
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microneedle transdermal patch loaded with iron(II) nanoparticles for non-invasive sustained delivery to combat anemia†
Bhavya
Surekha
a,
Parimal
Misra
a,
Anitha C.
Thippaiah
b,
Bindiganavale R.
Shamanna
b,
Aiswarya
Madathil
c and
Marina
Rajadurai
 *a
*a
aCenter for Innovation in Molecular and Pharmaceutical Sciences (CIMPS), Dr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad-500046, India. E-mail: marinasraj@gmail.com
bSchool of Medical Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad-500046, India
cSchool of Chemistry, University of Hyderabad Campus, Gachibowli, Hyderabad-500046, India
First published on 26th February 2024
Abstract
Anemia is a pressing global health issue, particularly impacting children and women of reproductive age. To combat iron deficiency anemia effectively, we propose a novel approach: a transdermal patch that delivers iron supplements via microneedles on a weekly basis. This patient-friendly patch aims to address the shortcomings of current treatments, such as low patient adherence, side effects, and poor oral iron bioavailability. Our innovation features a biodegradable polymer transdermal patch embedded with microneedles (100 in total) measuring approximately 340 μm in height and 50 μm in sharpness. These microneedles release iron in the form of ferrous sulfate nanoparticles (IS NPs) in a sustained manner over an extended period. Notably, the patch achieves an 80% drug release over 12 days, with an initial controlled burst release ranging from 10% to 30%. Crucially, this IS NP-loaded microneedle patch demonstrates exceptional drug release performance in vitro, tested on both artificial and cadaver porcine skin models. Notably, the burst release is below 10% when using porcine skin, and the drug release during the first 12 hours follows a near-linear pattern (0 order kinetics). These remarkable results provide a strong foundation for forthcoming in vivo studies, showing promise in the fight against iron deficiency anemia. To bolster our proposal, we conducted an “acceptance survey” among healthcare professionals and the intended beneficiaries to gauge the patch's suitability for the target demographic.
Introduction
Despite all advances in modern medical care, anemia remains an unsolved worldwide problem. According to the WHO, 40% of children, 37% of pregnant women and 30% of women of reproductive age are anemic globally.1,2 Among all, iron deficiency-induced anemia is the most prevalent.3 Although many medical interventions have been developed to prevent and treat iron deficiency anemia, there are still several gaps in today's treatment strategies. The major reasons why existing approaches are less efficient are: (i) poor bioavailability of iron in oral formulations, (ii) unpleasant side effects, and (iii) compliance issues. Apart from that, fear of needles is a very common problem in patients requiring intravenous iron supplementation.4 On the other hand, microneedle (MN) patches, which consists of an array of micrometer-sized needles placed on a base support, could help to overcome this problem as it provides fast and painless transdermal drug delivery.5–7 Moreover, loading 1 week – 12 days requirement of iron in a slow-release biodegradable MN patch would address the issues of bioavailability as well as unpleasant side-effects, at the same time eliminating the need for daily oral supplementary intake, and thus reducing non-compliance risks. There are several iron-containing topical patches available on the market (e.g. PatchMD, Iron Plus, The Friendly Patch, etc.); however, those patches are for daily use and only partially resolve the compliance issue.Developing a formulation with uniform daily sustained release for more than a few days is very challenging. Formulations based on safe and biodegradable polymers, such as poly(ethylene glycol) (PEG), poly lactic-co-glycolic acid (PLGA), polyvinyl alcohol (PVA), etc. are the most attractive choice. However, control of the initial drug burst release, common for such polymers, is not a trivial task.8 It can be achieved by careful choice of the polymeric components of the formulation (polymer type, molecular weight and ratio of polymers) in such a way that both processes affecting drug release (diffusion and erosion) are under tight control.
In addition, drug diffusion from the formulation into the media may be affected by its physical form and may vary significantly if the drug is loaded as a bulk powder or in the form of nanoparticles (NPs). In contrast to conventional formulations, NPs offer special benefits and have already emerged as a powerful drug delivery tool.9 In addition to the structurally optimized form, small size and high surface-to-volume ratio, NPs are expected to have significant therapeutic potential.10,11 Moreover, while preparing the formulation, the use of NPs allows more uniform distribution in the polymeric matrix and thus, better control over diffusion into media and reduced initial burst release.
In this work, we present a proof-of-concept for a biodegradable, sustained-release, MN trans-dermal patch loaded with iron for the prevention and treatment of anemia (Fig. 1). Model drug (ferrous sulfate heptahydrate, or iron sulfate, IS) is loaded in the form of NPs (IS NPs) into a biodegradable polymeric matrix which mediates uniform daily release of 80% of the loaded drug during 12 days with controlled initial burst release (10–30%). Microneedles (MNs) prepared from this polymeric mix are tested in vitro using both artificial and cadaver porcine skin with the help of the Franz diffusion apparatus. The promising results of this study give confidence that such MN-based iron delivery can help to overcome the gaps in existing schemes in the prevention and treatment of Iron deficiency anemia. The in vivo studies of such MNs are our next upcoming investigation.
 | ||
| Fig. 1 Scheme depicting the formulation, patch development and study. | ||
Results and discussion
In the formulation of a transdermal system designed for the prolonged release of micronutrients, we utilized ferrous sulfate heptahydrate (commonly known as iron sulfate, IS) as a representative drug. The IS was engineered into nanoparticles (NPs) and incorporated into soluble polymer films of diverse compositions. Notably, NPs offer distinct advantages over bulk materials, with improved solubility and permeability capturing significant attention. Furthermore, achieving a uniform distribution of NPs within a polymeric matrix is more attainable, thereby enhancing the consistency of sustained drug release.It is crucial to highlight that, based on our observations, IS NPs maintain their nanoparticulate form only when encased within the polymeric matrix within the MNs. Once the NPs exit the polymeric matrix and come into contact with aqueous media (due to polymer biodegradation or dissolution), they promptly dissolve, disintegrating into individual molecules. Consequently, in clinical scenarios, they pose no potential hazards.
Our research group developed a method for synthesizing IS nanoparticles (NPs) through the nano-precipitation of IS dissolved in water into acetonitrile (ACN) with vigorous stirring. The precipitated IS NPs were subsequently collected, washed with ACN, and dried. To achieve a uniform size distribution of the NPs, we explored different ratios of IS to ACN and various stirring rates. Optimal results, in terms of NP uniformity, were obtained by precipitating a 5 mM solution of IS in a 10-fold excess of ACN with stirring at 600 rpm.
Transmission electron microscopy (TEM) analysis revealed that the majority of the NPs had an average diameter of 50–70 nm (Fig. 2A and C). Some batches exhibited a few larger NPs and minor NP aggregation, possibly attributed to the absence of capping agents or stabilizers in our experimental setup. Selected area electron diffraction (SAED) analysis images (Fig. 2B) indicated a ring-like diffraction pattern, suggesting the amorphous nature of the NPs.
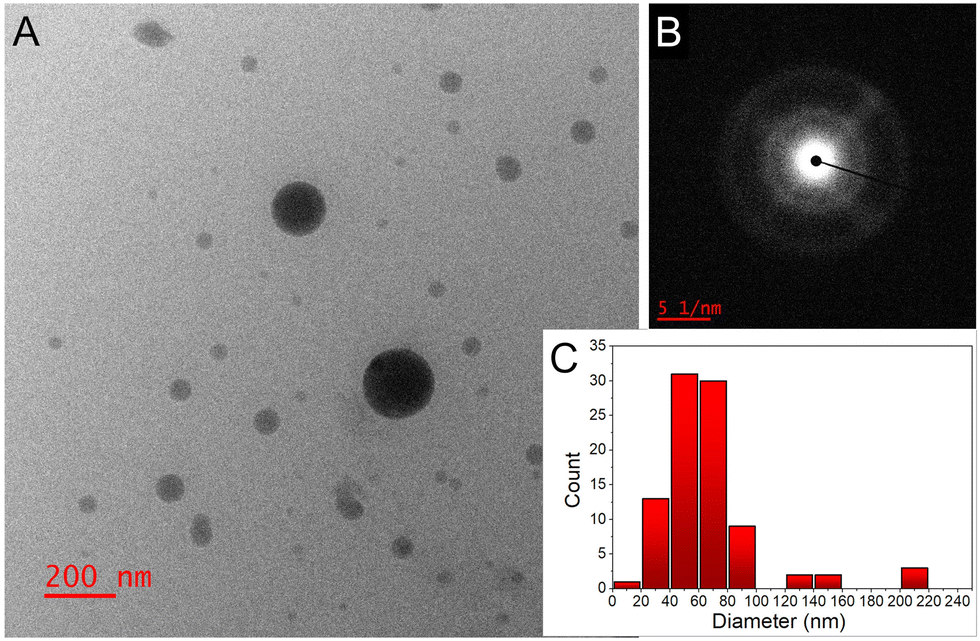 | ||
| Fig. 2 (A) TEM image, and (B) SAED pattern of the IS NPs; (C) NP size distribution histogram determined from the TEM images (total number of NPs was ∼100). | ||
In the pursuit of optimizing the long-term release of the drug from our formulation, we initially prepared soluble polymeric films loaded with IS NPs. Various polymers and NP contents were examined in this process. The tested polymers included poly lactic-co-glycolic acid (PLGA) 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, either alone or in combination with poly(ethylene glycol) (PEG) or polyvinyl alcohol (PVA) at different ratios ranging from 1:9 to 9:1 (some examples of polymeric compositions are provided in Table S1 in the ESI†). The loaded IS content varied between 5 and 40 wt%. The selection of these polymers was based on their non-cytotoxic, biodegradable properties and their capability to entrap therapeutics with a broad spectrum of properties and molecular weights. Among these polymers, PLGA stood out as particularly noteworthy for prolonged drug delivery systems, given its reputation as one of the most successful synthetic polymers in this regard. The rate of drug release is influenced by crucial parameters such as the polymer's crystallinity, glass transition temperature (Tg), and solubility. PLGA offers significant flexibility in manipulating these parameters through adjustments in the PLA/PGA ratio and molecular weight (MW).12
:50, either alone or in combination with poly(ethylene glycol) (PEG) or polyvinyl alcohol (PVA) at different ratios ranging from 1:9 to 9:1 (some examples of polymeric compositions are provided in Table S1 in the ESI†). The loaded IS content varied between 5 and 40 wt%. The selection of these polymers was based on their non-cytotoxic, biodegradable properties and their capability to entrap therapeutics with a broad spectrum of properties and molecular weights. Among these polymers, PLGA stood out as particularly noteworthy for prolonged drug delivery systems, given its reputation as one of the most successful synthetic polymers in this regard. The rate of drug release is influenced by crucial parameters such as the polymer's crystallinity, glass transition temperature (Tg), and solubility. PLGA offers significant flexibility in manipulating these parameters through adjustments in the PLA/PGA ratio and molecular weight (MW).12
In our effort to develop a sustained-release system lasting beyond 10 days, applicable to both hydrophilic and hydrophobic drugs with varying molecular weights, we opted for PLGA with a PLA:PGA ratio of 50:50 and high molecular weight (25–120 kDa). To fine-tune the dissolution rate and control the initial burst release, inherent in pure PLGA, we incorporated PEG and PVA as additives. Over 60 different compositions were systematically tested to identify the formulation possessing the desired properties (see some of the compositions in Table S1, ESI†). In this process, a fixed quantity of IS nanoparticles (NPs) was loaded into a blend of polymers and subsequently dried to form a film. These polymeric films were immersed in water with stirring at a consistent temperature to measure solubility rates and drug release at predefined time intervals. Table S2 (ESI†) provides a comprehensive summary of the tested polymeric compositions, with color-coded results corresponding to evaluated parameters such as sustained drug release (ensuring uniform daily release), low burst release (preferably below 50%), and dissolution rate (anticipating complete dissolution beyond 1 week).
Furthermore, Fig. 3A presents cumulative IS release graphs for a 14-day period from select compositions under evaluation. As anticipated, PLGA alone exhibited a prolonged dissolution rate but yielded a non-uniform drug release (Tables S1, S2 and Fig. 3A, ESI†), likely attributed to the porosity and reduced degree of crystallinity of the selected 50:50 ratio of PLA/PGA.13 Also, it is well-established that the majority of the drug-loaded PLGA formulations exhibit a common drawback: an initial uncontrolled burst of the drug. To address this issue, we incorporated other highly flexible and consequently more soluble polymers with the aim of adjusting both the diffusion (of drug, water, and polymer) and the erosion (of polymer) in the formulation, with the goal of achieving a more uniform release profile.8 Incorporating lower molecular weight PEG and PVA as additives (10–50%) enhanced drug release; however, it also accelerated the dissolution of the polymeric film, completing within 3–5 days. This rapid dissolution posed a challenge to our objective of creating a long-term formulation. Moreover, an escalation in the proportion of the PEG component (up to 70%) resulted in an increased burst release due to a heightened dissolution rate on days one and two. Hence, achieving a delicate equilibrium between an extended dissolution rate, a consistent drug release rate, and effective control over burst release became imperative.
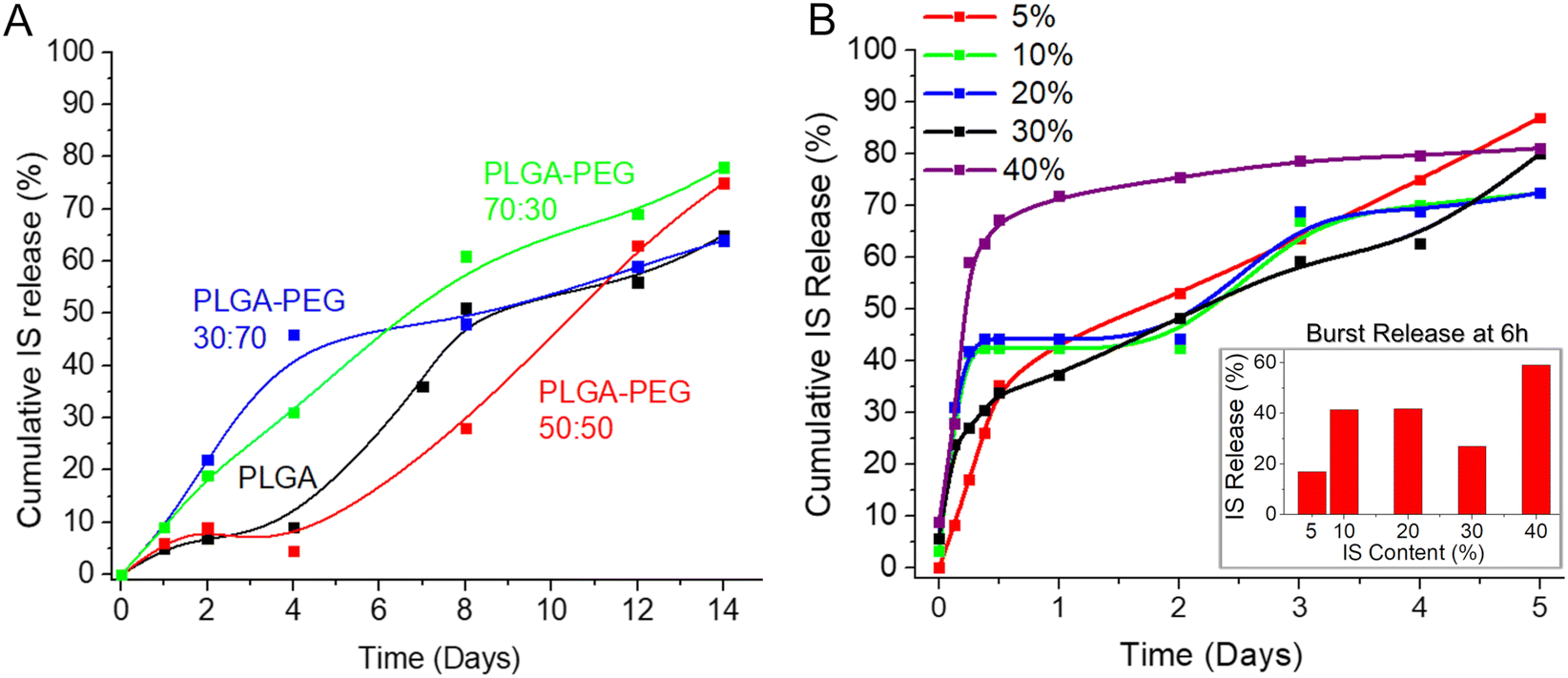 | ||
| Fig. 3 (A) IS release profile from different polymeric films immersed in water; (B) effect of the drug loading (wt%) on the drug release rate from IS-loaded films immersed in water. | ||
The solubility of the films and the release profile of the IS NPs were meticulously studied using a calorimetric method. To facilitate this, a calibration curve for iron quantification in the formulation was established employing the Spectroquant iron test kit (refer to ESI,† Fig. S1).
The optimal composition, characterized by low burst release, nearly uniform, and sustained drug release lasting over 1 week, was achieved by combining PLGA (Mw = 60 kDa) with the addition of 30% PEG (Mw = 10 kDa) (highlighted in the green box in Table S1 (ESI†) and represented by the green line in Fig. 3A). Subsequently, based on these favorable results, the formulation PLGA (60 kDa):PEG (10 kDa) = 70:30 was chosen for all subsequent studies. Despite the selected formulation (PLGA:PEG = 70:30) demonstrating nearly zero-order release kinetics, some minor burst release was observed within the first 6 hours. This phenomenon may be associated with the diffusion and migration of drug molecules toward the surface during fabrication and/or the quicker dissolution of polymeric films at thinner edges.
The quantity of the loaded drug significantly influences the drug release rate and profile.8 Consequently, we investigated various percentages of drug loading (5–40%) in the formulation (Fig. 3B). As observed in this study, an increase in drug loading resulted in a higher initial burst release, with almost 60% of the total loaded drug released within 6 hours when the loading was at 40 wt% of the total composition. Conversely, lower drug content proved effective in controlling burst release, with approximately 17% released in 6 hours when the drug loading was as low as 5 wt% of the total composition. However, such minimal drug loading renders the final product inefficient and costly. A loading of 30%, striking a balance between a reasonable amount of drug in the formulation and a relatively low burst release (approximately 26% after 6 hours), emerged as the optimal choice. Therefore, this loading percentage was selected for the final formulation and was consistently applied in all subsequent studies.
Given the perennial query of whether NP-based formulations surpass conventional formulations containing bulk materials, we undertook a comparison by loading nano-IS alongside bulk IS into the identical polymeric matrix. Two parameters were studied and compared for these formulations: distribution of IS in the polymeric matrix and drug release profile up to 24 h. In the case of bulk IS, high burst release of the drug was observed at 6 h; moreover the drug release was non-uniform in this case (red line, Fig. 4A). On the contrary, drug release in the case of nano-IS was much more uniform, although minor burst release was also observed (blue line, Fig. 4A). Photographs of the films obtained with the help of a transmission electron microscope (TEM) are presented in Fig. 4 (on the right). The images display the size, shape and distribution of IS in the polymeric films in the case of bulk IS loaded (Fig. 4B) and nano-IS loaded (Fig. 4C). Bulk IS is distributed unevenly in the polymeric matrix, with high aggregation near the edges of the film. The crystalline nature of the IS in this case is evident from the electron diffraction patterns shown in the inset (right low corner). On the other hand, an equal distribution of the IS in the polymeric film is observed in the case of the nano-material used for the formulation (Fig. 4C). Nanoparticles have an amorphous nature (see inset Fig. 4C) and well defined spherical shape with an average diameter of 50–70 nm. Some amount of the NPs is located very close to the edges and the surface of the film, and this explains minor drug burst release from the thin films discussed above.
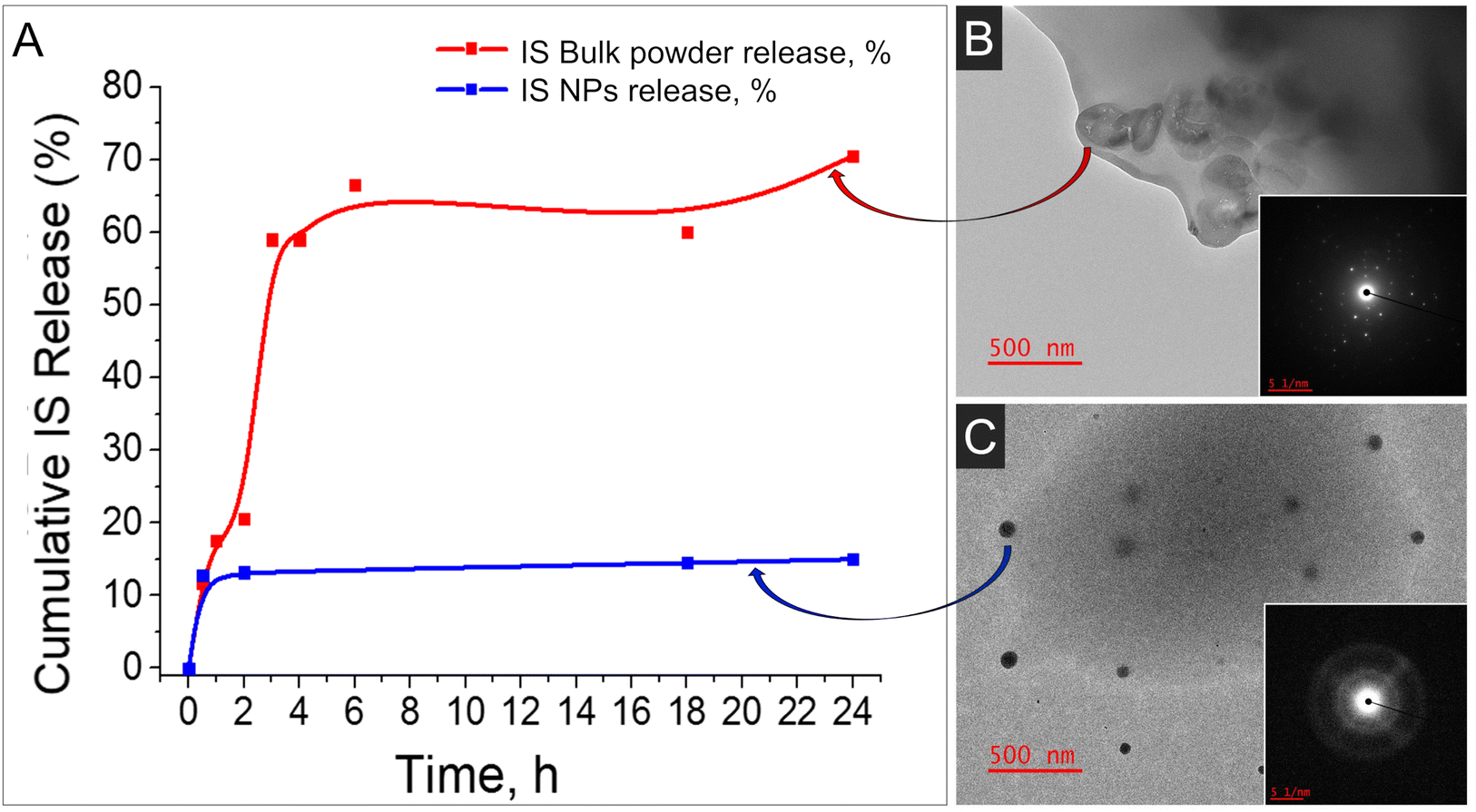 | ||
| Fig. 4 (A) Comparison of the drug release from the formulations with nano-IS with the bulk IS loaded in an identical polymeric matrix; (B) TEM image of the bulk IS loaded in a polymeric matrix; (C) TEM image of IS NPs loaded in the same polymeric matrix. Inset Figures show SAED analysis of the corresponding material. | ||
To summarize our findings, the best formulation for sustained long-term drug release with minimal initial burst release is a combination of PLGA (60 kDa): PEG(10 kDa) = 70:30, with 30 wt% of IS loaded in the form of NPs. This formulation can be used in the form of microneedles for convenient and hassle-free delivery of critical micronutrients, addressing the compliance issue at the same time.
To showcase the versatility of this formulation, we incorporated several widely utilized drugs into the same polymeric composition and examined the release profiles of these four novel formulations. Specifically, we chose four drugs intended for potential prolonged treatment – imipramine (for depression treatment), fenofibrate (for hypercholesterolemia treatment), haloperidol (for psychiatric conditions), and nimesulide (for inflammation treatment) – and loaded them into PLGA-PEG films in a manner similar to how we loaded IS. As illustrated in Fig. 5, each of the four drugs exhibited minimal burst release and maintained controlled sustained release for up to 10 days. However, optimization is required for each drug individually to attain optimal results and further reduce burst release. Nonetheless, this study establishes that the current composition extends beyond IS alone, demonstrating its successful application for various other drugs, even those with poor water solubility.
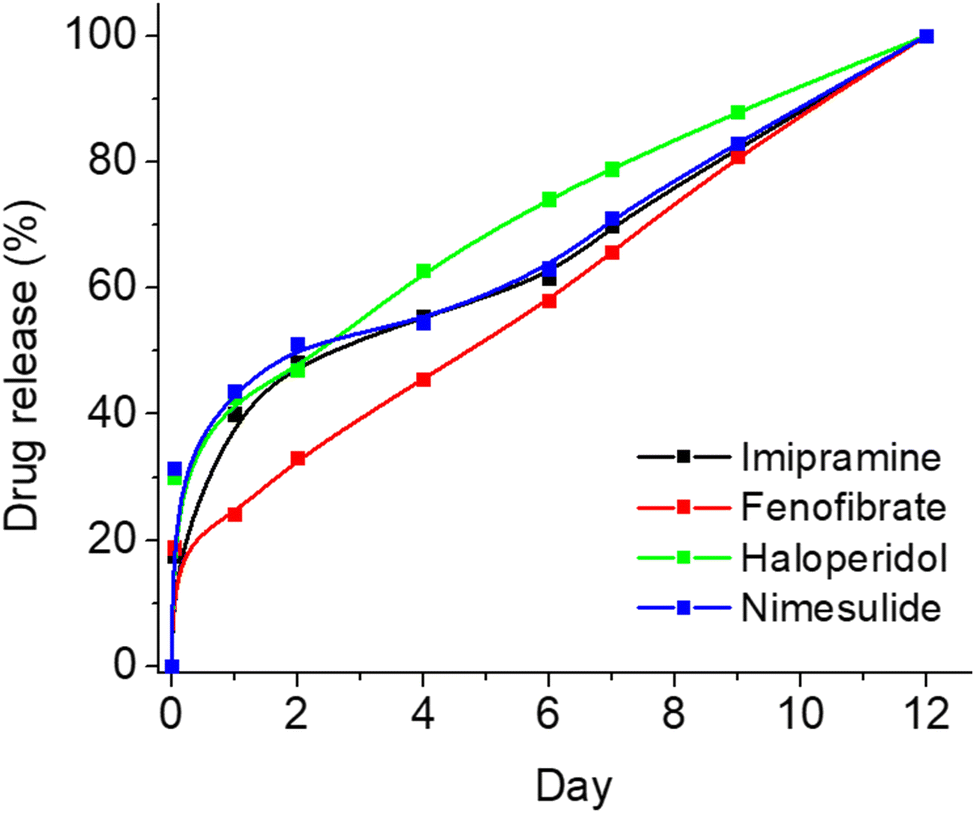 | ||
| Fig. 5 Effect of various drug loading on the release profile from the polymeric films. | ||
For the preparation of soluble MNs in our laboratory setting we selected the micro-molding process reported elsewhere.14 A commercially available master mold (Micropoint Technologies, Singapore) was chosen for fabrication of the MNs. Opting for a mold featuring MNs with a length of 500 μm, we anticipated the final MN length to be approximately 300–400 μm, which is sufficient to penetrate the skin layers of the stratum corneum and epidermis, without reaching the pain receptors in the dermis.15 The methodology for MN fabrication at small laboratory scale was improved and finalized in a step-by-step fashion (see Experimental section for step-by step procedure). The resulting patches exhibit uniformity and clarity, and are devoid of cracks and bubbles.
The prepared MN patches are presented in Fig. 6A–C. The optical microscope photograph reveals the uniformity and sharpness of the MNs. In addition to the MNs loaded with IS, coloured unloaded MNs were also fabricated for visual demonstration during face-to-face surveys (Fig. 6B). SEM images of the MNs are presented in Fig. 6E and F, showing expected dimensions of approximately 250 μm in width and 350 μm in length. While the SEM microphotograph may not depict sharp tips, the actual tip sharpness is approximately 5–10 μm. The MN tips exhibit some flexibility due to the chosen polymeric composition (PLGA:PEG mixture), selected based on drug release profiles. These polymers are inherently soft and elastic, with their rigidity potentially affected by elevated temperatures if stored at room temperature (RT). Nonetheless, the MNs remain sufficiently sharp to penetrate the skin, as demonstrated in Fig. S2 (ESI†), where an artificial skin model exhibits clearly visible holes made by the MNs throughout their full length. In the future, our focus will be on enhancing MN rigidity and storage conditions without compromising drug release properties. Meanwhile, the current formulation of MNs should be stored at lower temperatures (below 5 °C).
 | ||
| Fig. 6 Microneedle photographs and dimensions: (A) photograph of the IS loaded MN patch alone and (C) compared to human hair; (B) colored unloaded MNs; (D) optical microscope image of IS-loaded MNs; (E) and (F) SEM microphotograph of the IS-loaded MNs. | ||
In vitro evaluation of the MNs was executed using both artificial and porcine cadaver skin models. One of the good alternatives of an artificial skin model was introduced by Ryan F. Donnelly.16 He proposed the use of parafilm M (PF) as a model for MN insertion studies and to test drug permeation using dissolving MN arrays. Following this work, we adopted a similar model with some modifications (Fig. 7). For that, a single layer of PF was placed on a sheet of dental wax and then an MN array was inserted into the PF. Then, PF was folded around the baseplate of the MN array and thermally sealed, thus creating a hermetic “pouch”. This pouch was attached to a spatula and placed in the flask with water at RT for stirring using a magnetic stirrer (Fig. 7A). Samples were extracted at defined time intervals and replaced with an equal volume of water. In addition, we used the Franz diffusion apparatus for the drug release studies. In this case, MNs were also inserted into the PF as described above. The PF sheet with the MN arrays was placed and secured to the donor compartment of the diffusion cell and secured (Fig. 7B). Using a long needle, samples were extracted from the receptor compartment at defined time intervals and replaced with an equal volume of water. The sample was analysed using an optical detection technique and the drug release rate for each study was compared to drug release directly from films of the identical composition immersed in water. The results of all three studies are presented in Fig. 8.
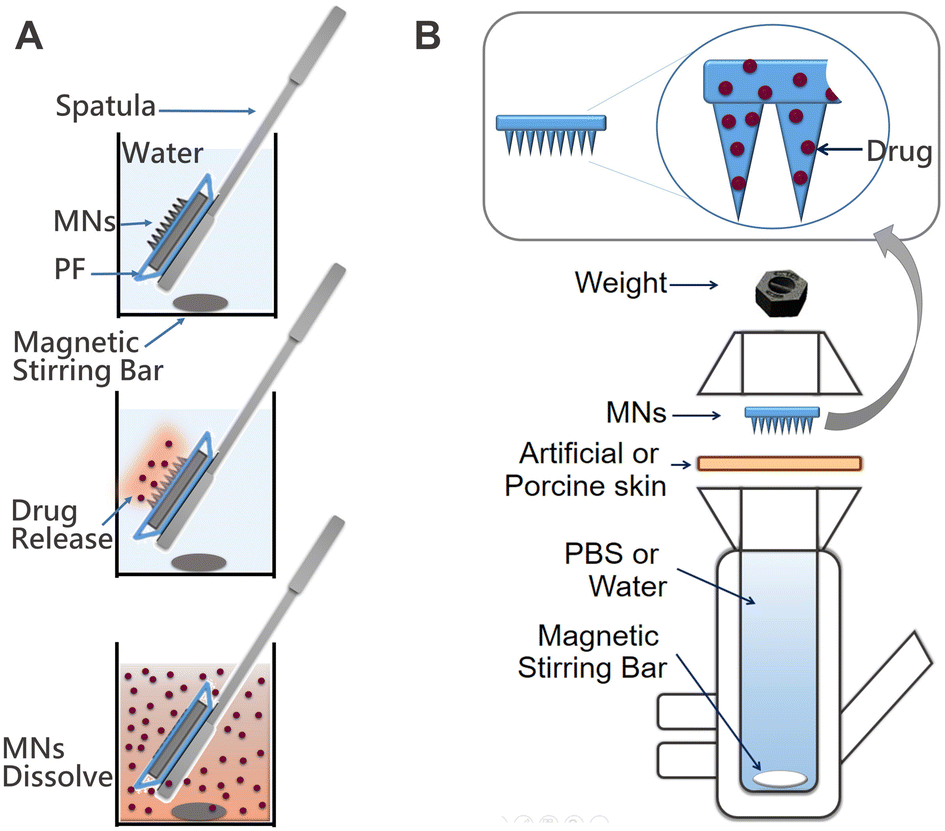 | ||
| Fig. 7 Diagrams of the set ups for the in vitro evaluation of drug release from MNs: (A) preparation and use of the hermetic “pouch” immersed in water; and (B) Franz diffusion cell system used for the permeation studies through artificial or porcine skin. | ||
 | ||
| Fig. 8 In vitro evaluation of drug release from the film and MNs: (A) IS-loaded film directly immersed in water (red line), IS MNs placed in a hermetic “pouch” and immersed in water (green line); (B) IS release from MNs on Franz diffusion apparatus through the artificial skin model (black line) and porcine skin (red line). | ||
As anticipated, the release rate is considerably faster for the IS-loaded films directly immersed in water (illustrated by the red line). This acceleration occurs because all sides of the film are exposed to water, facilitating drug release from the entire film surface. In contrast, when MNs are inserted into PF (as shown by the green line), the drug is initially released from the needles themselves and subsequently from the film through the holes created once the needles dissolve. In this model, water infiltrates the PF pocket, initiating the film's dissolution, resulting in a slightly slower drug release compared to the previous scenario.
To support this claim, we performed the following experiment: freshly prepared IS containing MNs were immersed in a 1% agarose gel. At defined time points, MNs were carefully extracted and examined using SEM to illustrate the degradation morphologies of both the needles and the patch surface. The agarose gel served as a straightforward in vitro transdermal model for MNs, simulating insertion into moist human skin. MNs were retrieved at specific time intervals: 1 h, 6 h, 12 h, 24 h, and 48 h after insertion into the agarose gel. The study results are depicted in Fig. 9A–J.
 | ||
| Fig. 9 (A)–(I) SEM images illustrating the degradation of the MNs after immersion in moist agarose gel. (J) Histogram demonstrating the decrease in height over time in moist media, determined from the aforementioned SEM images. | ||
Even after just 1 hour of immersion, mild degradation of the needles was observed (compared with unused needles in Fig. 6E). The needle size decreased from approximately 350 μm to around 220–290 μm, with the surface becoming more rough and slightly disrupted (Fig. 9A and B). Substantial changes were noted after 6 hours: the majority of needles were partially dissolved, and the maximum height observed was approximately 180 μm, accompanied by an increase in surface roughness (Fig. 9C and D). Similarly, needle height decreased further after 12 hours to 126–146 μm (Fig. 9E and F) and down to 75 μm at 24 hours (Fig. 9G and H), completely dissolving by 48 hours (Fig. 9I).
The most significant changes in dimensions and surface morphology occurred within the first 12 hours, explaining the minor initial drug burst release. After 24 hours, the majority of needles had already dissolved, and drug release occurred directly from the patch surface. An overall summary of needle degradation over time is presented in Fig. 9J as a histogram of needle height vs. time.
This observation aligns with another study conducted in our lab, where MN patches were immersed in water, and the rate of polymer dissolution (instead of the drug) was measured at predetermined time points. Absorbance intensity, measured using a UV-vis Spectrometer at λmax = 212 nm for PEG and 338 nm for PLGA, showed a faster increase in the first 6 hours, indicating the rapid dissolution of polymers from the MN surface after immersion in water. This supports the explanation of the initial biphasic burst release. In the following hours, the dissolution slowed down and stabilized, allowing for sustained drug release. (Refer to ESI,† Fig. S3 for details on polymer dissolution.)
In vitro studies employing porcine cadaver skin were conducted to closely simulate the future application environment (see Fig. 8B). The duration of this study was limited to 24 hours, as the properties of organic matter cannot be preserved over an extended period without undergoing changes. Freshly obtained cadaver porcine skin from the abdominal area was obtained and treated as described elsewhere.17 The in vitro data obtained using porcine skin closely correlates with the results obtained for artificial skin. As illustrated in Fig. 8B, the burst release is notably lower with porcine skin, and the release during the initial 12 hours follows a more linear pattern resembling 0-order kinetics. Overall, the release profile is superior with porcine skin, suggesting that the permeability of the natural material is more effectively controlled compared to the synthetic material. This model closely imitates an actual transdermal delivery application, providing confidence that a robust sustained release will be observed in vivo when MNs are inserted into live animal skin. In vivo studies are currently underway to confirm the required drug release behaviour.
As highlighted in the Introduction, the prevalence of malnutrition and anemia is alarmingly high, particularly in developing countries like India. In this context, we posit that IS-containing MNs hold significant social relevance, aiming to contribute to the enhancement of health conditions among individuals affected by anemia and pre-anemia. However, MN formulations are not yet widespread and may not be well understood, particularly among individuals with limited education residing in rural areas. This raises the question of whether IS-containing MNs would be readily accepted once manufactured.
To address this concern, we sought to conduct a brief acceptance survey among potential beneficiaries and doctors in India. The objective is to identify key acceptance criteria for the MN patch. The insights gained from the survey will not only contribute to gauging the potential acceptance of IS-containing MNs but will also inform our microneedle design process. Designing the microneedles based on end-user preferences, as revealed in the survey, is likely to enhance the prospects of success for this innovative product.
The survey was conducted in both on-line and face-to-face modes among 2 groups of potential stakeholders:
•Potential end users (95 residents: healthy women aged between 13 to 45 years from urban (60%) and rural (40%) areas), and
•Potential service providers – Health Care Professionals (100 residents, including Paediatricians, Gynaecologists, Dermatologists, Nutritionists and General Medicines).
Before conducting the survey, participants were provided with product information and subsequently briefed on a novel proposed transdermal delivery approach for iron supplementation. The educational session covered aspects such as safety, potential risks, any side effects, and the advantages of utilizing a patch for application compared to oral supplementation.
In summary, the survey results indicate that the majority of potential end-users from urban areas are well-informed about transdermal delivery and responded positively. A significant proportion (91.5%) viewed the patch as a valuable addition to existing interventions, with 63% expressing willingness to use it over other available treatments (additional details in ESI† Fig. S4). Conversely, women in rural areas exhibited hesitancy towards adopting the new delivery method. Notably, these women lacked awareness of how the patch could be applied and were introduced to the concept of transdermal delivery for the first time. Approximately 70% of them preferred traditional tablets, despite inconveniences and side effects, highlighting the need for further education and additional efforts to make the transdermal patch acceptable in such regions. Several concerns were raised, including potential pain during patch application, impact on cosmetic appearance, and waterproofing. However, these questions are deemed manageable during the manufacturing stage.
Contrary to the response from potential end-users, all of the surveyed doctors expressed a highly positive outlook and unanimously agreed that this is a very promising idea. The summarized results of the survey are provided in ESI† Tables S2 and S4, with an overall summary depicted in Fig. 10. A more extensive survey is currently in planning, aiming to encompass a broader population across various districts with a more detailed questionnaire. Upon completion, the comprehensive findings will be published elsewhere.
 | ||
| Fig. 10 Summary of the brief “acceptance survey” for the MN transdermal patch in India. | ||
Conclusions
This study introduces a proof-of-concept for a biodegradable, sustained-release microneedle (MN) transdermal patch loaded with iron sulfate, designed for the prevention and treatment of iron deficiency anemia. IS is incorporated in the form of NPs within a biodegradable polymeric matrix, facilitating a uniform daily release of 80% of the drug over 12 days, featuring controlled initial burst release below 30%. A comparative analysis of drug release from two formulation types—bulk conventional material versus NPs - revealed a distinct advantage for the NP-containing formulation. The evident difference lies in the controlled burst release observed with NPs, contrasting with the high burst release in the case of bulk drug, particularly during the initial 24 hours.Demonstrating the broader applicability of the developed formulation, four additional drugs were included for potential prolonged treatment across various diseases. These drugs, targeting depression (imipramine), hypercholesterolemia (fenofibrate), psychiatric conditions (haloperidol), and inflammation (nimesulide), exhibited minimal burst release and controlled sustained release over a 12-day period. This highlights the potential for new and convenient treatment approaches for chronic or life-long diseases.
IS-loaded MNs, derived from the identified polymeric blend, underwent an in vitro testing employing both artificial and cadaver porcine skin with the aid of the Franz diffusion apparatus. The study reveals that the initial burst release is effectively controlled, remaining below 20%. To further mitigate the initial burst release, we are contemplating the implementation of several strategies: (1) minor adjustments to the composition of the films; (2) the inclusion of an additional wash step for the films after fabrication; and (3) the application of an additional coating layer on the surface to safeguard IS NPs from immediate release. Presently, extensive in vivo studies of the MNs on Sprague Dawley rats are underway, constituting an ongoing investigation.
An abbreviated “acceptance survey” conducted in India has indicated a favourable reception among the target population. Specifically, 83% of potential end users express either a positive or neutral stance towards MN technology, while 91% of healthcare professionals exhibit positive or neutral sentiments.
Experimental section
Materials
The following chemicals were purchased from Sigma Aldrich: ferrous sulfate heptahydrate, poly(D,L-lactide-co-glycolide) 50:50 block copolymer (25, 60 and 120 kDa, PLGA), polyvinyl alcohol (PVA), sulforhodamine B sodium salt, DOWSILTM 184 elastomer kit (Polydimethylsiloxane, Dowsil, Batch No. H052J9A071) and Spectroquant iron test kit. Polyethylene glycol (2, 6, 10 and 20 kDa, PEG) was purchased from Alfa Aesar, Massachusetts, USA. Acetonitrile (ACN) was obtained from RANKEM Fine Chemicals, India. Microneedle master molds (10 × 10 array measuring B200 μm, H525 μm and P500 μm) were obtained from Micropoint Technologies, Singapore. Dental wax (Orthodontic Wax for Braces) was purchased from Dentosmile.
The following instruments were used for characterization: Eppendorf BioSpectrometer (Basic), Compound Microscope Leica DM E, Transmission Electron Microscope Tecnai G2FEI F12 (TEM, at an accelerating voltage of 120 kV, operating voltages are 20–200 kV, resolution 2.4 Å), and scanning electron microscope Philips XL30 ESEM (SEM, beam voltage 20 kV).
Experimental
Synthesis of IS nanoparticles
Ferrous sulfate heptahydrate (FeSO4⋅7H2O, iron sulfate, IS, 25 mg) was dissolved in nano-pure water (18 mL) to form a 5 mM solution. This IS aqueous solution was dropwise added to acetonitrile (ACN, 180 mL) using a 1 mL syringe or separating funnel attached with a 1 mL syringe under vigorous stirring (600 rpm) at 25 °C. After complete addition of IS solution, the suspension was left stirring for an hour. Precipitated IS NPs were collected by centrifugation at 4000 rpm for 10 min at 4 °C and washed with ACN thrice and dried under a nitrogen environment and stored at low temperature (below 0 °C).Preparation of polymeric films containing IS NPs
Polymers (PLGA, PEG and PVA) were co-dissolved at different ratios (see for example Table S1, ESI†) in ACN to form a polymer solution. A specific amount of IS NPs (5, 10 20, 30 or 40 wt%.) was added to the polymer solution and sonicated for 30 min to ensure uniform dispersion of the NPs in the polymeric matrix. 400 μL of the resulting final mixture was drop cast portion wise (100 μL at each time) on the clean surface of a Petri dish allowing each portion to dry for 1 h at RT to obtain the film with diameter = 13 mm and thickness ca. 700 μm. After addition of the last portion, the film was allowed to dry for 24 h at RT.Drug release study from the IS NP loaded films
The film was immersed in a tightly closed screw capped 15 mL Falcon tube filled with 7 mL of nanopure water. The release study was performed using an incubator at 37 °C and 180 rpm. The samples (0.3 mL) were collected at different time intervals and the same volume of fresh water was replaced after each sample collection. The sample was diluted (10 to 100 times) and 2 drops of reagent from a Spectroquant iron test kit were added from the container, mixed well and left to stand for 5 minutes to form the red violet complex. The absorbance of the above solution was measured at 563 nm using UV/Vis Spectrophotometer and the amount of released iron was determined with help of a calibration curve (ESI,† Fig. S1). On the last day of the study, the sample was ultra-sonicated for 30 min and the absorbance of the sample was recorded to obtain 100% drug release.Preparation of microneedles
In vitro evaluation of the MNs on Franz diffusion cell
For study using an artificial skin model, a single layer of parafilm M (PF, 2 cm × 4 cm) was placed on a sheet of dental wax and MNs were inserted and gently pressed with a finger to ensure full-surface contact. Then, PF was folded around the baseplate of the MN array, carefully disconnected from the wax, and placed on the donor compartment of the diffusion cell and secured with a weight (Fig. 7B). For study using porcine skin, freshly obtained cadaver porcine skin from the abdominal area was obtained and treated as described elsewhere.17 Briefly, porcine skin was shaved and sliced to a thickness of 1.2 mm, the subcutaneous fat was carefully removed. Following a heat separation process of 1 min in water (60 °C), the epidermis was peeled off the dermis and floated onto filter paper stratum corneum side uppermost. The skin samples were dried in a laminar air flow cabinet at ambient temperature and cut into 3 × 3 cm pieces. One such piece was placed on a sheet of dental wax and MNs were inserted and gently pressed with a finger to ensure full-surface contact. The skin with inserted MNs was carefully disconnected from the wax, and placed on the donor compartment of the diffusion cell and secured with a weight (Fig. 7B). In both cases, samples were extracted from the receptor compartment at defined time intervals and replaced with an equal volume of water. The samples were analyzed using reagent from a Spectroquant iron test kit as described above.Compliance with ethical standards
Information for the “acceptance survey” was gathered in compliance with current laws.Author contributions
BS and AM: NPs synthesis, experiments on drug release and MNs preparation; ACT and BRS: acceptance survey; PM: in vitro analysis; MR: ideation, experiments and writing manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their thanks to the support by the Department of Biotechnology, Government of India, grant No. BT/PR32386/MED/32/681/2019.References
- WHO Global Anaemia estimates, 2021 Edition. Available at: https://www.who.int/data/gho/data/themes/topics/anaemia_in_women_and_children.
- World Health Organization, Nutritional anemias: tools for effective prevention and control, Geneva, 2017. Available at: https://www.who.int/nutrition/publications/micronutrients/FNBvol29N1sep10.pdf.
- GBD 2021 Anaemia Collaborators. Prevalence, years lived with disability, and trends in anaemia burden by severity and cause, 1990–2021: findings from the Global Burden of Disease Study 2021. The Lancet Haematology, Published online July 31, 2023, DOI:10.1016/S2352-3026(23)00160-6.
- K. Alsbrooks and K. Hoerauf, PLoS One, 2022, 17(11), e0276814, DOI:10.1371/journal.pone.0276814.
- M. Avcil and A. Çelik, Micromachines, 2021, 12(11), 1321, DOI:10.3390/mi12111321.
- H. Zheng, X. Xie, H. Ling, X. You, S. Liang, R. Lin, R. Qiu and H. Hou, J. Mater. Chem. B, 2023, 11, 8327–8346 RSC.
- R. F. Donnelly and M. R. Prausnitz, Drug Delivery Transl. Res., 2024, 14(3), 573–580, DOI:10.1007/s13346-023-01430-8.
- J. Yoo and Y.-Y. Won, ACS Biomater. Sci. Eng., 2020, 6(11), 6053–6062 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley and R. M. Haley, et al. , Nat. Rev. Drug. Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. Di Stefano, Int. J. Mol. Sci., 2023, 24, 8194 CrossRef.
- J. M. Chan, P. M. Valencia, L. Zhang, R. Langer and O. C. Farokhzad, Methods Mol. Biol., 2010, 624, 163, DOI:10.1007/978-1-60761-609-2_11.
- I. J. Macha, B. Ben-Nissan, E. N. Vilchevskaya, A. S. Morozova, B. E. Abali, W. H. Müller and W. Rickert, Front. Bioeng. Biotechnol., 2019, 7, 37, DOI:10.3389/fbioe.2019.00037.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3(3), 1377–1397, DOI:10.3390/polym3031377.
- T. N. Tarbox, A. B. Watts and Z. Cui, et al. , Drug Delivery Transl. Res., 2018, 8, 1828–1843 CrossRef CAS PubMed.
- T. Waghule, G. Singhvi, S. K. Dubey, M. M. Pandey, G. Gupta, M. Singh and K. Dua, Biomed. Pharmacother., 2019, 109, 1249–1258 CrossRef CAS.
- E. Larrañeta, S. Stewart, S. J. Fallows, L. L. Birkhäuer, M. T. C. McCrudden, A. D. Woolfson and R. F. Donnelly, Int. J. Pharm., 2016, 497, 62–69 CrossRef PubMed.
- B. G. Aunera, C. Valentaa and J. Hadgraftb, J. Controlled Release, 2003, 89, 321–328 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01166f |
| This journal is © The Royal Society of Chemistry 2024 |