as photosensitizers in solar cell devices. Further, dye-sensitized solar cells (DSSCs) were fabricated using the synthesized SALPHEN and its metal complexes, and their photovoltaic parameters such as short circuit current (Isc), open circuit voltage (Voc), and fill factor (FF) were recorded, and the I–V curves of each DSSC were used to determine its power conversion efficiencies (η). The results show that metal ion coordination generally enhanced the power conversion efficiency of SALPHEN due to the increased π-character by metal chelation. According to the results, co-sensitization with Co(II)-SALPHEN was beneficial for a higher power conversion efficiency compared to the other complexes. In the metal complexes, the amino group from the ligand acts as an anchoring group and facilitates the easy transfer of electrons from the LUMO of SALPHEN to the conduction band of the TiO2 layer, and thus the metal complexes showed an increase in efficiency of 13.3%, 42.2%, and 15%, respectively, compared to SALPHEN (12%). Among the metal complexes, Co(II)-SALPHEN showed the greatest improvement in power conversion efficiency due to the increased photo-induced electron transfer. In addition, according to the DFT calculations, it was observed that the metal complexes showed a greater d orbital overlapping through the LUMO of the amino group from the ligand, which acts as an anchoring group, facilitating the easy transfer of electrons to the conduction band of the TiO2 layer. The obtained HOMO–LUMO gap for the complexes using the B3LYP, CAM-B3LYP functionals with the DGDZVP double-zeta basis set and B3LYP, B3PW91, and B3P86/6-311++(d,p) triple-zeta basis set showed a decrease, following the order of Co(II) > Fe(III) > Cu(II) in the excited state, suggesting that the Co(II)-complex is a potential sensitizer for DSSCs.Abbreviations
| SALPHEN | N,N-Bis(salicylimine)-o-phenyldiammine |
| DSSC | Dye-sensitized solar cell |
| η | Power conversion efficiency |
| MLCT | Metal-to-ligand charge transfer |
| LMCT | Ligand-to-metal charge transfer |
| MC | Metal-centered |
| TGA | Thermogravimetric analysis |
| FF | Fill factor |
| DFT | Density functional theory |
| TD-DFT | Time-dependent density functional theory |
| D | Electron donor |
| A | Electron acceptor |
| NIR | Near-infrared region |
| PCE | Power conversion efficiency |
| ICT | Intramolecular charge transfer |
| LHE | Light-harvesting efficiency |
| V oc | Open circuit voltage |
| ΔGinject | Driving force |
| HOMO | Highly occupied molecular orbital |
| LUMO | Low unoccupied molecular orbital |
| μ eff | Effective magnetic moments |
| DSC | Differential scanning calorimetry |
| FTO | Fluorine-doped tin oxide |
| CB | Conduction band |
| B3LYP | Hybrid functionals Becke, 3-parameter, Lee–Yang–Parr. |
| CAM-B3LYP | Coulomb attenuating method-hybrid functionals Becke, 3-parameter, Lee–Yang–Parr |
| B3PW91 | Exchange component of Perdew and Wang's 1991 functional |
| B3P86 | Hybrid functionals Becke, 3-parameter Perdew 86. |
| FF | Fill factor |
| Φ inject | Electron injection efficiency |
| η collect | Charge collection efficiency |
| ΔGinject | Driving force |
| RMSD | Root-mean square of deviation |
| NBO | Natural bond orbital |
| NTO | Natural transition orbitals |
| f | Oscillator strength |
| τ | Electron lifetime |
| GS | Ground state |
| ES | Excited state |
| μ | Dipole moment |
| Δα | Polarizability anisotropy |
| PCE | Power conversion efficiency |
| NIR | Near-infrared region |
| ICT | Intramolecular charge transfer |
| TGA | Thermogravimetric analysis |
| DSC | Differential scanning calorimetry |
| ε | Molar extinction coefficient |
| λ | Wavelength (nm) |
| ns | Half life |
| μ | Chemical potential |
| χ | Electronegativity |
| η | Chemical hardness |
| Cz-D2 dyes | 4,4′-(9-Octyl-9H-carbazole-3,6-diyl)bis(2,3-difluoro benzaldehyde) [Cz-2PhF2-CHO] |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Universidad Nacional Autónoma de México, Dirección General de Asuntos del Personal Académico, (Folio: IN202622) for the financial support. Also, acknowledge DGTIC-UNAM (Dirección General de Cómputo y de Tecnologías de Información y Comunicación, Universidad Nacional Autónoma de México) for the computational facilities and USAI (Unidad de Servicios de Apoyo a la Investigación, Facultad de Química, UNAM) for the analytical services. Eduardo Daniel Tecuapa Flores acknowledges COMECYT for the fellowship through the program “Investigadoras e Investigadores COMECYT EDOMÉX 2023” (FOLIO: ESYCA2023-11037). Jayanthi N acknowledge COMECYT (Consejo Mexiquense de Ciencia y Tecnología), México, for the financial support through “Financiamiento para Investigación de Mujeres Científica EDOMEX-FICDTEM-2021-01” (Folio: FICDTEM-2021-047) and Feria de Ciencias e Ingenierías de Estado de México, (Folio: 5385-2018).References
- J. Zhang, L. Xu and W.-Y. Wong, Energy materials based on metal Schiff base complexes, Coord. Chem. Rev., 2018, 355, 180–198, DOI:10.1016/j.ccr.2017.08.007.
- A. Tsaturyan, Y. Machida, I. Gozhikova and I. Shcherbakov, Binaphthyl-containing Schiff base complexes with carboxyl groups for dye sensitized solar cell: An experimental and theoretical study, J. Mol. Struct., 2018, 1162, 54–62, DOI:10.1016/j.molstruc.2018.02.082.
- A. Gencer Imer, R. H. B. Syan, M. Gülcan, Y. S. Ocak and A. tombak, The novel pyridine based symmetrical Schiff base ligand and its transition metal complexes: synthesis, spectral definitions and application in dye sensitized solar cells (DSSCs), J. Mater. Sci. Mater. Electron., 2018, 29, 898–905, DOI:10.1007/s10854-017-7986-z.
- P. S. Gangadhar, A. Jagadeesh, M. N. George, S. Prasanthkumar, S. Soman and L. Giribabu, Role of π-spacer in regulating the photovoltaic performance of copper electrolyte dye-sensitized solar cells using triphenylimidazole dyes, Mater. Adv., 2022, 3, 1231–1239, 10.1039/D1MA00852H.
- L. Zhang, J. M. Waddell, K. S. Low and X. Liu, Relating Electron Donor and Carboxylic Acid Anchoring Substitution Effects in Azo Dyes to Dye-Sensitized Solar Cell Performance, ACS Sustainable Chem. Eng., 2013, 1, 1440–1452, DOI:10.1021/sc400183t.
- W. Liu, S. H. Li, Y. C. Li, Y. Z. Yang, Y. Yu and S. P. Pang, Nitrogen-rich salts based on polyamino substituted N,N′-azo-1,2,4-triazole: a new family of high-performance energetic materials, J. Mater. Chem. A, 2014, 2, 15978–15986, 10.1039/C4TA03016H.
- M. M. Raikwar, K. K. Sonigara, D. S. Patil, H. Machhi, S. S. Soni and N. Sekar, Biphenyl-Amine-Based D–π–A′–π–A Sensitizers for DSSCs: Comparative Photo-Conversion Efficiency in Iodide/triiodide and Cobalt-Based Redox Electrolyte and DFT Study, ChemistrySelect, 2019, 4(24), 7371–7379 CrossRef CAS.
- N. Sinha and O. S. Wenger, Photoactive Metal-to-Ligand Charge Transfer Excited States in 3d6 Complexes with Cr0, MnI, FeII, and CoIII, J. Am. Chem. Soc., 2023, 145, 4903–4920, DOI:10.1021/jacs.2c13432.
- J. Lv, X. Wu, R. Wang, Y. Wu, S. Xu, F. Zhao and Y. Wang, Schiff base-type Cu(I) complexes containing naphthylpyridyl-methanimine ligands featuring higher light-absorption capability: Synthesis, structures, and photophysical properties, Polyhedron, 2022, 224, 116002, DOI:10.1016/j.poly.2022.116002.
- P. Mahadevi and S. Sumathi, Mini review on the performance of Schiff base and their metal complexes as photosensitizers in dye-sensitized solar cells, Synth. Commun., 2020, 50, 2237–2249, DOI:10.1080/00397911.2020.1748200.
- C. Gautam, D. Srivastava, G. Kociok-Kohn, S. W. Gosavi, V. K. Sharma, R. Chauthan and M. Muddassir, Copper(II) and cobalt(III) Schiff base complexes with hydroxy anchors as sensitizers in dye-sensitized solar cells (DSSCs), RSC Adv., 2023, 13, 9046–9054, 10.1039/D3RA00344B.
- Q. Gu, F. Chotard, J. Eng, A. P. M. Reponen, I. J. Vitorica-Yrezabal, A. W. Woodward and A. S. Romanov, Excited-State Lifetime Modulation by Twisted and Tilted Molecular Design in Carbene-Metal-Amide Photoemitters, Chem. Mater., 2022, 34, 7526–7542, DOI:10.1021/acs.chemmater.2c01938.
- A. Azaid, M. Raftani, M. Alaqarbeh, R. Kacimi, T. Abram, Y. Khaddam, D. Nebbch, A. Sbai, T. Lakhlifi and M. Bouchrine, New organic dye-sensitized solar cells based on the D–A–π–A structure for efficient DSSCs: DFT/TD-DFT investigations, RSC Adv., 2022, 12, 30626–30638, 10.1039/D2RA05297K.
- M. A. K. L. Dissanayake, K. Umair, G. K. R. Sanadeera and J. M. K. W. Kumari, Effect of electrolyte conductivity, co-additives and mixed cation iodide salts on efficiency enhancement in dye sensitized solar cells with acetonitrile-free electrolyte, J. Photochem. Photobiol., A, 2021, 415, 113308, DOI:10.1016/j.jphotochem.2021.113308.
- D. A. Chalkias, C. Charalampopoulos, A. K. Andreopoulou, A. Karavioti and E. Stathatos, Spectral engineering of semi-transparent dye-sensitized solar cells using new triphenylamine-based dyes and an iodine-free electrolyte for greenhouse-oriented applications, J. Power Sources, 2021, 496, 229842, DOI:10.1016/j.jpowsour.2021.229842.
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Gratzel and W. Zhu, High-conversion-efficiency organic dye-sensitized solar cells: molecular engineering on D–A–π-A featured organic indoline dyes, Energy Environ. Sci., 2012, 5, 8261–8272, 10.1039/C2EE22108J.
- M. Shahin, A. M. Alazaly, M. A. Ismail and A. A. Abdel-Shafi., Effect of the π-bridge structure on the intramolecular charge transfer of push–pull 2-phenylthiophene and 2-(furan-2-yl)pyridine derivatives, J. Mol. Liq., 2023, 378, 121624, DOI:10.1016/j.molliq.2023.121624.
- M. Cao, L. Wang, H. Gao, H. Jiang and H. Song, Intrinsic influence of selenium substitution in thiophene and benzo-2,1,3-thiadiazole on the electronic structure, excited states and photovoltaic performances evaluated using theoretical calculations, New J. Chem., 2023, 47, 1797–1807, 10.1039/D2NJ04490K.
- K. Basuroy, J. D. J. Velazquez-Garcia, D. Storozhuk, R. Henning, D. J. Gosztola, S. Thekku Veedu and S. Techert, Axial vs equatorial: Capturing the intramolecular charge transfer state geometry in conformational polymorphic crystals of a donor–bridge–acceptor dyad in nanosecond-time-scale, J. Chem. Phys., 2023, 158, 054304, DOI:10.1063/5.0134792.
- K. Liu, J. Zhang, Q. Shi, L. Ding, T. Liu and Y. Fang, Precise Manipulation of Excited-State Intramolecular Proton Transfer via Incorporating Charge Transfer toward High-Performance Film-Based Fluorescence Sensing, J. Am. Chem. Soc., 2023, 145, 7408–7415, DOI:10.1021/jacs.2c13843.
- A. Saha and B. Ganguly, A DFT study to probe homo-conjugated norbornylogous bridged spacers in dye-sensitized solar cells: an approach to suppressing agglomeration of dye molecules, RSC Adv., 2020, 10, 15307–15319, 10.1039/C9RA10898J.
- Q. Alkhatib, W. Helal and A. Marashdeh, Accurate predictions of the electronic excited states of BODIPY based dye sensitizers using spin-component-scaled double-hybrid functionals: a TD-DFT benchmark study, RSC Adv., 2022, 12, 1704–1717, 10.1039/D1RA08795A.
- M. R. S. A. Janjua, M. U. Khan, M. Khalid, N. Ullah, R. Kalgaonkar, K. Alnoami, N. Baqader and S. Jamil, Theoretical and Conceptual Framework to Design Efficient Dye-Sensitized Solar Cells (DSSCs): Molecular Engineering by DFT Method, J. Cluster Sci., 2021, 32, 243–253, DOI:10.1007/s10876-020-01783-x.
- F. A. Al-Temimei and A. H. Omran Alkhayatt, A DFT/TD-DFT investigation on the efficiency of new dyes based on ethyl red dye as a dye-sensitized solar cell light-absorbing material, Optik, 2020, 208, 163920, DOI:10.1016/j.ijleo.2019.163920.
- B. Annaraj, S. Pan, M. A. Neelekantan and P. K. Chattaraj, DFT study on the ground state and excited state intramolecular proton transfer of propargyl arm containing Schiff bases in solution and gas phases, Comput. Theor. Chem., 2014, 1028, 19–26, DOI:10.1016/j.comptc.2013.11.018.
- C. Liu, D. Zhang, M. Gao and S. Liu, DFT studies on second-order nonlinear optical properties of a series of axially substituted bis(salicylaldiminato) zinc(II) Schiff-base complexes, Chem. Res. Chin. Univ., 2015, 31, 597–602, DOI:10.1007/s40242-015-5004-7.
- A. Bourouina and M. Rekhis, Structural and electronic study of iron-based dye sensitizers for solar cells using DFT/TDDFT, J. Mol. Model., 2017, 23, 1–9, DOI:10.1007/s00894-017-3478-6.
- T. B. Raju, J. V. Vaghasiya, M. A. Afroz, S. S. Soni and P. K. Iyer, Effect of mono- and di-anchoring dyes based on o,m-difluoro substituted phenylene spacer in liquid and solid state dye sensitized solar cells, Dyes Pigm., 2020, 174, 108021 CrossRef CAS.
- N. N. Ayare, S. Sharma, K. K. Sonigara, J. Prasad, S. S. Soni and N. Sekar, Synthesis and computational study of coumarin thiophene-based D-π-A azo bridge colorants for DSSC and NLOphoric application, J. Photochem. Photobiol., A, 2020, 394, 112466 CrossRef CAS.
- S. Samiee and P. Hossienpour, Tuning the electronic and optical properties of Pt(diimine)(dithiolate) complexes through different anchoring groups; A DFT/TD-DFT study, Inorg. Chim. Acta, 2019, 494, 13–20, DOI:10.1016/j.ica.2019.05.006.
- M. P. Balanay and D. H. Kim, DFT/TD-DFT molecular design of porphyrin analogues for use in dye-sensitized solar cells, Phys. Chem. Chem. Phys., 2008, 10, 5121–5127, 10.1039/B806097E.
- K. I. Ansari, S. Kasiri, J. D. Grant and S. S. Mandal, Fe(III)-Salen and Salphen Complexes Induce Caspase Activation and Apoptosis in Human Cells, SLAS Discovery, 2011, 16, 26–35, DOI:10.1177/1087057110385227.
- S. M. Elbert and M. Mastalerz., Metal Salen- and Salphen-Containing Organic Polymers: Synthesis and Applications, Org. Mater., 2020, 02, 182–203, DOI:10.1055/s-0040-1708501.
- H. Hirohiko, M. Ito and K. Araki, Twin Salphen: Asymmetric Heterodinuclear Complexes {MaMbL|Ma, Mb = Ni, Cu, Zn} of a Symmetrically Fused Salphen Ligand, Inorg. Chem., 2009, 48, 10703–10707, DOI:10.1021/ic901937p.
- M. A. Sahudin, M. S. Su’ait, L. L. Tan, Y. H. Lee and N. H. Abd Karim, Zinc(II) salphen complex-based fluorescence optical sensor for biogenic amine detection, Anal. Bioanal. Chem., 2019, 411, 6449–6461, DOI:10.1007/s00216-019-02025-4.
- N. H. Abd Karim, O. Mendoza, A. Shivaligam, A. J. Thompson, S. Ghosh, M. K. Kuimova and R. Villar, Salphen metal complexes as tunable G-quadruplex binders and optical probes, RSC Adv., 2014, 4, 3355–3363, 10.1039/C3RA44793F.
- M. A. Sahudin, M. S. Su’ait, L. L. Tan, Y. H. Lee and N. H. Karim, Zinc(II) salphen complex-based fluorescence optical sensor for biogenic amine detection, Anal. Bioanal. Chem., 2019, 411, 6449–6461, DOI:10.1007/s00216-019-02025-4.
- A. Saunderson, A permanent magnet Gouy balance, Phys. Educ., 1968, 3, 272 CrossRef.
- A. Earnshaw, Introduction to Magnetochemistry, Academic Press, New York, USA, 1968 Search PubMed.
- S. A. M. Sukri, L. Y. Heng and N. H. A. Karim, Synthesis and characterisation of platinum (II) salphen complex and its interaction with calf thymus DNA, AIP Conf. Proc., 2014, 1614, 419–426, DOI:10.1063/1.4895234.
- M. Frisch, F. Clemente, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennuci, G. A. Petterson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchain, A. F. Izmaylov, J. Boino and G. Zhe, Gaussian 9., 2009, 20–44 Search PubMed.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777, DOI:10.1063/1.1558471.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- K. Kim and K. D. Jordan., Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer, J. Phys. Chem., 1994, 98, 10089–10094 CrossRef CAS.
- S. Kurth and J. P. Perdew., Density-functional correction of random-phase-approximation correlation with results for jellium surface energies, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(16), 10461–10468 CrossRef CAS.
- C. Sosa, J. Andzelm, B. C. Elkin and E. Wimmer, A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds, J. Phys. Chem., 1992, 96, 6630–6636 CrossRef CAS.
- A. D. Becke, Density functional thermochemistry. I. The effect of the exchange only gradient correction, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS.
- A. D. Becke, Density-fnnctional exchange-energy approximation with correct asymptotic behaviour, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; L. Goerigk and S. Grimme, Accurate Dispersion-Corrected Density Functionals for General Chemistry Applications, Model. Mol. Prop., 2011, 3–16 Search PubMed.
- L. Freitag and L. González, Theoretical Spectroscopy and Photodynamics of a Ruthenium Nitrosyl Complex, Inorg. Chem., 2014, 53, 6415–6426, DOI:10.1021/ic500283y.
- L. E. Roy, G. Scalmani, R. Kobayashi and E. R. Batista, Theoretical studies on the stability of molecular platinum catalysts for hydrogen production, Dalton Trans., 2009, 6719–6721, 10.1039/B911019B.
- E. Varathan, D. Vijay and V. Subramanian, Rational Design of Carbazole- and Carboline-Based Ambipolar Host Materials for Blue Electrophosphorescence: A Density Functional Theory Study, J. Phys. Chem. C, 2014, 118, 21741–21754, DOI:10.1021/jp500665k.
- J. Conradie, Density functional theory calculated data of different electronic states and bond stretch isomers of tris(trifluoroacetylacetonato)-manganese(III), Data Brief, 2019, 27, 104758, DOI:10.1016/j.dib.2019.104758.
- H. C. Chang, Y. S. Wang, Y. T. Lee and H. C. Chang, Studying protonated ion hydration by infrared spectroscopy of size-selected NH4 + (H2O)n clusters in a free jet expansion, Int. J. Mass Spectrom., 1998, 179, 91–102, DOI:10.1016/S1387-3806(98)14065-4.
- L. Guofa, S. Tongshun and Z. Yongnian, Infrared and Raman spectra of complexes about rare earth nitrate with Schiff base from o-vanillin and 1-naphthylamine, J. Mol. Struct., 1997, 412, 75–81, DOI:10.1016/S0022-2860(97)00026-4.
- E. Bouaziz, C. B. Hassen, N. Chniba-Boudjada, A. Daoud, T. Mhiri and M. Boujelbene, Crystal structure, Hirshfeld surface analysis, vibrational, thermal behavior and UV spectroscopy of (2,6-diaminopyridinium) dihydrogen arsenate, J. Mol. Struct., 2017, 1145, 121–131, DOI:10.1016/j.molstruc.2017.05.043.
- O. Cozar, N. Leopold, C. Jelic, V. Chis, L. David, A. Mocanu and M. Tomoaia-Cotişel, IR, Raman and surface-enhanced Raman study of desferrioxamine B and its Fe(III) complex, ferrioxamine B, J. Mol. Struct., 2006, 788, 1–6, DOI:10.1016/j.molstruc.2005.04.035.
- A. Espina, M. Vega Cañamares, Z. Jurašeková and S. Sanchez-Cortes, Analysis of Iron Complexes of Tannic Acid and Other Related Polyphenols as Revealed by Spectroscopic Techniques: Implications in the Identification and Characterization of Iron Gall Inks in Historical Manuscripts, ACS Omega, 2022, 7, 27937–27949, DOI:10.1021/acsomega.2c01679.
- M. A. Diab, G. G. Mohamed, W. H. Mahmoud, A. Z. EL-Sonbati, S. M. Morgan and S. Y. Abbas, Inner metal complexes of tetradentate Schiff base: Synthesis, characterization, biological activity and molecular docking studies, Appl. Organomet. Chem., 2019, 33, e4945, DOI:10.1002/aoc.4945.
- A. Al-fahdawi, H. Mohammed and D. Zaidan, Synthesis, Characterization and Evaluation of fungial Activity of Copper(II) and Cobalt(II) Complexes with Imine Glutaraldehyde Derivatives, IOP Conf. Ser.: Mater. Sci. Eng., 2021, 1058, 012078, DOI:10.1088/1757-899X/1058/1/012078.
- C. A. Huerta Aguilar, J. Narayanan, J. A. Balderas Lopez, M. Flores-Alamo, E. F. Velazquez Contreras, K. A. Lopez Gastelum, F. Rocha Alonzo, C. B. Palacios Cabrera and A. J. Santiago Cuevas., Enhanced photocatalytic degradation of 2-thiobenzimidazole by the tris(8-quinolinolato)cobalt(III) complex through peroxide adduct formation: theoretical and experimental investigations, New J. Chem., 2020, 44, 4786–4797, 10.1039/c9nj04027g.
- G. Ting, Y. Yang, W.-B. Sun, G.-M. Li, G.-F. Hou and P.-F. Yan., Syntheses, structure and near-infrared (NIR) luminescence of Er2, Yb2, ErYb of homodinuclear and heterodinuclear lanthanide(III) complexes based on salen ligand, CrystEngComm, 2013, 15, 6213–6220, 10.1039/C3CE40714D.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, J. Wiley and Sons, New York, 5th edn, 1997 Search PubMed.
- M. H. Soliman and G. G. Mohamed, Preparation, spectroscopic and thermal characterization of new metal complexes of verlipride drug. In vitro biological activity studies, Spectrochim. Acta, Part A, 2012, 91, 11–17, DOI:10.1016/j.saa.2012.01.021.
- M. Moreno-Alvero, F. Luna-Giles, F. J. Barros-Garcia, E. Viñuelas-Zahínos and M. C. Fernandez-Calderon, Cobalt(II) complexes derived from a 2-aminobenzimidazole-thiazoline ligand: Synthesis, characterization, crystal structures and antimicrobial activity studies, Polyhedron, 2021, 207, 115390, DOI:10.1016/j.poly.2021.115390.
- G. Sciortino, J.-D. Maréchal, I. Fábián, N. Lihi and E. Garribba., Quantitative prediction of electronic absorption spectra of copper(II)–bioligand systems: Validation and applications, J. Inorg. Biochem., 2020, 204, 110953, DOI:10.1016/j.jinorgbio.2019.110953.
- S. Y. Shaban, A. E.-M. M. Ramadan, M. M. Ibrahim, F. I. Elshami and R. van Eldik, Square planar versus square pyramidal copper(II) complexes containing N3O moiety: Synthesis, structural characterization, kinetic and catalytic mimicking activity, Inorg. Chim. Acta, 2019, 486, 608–616, DOI:10.1016/j.ica.2018.11.024.
- A. A. Siddiqi, N. Shahab, N. Lutfullah and Y. Chebude., Template synthesis of symmetrical transition metal dithiocarbamates, J. Braz. Chem. Soc., 2006, 17, 107–112, DOI:10.1590/S0103-50532006000100016.
- F. Gao, I. Y. Wang, J. Zhang, D. Shi, M. Wang, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel., A new heteroleptic ruthenium sensitizer enhances the absorptivity of mesoporous titania film for a high efficiency dye-sensitized solar cell, Chem. Commun., 2008, 2635–2637, 10.1039/B802909A.
- A. De Candi, J. P. Marcolongo and L. D. Slep, A new ruthenium nitrosyl species based on a pendant-arm 1,4,8,11-tetraazacyclotetradecane (cyclam) derivative: An experimental and theoretical study, Polyhedron, 2007, 26, 4719–4730, DOI:10.1016/j.poly.2007.04.038.
- W. Yang, R. G. Parr and R. Pucci., Electron density, Kohn–Sham frontier orbitals, and Fukui functions, J. Chem. Phys., 1984, 81, 2862–2863, DOI:10.1063/1.447964.
- A. Siiskonen and A. Priimagi, Benchmarking DFT methods with small basis sets for the calculation of halogen-bond strengths, J. Mol. Model., 2017, 23, 50, DOI:10.1007/s00894-017-3212-4.
- S. Alagawani, V. Vasilyev and F. Wang, Optical spectra of EGFR inhibitor AG-1478 for benchmarking DFT functionals, Electron. Struct., 2023, 5, 023011, DOI:10.1088/2516-1075/ace015.
- M. Ermrich and D. Opper, XRD for the analyst Getting acquainted with the principles, PANalytical B.V., Almelo, The Netherlands, 2013, vol. 2 Search PubMed.
- O. Prakash, L. Lindh, N. Kaul, N. Rosemann, I. B. Losada, C. Johnson, P. Chábera, A. Ilic, J. Schwarz, A. K. Gupta, J. Uhlig, T. Ericsson, L. Häggström, P. Huang, J. Bendix, D. Strand, A. Yartsev, R. Lomoth, P. Persson and K. Wärnmark, Photophysical Integrity of the Iron(III) Scorpionate Framework in Iron(III)–NHC Complexes with Long-Lived 2LMCT Excited States, Inorg. Chem., 2022, 61, 17515–17526, DOI:10.1021/acs.inorgchem.2c02410.
- R. Shah, T. M. Habeebullah, F. Saad, I. Althagafi, A. Y. Al-dawood, A. M. Al-Solimy, Z. A. Al-Ahmed, F. Al-Zahrani, T. A. Farghaly and N. E.-M. Shah, Characterization of new Co(II) complexes and photographic monitoring for their toxic impact on breast cancer cells according to simulation study, Appl. Organomet. Chem., 2020, 34, e5886, DOI:10.1002/aoc.5886.
- P. Gull and A. A. Hashmi, Synthesis, XRD and spectroscopic characterization of pharmacologically active Cu(II) and Zn(II) complexes, J. Mol. Struct., 2017, 1139, 264–268, DOI:10.1016/j.molstruc.2017.03.053.
- O. Al Rahal, B. M. Kariuki, C. E. Hughes, P. A. Williams, X. Xu, S. Gaisford, D. Iuga and K. D. M. Harris, Unraveling the Complex Solid-State Phase Transition Behavior of 1-Iodoadamantane, a Material for Which Ostensibly Identical Crystals Undergo Different Transformation Pathways, Cryst. Growth Des., 2023, 23, 3820–3833, DOI:10.1021/acs.cgd.3c00223.
- Z. Cao, Y. Yim, W. Yang, G. Zhao, Y. Zhou, Y. Peng, W. Wang, W. Zhou and D. Tang, Amorphous Co–Pi anchored on CdSe/TiO2 nanowire arrays for efficient photoelectrochemical hydrogen production, J. Mater. Sci., 2019, 54, 3284–3293, DOI:10.1007/s10853-018-3079-5.
- L. Tabrizi, D. Q. Dao and T. A. Vu, Experimental and Theoretical Evaluation on the Antioxidant Activity of a Copper (II) Complex Based on Lidocaine and Ibuprofen Amide-Phenanthroline Agents, RSC Adv., 2019, 9, 3320–3335, 10.1039/C8RA09763A.
- W. Bury, E. Krajewska, M. Dutkiewicz, K. Sokołowski, I. Justyniak, Z. Kaszkur, K. J. Kurzydłowski, T. Płociński and J. Lewiński, tert-Butylzinc hydroxide as an efficient predesigned precursor of ZnO nanoparticles, Chem. Commun., 2011, 47, 5467–5469, 10.1039/C1CC11328C.
- C.-H. Lin, C. H. Tsai, F.-G. Tseng, Y.-Y. Yu, H.-C. Wu and C.-K. Hsieh, Low-Temperature Thermally Reduced Molybdenum Disulfide as a Pt-Free Counter Electrode for Dye-Sensitized Solar Cells, Nanoscale Res. Lett., 2015, 10, 446, DOI:10.1186/s11671-015-1156-0.
- Y. Meng and A. J. Bard, Measurement of Temperature-Dependent Stability Constants of Cu(I) and Cu(II) Chloride Complexes by Voltammetry at a Pt Ultramicroelectrode, Anal. Chem., 2015, 87, 3498–3504, DOI:10.1021/acs.analchem.5b00052.
- S. S. Magendran, F. S. A. Khan, N. M. Mubarak, M. Vaka, R. Walvekar, M. Khalid, E. C. Abdullah, S. Nizamuddin and R. R. Karri, Synthesis of organic phase change materials (PCM) for energy storage applications: A review, Nano-Struct. Nano-Objects, 2019, 20, 00399, DOI:10.1016/j.nanoso.2019.100399.
- Z. Du, A. Qiao, H. Zhou, W. M. Winters, J. Zhu, G. He, I. Parkin, H. Tao and Y. Yue., The glass transition in the high-density amorphous Zn/Co-ZIF-4, Chem. Commun., 2023, 59, 11871–11874, 10.1039/D3CC02492J.
- K. Chaitanya, X.-H. Ju and B. M. Heron, Theoretical study on the light harvesting efficiency of zinc porphyrin sensitizers for DSSCs, RSC Adv., 2014, 4, 26621–26634, 10.1039/C4RA02473G.
- I. Chung, B. Lee, J. He, R. Chang. and M. Kanatzidis, All-solid-state dye-sensitized solar cells with high efficiency, Nature, 2012, 485, 486–489, DOI:10.1038/nature11067.
- C. Klein, K. NAzeeruddin, D. Di Censo, P. Liska and M. Grätzel, Amphiphilic Ruthenium Sensitizers and Their Applications in Dye-Sensitized Solar Cells, Inorg. Chem., 2004, 43, 4216–4226, DOI:10.1021/ic049906m.
- Z. Yu, X. Zhang, H. Zhang, Y. Huang, Y. Li, X. Zhang and Z. Gan, Improved power conversion efficiency in radial junction thin film solar cells based on amorphous silicon germanium alloys, J. Alloys Compd., 2019, 803, 260–264, DOI:10.1016/j.jallcom.2019.06.276.
- T. N. Murakami, N. Koumura, N. Kimura and S. Mori, Structural Effect of Donor in Organic Dye on Recombination in Dye-Sensitized Solar Cells with Cobalt Complex Electrolyte, Langmuir, 2014, 30, 2274–2279, DOI:10.1021/la4047808.
- M. S. Khan, H. U. Haq, S. Abbasi, S. Z. Syeda and M. Arshad., Theoretical designing of selenium heterocyclic non-fullerene acceptors with enhanced power conversion efficiency for organic solar cells: a DFT/TD-DFT-based prediction and understanding, J. Mol. Model., 2022, 28, 228, DOI:10.1007/s00894-022-05225-5.
- J. Preat, Photoinduced Energy-Transfer and Electron-Transfer Processes in Dye-Sensitized Solar Cells: TDDFT Insights for Triphenylamine Dyes, J. Phys. Chem. C, 2010, 114, 16716–16725, DOI:10.1021/jp1050035.
- R. M. Pallares, X. Su, S. Lim and N. T. K. Thanh, Fine-tuning of gold nanorod dimensions and plasmonic properties using the Hofmeister effects, J. Mater. Chem. C, 2016, 4, 53–61, 10.1039/C5TC02426A.
Footnote |
| † Electronic supplementary information (ESI) available: Optical properties of SALPHEN by DFT, Fukui indexes, dipole moment, polarizability and hyperpolarizability for SALPHEN at gaseous state, bond lengths and bond angles resulted in ground state and excited state for SALPHEN and its complexes, using functional B3LYP, CAM-B3LYP/DGDZVP double-zeta basis set and functionals B3LYP, B3PW91, B3P86/6-311++(d,p) triple-zeta basis set at level of theory, the optimized geometrical data of SALPHEN and its complexes at gaseous state, condensed Mulliken charges of SALPHEN with its metal complexes at gaseous state, TD-DFT spectral data of electronic transitions SALPHEN at gaseous state and it is complexes, optimized structure of metal complexes, theoretical FTIR spectra of metal complexes, TD-DFT spectra of SALPHEN using different functionals at gaseous state, TD-DFT spectra of SALPHEN and its complexes using functional B3LYP/DGDZVP and B3PW91/6-311++G(d,p) basis set at gaseous state, in ground state and in excited state. See DOI: https://doi.org/10.1039/d3ma00982c |
| This journal is © The Royal Society of Chemistry 2024 |