DOI:
10.1039/D4MA00059E
(Paper)
Mater. Adv., 2024,
5, 3281-3288
Halogen substituted tetraphenylethylene AIEgens: facile single-crystal-to-single-crystal transformation, polymorphism and mechanofluorochromism†
Received
19th January 2024
, Accepted 17th February 2024
First published on 19th February 2024
Abstract
The molecular structure and supramolecular organization of fluorophores play an important role in generating solid-state fluorescent materials and understanding the mechanism of fluorescence switching/tuning. Molecules that retain the single crystalline nature after applying external stimuli can provide a deep insight into precise control over fluorescence modulation. Herein, we reported the synthesis of eight halogen-substituted tetraphenylethylene (TPE) aggregation-induced emissive (AIEgens) derivatives and investigated the impact of the halogens (F, Cl, Br, and I) on polymorphism and topochemical structural transformation. TPE derivatives with F showed the highest fluorescence (Φf = 58–91%) and heavy atom substitution (I) reduced the fluorescence (Φf = 14–16%). Solid-state structural studies revealed the formation of polymorphs in ClTPE and BrTPE derivatives whereas FTPE and ITPE did not show any polymorphs. The PXRD and DSC studies were further performed to substantiate polymorph formation, which revealed that the formation of stronger intermolecular interactions in FTPE prevented polymorph formation. Importantly, the ClTPE polymorph exhibited facile single-crystal-to-single-crystal structural transformation upon heating/solvent exposure. The formation of polymorphs, structural transformation and crystal growth characteristics from different solvents produced tuneable and switchable fluorescence. A twisted non-planar TPE unit was also prepared to demonstrate mechanical pressure-induced reversible fluorescence switching. The present work attempted to explore the structural design principles for generating polymorphs and facilitating topochemical transformations in AIEgens, which will enhance our comprehensive understanding of the mechanism underlying fluorescence switching/tuning.
Introduction
Organic solid-state fluorescent materials with tunable and switchable fluorescence are considered to be potential materials for various applications including organic light-emitting semiconductors, information storage, sensors and bioimaging.1–7 The fluorescence efficiency of organic molecules in the solid state is strongly influenced by different factors such as molecular motions, conjugation, molecular structures, functional groups, and intermolecular interactions.8–11 Particularly, fluorophore structures and supramolecular interactions controlled molecular organization exerting a strong impact on the fluorescence efficacy as well as fluorescence switching and tuning.10–13 For example, the strong fluorescence of aromatic conjugated rigid molecules was significantly quenched due to the aggregation-caused quenching effect in the solid state.14 In contrast, aggregation-induced strongly enhanced emission was observed for a class of non-planar organic fluorophores that hindered strong aromatic π–π stacking in the solid state.15,16 Structure–property mechanistic studies suggested that intermolecular interactions facilitated molecular rigidification and restriction of intramolecular rotation, suppression of Kasha's rule of fluorescence and excimer formation and restricted access to conical intersection could account for aggregation-induced emission (AIE).17,18 Engineering intermolecular interactions with subtle structural change and polymorphism by using conformationally flexible fluorophores resulted in tunable fluorescence.19–22 For instance, nitrophenyl substitution with the tetraphenylethylene (TPE) core restricted the intersystem crossing process and produced on–off reversible mechanofluorochromic materials.23 Discrete dimers with weak π–π interactions in benzothiazole molecules exhibited a mechanical pressure-induced remarkable fluorescence shift and high sensitivity.24 Switching molecular packing between crystalline and amorphous phases under external stimuli is the most common method attributed to fluorescence control.25–27 However, the lack of clear molecular organization information about the amorphous phase makes it difficult to elucidate the mechanism of fluorescence modulation. On the other hand, achieving reversible crystal-to-crystal transition at the molecular level by polymorphs is a promising strategy for understanding fluorescence switching mechanisms.28,29 But weak intermolecular interaction driven organic molecular systems rarely preserve the single crystalline nature and exhibit reversible structural transformation.30
Halogen bonding has been widely employed as a supramolecular non-covalent interaction to achieve desired molecular assembly in liquid crystals and electronic and magnetic materials.31–33 Halogens in organic fluorophores can regulate the intermolecular interaction that might facilitate the improvement of solid state fluorescence.34–39 Br⋯Br interaction in highly planar enaminone fluorophores blocked nonradiative pathways efficiently and facilitated radiative processes.40 Br substitution in phenothiazine-based benzoxazole derivatives disrupted π–π stacking and produced stronger mechanofluorochromism.41 Halogen substitution in carbazole-based derivatives showed polymorphism and substituent dependent enhancement.42 Furthermore, heavy halogens such as Br and I can increase spin–orbit coupling via heavy atom effects that efficiently quench singlet excited states and promote singlet-to-triplet intersystem crossing and vice versa.43,44 Heavy atom effects and halogen interactions were included to develop interesting phosphorescent materials.45–47 The insignificant impact of halogen substitution as well as strong quenching by heavy atom substitution on fluorescence has also been reported.48–50 These varied responses and fluorescence modulation by controlling intermolecular interactions deserve more investigation with halogen-substituted fluorophores to better establish the structure–property relationship. In this study, we have synthesized a series of TPE-based donor–acceptor derivatives (Fig. 1) and explored the impact of halogen atoms and substitution positions on the molecular arrangements, single-crystal to single-crystal topochemical structural transformation polymorphism and mechanofluorochromism. Although halogen substitution did not have a significant impact on the fluorescence shift, the fluorescence efficiency was gradually reduced from F to I. DSC and powder XRD (PXRD) studies of Cl and Br substituted compounds further demonstrated the interesting polymorphism and single-crystal to single-crystal structural transformation.
 |
| | Fig. 1 Molecular structure of halogen substituted TPE derivatives. | |
Results and discussion
TPE-halogen donor–acceptor derivatives were synthesized by a condensation reaction between TPE-aldehyde and the corresponding halogen-substituted phenylacetonitrile (details in Scheme S1, ESI†). All the molecules exhibited typical aggregation-induced emission in the solid state as in previous reports.28,51,52 Crystallization of halogen-substituted TPE derivatives was attempted using different solvents in order to obtain polymorphs for the further topochemical structural transformation study (the procedure for crystal growth is shown in Fig. S1 and S2, ESI†). Table 1 summarizes the crystallization solvent lists, polymorph details, and related fluorescence properties. TPE derivatives with halogen atoms showed strongly enhanced solid-state fluorescence. Fluorescence emission wavelength λmax did not vary significantly with the variation of the halogen atom. Fluorine-substituted FTPE molecules exhibited the highest fluorescence efficiency of 91% whereas other heavy atoms such as Br and I substitution reduced the fluorescence with a quantum yield of 14–16%. The solvents of crystallization influenced the fluorescence λmaxvia producing polymorphs or due to crystal habits. The fluorescence λmax of these crystals can be visualized with color changes shown in fluorescence images (Fig. S3, ESI†).
Table 1 Fluorescence emission wavelength (λmax) and absolute quantum yield (Φf) of crystals grown in various solvents
| Compounds |
Solvents for crystallization |
Polymorphs |
λ
max (nm) |
Φ
f (%) |
Compounds |
Solvents for crystallization |
Polymorphs |
λ
max (nm) |
Φ
f (%) |
| “—” means that the compound had poor crystallinity in the corresponding solvents and no single crystal was obtained. |
|
3FTPE
|
CH3CN |
Polymorph-1 |
490 |
58 |
4FTPE
|
CH3CN |
— |
— |
— |
| MeOH |
|
478 |
66 |
MeOH |
Polymorph-1 |
493 |
66 |
| DMF |
|
483 |
81 |
DMF |
488 |
91 |
| DMSO |
|
487 |
73 |
DMSO |
491 |
80 |
|
3ClTPE
|
CH3CN |
Polymorph-1 |
494 |
29 |
4ClTPE
|
CH3CN |
Polymorph-1 |
491 |
51 |
| MeOH |
|
499 |
25 |
MeOH |
Polymorph-2 |
503 |
34 |
| DMF |
— |
— |
— |
DMF |
497 |
41 |
| DMSO |
— |
— |
— |
DMSO |
494 |
43 |
|
3BrTPE
|
CH3CN |
Polymorph-1 |
497 |
39 |
4BrTPE
|
CH3CN |
Polymorph-1 |
487 |
47 |
| MeOH |
|
495 |
38 |
MeOH |
Polymorph-2 |
509 |
33 |
| DMF |
— |
— |
— |
DMF |
496 |
36 |
| DMSO |
Polymorph-2 |
492 |
50 |
DMSO |
496 |
37 |
|
3ITPE
|
CH3CN |
Polymorph-1 |
492 |
16 |
4ITPE
|
CH3CN |
—
|
—
|
—
|
| MeOH |
|
492 |
16 |
MeOH |
—
|
—
|
—
|
| DMF |
|
503 |
14 |
DMF |
—
|
—
|
—
|
| DMSO |
—
|
—
|
—
|
DMSO |
—
|
—
|
—
|
Single crystal structural analysis of halogen substituted TPE derivatives showed varying intermolecular interactions and molecular packing in the crystal lattice. It is noted that the TPE core unit displayed a twisted molecular conformation. However, the TPE phenyl donor and phenylacetonitrile acceptor adopted a coplanar/twisted conformation depending on the substitution (Table S1, ESI†). FTPE and ITPE exhibited a coplanar conformation, while ClTPE and BrTPE adopted a coplanar/twisted conformation. The extent of twisting was also significantly influenced by the halogen. 3FTPE crystals grown from different solvents produced the same structure and packing (Fig. S4(a) and (c), ESI†). H-bonding interaction between F and phenyl hydrogen produced a dimer that was further linked by cyano nitrogen H-bonding with phenyl hydrogen (Fig. S4b, ESI†). Furthermore, similar PXRD patterns of 3FTPE crystals ruled out any polymorphism (Fig. S5a, ESI†). DSC studies of 3FTPE crystals exhibited only a melting point of 174 °C (Fig. S5b, ESI†). 4FTPE contained four molecules in the asymmetric unit (Fig. S6a, ESI†). F intermolecular interaction with phenyl hydrogen in the crystal lattice of 4FTPE produced a helical network structure (Fig. S6b, ESI†). The molecular packing diagram exhibited opposite molecular arrangement. 4FTPE crystals grown from different solvents did not show any polymorphism that was further confirmed by PXRD and DSC studies (Fig. S7, ESI†). 3ITPE also did not produce any polymorphic structures (Fig. S8, ESI†). Intermolecular interactions between I and the carbon on the phenyl group connected the crystal network (Fig. S9b, ESI†). It should be noted that our attempt with 4ITPE for obtaining quality crystals was not successful.
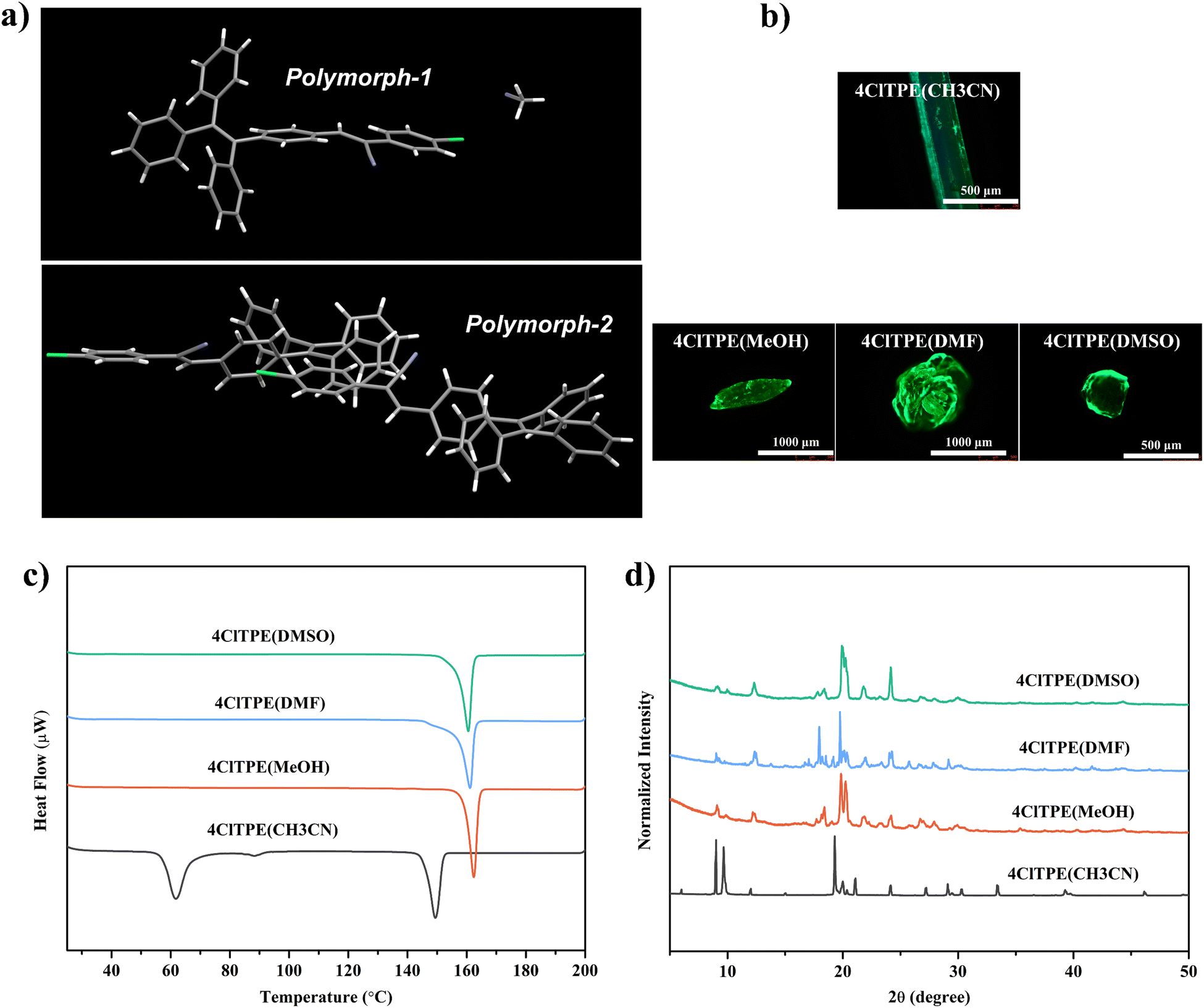
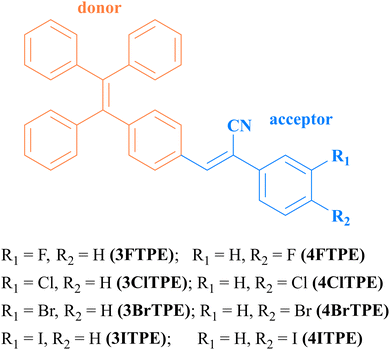
Although 3ClTPE crystals did not show any polymorphism (Fig. S10, ESI†), the TPE phenyl donor and phenyl acetonitrile acceptor exhibited coplanar and slightly twisted molecular conformations. The asymmetric unit of 3ClTPE contained two molecules in the crystal lattice (Fig. S11a, ESI†). The weak intermolecular interactions between Cl and phenyl hydrogen resulted in a cyclic structure (Fig. S11b, ESI†). Molecular packing revealed opposite molecular organization (Fig. S11c, ESI†). However, two different crystal structures of 4ClTPE were obtained. 4ClTPE crystals obtained from CH3CN revealed inclusion of solvent molecules in the crystal lattice (Fig. 2a(polymorph-1)). The weak intermolecular interactions between acetonitrile nitrogen and phenylacetonitrile hydrogen stabilize the solvent molecules in the crystal lattice (Fig. S12b, ESI†). The inclusion of acetonitrile hindered close interaction between 4ClTPE molecules. On the other hand, CH3OH/DMF/DMSO grown crystals of 4ClTPE showed polymorphism. The asymmetric unit containing two molecules did not include any solvent molecules (Fig. 2a(polymorph-2)). Cl halogen interaction with cyano nitrogen and C–H⋯π interactions between TPE units produced a network structure (Fig. S12d. ESI†). Molecules adopted opposite molecular packing in the solid state (Fig. S12e, ESI†). PXRD of 4ClTPE crystals also confirmed the formation of polymorphs in CH3CN and CH3OH/DMF/DMSO (Fig. 2d). DSC studies of 4ClTPE crystals grown from acetonitrile showed a clear phase transition at 57 °C before melting at 146 °C (Fig. 2c). However, CH3OH/DMF/DMSO grown 4ClTPE crystals melted at 157 °C without any phase transition. Interestingly, bromo-substituted 3BrTPE and 4BrTPE produced fluorescent polymorphs. 3BrTPE crystals grown from DMSO showed inclusion of solvent molecules in the crystal lattice (Fig. S13c, ESI†). But CH3CN/CH3OH grown crystals did not show any solvent molecule in the crystal lattice (Fig. S13a, ESI†). The inclusion of DMSO led to higher molecular twisting between the TPE phenyl donor and the phenyl acetonitrile acceptor whereas a coplanar conformation was observed in CH3CN grown crystals. PXRD and DSC studies further supported the formation of polymorphs in 3BrTPE crystals (Fig. S14, ESI†). The crystals grown from DMSO showed a phase transition at 88 °C before melting at 193 °C. In contrast, CH3CN/CH3OH grown crystals melted at 161 °C and did not show any phase transition. Intermolecular interactions between cyano nitrogen and phenylacetonitrile hydrogen produced a network structure in the crystal lattice (Fig. S13b, ESI†). The adjacent molecules adopted opposite molecular orientation in DMSO grown crystals whereas the same side orientation was observed in CH3CN/CH3OH grown crystals (Fig. S13(d) and (b), ESI†). However, the next layer in CH3CN/CH3OH grown crystals displayed the opposite orientation. Similar to 4ClTPE, 4BrTPE crystals grown from CH3CN included the solvent molecule in the crystal lattice (Fig. S15a, ESI†). Crystals grown from CH3OH/DMF/DMSO showed the same structure without any solvent molecules (Fig. S15c, ESI†). PXRD also supported the polymorphism by showing different patterns for crystals grown from CH3CN (Fig. S16a, ESI†). DSC studies showed clear phase transition at 51 °C before melting at 172 °C for crystals grown from CH3CN (Fig. S16b, ESI†). The other polymorph showed only a melting point of 172 °C. The molecular conformation and packing of 4BrTPE crystals were also similar to those of 4ClTPE crystals (Fig. S12 and S15, ESI†). The donor and acceptor units adopted a nearly coplanar conformation in CH3CN polymorphs whereas a twisted conformation was observed in polymorphs obtained from CH3OH/DMF/DMSO. Through single crystal structure analysis of these TPE derivatives, we found that the interaction of halogen atoms with other molecules plays an important role in the formation of crystals. Under the same crystallization conditions, TPE derivatives substituted by F with the smallest atomic radius and the strongest electronegativity exhibited good crystallinity and were conducive to producing crystals of the same structure. However, compounds substituted by I, which has the largest atomic radius and the weakest electronegativity, had poor crystallinity and not even crystals could be obtained. Only ClTPE and BrTPE tended to produce polymorphs. Crystal habits and polymorphism halogen substituted TPE derivatives contributed to fluorescence modulation.
 |
| | Fig. 2 Molecular conformation (a), fluorescence images (b), DSC analysis (c) and PXRD analysis (d) of 4ClTPE crystals. | |
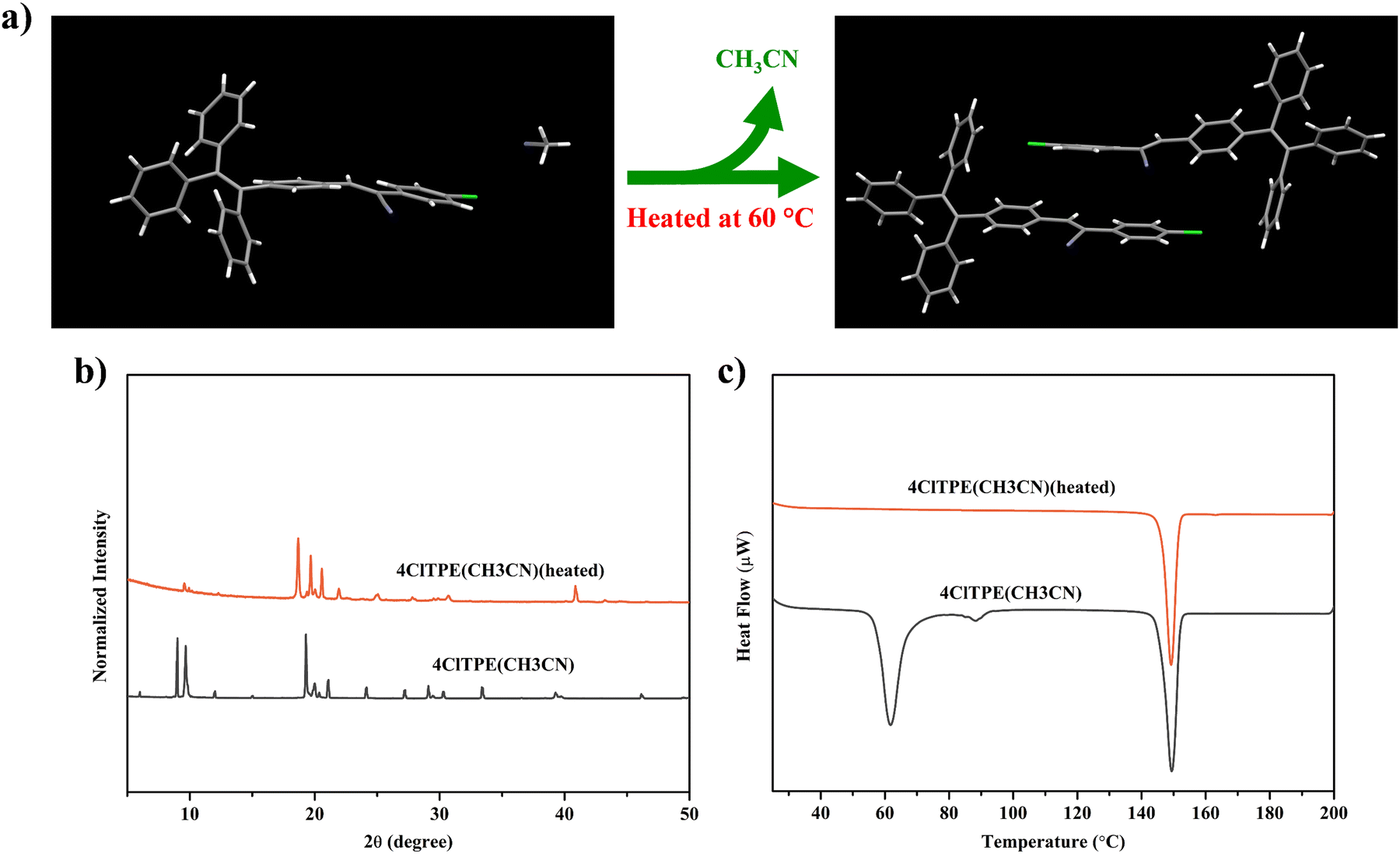
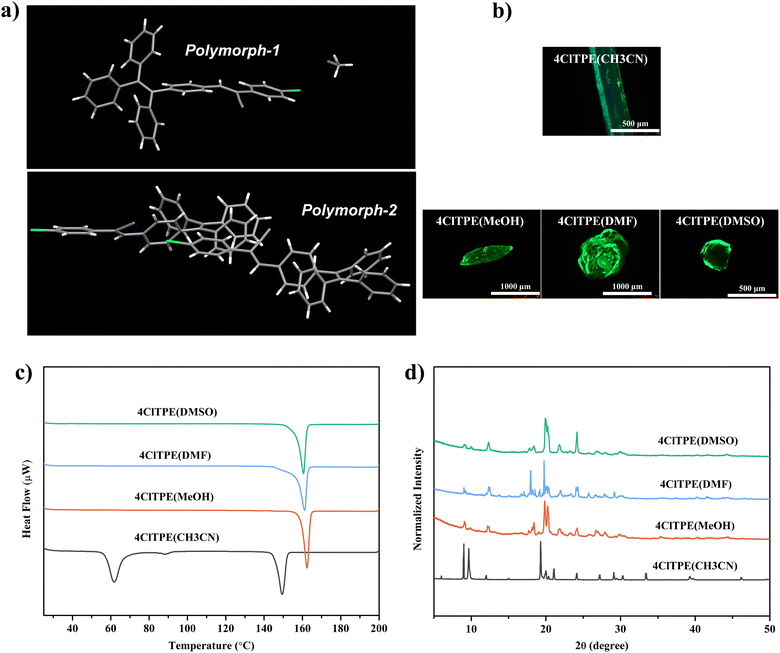
Furthermore, the polymorphic crystals were explored for single-crystal-to-single-crystal structural transformation by heating. 4ClTPE crystals grown from CH3CN that included solvent molecules in the crystal lattice lost the solvent molecules upon heating. Interestingly, the crystals retained their single crystalline nature. Single crystal analysis confirmed the removal of solvent molecules (Fig. 3a). Intermolecular interactions between cyano nitrogen and phenylacetonitrile hydrogen connected the supramolecular dimers. After heating, PXRD of the treated sample showed clearly different patterns and the phase transition peak in DSC has also disappeared (Fig. 3(b) and (c)). In contrast, the solid state fluorescence of 4ClTPE before and after heating did not show any significant variation (Fig. S18(a), (c), ESI† and Table 2) although polarized optical microscopy images exhibited clear differences before and after heating the crystals (Fig. S18b, ESI†). The fluorescence efficiency of the 4ClTPE solid was slightly reduced after heating (Φf = 45% (after heating) and 51% (before heating))(Table 2). Comparing the molecular conformation and packing of the crystals before and after heating (Fig. S12b and S17c, ESI†), it was found that there was no significant change. Just as the acetonitrile molecules disappeared, the face-to-face stacking became tighter. This should be the reason why the fluorescence λmax did not change but the fluorescence efficiency decreased after the crystals were heated.53 Heating of 3BrTPE crystals grown from DMSO also produced different PXRD patterns and indicated structural conversion (Fig. S19d, ESI†). DSC of heated 3BrTPE crystals did not show any phase transition that further supported the structural transformation (Fig. S19c, ESI†). However, the crystals lost their single crystalline character and hence structural studies could not be performed. The solid state fluorescence of 3BrTPE showed clear modulation before and after heating (Fig. S19(a) and (b), ESI† and Table 2). Before heating, it showed fluorescence at 492 nm that was shifted to 506 nm after heating. Similarly, 4BrTPE crystals grown from CH3CN also exhibited structural transformation upon heating. PXRD showed different patterns after heating and the phase transition in 4BrTPE CH3CN also disappeared in DSC charts (Fig. S20(d) and (c), ESI†). However, similar to 4ClTPE, 4BrTPE crystals grown from CH3CN did not show any significant fluorescence modulation before and after heating (Fig S20(a) and (b), ESI†). The loss of single crystalline nature hindered single crystal structural studies.
 |
| | Fig. 3 Single crystal-to-single crystal transition (a), PXRD analysis (b) and DSC analysis (c) of the 4ClTPE (CH3CN) crystal under thermal stimulation. | |
Table 2 Fluorescence emission wavelength (λmax) and absolute quantum yield (Φf) of crystals (4ClTPE(CH3CN) and 3BrTPE(DMSO)) before and after thermal stimulation
| Compounds |
Crystals |
λ
max (nm) |
Φ
f (%) |
|
4ClTPE
|
4ClTPE(CH3CN) |
491 |
51 |
| 4ClTPE(CH3CN)(heated) |
490 |
45 |
|
3BrTPE
|
3BrTPE(DMSO) |
492 |
50 |
| 3BrTPE(DMSO)(heated) |
506 |
39 |
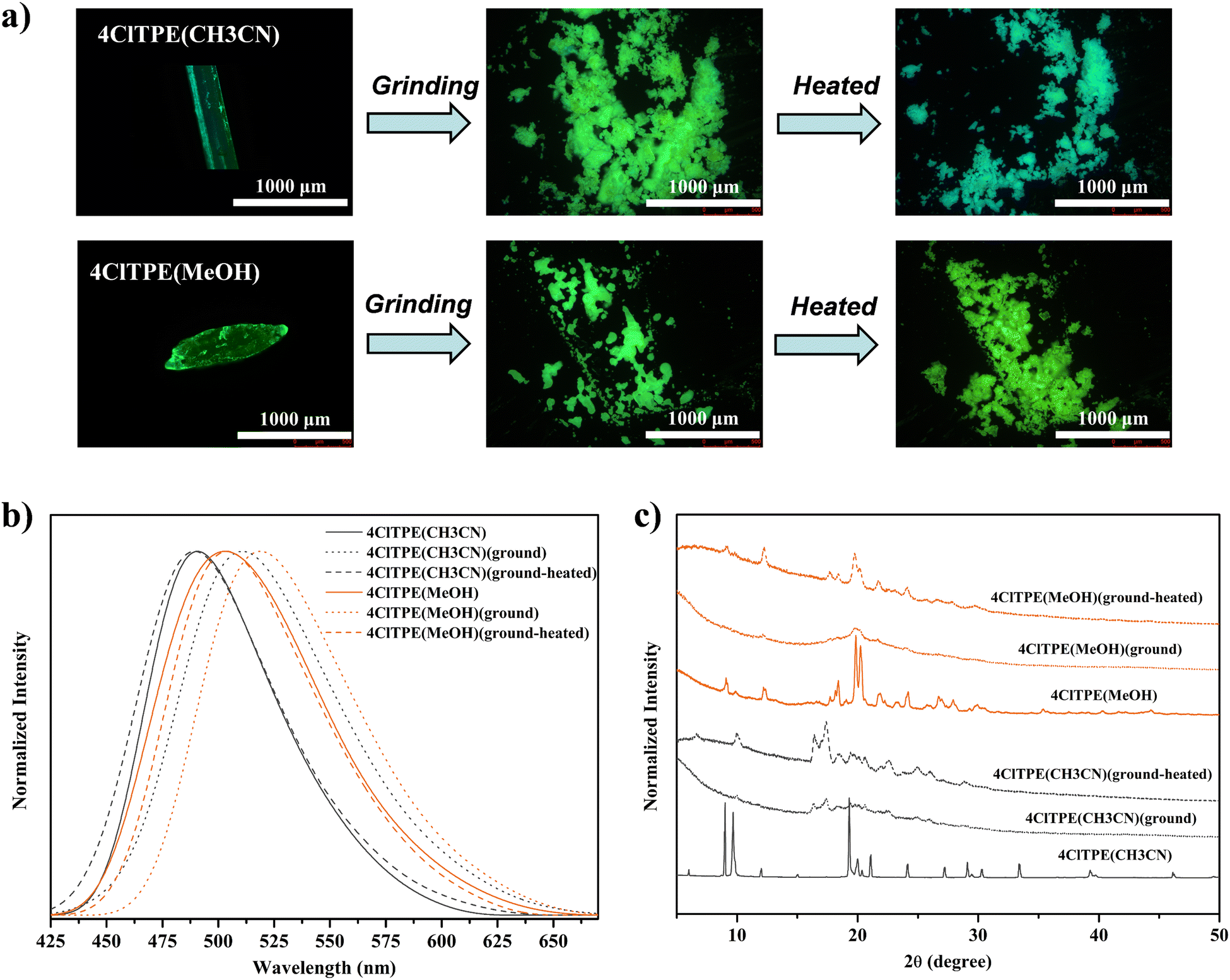
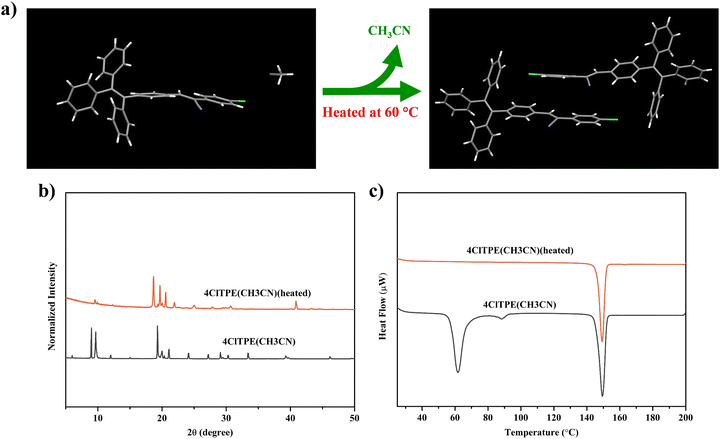
TPE derivatives with a twisted non-planar molecular structure are known to exhibit stimuli-induced reversible fluorescence switching.54–564ClTPE and 3BrTPE crystals were chosen as representative examples for demonstrating mechanical pressure induced reversible fluorescence switching. The fluorescence of 4ClTPE crystals grown from CH3CN showed a red shift from 491 to 511 nm upon crushing whereas heating reversed the fluorescence to 489 nm (Fig. 4(a), (b) and Table 3). Similarly, 3BrTPE crystals grown from CH3CN showed a fluorescence change from 497 to 514 nm upon crushing and the fluorescence was reversed to 492 nm upon heating (Fig. S22b, ESI† and Table 3). Fluorescence images also showed a clear colour change upon crushing and heating (Fig S22a, ESI†). In general, reversible fluorescence switching was attributed to the transformation of the crystalline phase to the amorphous phase.57–59 The sharp PXRD peaks for 4ClTPE and 3BrTPE indicated the crystalline nature of the compounds (Fig. 4c and Fig. S22c, ESI†). However, the peak intensities were significantly reduced after crushing which suggested conversion of the crystalline to the amorphous/partial amorphous phase. The crystallinity was regained upon heating. Hence PXRD indicated reversible phase transformation from the crystalline to the amorphous phase and vice versa that contributed to reversible fluorescence switching.
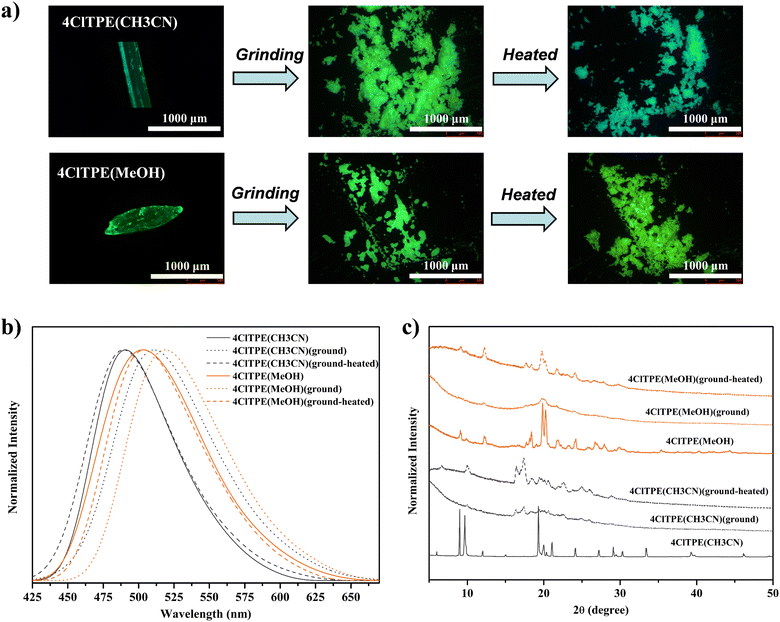 |
| | Fig. 4 Mechanofluorochromism of 4ClTPE crystals in grinding and subsequent heating. Fluorescence images (a), fluorescence emission spectra (λex = 370 nm) (b) and PXRD analysis (c). | |
Table 3 Fluorescence emission wavelength (λmax) and absolute quantum yield (Φf) of mechanofluorochromism of 4ClTPE and 3BrTPE crystals
| Compounds |
Crystals |
λ
max (nm) |
Φ
f (%) |
Crystals |
λ
max (nm) |
Φ
f (%) |
|
4ClTPE
|
4ClTPE(CH3CN) |
491 |
51 |
4ClTPE(MeOH) |
503 |
34 |
| 4ClTPE(CH3CN)(ground) |
511 |
40 |
4ClTPE(MeOH)(ground) |
519 |
46 |
| 4ClTPE(CH3CN)(ground-heated) |
489 |
52 |
4ClTPE(MeOH)(ground-heated) |
504 |
27 |
|
3BrTPE
|
3BrTPE(CH3CN) |
497 |
39 |
3BrTPE(DMSO) |
492 |
50 |
| 3BrTPE(CH3CN)(ground) |
514 |
45 |
3BrTPE(DMSO)(ground) |
506 |
52 |
| 3BrTPE(CH3CN)(ground-heated) |
492 |
45 |
3BrTPE(DMSO)(ground-heated) |
499 |
57 |
Conclusions
In summary, eight halogen-substituted TPE derivatives were synthesized and demonstrated halogen-dependent polymorphism, topochemical conversion, and solid-state fluorescence. Halogen substitution did not alter the fluorescence emission wavelength significantly. But the fluorescence efficiency was strongly influenced by halogen. FTPE exhibited highly enhanced solid-state fluorescence compared to ITPE compounds. ClTPE and BrTPE derivatives produced polymorphism depending on the solvents of crystallization. The formation of polymorphs was further confirmed by PXRD and DSC studies. Interestingly, polymorphs including solvent molecules exhibited topochemical conversion upon heating the crystals and retained their single crystalline nature. Single crystal structural studies, PXRD, and DSC analysis indicated the formation of a new polymorphic structure. Halogen-substituted TPE derivatives showed reversible mechanofluorochromism upon crushing and heating. PXRD studies indicated that reversible phase transformation between crystalline and amorphous contributed to reversible fluorescence switching. The present work attempted to explore the structural design principles for generating polymorphs and facilitating topochemical transformations in AIEgens will enhance the comprehensive understanding of the mechanism underlying fluorescence switching/tuning.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the support from the Banavali Green and Sustainable Chemistry Fund in Arts and Science at the University of Missouri – St Louis, USA. Dr Savarimuthu Philip Anthony from SASTRA Deemed University, India, is greatly acknowledged for his contribution at the initial stage of this work.
Notes and references
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef PubMed.
- H. Wang, X. Ji, Z. A. Page and J. L. Sessler, Mater. Chem. Front., 2020, 4, 1024–1039 RSC.
- Y. Wang, X. Tan, Y.-M. Zhang, S. Zhu, I. Zhang, B. Yu, K. Wang, B. Yang, M. Li, B. Zou and S. X.-A. Zhang, J. Am. Chem. Soc., 2015, 137, 931–939 CrossRef CAS PubMed.
- Z. Peng, X. Feng, B. Tong, D. Chen, J. Shi, J. Zhi and Y. Dong, Sens. Actuators, B, 2016, 232, 264–268 CrossRef CAS.
- K. Li, T.-B. Ren, S. Huan, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2021, 143, 21143–21160 CrossRef CAS.
- D. Su, C. L. Teoh, L. Wang, X. Liu and Y.-T. Chang, Chem. Soc. Rev., 2017, 46, 4833–4844 RSC.
- F. Gao, R. Du, C. Han, J. Zhang, Y. Wei, G. Lu and H. Xu, Chem. Sci., 2019, 10, 5556–5567 RSC.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H.-Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed.
- P. S. Hariharan, D. Moon and S. P. Anthony, J. Mater. Chem. C, 2015, 3, 8381–8388 RSC.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922–3928 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168–6173 CrossRef CAS PubMed.
- B. Zhang, J. Jiang, W. Wang, Q. Tu, R. Yu, J. Wang and M.-S. Yuan, Mater. Chem. Front., 2022, 6, 613–622 RSC.
- J. N. Zhang, H. Kang, N. Li, S. M. Zhou, H. M. Sun, S. W. Yin, N. Zhao and B. Z. Tang, Chem. Sci., 2017, 8, 577–582 RSC.
- P. S. Hariharan, G. Parthasarathy, A. Kundu, S. Karthikeyan, Y. Sagara, D. Moon and S. P. Anthony, Cryst. Growth Des., 2018, 18, 3971–3979 CrossRef CAS.
- W. Zhao, Z. He, Q. Peng, J. W. Y. Lam, H. Ma, Z. Qiu, Y. Chen, Z. Zhao, Z. Shuai, Y. Dong and B. Z. Tang, Nat. Commun., 2018, 9, 3044 CrossRef PubMed.
- Q. Luo, C. Lv, H. Sheng, F. Cao, J. Sun, C. Zhang, M. Ouyang, B. Zou and Y. Zhang, Adv. Opt. Mater., 2020, 8, 1901836 CrossRef CAS.
- S. Guo, G. Zhang, L. Kong, Y. Tian and J. Yang, Chem. – Eur. J., 2020, 26, 3834–3842 CrossRef CAS.
- J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC.
- Y. Chen, B. Wang, Y. Lei, Y. Zhou, Y. Guo, M. Liu, W. Gao, X. Huang and H. Wu, Mater. Chem. Front., 2022, 6, 459–465 RSC.
- P. S. Hariharan, C. Pan, S. Karthikeyan, D. Xie, A. Shinohara, C. Yang, L. Wang and S. P. Anthony, Dyes Pigm., 2020, 174, 108067 CrossRef CAS.
- C. Ge, J. Liu, X. Ye, Q. Han, L. Zhang, S. Cui, Q. Guo, G. Liu, Y. Liu and X. Tao, J. Phys. Chem. C, 2018, 122, 15744–15752 CrossRef CAS.
- Y. Liu, A. Li, S. Xu, W. Xu, Y. Liu, W. Tian and B. Xu, Angew. Chem., Int. Ed., 2020, 59, 15098–15103 CrossRef CAS PubMed.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS PubMed.
- P. Rajamalli, P. Gandeepan, M.-J. Huang and C.-H. Cheng, J. Mater. Chem. C, 2015, 3, 3329–3335 RSC.
- C. Ma, X. Zhang, Y. Yang, Z. Ma, L. Yang, Y. Wu, H. Liu, X. Jia and Y. Wei, Dyes Pigm., 2016, 129, 141–148 CrossRef CAS.
- Y. Qi, Y. Wang, Y. Yu, Z. Liu, Y. Zhang, G. Du and Y. Qi, RSC Adv., 2016, 6, 33755–33762 RSC.
- A. D’Aléo, A. Saul, C. Attaccalite and F. Fages, Mater. Chem. Front., 2019, 3, 86–92 RSC.
- Z. Zhou, Q. Liu, X. Chen, G. Xu, S. Wang, Y. Tu, J. Zhang, X. Zheng, J. Xiang, X. Feng, Y. Zhang, S. Xie, Z. Zeng and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2009024 CrossRef CAS.
- Y. Chen, Y. Zhou, Z. Wang, M. Wang, W. Gao, Y. Zhou, M. Liu, X. Huang and H. Wu, CrystEngComm, 2019, 21, 4258–4266 RSC.
- H. Li, H. Shu, Y. Liu, X. Wu, H. Tian, H. Tong and L. Wang, Adv. Opt. Mater., 2019, 7, 1801719 CrossRef.
- P. Xue, B. Yao, J. Sun, Q. Xu, P. Chen, Z. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC.
- P. Gayathri, S. Karthikeyan, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, CrystEngComm, 2019, 21, 6604–6612 RSC.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- K. Kanosue and S. Ando, ACS Macro Lett., 2016, 5, 1301–1305 CrossRef CAS PubMed.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- S. Gan, S. Hu, X.-L. Li, J. Zeng, D. Zhang, T. Huang, W. Luo, Z. Zhao, L. Duan, S.-J. Su and B. Z. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 17327–17334 CrossRef CAS PubMed.
- H. Shi, Z. An, P.-Z. Li, J. Yin, G. Xing, T. He, H. Chen, J. Wang, H. Sun, W. Huang and Y. Zhao, Cryst. Growth Des., 2016, 16, 808–813 CrossRef CAS.
- W. A. Morris, T. Liu and C. L. Fraser, J. Mater. Chem. C, 2015, 3, 352–363 RSC.
- A. Ghodbane, N. Saffon, S. Blanc and S. Fery-Forgues, Dyes Pigm., 2015, 113, 219–226 CrossRef CAS.
- J. Zhou, L. Liu, Y. Pan, Q. Zhu, Y. Lu, J. Wei, K. Luo, Y. Fu, C. Zhong, Y. Peng and Z. Song, Chem. – Eur. J., 2018, 24, 17897–17901 CrossRef CAS PubMed.
- M. Qayyum, T. Bushra, Z. A. Khan, H. Gul, S. Majeed, C. Yu, U. Farooq, A. J. Shaikh and S. A. Shahzad, ACS Omega, 2021, 6, 25447–25460 CrossRef CAS PubMed.
- L. Wang, R. Zhang, Z. Huang, S. Guo, J.-X. Yang and L. Kong, Dyes Pigm., 2022, 197, 109909 CrossRef CAS.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- Y. Yin, Z. Chen, C. Fan, G. Liu and S. Pu, ACS Omega, 2019, 4, 14324–14332 CrossRef CAS PubMed.
- Z. Qiu, Z. Yang, W.-C. Chen, L. Xing, S. Hu, S. Ji, Q. Yang, N. Cai, X. Ouyang and Y. Huo, J. Mater. Chem. C, 2020, 8, 4139–4147 RSC.
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- Z. Zhang, Z. Wu, J. Sun, B. Yao, P. Xue and R. Lu, J. Mater. Chem. C, 2016, 4, 2854–2861 RSC.
- S. Zhang, Y. Huang, L. Kong, X. Zhang and J. Yang, Dyes Pigm., 2020, 181, 108574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, MS, absorption, and fluorescence spectra, DSC and PXRD, single crystal structural packing, and fluorescence images. See DOI: https://doi.org/10.1039/d4ma00059e |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chengjun
Pan
a,
Chengjun
Pan