DOI:
10.1039/D3RA05098J
(Review Article)
RSC Adv., 2023,
13, 26484-26508
Emerging developments in dye-sensitized metal oxide photocatalysis: exploring the design, mechanisms, and organic synthesis applications
Received
27th July 2023
, Accepted 22nd August 2023
First published on 4th September 2023
Abstract
In the present day, the incorporation of environmentally conscious practices in the realm of photocatalysis holds a prominent position within the domain of organic synthesis. The imperative to tackle environmental issues linked to catalysts that cannot be recycled, generation of waste, byproducts, and challenges in achieving reaction selectivity during organic synthesis are more crucial than ever. One potential solution involves the integration of recyclable nanomaterials with light as a catalyst, offering the possibility of achieving sustainable and atom-efficient transformations in organic synthesis. Metal oxide nanoparticles exhibit activation capabilities under UV light, constituting a small percentage (4–8%) of sunlight. However, this method lacks sufficient environmental friendliness, and the issue of electron–hole recombination poses a significant hurdle. To tackle these challenges, multiple approaches have been proposed. This comprehensive review article focuses on the efficacy of dyes in enhancing the capabilities of heterogeneous photocatalysts, offering a promising avenue to overcome the constraints associated with metal oxides in their role as photocatalysts. The article delves into the intricate design aspects of dye-sensitized photocatalysts and sheds light on their mechanisms in facilitating organic transformations.
 Dana A. Kader | Dana A. Kader received his Master's degree in Chemistry from the Eastern Mediterranean University (EMU), Cyprus, in 2014. He is currently in the final stages of his PhD studies in Organic Chemistry at the University of Sulaimani, College of Science, Chemistry Department, in Sulaimani-Iraq. His research interests primarily revolve around the design of heterogeneous photocatalysts for organic transformations and the degradation of organic pollutants. |
 Sewara J. Mohammed | Sewara J. Mohammed received his master's degree in 2014 from the Department of Chemistry, College of Science at Sulaimani University. Currently, he is in the final stages of his PhD studies in the same department and university. His current interests involve researching the synthesis of carbon nanomaterials and their applications in enhancing the optical and electrical properties of polymer materials. |
1. Introduction
As organic transformations progress and evolve, the potential for environmental pollution also rises due to the utilization of extensive catalysts such as strong acids,1 strong bases,2 metals,3 and heavy metals,4 which result in the generation of byproducts. Researchers persist in seeking alternatives to improve these issues and mitigating the associated risks. Within dye-sensitized photocatalysis, dyes assume a photosensitizer role by absorbing photons and conveying their energy to the metal oxide photocatalyst.5 This transfer of energy results in the creation of electron–hole pairs, which actively engage in diverse chemical reactions such as photocatalytic degradation of pollutants,6 organic transformations,7 water splitting,8 carbon dioxide reduction,9 electrochemical reactions,10 environmental remediation,11 electrocatalysis,12 fuel production,13 photodegradation of pharmaceuticals,14 and green chemistry applications.15 The efficiency and performance of dye-sensitized systems heavily depend on the design and selection of the sensitizer dye, making it a crucial research area within the realm of photovoltaics and photocatalysis.16
Recent extensive reviews have thoroughly discussed the remarkable potential of organic dyes, iridium, and ruthenium metal complexes as homogeneous photocatalysts.17,18 These catalysts have successfully facilitated diverse organic transformations19,20 under mild conditions, utilizing visible light as the activating source. However, one significant drawback is their high cost, prompting chemists to explore alternative options and seek more affordable catalysts actively.
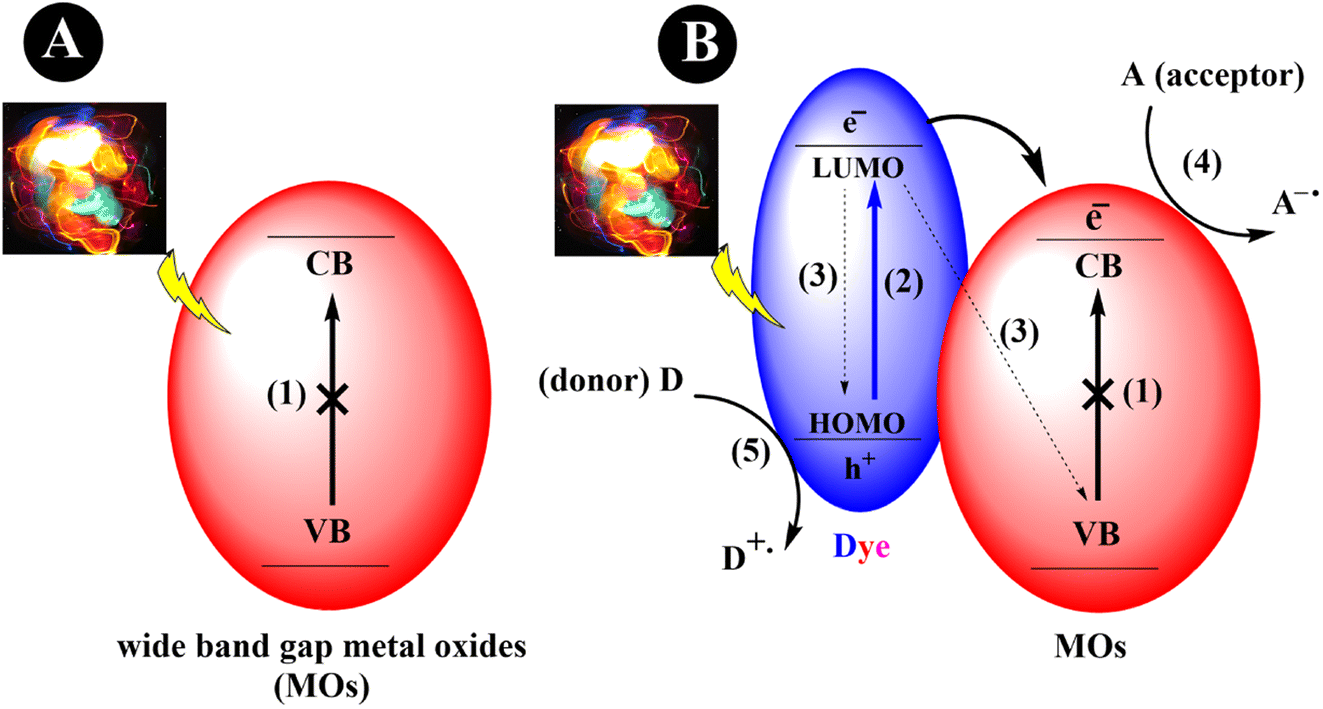
The process of dye sensitization involves the binding or adsorption of a dye or sensitizer, typically an organic dye, onto the metal oxide surface.21 When the dye is exposed to visible light, excitation of electrons from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) will occur, creating a vacant space (hole) in the HOMO. Consequently, this leads to the formation of electron–hole pairs. Following excitation, the electrons are transferred to the CB of the MOs. These electrons can further be transferred to an electron acceptor, such as a substrate, thereby indirectly extending the visible light photoresponse of wide bandgap semiconductors. Simultaneously, the oxidized form of the dye can be regenerated through the presence of a sacrificial electron donor (Scheme 1).
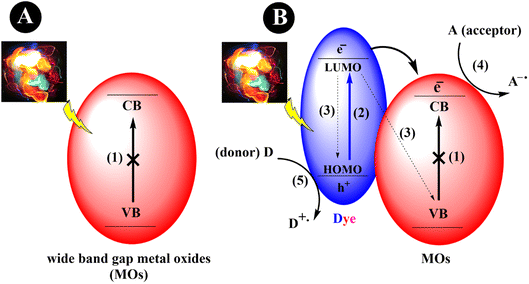 |
| | Scheme 1 An exemplary depiction of MOs photocatalytic mechanisms. (a) Wide bandgap metal oxide nanoparticles (1) cannot be activated by visible light. (b) Dye-sensitized metal oxide nanoparticles that can be activated under visible light irradiation. (2) Excitation of an electron from HOMO to LUMO. (3) Recombination of the generated electron–holes. (4) Reduction cycle, and (5) oxidation cycle. | |
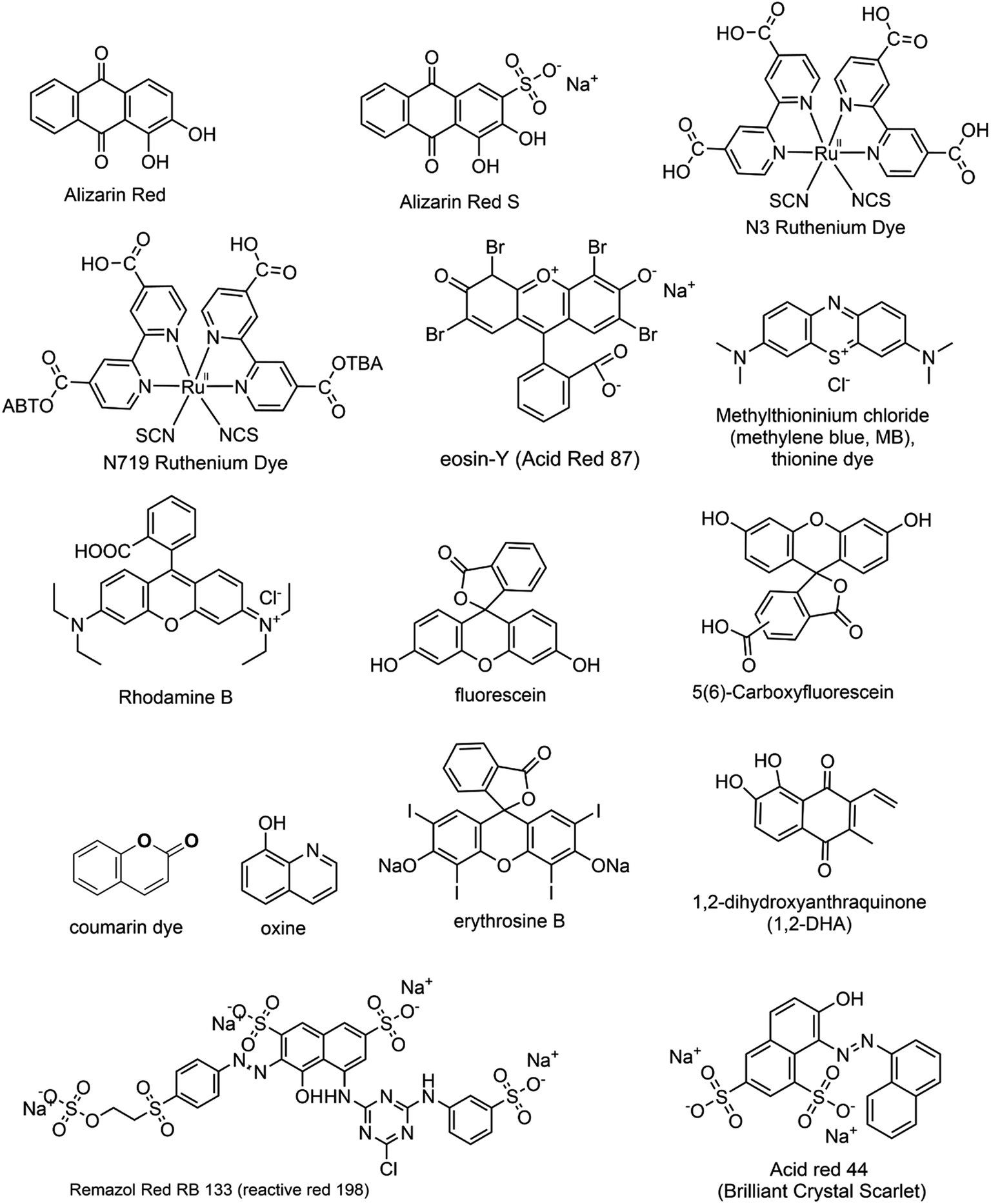
While dye sensitization offers several advantages, there are certain fundamental criteria to consider when selecting a suitable dye. These requisites include: (i) Broad Absorption Range: the dye should have a wide absorption range in the visible region to effectively harness visible light. (ii) Energy Level Alignment: the LUMO of the dye should be more negative than the conduction band (CB) of the semiconductor, while its HOMO should be more positive than the redox potential of the sacrificial electron donor. (iii) Photostability: the dye should exhibit high photostability to withstand prolonged exposure to light. (iv) Extended Excited State Lifetimes: the dye should possess extended excited state lifetimes for efficient electron transfer processes. These factors in selecting an appropriate dye are crucial for achieving optimal performance in dye-sensitized systems.22,23 Fig. 1 presents an array of diverse dye structures. These dyes have been utilized to sensitize metal oxide nanoparticles distinguished by their wide bandgap. Consequently, these sensitized nanoparticles become capable of being activated under visible light exposure.24–30
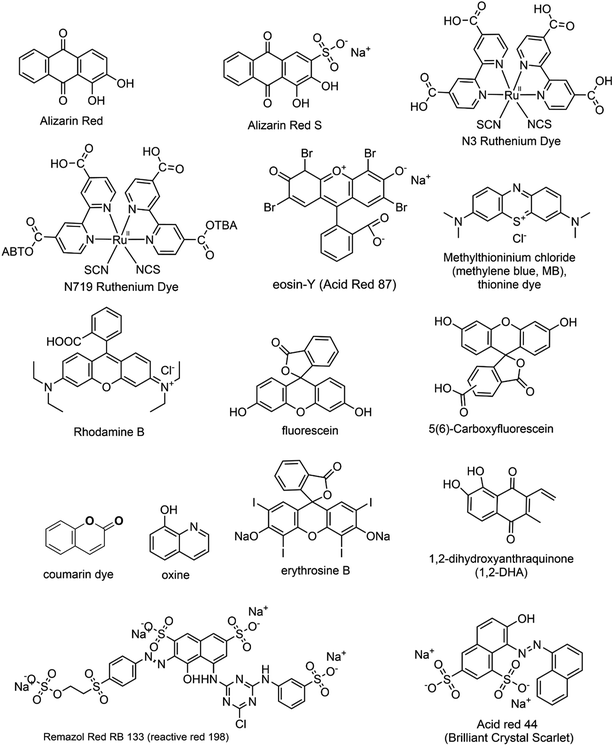 |
| | Fig. 1 The structural representations of several commonly used dyes employed as photosensitizers for the visible light activation of metal oxide nanoparticles. | |
This review article centers on the latest progress in utilizing visible light-induced dye-sensitized heterogeneous photocatalysis for environment-friendly organic transformations. It categorizes dyes employed for nanoparticle sensitization and delves into catalyst design, pathways of reaction mechanisms, and the distinct utility of dye-synthesized nanoparticles, especially in the context of organic transformations conducted under green conditions from 2008 to 2023. Based on the thorough research by the authors, this review article is positioned as a pioneering contribution in the current times. It comprehensively addresses all facets of dye-sensitized metal oxide design, delves into their applications in organic transformations, and offers in-depth insights into their underlying mechanisms. This review article not only fills the gap in the existing literature but also provides an excellent avenue for young researchers interested in this field to explore and engage.
2. Applications of dye-sensitized metal oxide nanoparticles in organic synthesis
2.1. Alizarin red (AR) and alizarin red S (ARS) dye-sensitized metal oxide nanoparticles
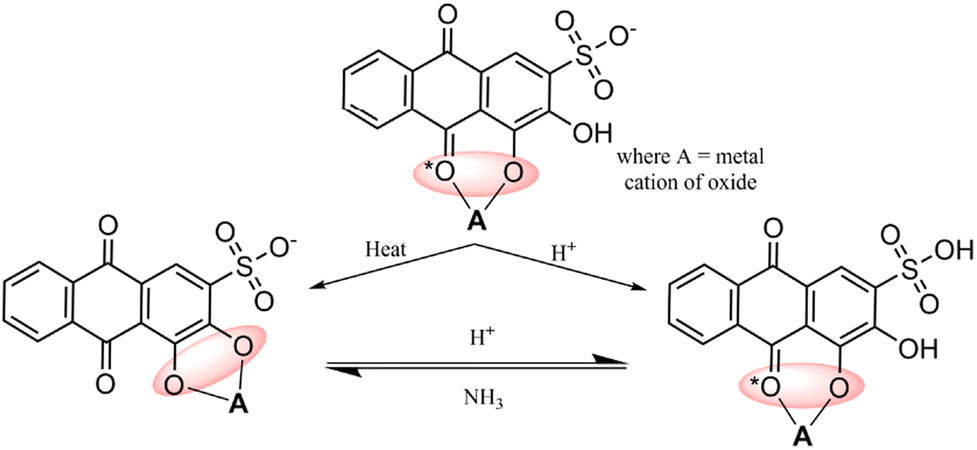
Alizarin Red S, an anthraquinone dye, features an anthraquinone core with one sulfonate group (SO3H) and two hydroxyl groups (OH) attached. Known for its red hue, it is a versatile dye and indicator. With robust absorption within the 500–600 nanometer range,31 it is applied in visible-light sensitization processes. It enters higher energy states upon absorbing photons, enabling efficient electron injection into metal oxide conduction bands and promoting charge separation for enhanced photocatalysis. ARS displays stability under specific conditions, which is ideal for prolonged sensitization. It binds to metal oxide surfaces via phenolic (OH) groups or phenolic (OH) and carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups (see Fig. 2),32 fostering a stable dye-metal oxide junction crucial for effective charge separation. Due to the remarkable characteristics exhibited by Alizarin Red (AR) and Alizarin Red S (ARS),33–35 these dyes have found extensive application as dye sensitizers for activating nanoparticles with wide bandgaps. This article highlights the latest advancements and applications of these dyes in organic synthesis.
O) groups (see Fig. 2),32 fostering a stable dye-metal oxide junction crucial for effective charge separation. Due to the remarkable characteristics exhibited by Alizarin Red (AR) and Alizarin Red S (ARS),33–35 these dyes have found extensive application as dye sensitizers for activating nanoparticles with wide bandgaps. This article highlights the latest advancements and applications of these dyes in organic synthesis.
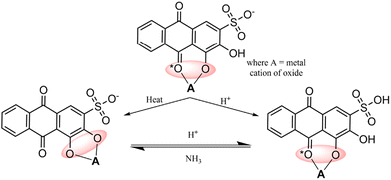 |
| | Fig. 2 Possible interactions between alizarin red S (ARS) and metal oxides. Reproduced from ref. 32 with permission, copyright 2019, Royal Society of Chemistry. | |
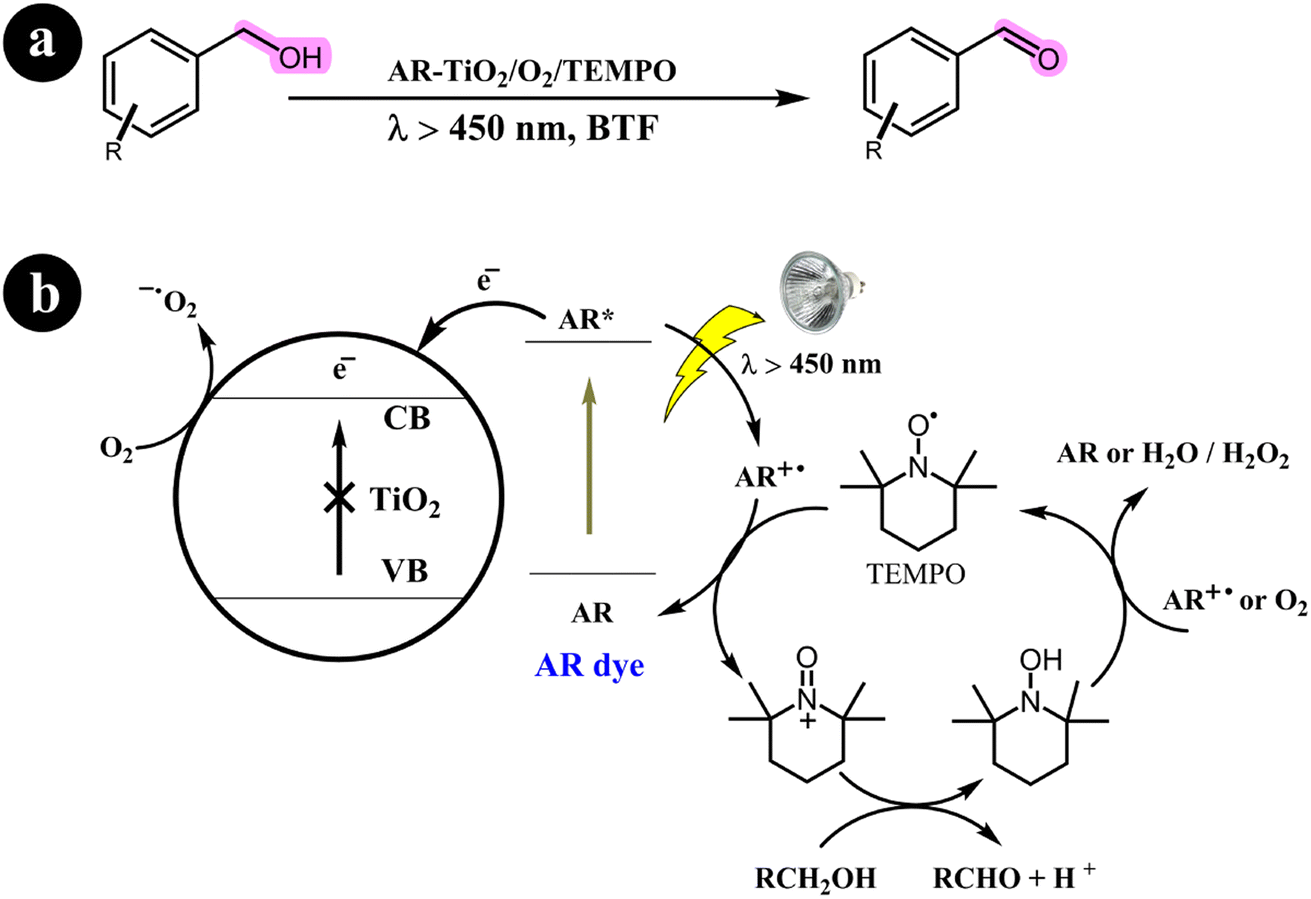
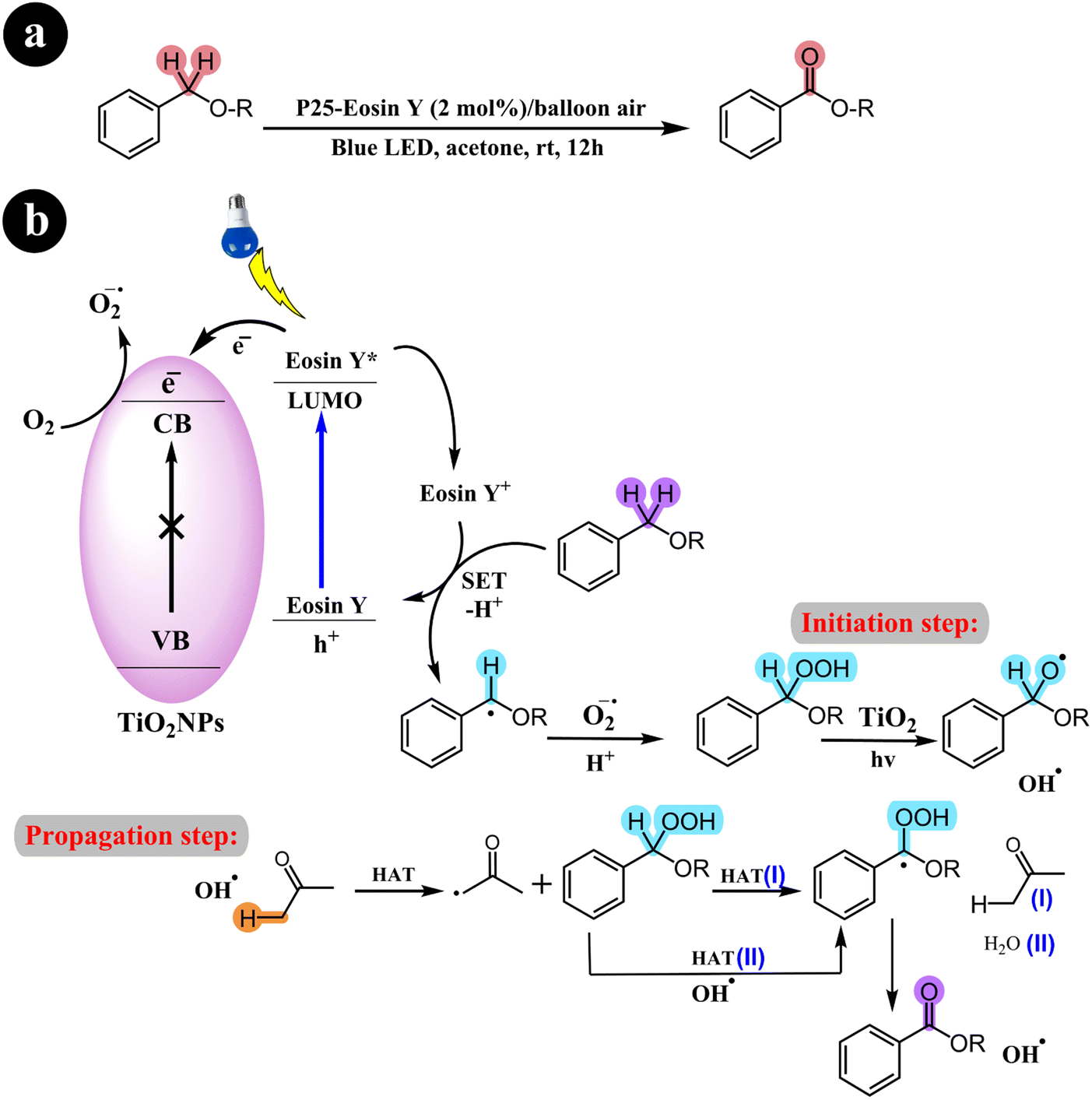
2.1.1. Oxidation of alcohols to aldehydes and ketones. Miao Zhang and colleagues (2008)36 have extensively documented the aerobic oxidation of alcohols, producing aldehydes or ketones with outstanding selectivity. The conversion rates observed range from 43% to 100%. This remarkable transformation is facilitated through the use of a visible light photocatalysis system employing dye-sensitized TiO2 and TEMPO. Benzo trifluoride (BTF) is employed as the solvent, while alizarin red acts as the photosensitizer. For a comprehensive overview of the mechanistic pathway and conversion reaction, refer to Scheme 2.
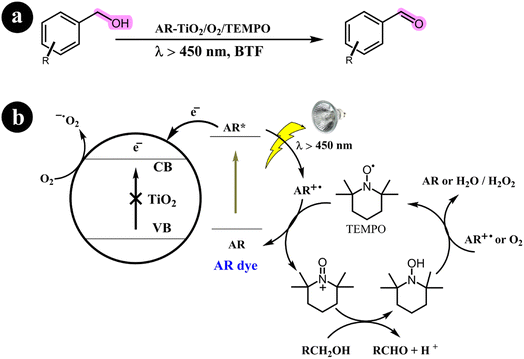 |
| | Scheme 2 The illustration of (a) the conversion of alcohols to their corresponding aldehydes and ketones and (b) the catalyst design and working mechanism. Reproduced with permission from ref. 36. Copyright 2008, John Wiley and Sons. | |
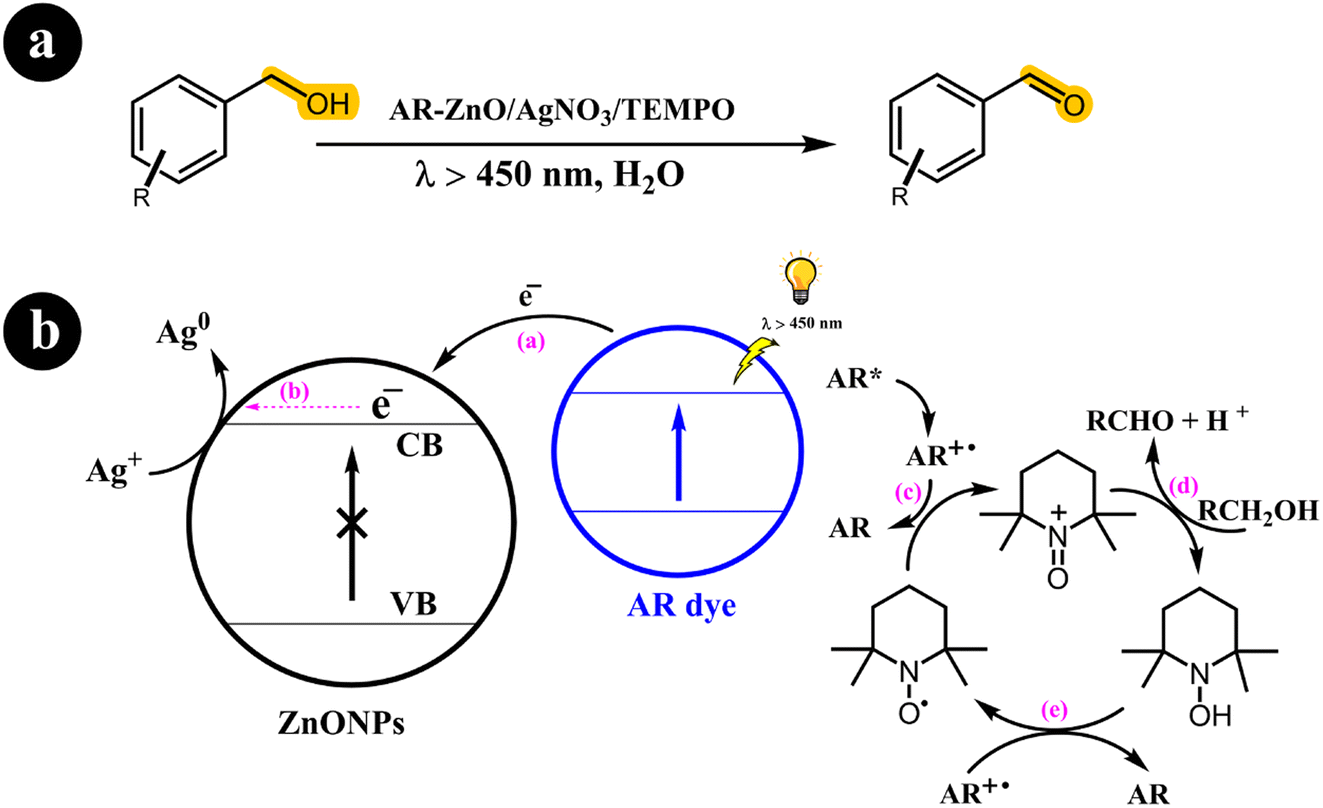
In their research publication, Vineet Jeena and Ross S. Robinson (2012) introduced an innovative method for converting alcohols into aldehydes (Scheme 3). Their approach involved employing a dye-sensitized metal oxide, silver salt, and TEMPO system in an aqueous solution.37
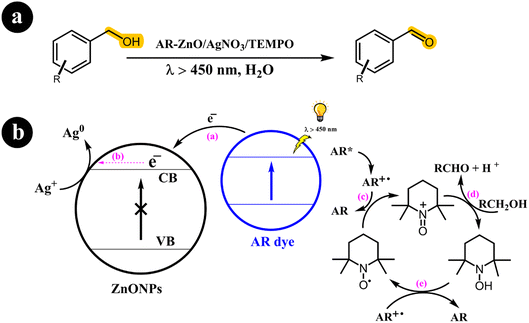 |
| | Scheme 3 The illustration of (a) reaction conditions for the conversion of alcohols to their corresponding carbonyl compounds and (b) the catalyst design and working mechanism. Reproduced from ref. 37 with permission, copyright 2012, Royal Society of Chemistry. | |
Sensitizing the semiconductor with alizarin red (AR) enabled activation via visible light exposure. Remarkably, they observed that, under these specific conditions, the dye, rather than the semiconductor, undergoes activation, effectively preventing the generation of hydroxyl radicals (OH). When illuminated by visible light, the activated dye transfers an electron to the conduction band (CB) of the semiconductor. Silver ions promptly utilize this electron, causing their reduction to metallic silver. Concurrently, the activated dye reverts to its original state, oxidizing TEMPO from its radical form to an oxoammonium salt. This oxoammonium salt acts as the active oxidizing agent for TEMPO, facilitating the conversion of the alcohol to its corresponding carbonyl derivative. As a result, hydroxylamine is produced, which can regenerate the TEMPO radical through oxidation by the dye radical.
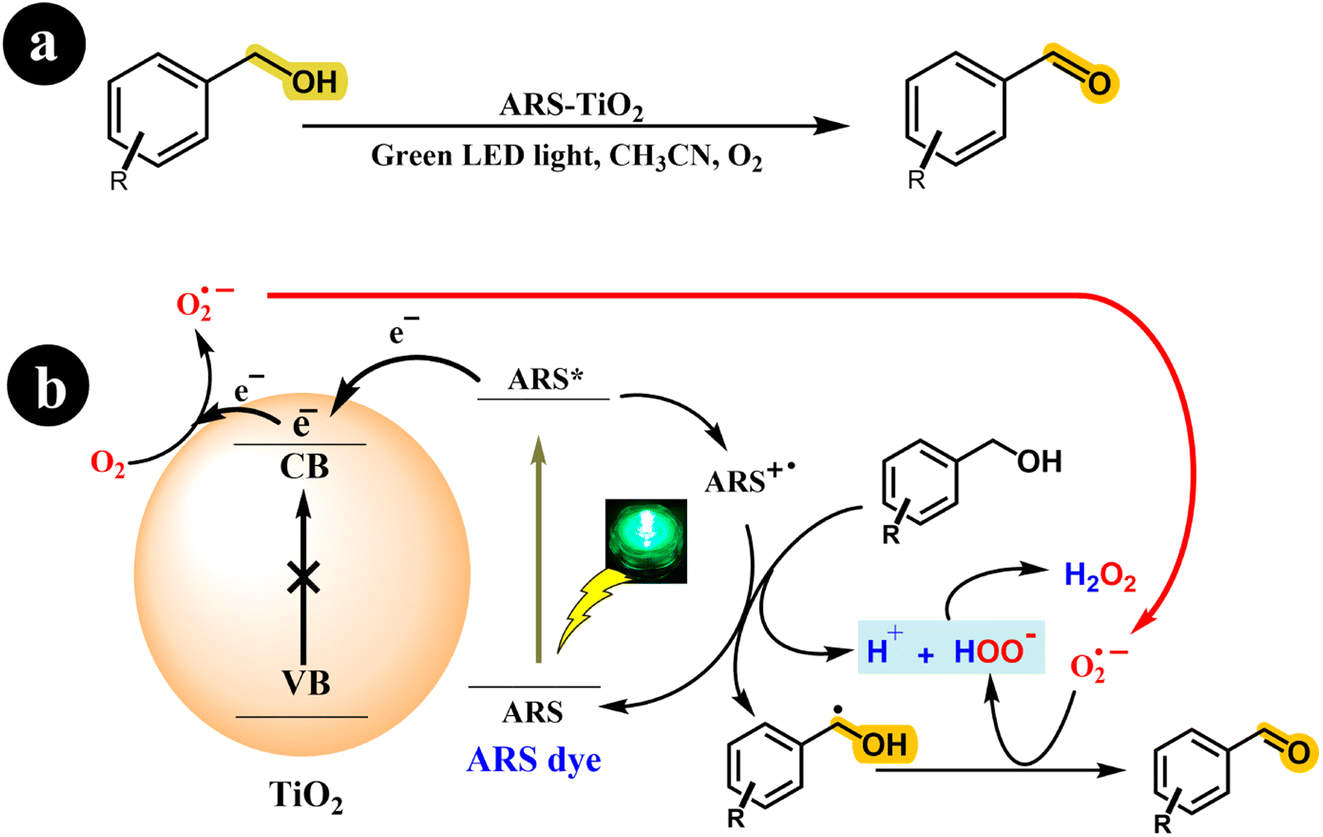
Investigating photoredox catalysts without a redox mediator, such as TEMPO, for the aerobic oxidation of alcohols to ketones and aldehydes presents an intriguing research avenue. Recently, Xia Li and his research team (2018) developed an ARS-TiO2 photocatalyst and effectively employed it to synthesize aldehydes and ketones from alcohols. This transformation was achieved by subjecting the reaction mixture to green LED light irradiation and atmospheric O2.38 Interestingly, their study revealed that rutile TiO2 exhibited a modest 10% conversion of benzyl alcohol, while anatase TiO2 displayed remarkable conversion rates and selectivity. The researchers conducted a series of experiments to gain further insights into the reaction mechanism, elucidating a possible mechanism depicted in Scheme 4.
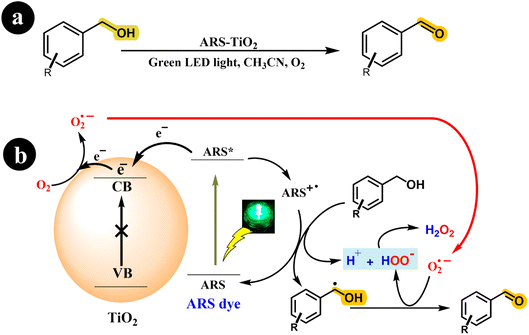 |
| | Scheme 4 (a) The reaction conditions and (b) the proposed mechanism for the selective oxidation of benzyl alcohol with air using dye-sensitized TiO2 photocatalysis under visible light irradiation. Reproduced with permission from ref. 38, copyright 2018, Elsevier. | |
To improve upon the previous findings of Ross S. Robinson and Timothy M. Underwood (2019), a catalysis system (ARS/ZnO/TEMPO/Ag–Na) was developed to facilitate the conversion of alcohols to aldehydes under visible light exposure.32 The researchers successfully demonstrated a modified procedure that significantly reduced the required amount of silver nitrate from 18 to 1 equivalent. Despite this reduction, the catalyst maintained its remarkable photocapacity, effectively mediating the oxidation of alcohols to aldehydes under visible light irradiation. The conversions achieved in the experiments surpassed 99%, highlighting the efficacy of this refined approach (Table 1).
Table 1 Utilization of alizarin red and alizarin red S dyes as photosensitizers in oxidizing alcohols to aldehydes and ketones under diverse experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
AR-TiO2/O2/TEMPO |
λ > 450 nm |
BTF |
10–22 |
7–80 |
Not given |
93–99 |
36 |
| 2 |
AR-ZnO/AgNO3/TEMPO |
λ > 450 nm |
H2O |
2 |
Not given |
12–99 |
80–100 |
37 |
| 3 |
AR-TiO2/O2 |
Green LEDs |
CH3CN |
2 |
45–100 |
Not given |
97–99 |
38 |
| 4 |
ARS-ZnO/TEMPO/Ag |
λ > 450 nm |
H2O |
7 |
10–99 |
Not given |
Not given |
32 |
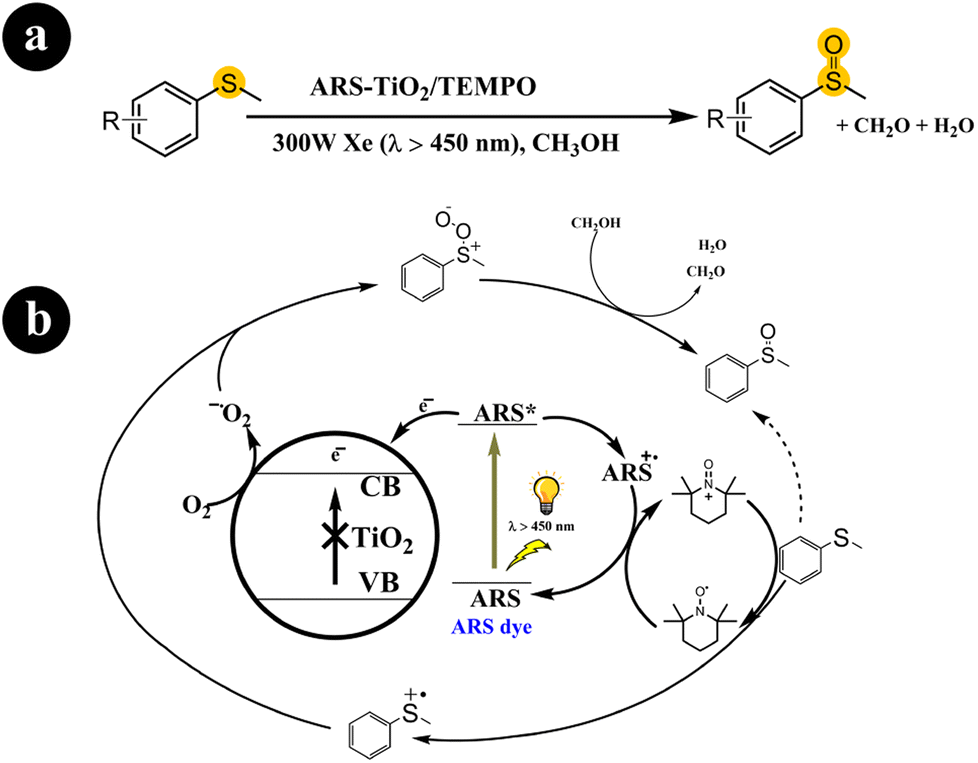
2.1.2. Oxidation of sulfides to sulfoxides. Sulfoxides play a crucial role as fundamental components in organic synthesis and have proven valuable across diverse application fields.39 Xianjun Lang and colleagues (2016) conducted a study where they successfully achieved the oxidation of organic sulfides to obtain desired products (sulfoxides) under mild conditions.40 They employed molecular oxygen as the oxidizing agent and harnessed visible light to initiate the catalytic process. They utilized dye-sensitized ARS in conjunction with TiO2 nanoparticles to facilitate the reaction. In their experimental setup, they prepared a photocatalyst that responded to visible light by binding ARS to the surface of TiO2 nanoparticles. This enabled the oxidation of organic sulfide molecules using O2, with the assistance of TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl) as a redox mediator and stabilizer. Xianjun Lang and colleagues conducted several control experiments to understand the underlying reaction mechanism better. Through these experiments, they observed that when the catalyst was exposed to visible light with a wavelength greater than 450 nm, ARS was excited, forming an excited state ARS*.Subsequently, a charge transfer occurred from ARS* to TiO2, causing an electron to be injected into the conduction band of TiO2 and forming ARS˙+. The electron in the conduction band of TiO2 facilitated the conversion of O2 to O2˙− (superoxide) while ARS+ oxidized TEMPO, generating TEMPO+. Concurrently, TEMPO+ interacted with thioanisole through a single-electron transfer process, forming an S-centered free-radical cation. At the same time, TEMPO+ converted to its initial form (TEMPO). The S-centered free-radical cation exhibited a preference for reacting with the O2˙− species formed in the conduction band of TiO2, leading to the production of sulfide peroxide and allowing for the subsequent formation of methyl phenyl sulfoxide in the presence of methanol as the solvent. For further details regarding the reaction conditions and mechanistic pathway, refer to Scheme 5.
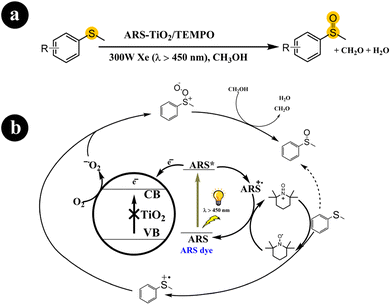 |
| | Scheme 5 (a) The reaction conditions for this process include specific parameters such as the type of catalyst (ARS-sensitized TiO2), the presence of a co-catalyst (TEMPO), the use of visible light as the energy source, and the involvement of molecular oxygen (aerobic conditions). (b) One potential mechanistic route for the visible light photocatalytic aerobic oxidation of thioanisole involves ARS-sensitized TiO2 and TEMPO. Reproduced with permission from ref. 40. Copyright 2016, John Wiley and Sons. | |
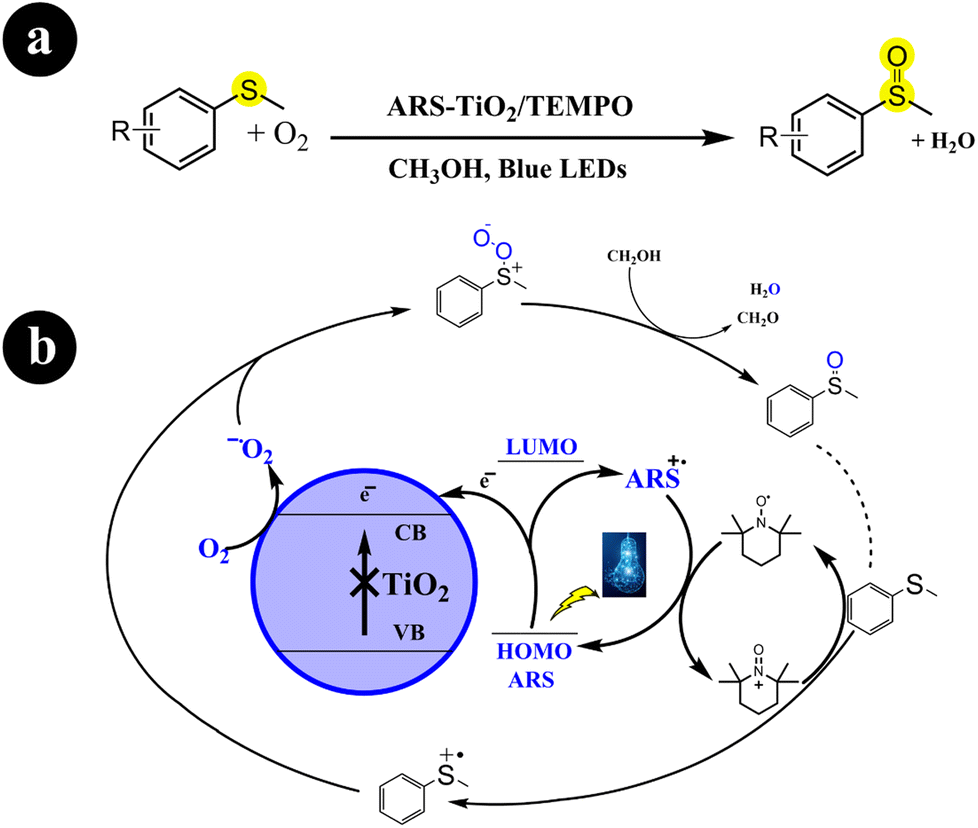
In two different studies conducted by Huimin Hao and colleagues (2018),41 Wan Ru Leow and Xiaodong Chen (2019),42 ARS-TiO2 photocatalyst was developed by creating a surface complex between TiO2 and alizarin red S dye. Subsequently, this fabricated catalyst was employed to catalyze the oxidation of sulfides to sulfoxides using molecular oxygen, facilitated by visible light irradiation. Their research effectively enhanced the selectivity of sulfoxide synthesis from sulfides by implementing an aerobic oxidation system composed of ARS-TiO2/TEMPO/O2, which was activated under blue light irradiation. Moreover, they discovered that the choice of solvent and the presence of TiO2 are crucial factors that greatly enhance the selectivity. For enhanced comprehension of reaction conditions and mechanistic pathways, refer to Scheme 6.
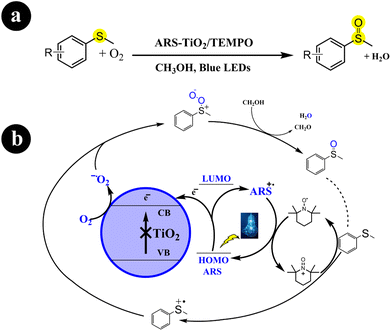 |
| | Scheme 6 (a) Reaction conditions and (b) possible mechanism for the oxidation of sulfides to sulfoxides by ARS-TiO2 under blue LED irradiation. Reproduced with permission from ref. 41. Copyright 2018, John Wiley and Sons. | |
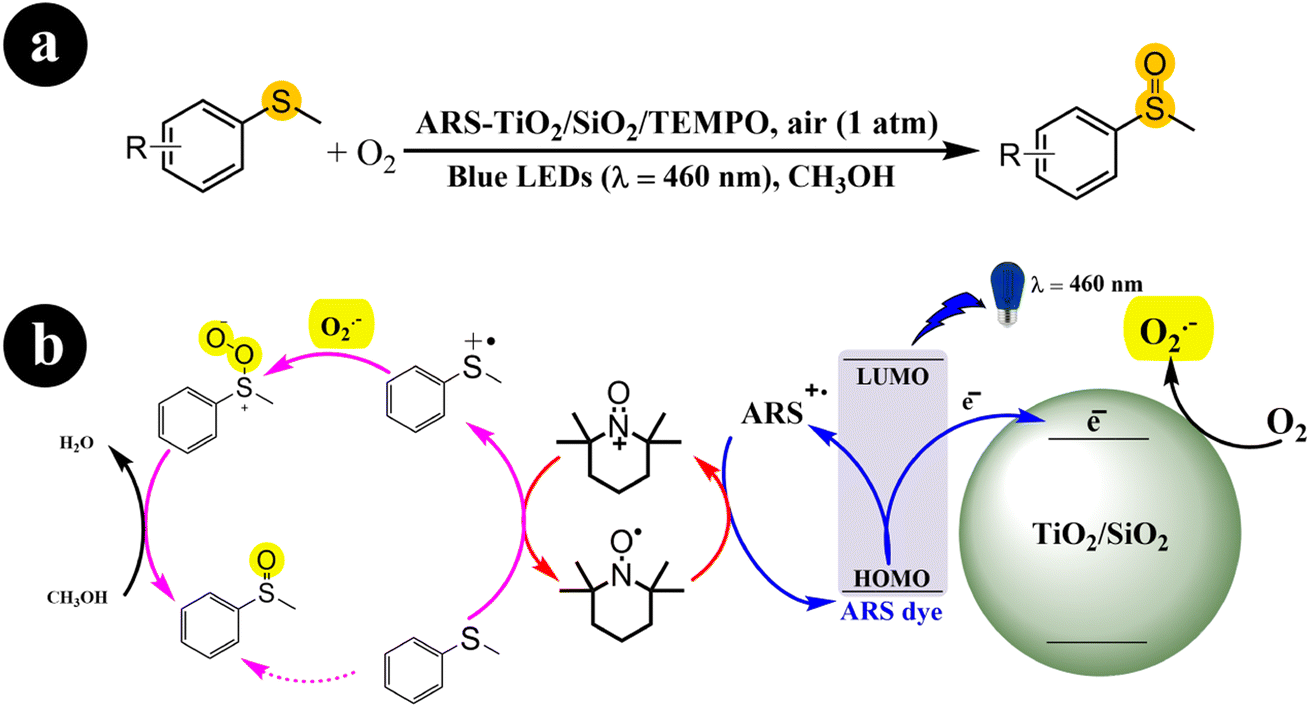
Fengwei Huang et al. (2021) developed a photocatalysis system (ARS-TiO2/SiO2) in collaboration with TEMPO. This system demonstrated exceptional conversion rates and selectivity for the aerobic oxidation of organic sulfides when exposed to visible light irradiation.43 The authors demonstrated the functionalities of ARS, TEMPO, and TiO2/SiO2 in their study. ARS acted as a visible light harvester, enabling efficient light absorption. TEMPO functioned as a redox mediator, facilitating the oxidation reactions. TiO2/SiO2 played a crucial role in improving the selectivity of the reactions. The researchers proposed a viable mechanism for the process (Scheme 7). It begins with the excitation of electrons from the dye by blue LEDs, which are then directly injected into the conduction band of TiO2/SiO2. Simultaneously, the dye (ARS) loses electrons and transforms into positive free radicals (ARS˙+). These free radicals are restored to a neutral state by reacting with TEMPO, producing TEMPO+. The TEMPO+ species activates methyl phenyl sulfide, converting it into a sulfur radical cation. In addition, a parallel process takes place where the previously formed charged sulfur radical reacts with O2˙− generated from the interaction between O2 and the previously injected electrons. This reaction leads to the formation of a persulfoxide compound. Finally, with the assistance of the solvent, methyl phenyl sulfoxide is produced as the end product of the process.
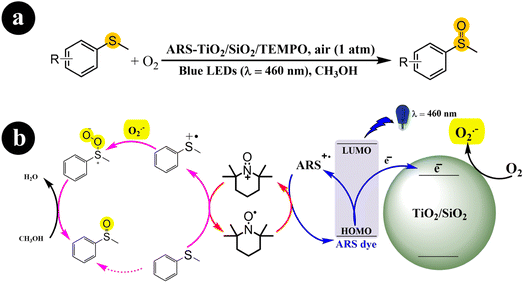 |
| | Scheme 7 (a) Reaction conditions for the aerobic oxidation of sulfides and (b) the proposed mechanism for ARS-TiO2/SiO2/TEMPO co-catalysis under blue LEDs irradiation. Reproduced with permission from ref. 43, copyright 2021, Elsevier. | |
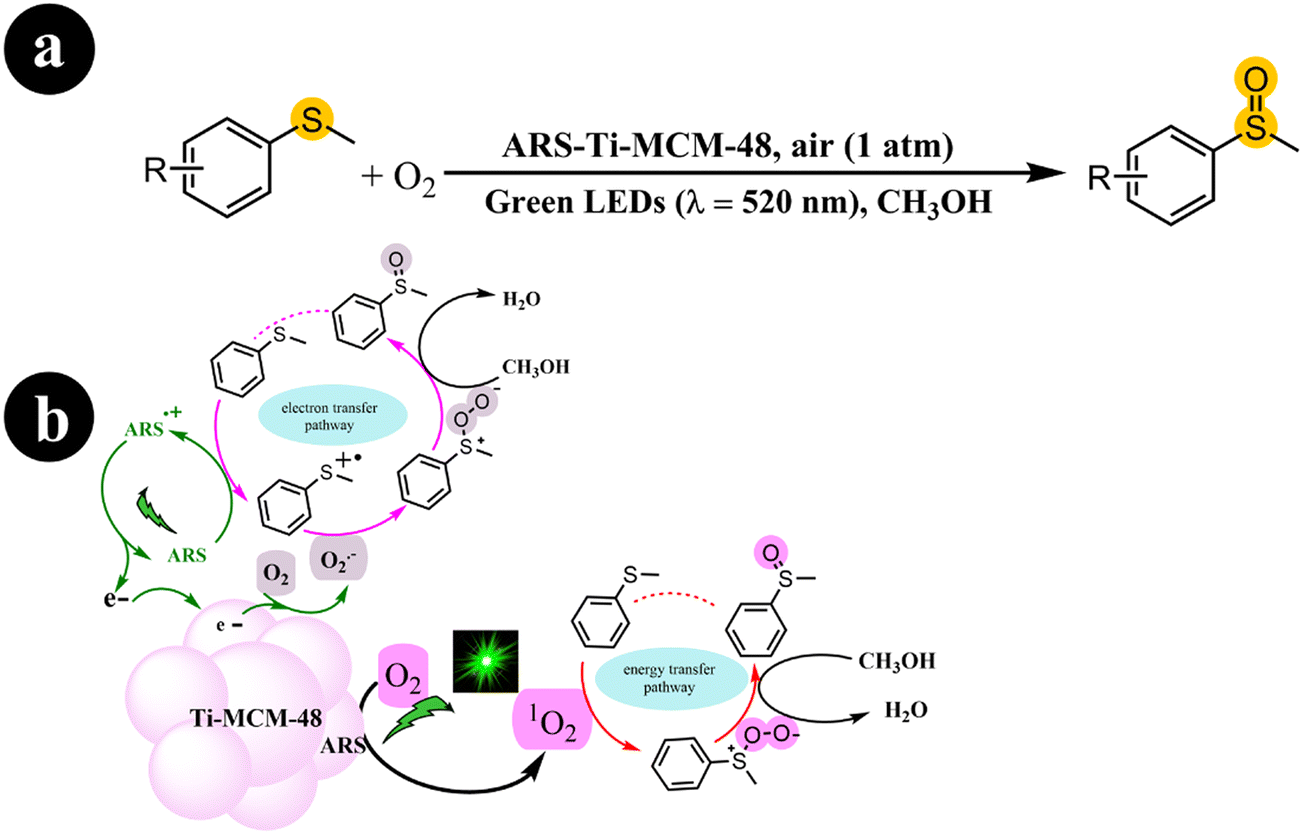
In another work, Fengwei Huang et al. (2022) developed a photocatalyst called ARS-Ti-CMS-48, demonstrating activity under visible light. This was achieved by incorporating ARS into Ti-CMS-48. The synthesis process involved the initial preparation of Ti-MCM-48, characterized by its well-structured cubic mesostructured and evenly distributed Ti sites. Subsequently, ARS was embedded into Ti-MCM-48, forming a visible light-responsive photocatalyst known as ARS-Ti-MCM-48.44
The photocatalyst obtained was subjected to green light activation for the selective aerobic oxidation of sulfides to sulfoxides. The authors demonstrated that the activation of O2 involved both energy transfer and electron transfer pathways. In addition, they emphasized the significant role of Ti sites in this process. The authors further illustrated that embedding ARS into the mesopores of Ti-MCM-48 effectively reduced the occurrence of photobleaching. The researchers conducted a series of control experiments to provide further evidence and elucidate the mechanistic pathway. Based on the obtained findings, they proposed a mechanism, as depicted in Scheme 8. The process commences by exciting ARS, resulting in the generation of ARS˙+ and the release of an electron (e−). Subsequently, sulfide reacts with ARS˙+, causing its return to the ground state and giving rise to a sulfur radical cation. Meanwhile, the adsorbed O2 molecules on the photocatalyst undergo reduction to form O2˙− due to electron migration originating from the photoexcitation process. With the involvement of O2˙−, the sulfur radical cation undergoes conversion into a persulfoxide, ultimately transitioning into a sulfoxide with the assistance of CH3OH. Furthermore, alongside the aforementioned pathway, the direct engagement of singlet oxygen (1O2) through an energy transfer pathway over ARS-Ti-MCM-48 also contributes to the formation of persulfoxide from sulfide. This involvement of 1O2 facilitates the subsequent transformation of the persulfoxide, leading to the desired product of sulfoxide (Table 2).
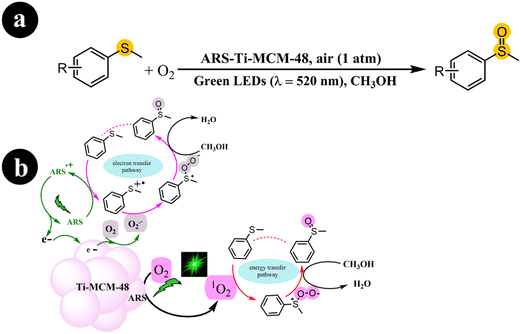 |
| | Scheme 8 (a) Reaction conditions for the aerobic oxidation of sulfides and (b) the proposed mechanism for ARS-Ti-MCM-48 catalysis under green LEDs irradiation. Reproduced with permission from ref. 44, copyright 2022, Elsevier. | |
Table 2 Utilization of alizarin red and alizarin red S dyes as photosensitizers in oxidizing sulfides to sulfoxides under diverse experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
ARS-TiO2/O2/TEMPO |
Xe lamp (λ > 450 nm) |
CH3OH |
2.5–12 |
57–84 |
Not given |
79–98 |
40 |
| 2 |
AR-TiO2/O2/TEMPO |
Blue LEDs |
CH3OH |
0.7–4 |
78–96 |
Not given |
82–95 |
41 |
| 3 |
ARS-TiO2/SiO2/TEMPO/air (1 atm) |
Blue LEDs (λ = 460 nm) |
CH3OH |
0.6–2.9 |
90–94 |
Not given |
89–98 |
43 |
| 4 |
ARS-Ti-MCM-48/air (1 atm) |
Green LEDs (λ = 520 nm) |
CH3OH |
0.6–2.4 |
51–94 |
Not given |
94–99 |
44 |
2.1.3. Oxidation of amines to imines. The selective aerobic oxidation of amines to imines represents a significant advancement in the field of organic synthesis, offering a more efficient and sustainable approach for the preparation of imines, which are valuable intermediates in pharmaceuticals,45 agrochemicals,46 and other fine chemical applications.In their study, Zhan Wang and Xianjun Lang (2018) presented a method that enables the selective oxidation of amines using the conduction band electrons of TiO2 as the sole catalyst for O2 activation and the oxidation reaction.47 The process takes place under visible light irradiation, utilizing dye molecules (ARS) bound to TiO2 to capture energy and inject electrons into the TiO2 conduction band, forming dye radical cations. Charge transfer from these dye radical cations to the amines leads to the conversion of TEMPO to TEMPO+, which acts as an oxidant capable of directly oxidizing amines to imines through a two-electron process. The regeneration of TEMPO is achieved by oxidizing TEMPO-H with superoxide, which is generated by the interaction between the available electrons in the system and molecular oxygen. Each component in the catalysis system plays a specific role: the ARS dye enables visible light absorption, TiO2 activates O2, and TEMPO facilitates oxidative transformations. For further information regarding reaction conditions and mechanistic insights, refer to Scheme 9.
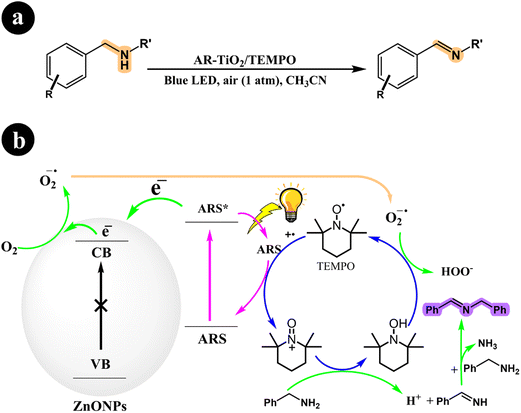 |
| | Scheme 9 (a) Reaction conditions for the aerobic oxidation of amines to imines by ARS-TiO2 using O2 with TEMPO under blue light irradiation and (b) mechanistic pathway. Reproduced with permission from ref. 47, copyright 2018, Elsevier. | |
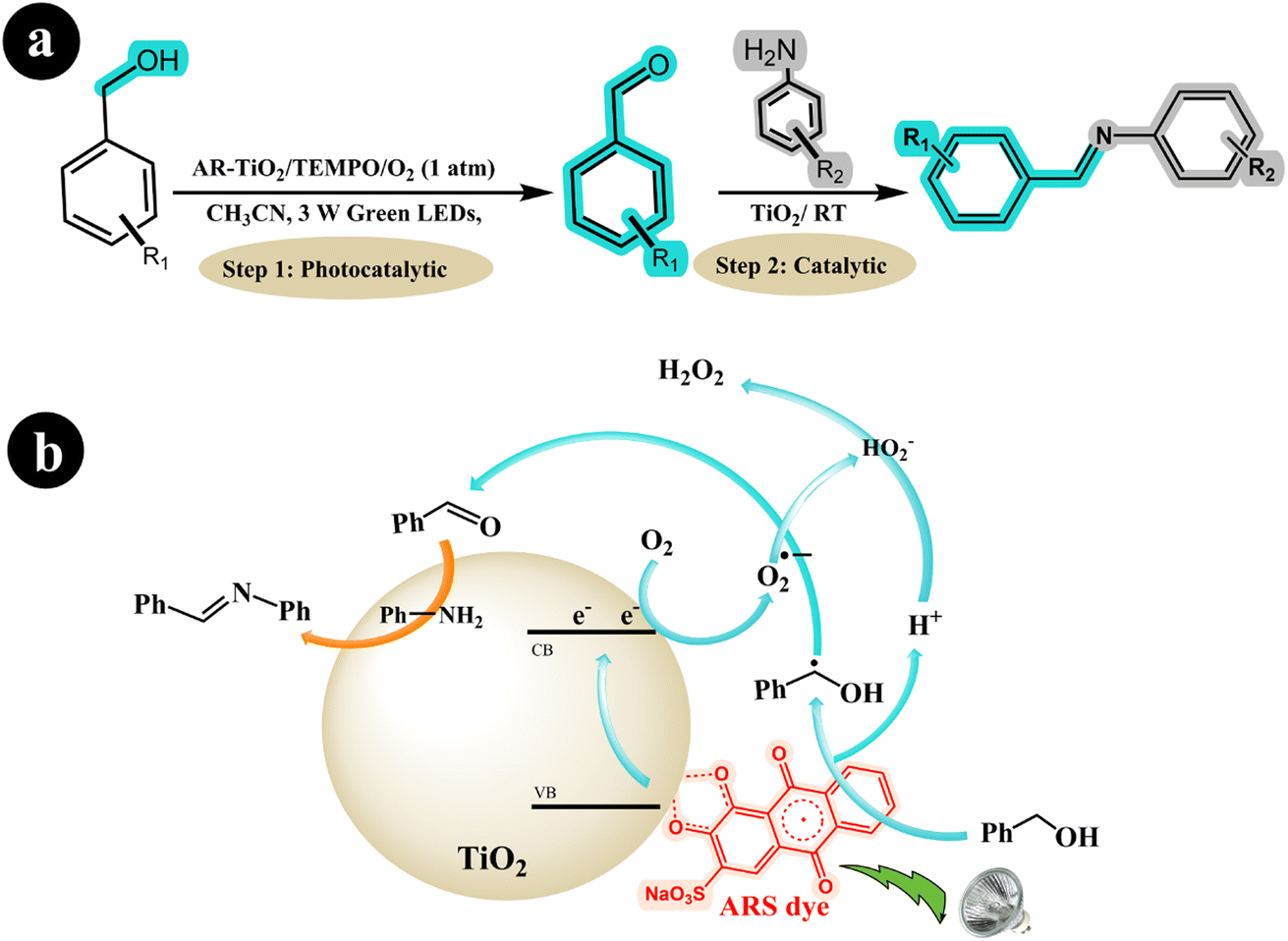
In a separate study, Hui Xu et al. (2019) employed ARS-TiO2 to synthesize imines from amines and alcohols, utilizing oxygen as an oxidizing agent while irradiating the reaction mixture with green LED light.48 The research group made a groundbreaking discovery by developing a one-pot protocol for the photoredox synthesis of imines from alcohols and amines. The protocol consisted of two steps: In the first step, highly selective oxidation of the alcohol to the corresponding aldehyde was achieved using the ARS-TiO2/O2/Green LED light photocatalysis system. Then, the formed aldehydes were coupled with amines in the second step using a TiO2 catalyst. This innovative approach allowed for the efficient and direct synthesis of imines in a single reaction vessel. In their study, the researchers harnessed the catalytic and photocatalytic properties of TiO2 to broaden the applicability of imines. According to the authors, the reaction mechanism can be described as follows: When exposed to visible light, the excited ARS* species transfers an electron to the conduction band (CB) of TiO2. This electron then reacts with O2, converting it into a superoxide radical anion (O2˙−). Simultaneously, the ARS species is regenerated from ARS* by accepting an electron from the alcohol, forming a benzyl alcohol radical and releasing a proton (H+). Subsequently, the radical is further oxidized to the desired aldehyde with the assistance of the O2˙− species. Following this step, the Lewis acid properties of the TiO2 surface facilitate the formation of imines from the amines and aldehydes (see Scheme 10).
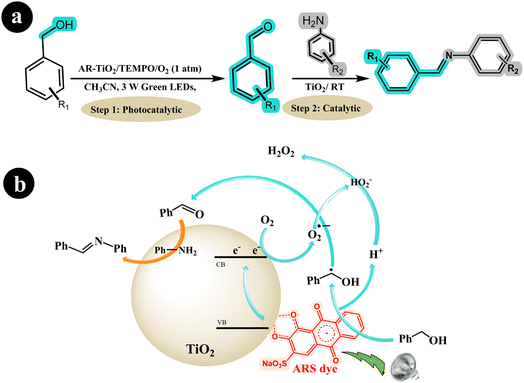 |
| | Scheme 10 (a) The reaction conditions and (b) the proposed mechanism for synthesizing imines from amines and alcohols with air using dye-sensitized TiO2 photocatalysis under green light irradiation. Reproduced with permission from ref. 48, copyright 2019, Elsevier. | |
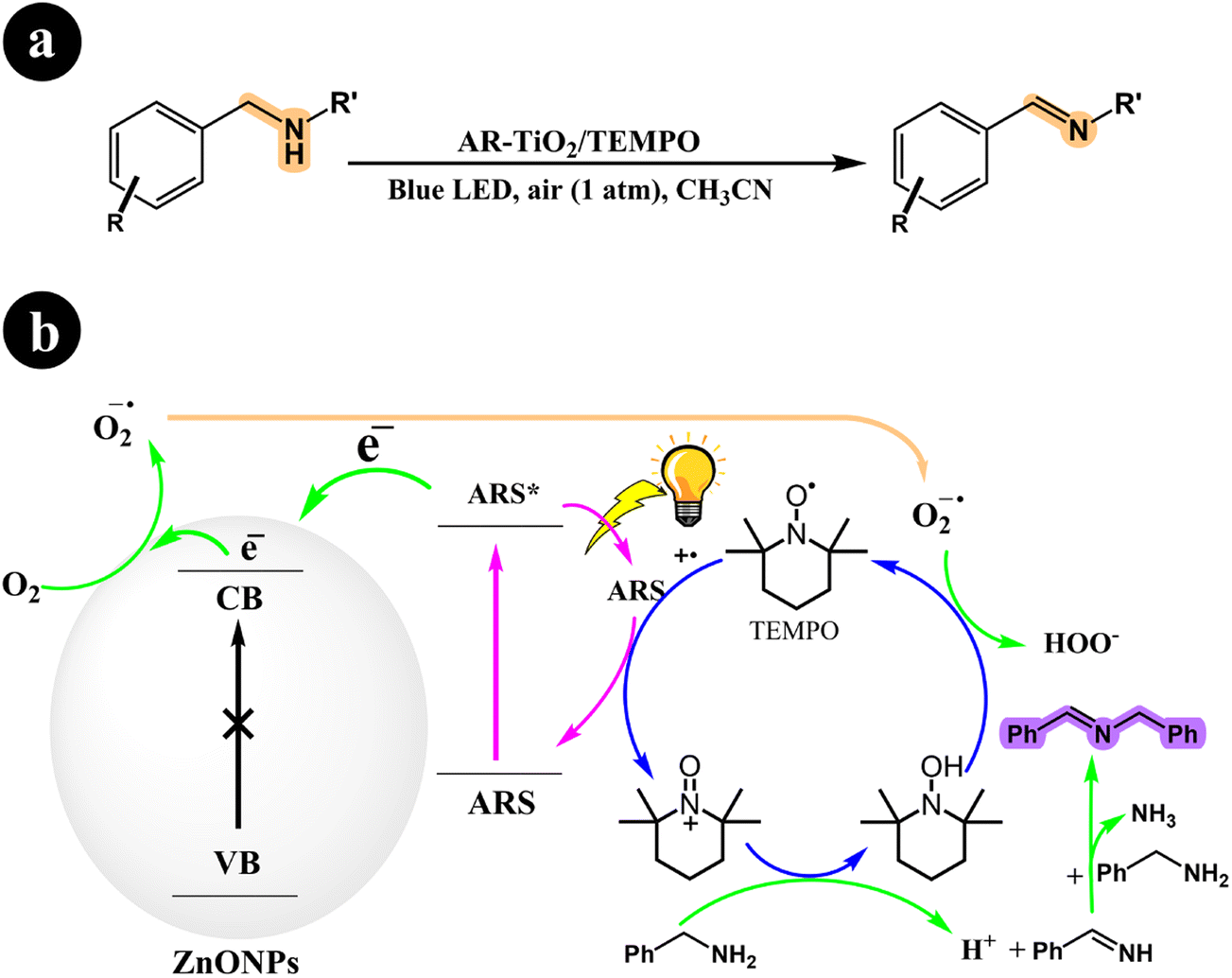
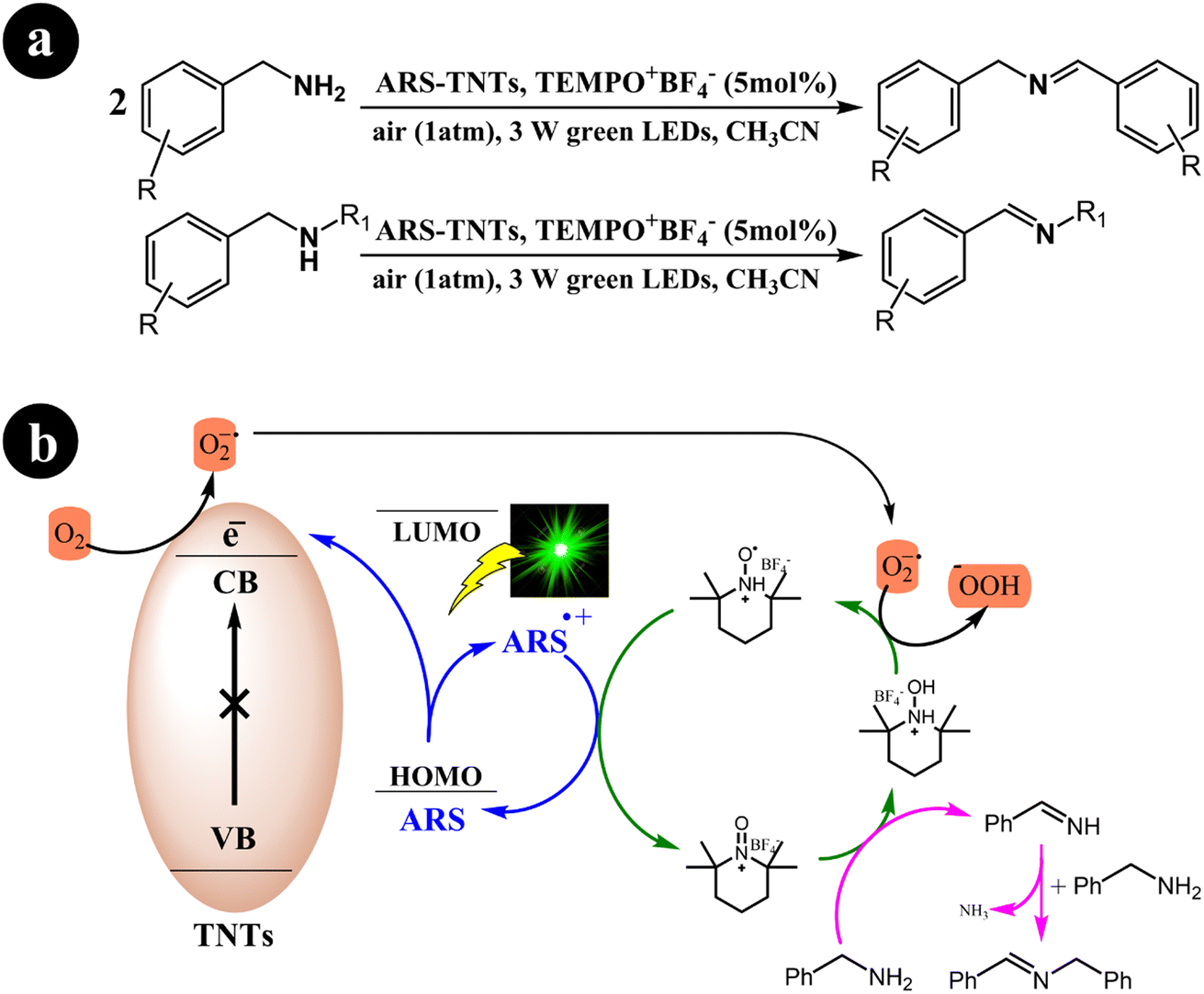
In their research, Jun Zhou et al. (2021) devised a collaborative system employing ARS, TiO2 nanotubes (TNTs), and an additional redox mediator to convert primary/secondary amines to imines.49 The authors emphasized that the crucial factor for successful cooperative photocatalysis lies in the electron transfer between the oxidatively quenched ARS-TNTs and the redox mediator. Utilizing green light, they effectively established a selective aerobic oxidation process for primary and secondary amines through the cooperative photocatalysis of ARS-TNTs with 2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate (TEMPO+BF4−). Significantly, the photocatalytic performance of anatase TNTs exhibited an impressive improvement of approximately 2.2 times compared to the P25 TiO2 precursor. The authors demonstrated that the enhanced polarity of TEMPO+BF4− rendered it more efficient in facilitating electron transfer when compared to (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO). Consequently, TEMPO+BF4− achieved over 1.5 times the activity of TEMPO in the selective aerobic conversions of amines.
The researchers proposed a catalytic process mechanism (Scheme 11) initiated by the excitation of ARS through green light irradiation. When ARS is adsorbed on the TNTs' surface, it can directly inject electrons from its HOMO into the conduction band of the TNTs. This results in the formation of a radical cation known as ARS˙+. Subsequently, the electrons in the conduction band of TNT facilitate the conversion of O2 into O2˙−. In addition, the introduced TEMPO+BF4− plays a crucial role in the oxidation of benzylamine to benzylideneamine through a direct two-electron transfer process, leading to the formation of TEMPOH2+BF4−. The desired product, N-benzylidenebenzylamine, is generated by coupling the previously formed benzylideneamine with the unreacted benzylamine. Finally, O2˙− oxidizes TEMPOH2+BF4− to yield TEMPOH+BF4−, simultaneously promoting the transition of the ARS radical cation to its ground state.
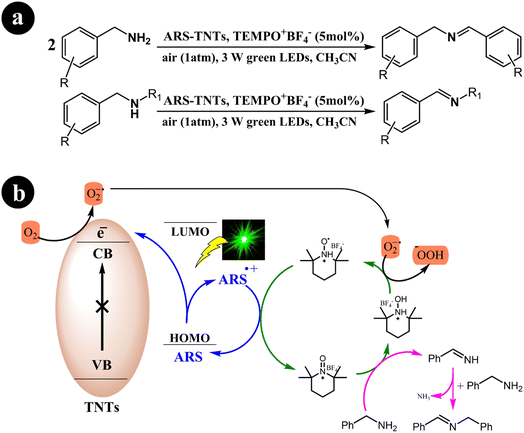 |
| | Scheme 11 (a) reaction conditions for the aerobic oxidation of primary and secondary amines, and (b) mechanistic study for the co-catalyst utilized under green light irradiation. Reproduced with permission from ref. 49, copyright 2021, Elsevier. | |
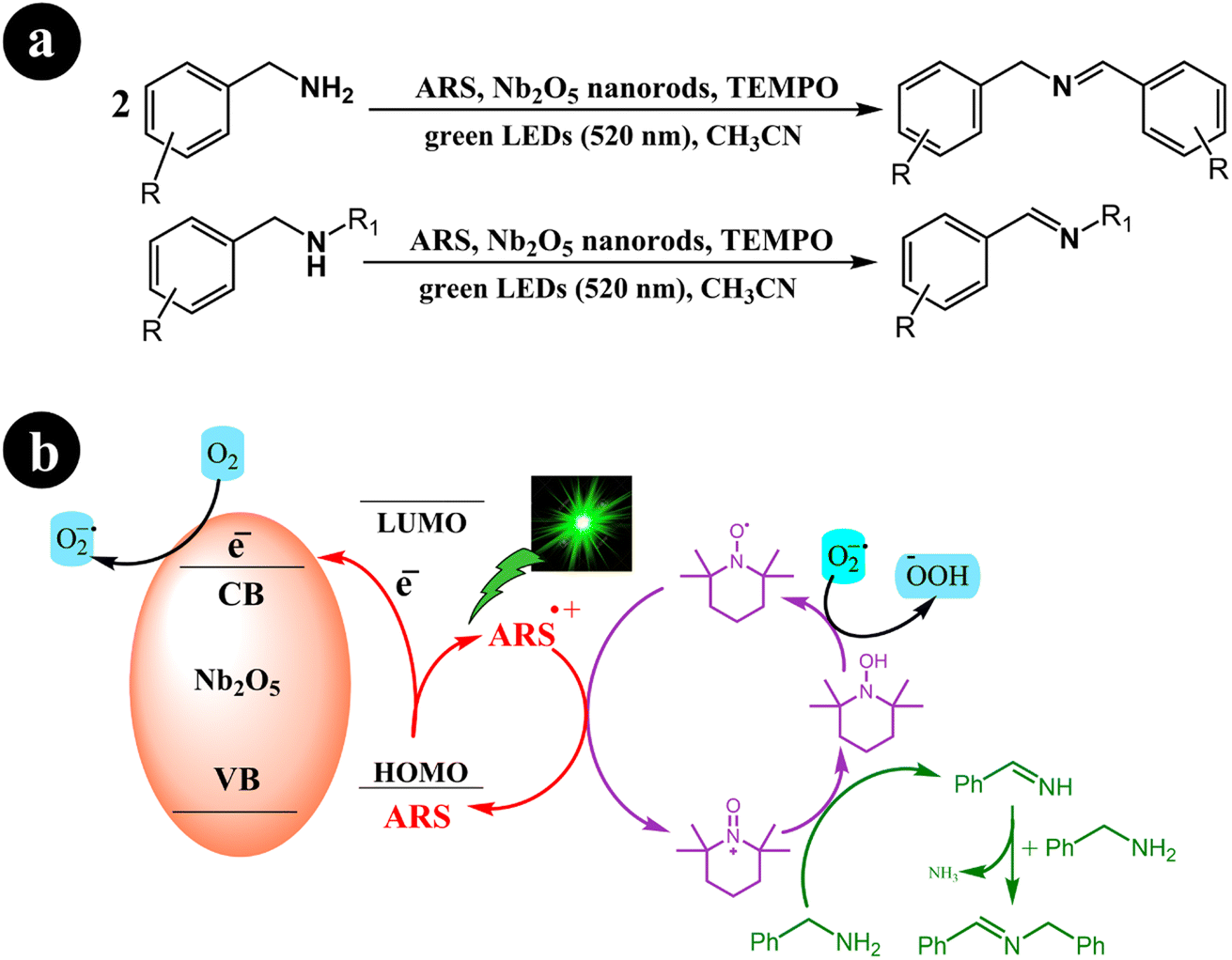
Xiaoming Ma et al. (2021) synthesized one-dimensional niobium pentoxide (1D) Nb2O5 nanorods. These nanorods were combined with ARS to create a highly efficient visible light photocatalyst known as ARS-Nb2O5. The researchers utilized a fabricated dye-synthesized photocatalyst with TEMPO for the selective aerobic oxidation of amines into imines. This process was carried out under green light irradiation and achieved an exceptional selectivity of 95% within 2 h. In their study, the authors compared the performance of the 1D Nb2O5 nanorods with that of 3D hierarchical Nb2O5. The results demonstrated that the 1D structure outperformed the 3D hierarchical Nb2O5, yielding approximately twice the desired outcome.50 The researchers proposed a mechanism to convert amines into imines, considering the optimal conditions described in Scheme 12. This proposed mechanism initiates the process by exposing the system to green light irradiation. During this step, the electron is directly transferred from the HOMO of the dye to the conduction band of Nb2O5 nanorods. This electron transfer leads to the creation of a radical cation (ARS˙+). Subsequently, ARS˙+ undergoes electron transfer with TEMPO at the interface of Nb2O5 nanorods, resulting in the regeneration of its ground state (ARS). At the same time, TEMPO undergoes a conversion into TEMPO+. Following these steps, TEMPO+ is an oxidizing agent for benzylamine, facilitating its conversion into benzylideneamine through a two-electron transfer process. Simultaneously, TEMPO+ experiences reduction and transforms into TEMPOH by accepting a hydrogen atom from benzylamine's Cα–H bond. Afterward, benzylideneamine undergoes coupling with unreacted benzylamine, leading to the desired product, N-benzylidenebenzylamine. In addition, the electrons present in the conduction band are captured by O2, resulting in the generation of O2˙−. This species can then combine with TEMPOH to regenerate TEMPO (Table 3).
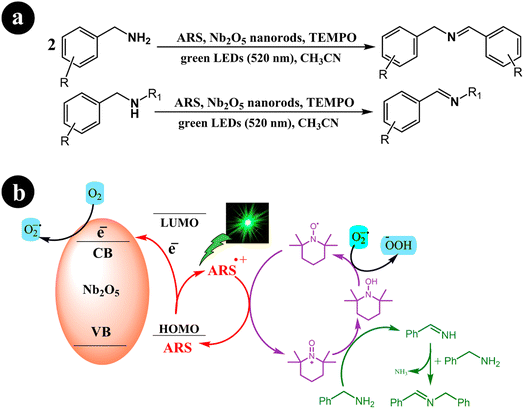 |
| | Scheme 12 (a) Reaction conditions for the aerobic oxidation of amines and (b) proposed mechanism for ARS-Nb2O5/TEMPO co-catalysis under green LEDs irradiation. Reproduced with permission from ref. 50, copyright 2021, Elsevier. | |
Table 3 Utilization of alizarin red and alizarin red S dyes as photosensitizers in the process of amine to imine conversion under diverse experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
ARS-TiO2/TEMPO/air (1 atm) |
Blue LEDs |
CH3CN |
1.5–5.5 |
54–99 |
Not given |
61–98 |
47 |
| 2 |
AR-TiO2/O2/TEMPO |
Green LEDs |
CH3CN |
0.5–3 |
Not given |
66–98 |
Not given |
48 |
| 3 |
ARS-TNTs/TEMPO+BF4−/air (1 atm) |
Green LEDs |
CH3CN |
0.6–2 |
64–97 |
Not given |
77–99 |
49 |
| 4 |
ARS-Nb2O5/TEMPO/aerobic O2 |
Green LEDs (λ = 520 nm) |
CH3CN |
1.5–2 |
61–99 |
Not given |
79–99 |
50 |
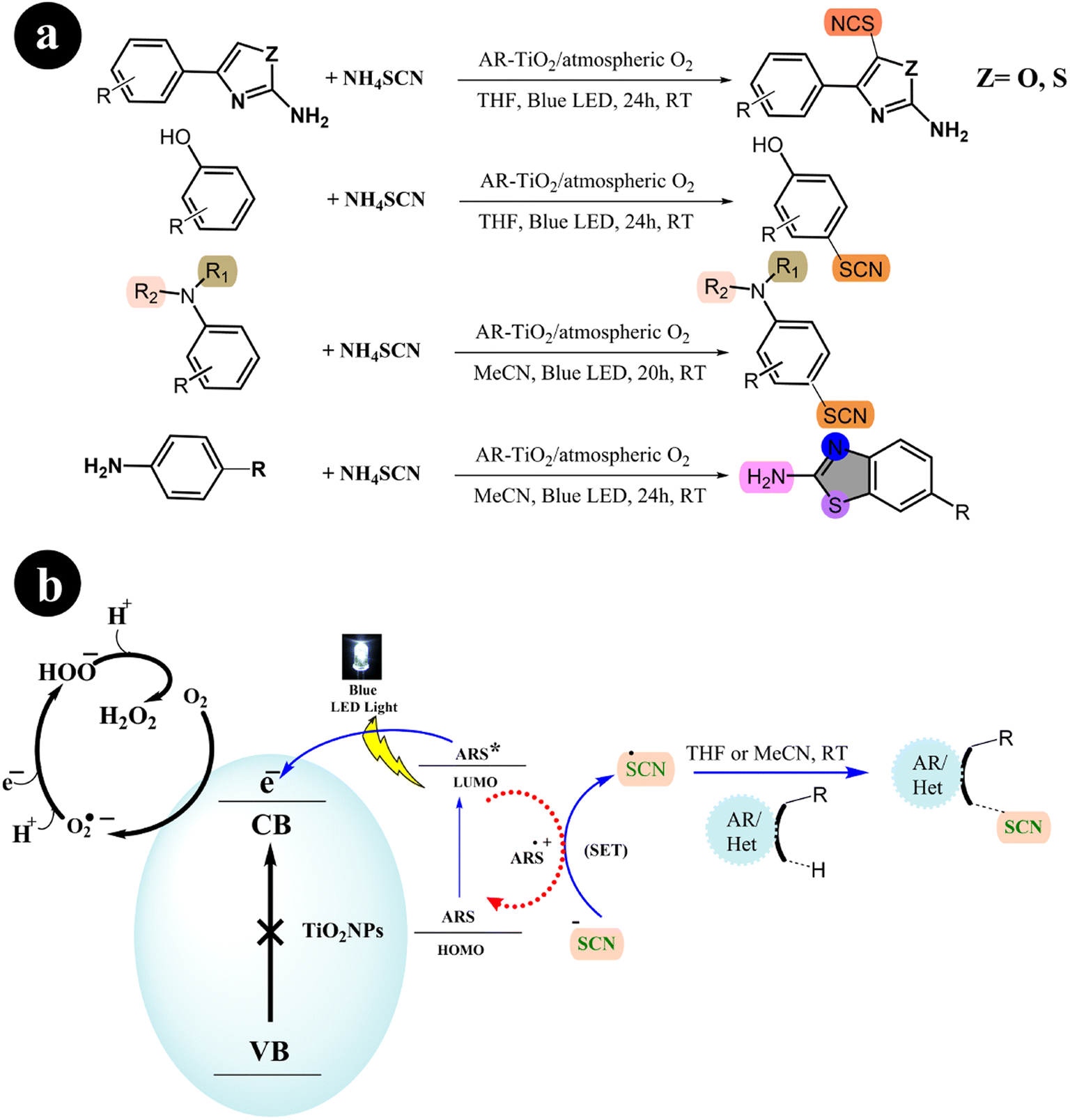
2.1.4. Thiocyanation and cyclization reaction. In their study, Mehdi Koohgard and colleagues (2020) successfully enhanced the photocatalytic performance of TiO2 nanoparticles by incorporating ARS as a sensitizer. This modification resulted in the creation of a photocatalyst (ARS-TiO2).25 The process of creating this catalyst was relatively straightforward: a mixture of ARS and TiO2 was stirred together at room temperature in the absence of light. Once synthesized, the catalyst exhibited activation capabilities when exposed to visible light. The researchers then utilized the fabricated catalyst to selectively modify sp2 C–H bonds through thiocyanation and cyclization reactions (Scheme 13). To achieve this, blue LED light was used as the irradiation source. The team found that their method could be easily scaled up and was compatible with various functional groups, demonstrating its versatility in practical applications (Table 4).
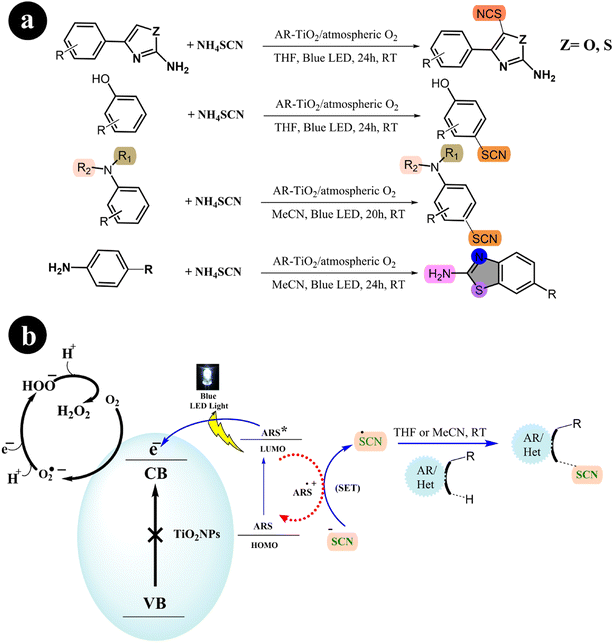 |
| | Scheme 13 (a) Reaction conditions and (b) catalytic activation of sp2 C–H bonds through thiocyanation and cyclization reactions facilitated by ARS-TiO2 under blue LED irradiation. Reproduced from ref. 25, with permission, copyright 2020, Royal Society of Chemistry. | |
Table 4 Utilization of alizarin red S dye as a photosensitizer in the thiocyanation of different substrates under specific experimental conditions
| Catalyst design |
Substrates |
Light source |
Solvent |
T [h] |
Yield [%] |
Ref. |
| ARS-TiO2/atmospheric O2 |
2-Amino-4-phenylthiazole derivatives |
Blue LEDs |
THF |
24 |
73–97 |
25 |
| Heterocyclic substrates containing nitrogen atom |
Blue LEDs |
THF |
24 |
0–89 |
| Phenol derivatives |
Blue LEDs |
THF |
24 |
0–92 |
| Aniline derivatives |
Blue LEDs |
CH3CN |
20 |
45–96 |
| 2-Aminobenzothiazole derivatives |
Blue LEDs |
CH3CN |
24 |
56–82 |
| Indole and pyrrole derivatives |
Blue LEDs |
THF |
20 |
77–98 |
2.2. Ruthenium dye complexes-synthesized metal oxide nanoparticles
Despite their significant cost, the exceptional optical properties of ruthenium dyes have spurred numerous researchers to utilize them as photosensitizers across various fields, including solar cells51 and photocatalysts.52 These dyes' remarkable attributes collectively amplify their effectiveness in sensitizing metal oxide surfaces for diverse applications, their absorption range, adjustability, prolonged excited state duration, redox characteristics, stability, and interaction with metal oxides.53
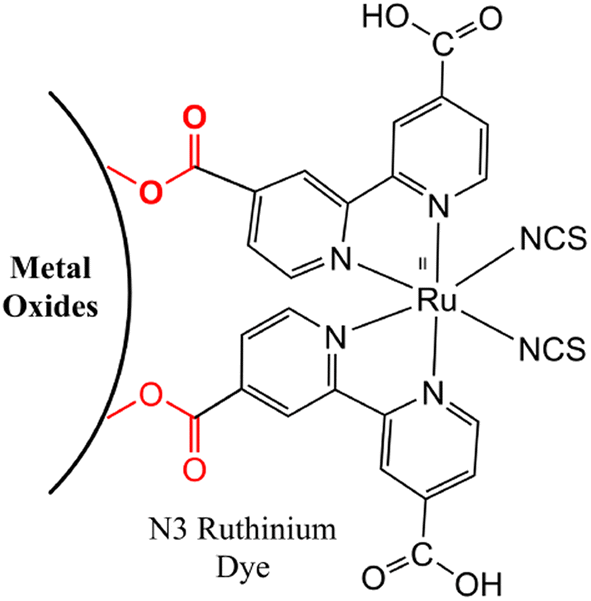
These dyes play a central role in capturing light energy and accelerating the conversion of photons into electrical current. Ruthenium dyes can bond with the surfaces of metal oxides through carboxylic acid groups, forming ester linkages (Fig. 3) that establish a strong connection between the dye and the metal oxide surface while facilitating effective electronic communication between the two components.54
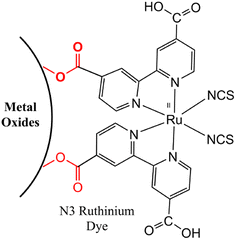 |
| | Fig. 3 Conjugation strategy between ruthenium dyes with the surface of metal oxides through ester linkages. Adapted with permission from ref. 54. Copyright 2006, John Wiley and Sons. | |
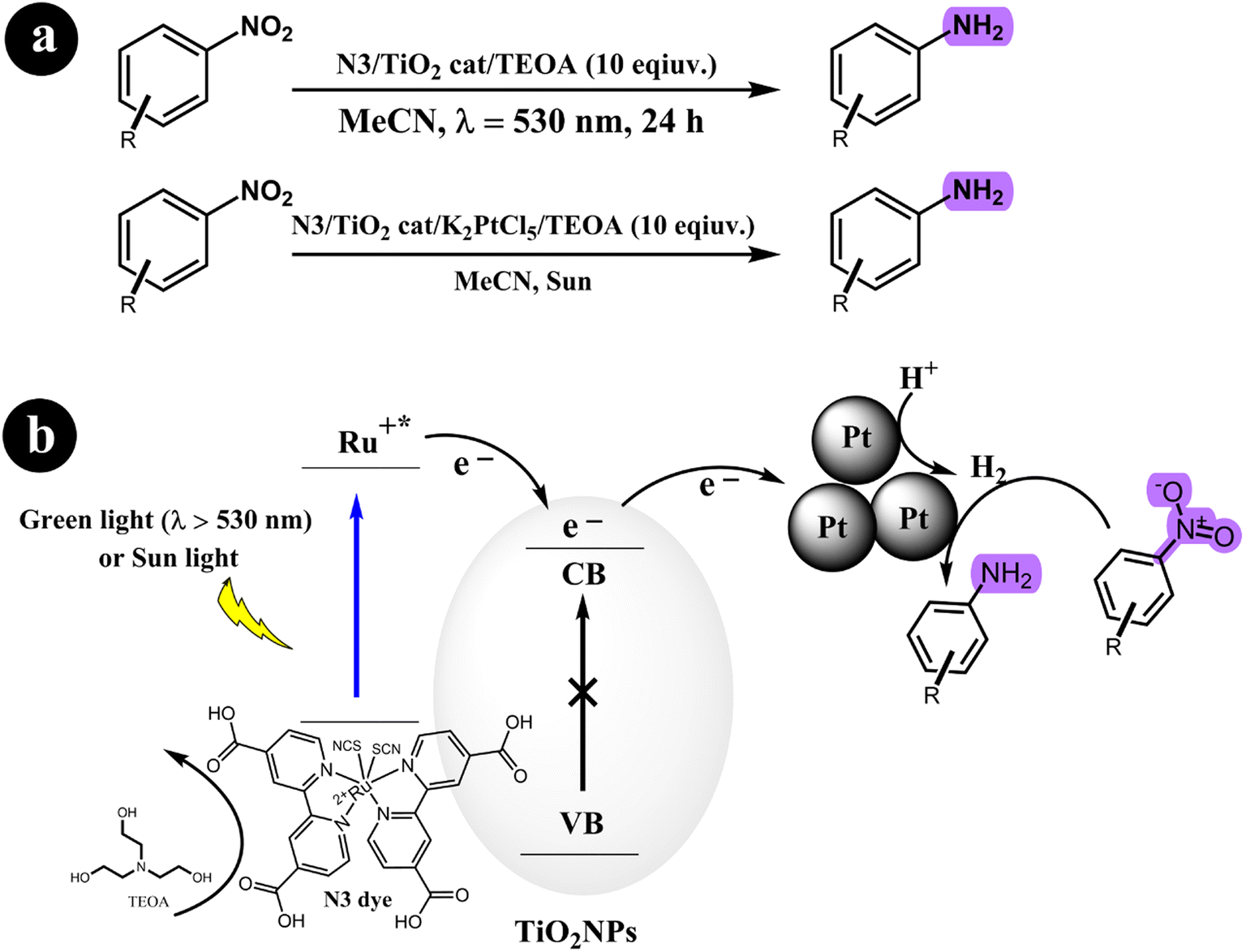
2.2.1. Reduction of nitroarenes to anilines. Stefan Fuldner et al. (2010)55 devised a novel method for reducing nitroarenes to anilines using a photocatalytic system. The system employed a ruthenium complex as a photosensitizer to activate titanium dioxide, forming Ru-sensitized TiO2 photocatalysts. This activation process was achieved by subjecting the photocatalysts to either green LED light or sunlight irradiation. To enhance the catalytic activity, minute quantities of transition metal salts were utilized as co-catalysts. These co-catalysts facilitated the formation of metal nanoparticles with a precise size distribution under specific experimental conditions. The resulting catalyst exhibited excellent performance in various organic synthesis reactions. Their study demonstrated that unmodified TiO2 resulted in a low conversion rate of approximately 2% for nitrobenzene when the reaction mixture was exposed to a light source with a wavelength of 530 nm. However, when TiO2 was modified with N3 without including transition metals, the conversion rate increased significantly to 39%. The researchers observed a remarkable enhancement in the catalytic activity by incorporating a small quantity of transition metal salts, achieving a conversion rate of up to 99%. Scheme 14 depicts the proposed mechanism for converting nitroarenes to anilines under the specified reaction conditions.
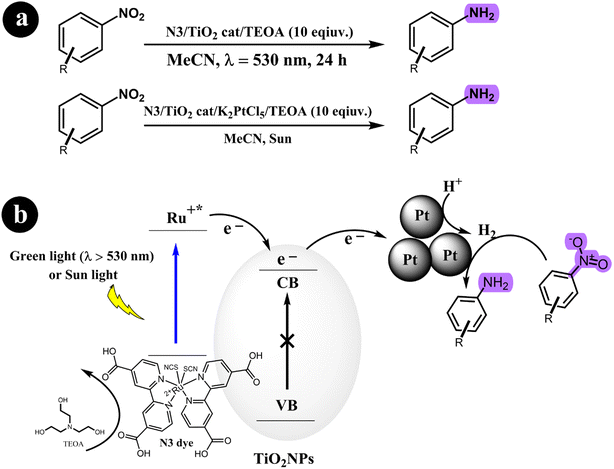 |
| | Scheme 14 (a) Reaction condition for the conversion of nitroarenes to anilines and (b) the proposed mechanism involving the reduction of nitroarenes using a green-light/sun light mediated process facilitated by dye-sensitized TiO2 and transition metal clusters. TEOA (triethanolamine) is also involved in the reaction. Reproduced from ref. 55 with permission, copyright 2010, Royal Society of Chemistry. | |
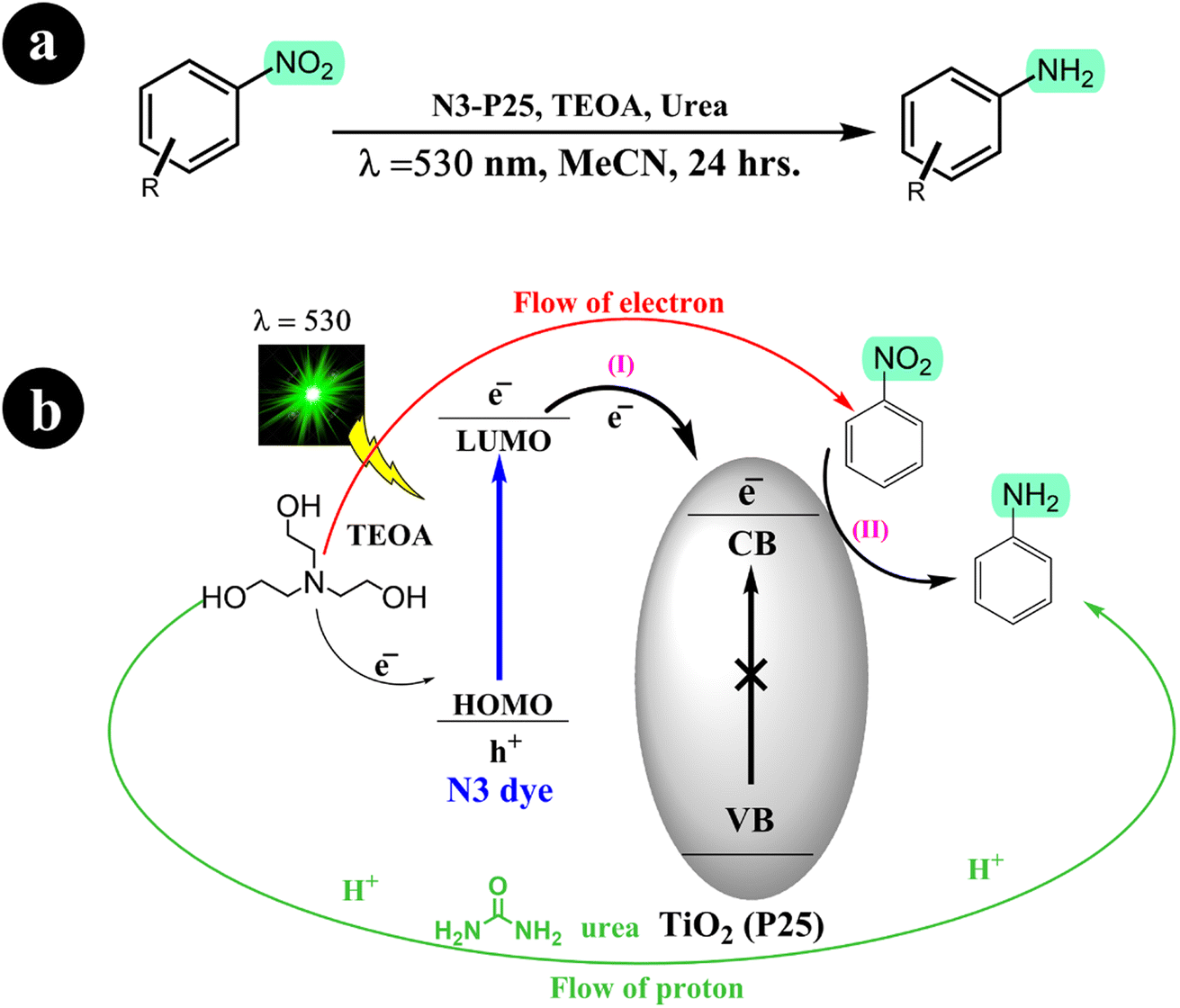
Building on their previous research concerning the reduction of nitroarenes to aminoarenes, Stefan Fuldner and his research team embarked on a new study involving the development of N3 dye-sensitized titanium dioxide (TiO2) for the conversion of nitroarenes into aminoarenes (2011).56 They modified commercially available TiO2 particles (P25) by incorporating the ruthenium complex N3. Utilizing green LED light (530 ± 10 nm) as the catalyst activation source, they successfully facilitated the photoreduction of nitroarenes in an acetonitrile solution with triethanolamine (TEOA), serving as the sacrificial electron donor. Notably, the catalytic system exhibited exceptional selectivity, producing solely the corresponding aminoarenes without any unwanted byproducts. In addition, the researchers observed that adding a small quantity of urea to the reaction mixture significantly enhanced the conversion rate. For more comprehensive information regarding the reaction conditions and mechanism, refer to Scheme 15.
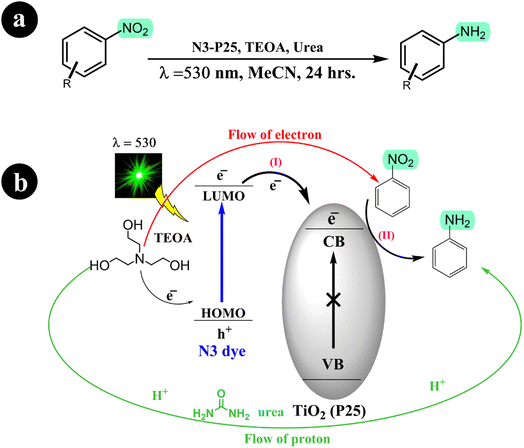 |
| | Scheme 15 (a) The conversion reaction and the conditions for the catalysis system; (b) the reduction of nitrobenzene through photocatalysis using dye-sensitized TiO2 is achieved in the presence of urea, which acts as a mediator for proton transfer. Reproduced from ref. 56 with permission, copyright 2011, Royal Society of Chemistry. | |
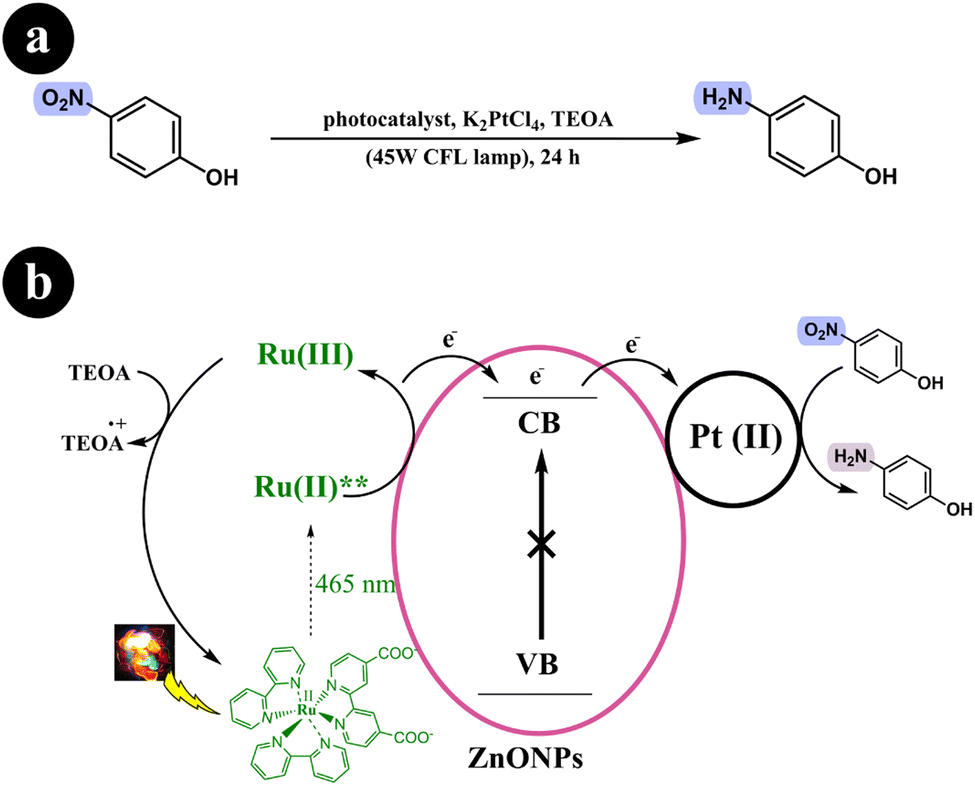
Sumana Bhar and Rajakumar Ananthakrishnan (2015) conducted a study to explore a photocatalytic system tailored to reduce p-nitrophenol using visible light. To achieve this, they utilized a hybrid catalyst comprising sensitized zinc oxide nanoparticles (ZnONPs) combined with a ruthenium(II)-complex, complemented by platinum(II) as a co-catalyst. The researchers conducted various parameter studies, including examining different concentrations of Pt(II) ions, to enhance the outcomes. The study yielded promising results, with a maximum reduction percentage of 55–60%.52 A series of experiments were conducted to gain insights into the mechanistic pathway. The findings revealed that the photoreduction of p-nitrophenol occurs via an electron transfer process. The researchers documented this significant observation as a part of their study (Table 5).
Table 5 Application of ruthenium-dye complexes as photosensitizers for the reduction of nitroarenes to their corresponding amines, conducted under various experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
N3-TiO2/TEOA/K2PtCl6 |
Green LEDs (λ > 530 nm) |
CH3CN |
24 |
39–99 |
6–91 |
Not given |
55 |
| Sun |
CH3CN |
11 |
4–80 |
Not given |
Not given |
| 2 |
N3-P25/TEOA/urea |
Green LEDs (λ > 530 nm) |
CH3CN |
24 |
Up to 99 |
Not given |
Up to 99 |
56 |
| 3 |
ZnO–Ru(II)/Pt(II)/TEOA |
CFL |
CH3CN |
24 |
55–60 |
Not given |
Not given |
52 |
Based on the control experiments conducted in their study, a proposed mechanism was formulated. Upon visible light irradiation, the Ru-dye absorbs green light and becomes excited, forming an excited singlet state. This excited singlet state undergoes inter-system crossing (ISC), forming a stable triplet state. Subsequently, the excited dye molecule injects electrons into the conduction band of ZnONPs. These electrons are then transferred to the surface of Pt(II), which facilitates the reduction of p-nitrophenol by abstracting a proton from the sacrificial donor, ultimately producing H2. To complete the catalytic cycle, the sacrificial donor, TEOA, reduces Ru(III) back to its initial Ru(II) state (see Scheme 16).
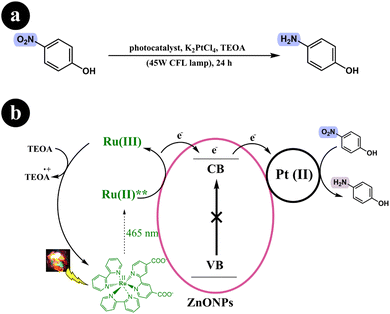 |
| | Scheme 16 Reaction conditions and (b) diagrammatic representation that elucidates the potential mechanism underlying the photoreduction process of p-nitrophenol into p-aminophenol. Reproduced from ref. 52, with permission, copyright 2015, Royal Society of Chemistry. | |
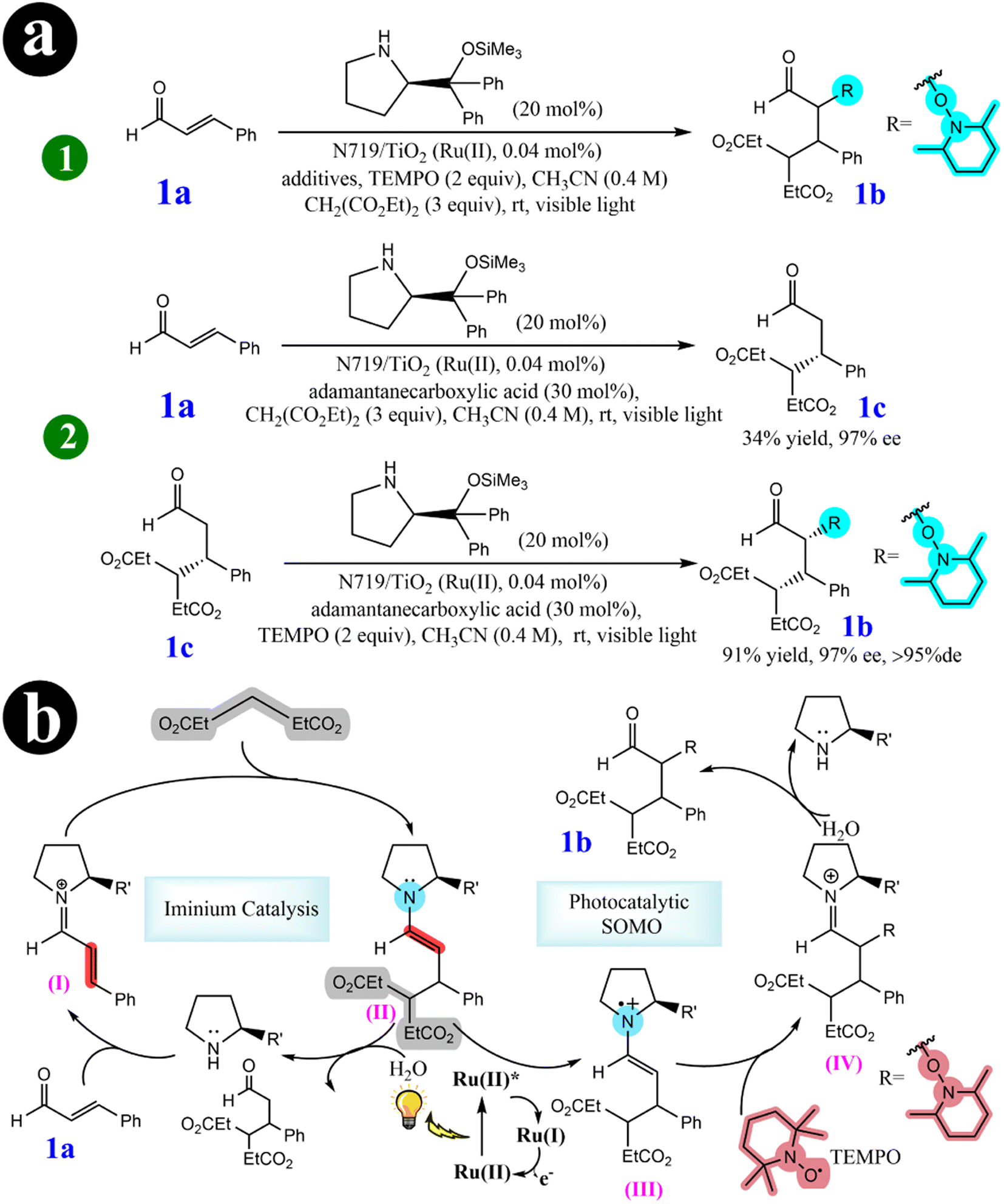
2.2.2. Tandem Michael addition/oxyamination of aldehydes. In their pursuit of advancing green organocatalytic processes, Hyo-Sang Yoon and his research group (2012)57 have been actively involved in developing ecofriendly oxidation protocols in enamine-mediated organocatalytic reactions. Their extensive investigations have made a significant breakthrough by identifying N719-sensitized TiO2 as an effective catalyst for organo photoreactions. This catalytic system triggers highly diastereoselective and enantioselective tandem iminium/SOMO (singly occupied molecular orbital) reactions, forming R,β-substituted aldehydes. These reactions are noteworthy because they occur under cost-effective, nontoxic, and visible light-induced photocatalytic conditions, underscoring their environmentally sustainable characteristics. To achieve a tandem iminium-SOMO reaction, researchers explored the combination of Michael additions of malonates to R,β-unsaturated aldehydes, followed by the R-oxyamination of the resulting β-substituted aldehydes, all in a single reaction pot under visible light. A series of experiments were conducted to investigate the influence of the dye-sensitized TiO2 photocatalysis system. Initially, the reaction mixture was catalyzed solely by the N719 dye, resulting in a maximum conversion rate of 62%. However, when TiO2 was used as the catalyst alone, only 11% of the desired product was obtained. The most promising outcome (yield = 80%) was observed when N719 dye was combined with TiO2. This highlighted the synergistic effect of the combined catalysts. Researchers have presented a highly efficient multicatalytic system for the tandem Michael addition/oxyamination of aldehydes under organophotocatalytic conditions. A chiral amine organocatalyst in conjunction with N719/TiO2 under visible light achieved favorable results. For a comprehensive understanding of the reaction conditions and the mechanistic pathway involved, refer to Scheme 17 and (Table 6).
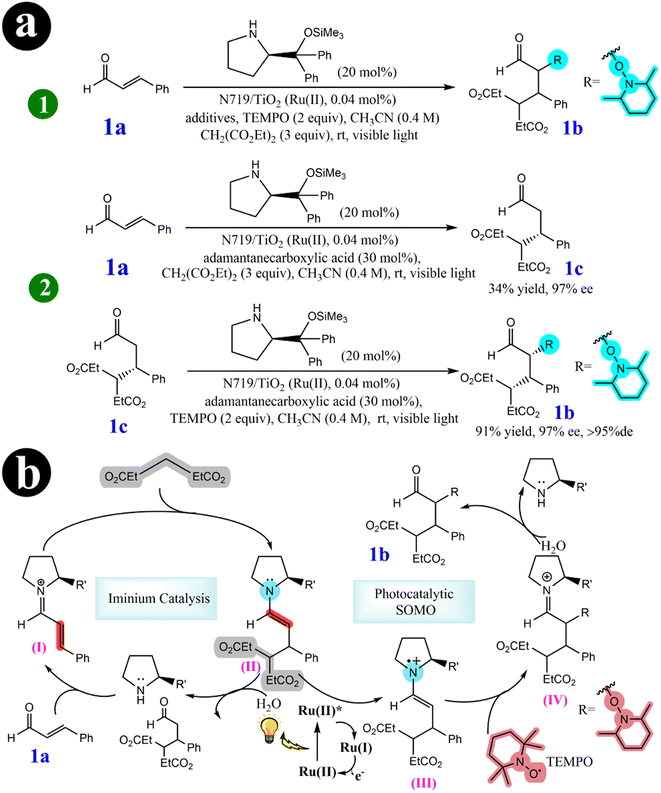 |
| | Scheme 17 Elucidation of (a) the tandem iminium/SOMO reaction is investigated using photocatalysts (1), and stepwise reactions are explored under organophotoreaction conditions (2). (b) The mechanism for the tandem Michael addition/oxyamination reaction. Reproduced with permission from ref. 57. Copyright 2012, American Chemical Society. | |
Table 6 Application of ruthenium-dye complexes as photosensitizers for the Michael addition/oxyamination of aldehydes, conducted under specific experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
N719-TiO2/TEPO/CH2(CO2Et)2 |
Visible light (8 W cool white fluorescent tubes) |
CH3CN |
18–21 |
Not given |
30–80 |
90–99 |
57 |
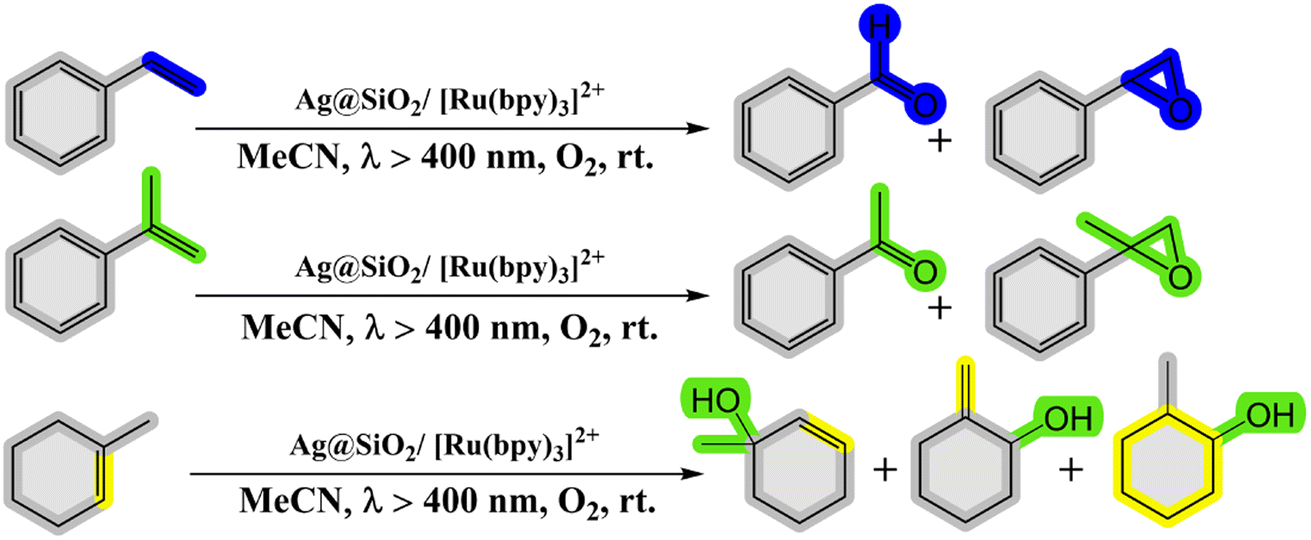
2.2.3. Oxidation of alkenes. Kohsuke Mori and his team (2010) have made significant strides in developing a remarkably efficient system for selectively oxidizing alkenes to yield epoxides and ketones. This system employs a photocatalytic configuration comprising a Ru(bpy)32+(bpy-2,2-bipyridine) complex supported on SiO2-encapsulated silver nanoparticles (Ag@SiO2). Operating under visible light and molecular oxygen, this setup exhibits groundbreaking advancements by harnessing the phenomenon of Localized Surface Plasmon Resonance (LSPR) demonstrated by the silver nanoparticles. The dye employed in the system effectively absorbs visible light, creating an excited singlet metal-to-ligand charge-transfer state (1MLCT). This state seamlessly undergoes intersystem crossing with exceptional efficiency, transitioning into a triplet MLCT state (3MLCT). The researchers successfully demonstrated that the Ag@SiO2/[Ru(bpy)3]2+ system, when subjected to visible light irradiation in the presence of oxygen at room temperature, acts as an exceptional photocatalyst for selectively oxidizing alkene derivatives into alcohols, aldehydes, ketones, and epoxides58 (as depicted in Scheme 18 and Table 7).
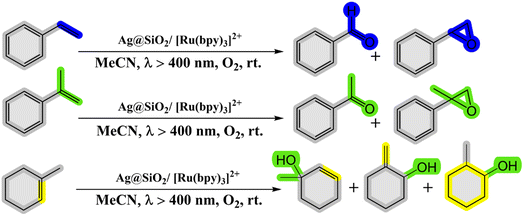 |
| | Scheme 18 Reaction condition of alkenes to the corresponding oxygenated products. Reproduced with permission from ref. 58. Copyright 2010, John Wiley and Sons. | |
Table 7 Application of ruthenium–dye complex as a photosensitizer for the oxidation of styrene derivatives, conducted under specific experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
Ag@SiO2/[Ru(bpy)3]2+ |
Xe lamp (55 W, λ > 400 nm) |
CH3CN |
24 |
Not given |
Not given |
Not given |
58 |
2.3. Eosin Y dye and 5(6)-carboxyfluorescein [5(6)-FAM]-synthesized metal oxide nanoparticles
Dyes incorporating carboxylic acid and catechol groups can establish complexes with metal oxides,59 leading to interactions between the metal oxide and the dye. These interactions primarily occur through electrostatic attractions between the anionic segments of the dyes and the cationic regions (metal ions) present on the surface of the metal oxides. Within this particular category, examples such as 5(6)-carboxyfluorescein, distinguished by the inclusion of hydroxyl (OH) and carboxylic acid groups, as well as eosin Y, characterized by carboxylic acid groups, fall into this classification. Consequently, these dyes emerge as promising contenders for functioning as photosensitizers, enabling the activation of wide-bandgap metal oxides upon exposure to visible light.
2.3.1. Oxidation of benzyl ethers to benzoates. In an impressive advancement, Li Ren and his research group (2017) successfully devised an effective catalysis system that enables the aerobic oxidation of benzyl ethers to benzoates.This breakthrough was achieved using eosin Y-sensitized titanium dioxide under visible light and molecular oxygen conditions.60 The catalytic system exhibits numerous notable advantages, including its simplicity of setup, affordability, and ability to accommodate a wide range of substrates. In addition, the team proposed a logical mechanistic pathway for the reaction, which involves a photoinduced, radical-based two-step process mediated by an isolable peroxide intermediate. This innovative development paves the way for exciting opportunities in the field of catalysis (refer to Scheme 19) and (Table 8).
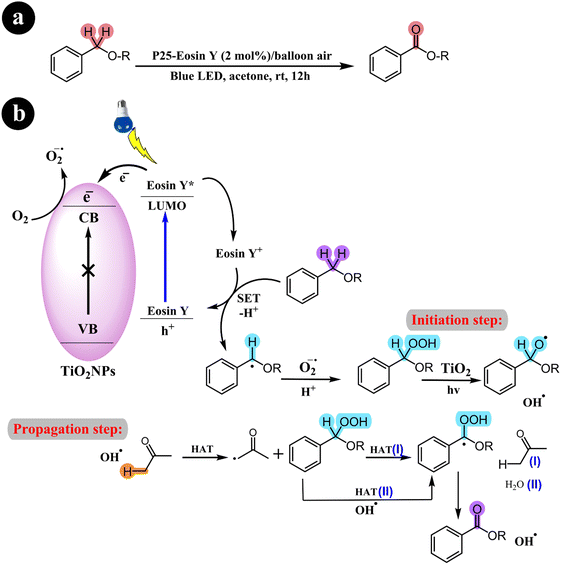 |
| | Scheme 19 (a) Catalytic system, reaction conditions and (b) a conceivable mechanism for the aerobic oxidation process of benzyl ethers. Reproduced with permission from ref. 60. Copyright 2017, American Chemical Society. | |
Table 8 Application of eosin Y dye as a photosensitizer for the selective aerobic oxidation of benzyl ethers to benzoates, conducted under specific experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
P25-eosin Y/balloon air |
Blue LED |
Acetone |
12 |
100 |
54–92 |
Not given |
60 |
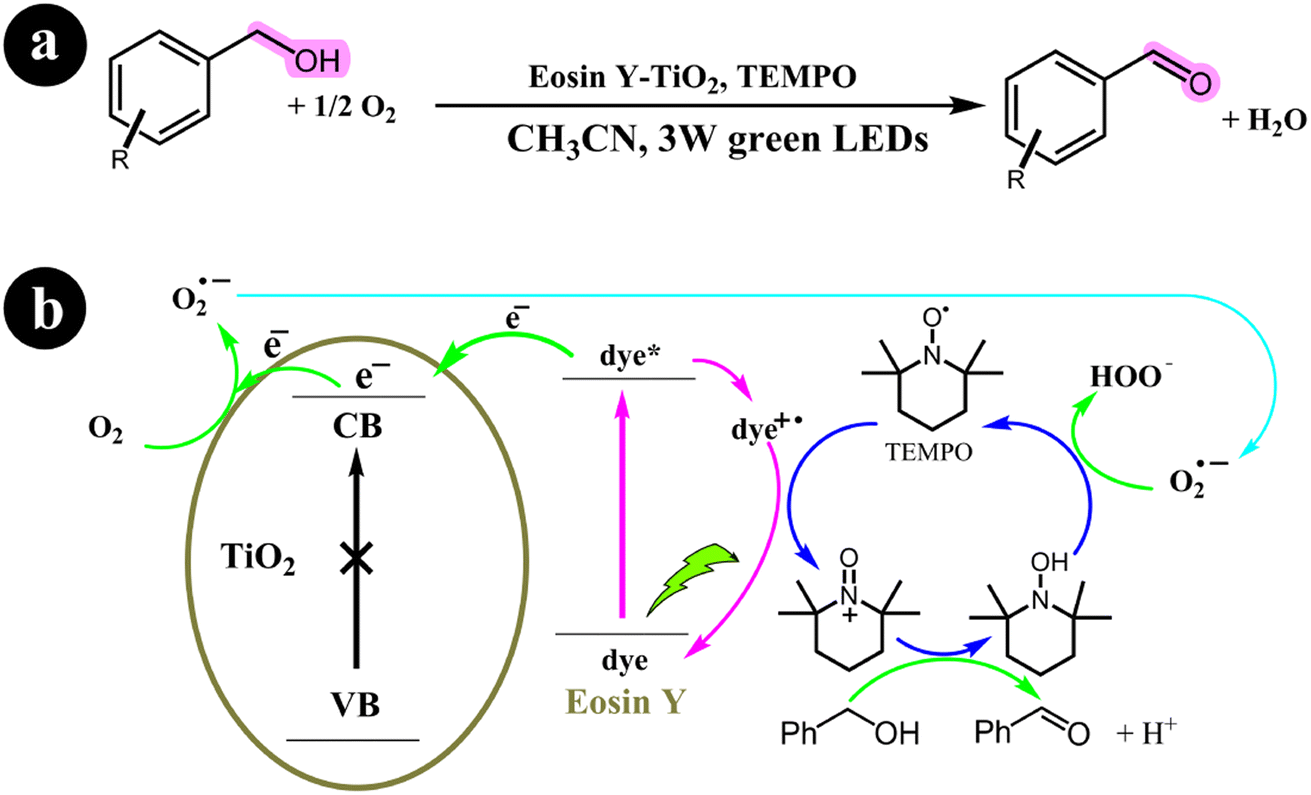
2.3.2. Oxidation of alcohols. Yichi Zhang and colleagues (2017) undertook a research study focusing on developing a technique that employed dye-sensitized TiO2 with TEMPO as a co-catalyst. Their primary aim was to oxidize alcohols and successfully synthesize carbonyl compounds with a remarkable selectivity of 98%. This process was achieved under mild reaction conditions, utilizing molecular oxygen as a green oxidizing agent and green light irradiation. To enhance the photocatalytic performance of TiO2, they utilized Eosin Y as a photosensitizer, leading to significant improvements in the overall efficiency of the process. The conversion rates achieved ranged from 14% to 99% for the targeted product.61 The reaction conditions and the mechanistic pathway are shown in Scheme 20.
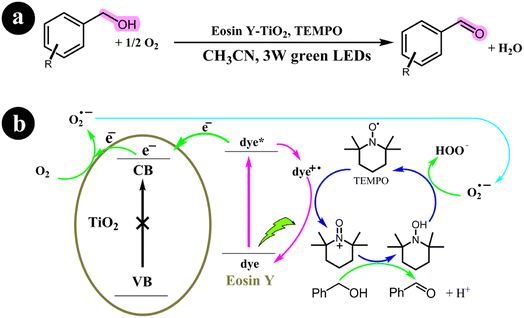 |
| | Scheme 20 (a) Reaction conditions and (b) photocatalytic aerobic oxidation of alcohols for the eosin Y-sensitized TiO2 nanoparticles. Reproduced from ref. 61, with permission, copyright 2017, Royal Society of Chemistry. | |
In a related investigation, Ningning Wang et al. (2019) devised a photocatalysis system by creating a dye-based catalyst composed of 5(6)-carboxyfluorescein [5(6)-FAM], CH3O-TEMPO, and TiO2. This catalyst 5(6)-FAM-TiO2/CH3O-TEMPO was applied to the aerobic oxidation of alcohols, utilizing green LED light for irradiation. The process resulted in the production of aldehydes and ketones with remarkable selectivity and high yields. Notably, the researchers opted to employ CH3O-TEMPO instead of TEMPO alone and observed that CH3O-TEMPO displayed enhanced effectiveness in the oxidation process (Table 9).62
Table 9 Application of eosin Y dye as a photosensitizer for the selective aerobic oxidation of alcohols to the corresponding aldehydes and ketones, conducted under different experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
TiO2-eosin Y/TEMPO/O2 |
Green LEDs |
CH3CN |
1–4 |
14–99 |
Not given |
50–100 |
61 |
| |
TiO2-5(6)-FAM/CH3O-TEMPO/aerobic O2 |
Green LEDs |
CH3CN |
1–2 |
21–100 |
Not given |
26–100 |
62 |
2.4. Thionine dye (TH)-sensitized metal oxide nanoparticles for the oxidation of alcohols
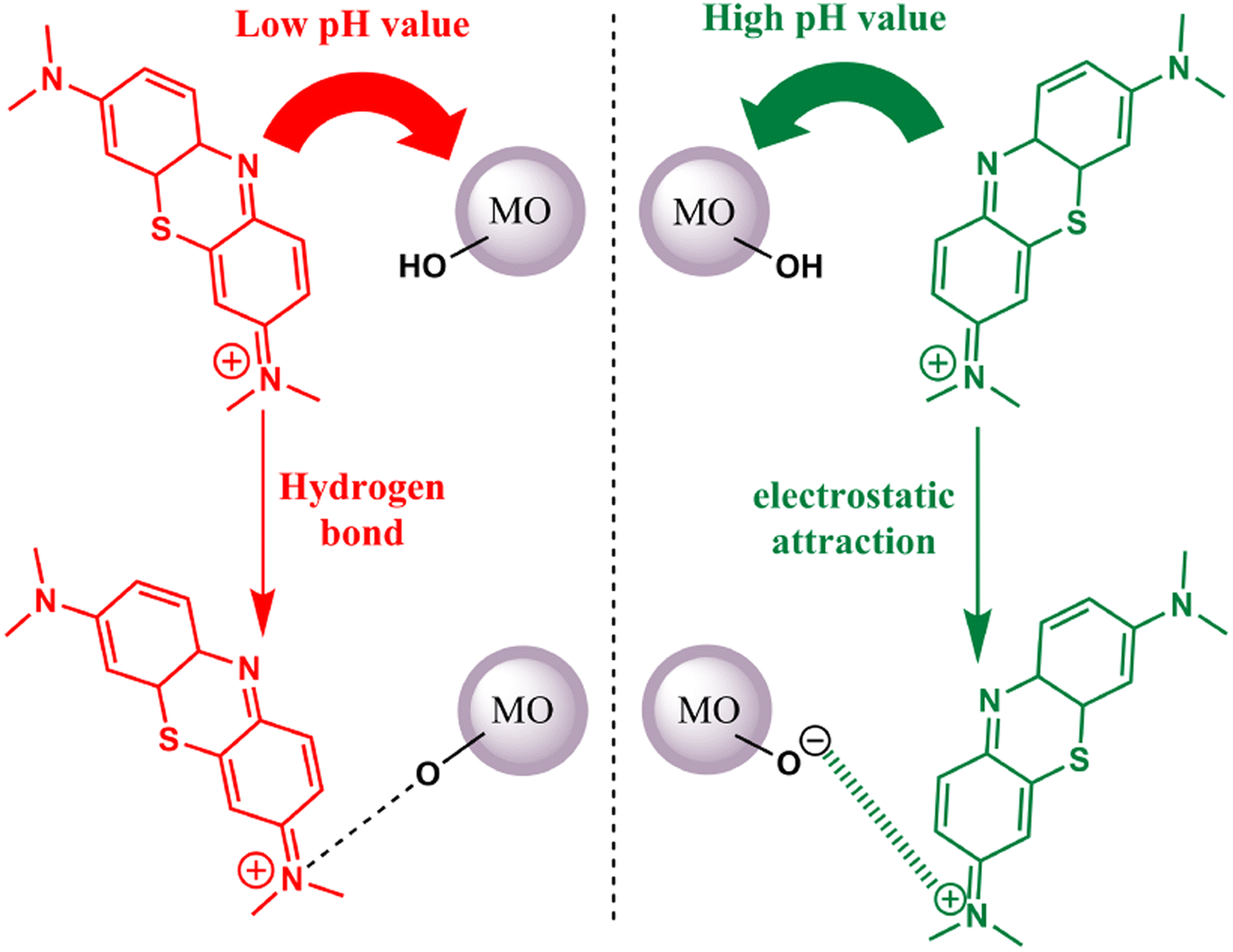
Thionine dye (TH), also known as Methylene Blue (MB), has been employed as a photosensitizer for the activation of metal oxides in the presence of visible light. The binding mechanism between this dye and metal oxides can be attributed to either hydrogen bonding or electrostatic attraction. Notably, the binding process is pH-sensitive. Under low pH conditions, both the metal oxides and thionine dye carry a negative surface charge. In this scenario, the primary mode of adsorption is governed by hydrogen bonding. This interaction is formed through the nitrogen atoms present in MB and the hydroxyl groups on the surface of the metal oxides. Conversely, the metal oxide surface remains negatively charged when the pH is elevated. Consequently, the cationic thionine dye can undergo robust adsorption onto the metal oxide surface, driven by electrostatic attraction63 (refer to Fig. 4).
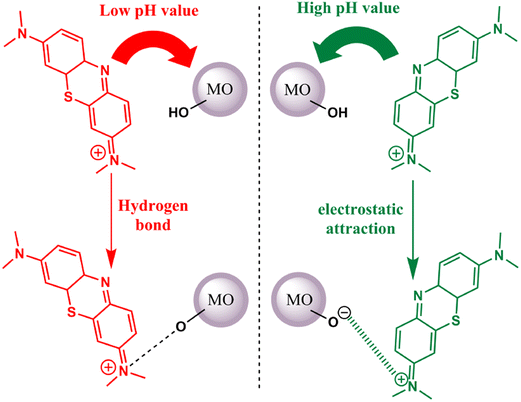 |
| | Fig. 4 Possible interactions between the TH dye and metal oxides at low pH (hydrogen bonding) and high pH (electrostatic attraction). Adapted with permission from ref. 63, copyright 2018, Elsevier. | |
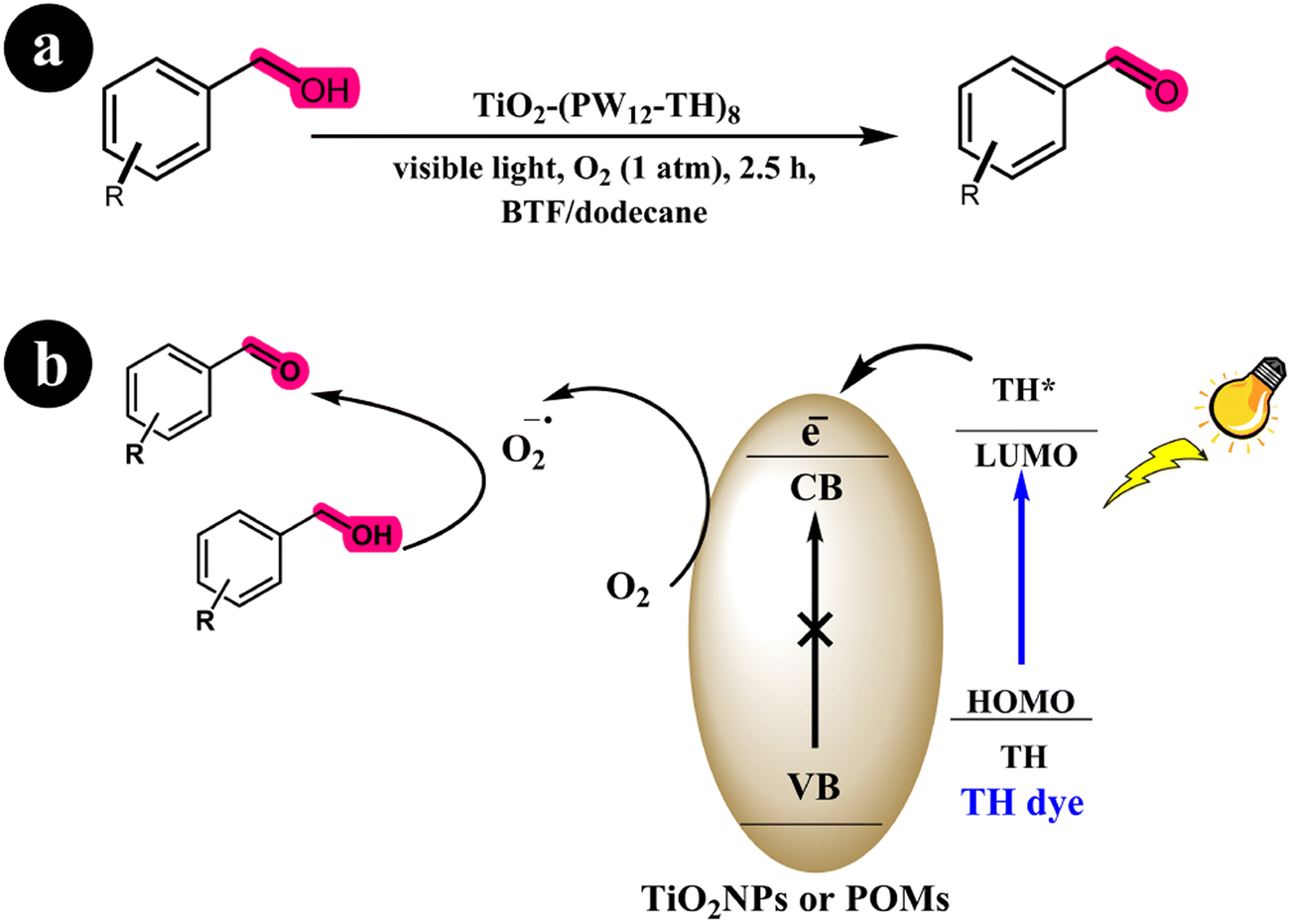
Xue Yang and colleagues (2017) developed a dye-sensitized photocatalyst known as TiO2-(PW12-TH)8, employing the utilization of thionine dye (TH) and PW12O403− (referred to as PW12). This newly synthesized catalyst was employed to selectively oxidize various substituted alcohols to aldehydes, utilizing visible light irradiation. According to their findings, the photocurrent response exhibited by the catalyst exceeded that of SiO2@(PW12-TH)8, P25-(PW12-TH)8, and TiO2/TH.64 This enhanced performance of the novel catalyst can be attributed to its more effective transfer and prolonged lifetime of the excited species stimulated by photon energy. Through multiple control experiments, the researchers demonstrated that the principal reactive components in the proposed system were electrons and superoxide radicals. Moreover, the selectivity of the catalyst was attributed to the absence of holes and hydroxyl radicals. The researchers utilize oxygen as the oxidation molecule. Oxygen is readily available and cost-effective. An additional advantage of this system is that the only reaction side product is water (H2O), thereby generating no secondary pollution. The researchers initially described the absorption of visible light energy by the TH dye to elucidate the reaction mechanism. This absorption led to excitation from the HOMO to the LUMO of the dye, triggering an electron transfer from the LUMO of the dye to the conduction band (CB) of TiO2 or polyoxometalates (POMs). Subsequently, the transferred electron functioned as a reducing agent, facilitating the reduction of molecular oxygen to superoxide radical species. Ultimately, this process resulted in the conversion of alcohols into their corresponding aldehydes (see Scheme 21) and (Table 10).
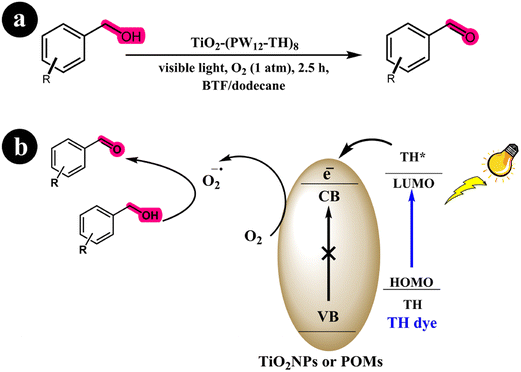 |
| | Scheme 21 (a) Reaction conditions for the aerobic oxidation of aryl alcohols to aldehydes by TiO2-(PW12-TH)8 under Blue light irradiation; (b) mechanistic pathway. Reproduced with permission from ref. 64, copyright 2017, Elsevier. | |
Table 10 Application of thionine (TH) dye as a photosensitizer for the selective aerobic oxidation of alcohols, conducted under specific experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
TiO2-(PW12-TH)8/O2 |
Visible light |
BTF/dodecane |
2.5 |
17.1–61.3 |
Not given |
>99 |
64 |
2.5. Rhodamine B (RhB) and fluorescein (Fl) dyes-sensitized metal oxide nanoparticles for the reduction of ketones
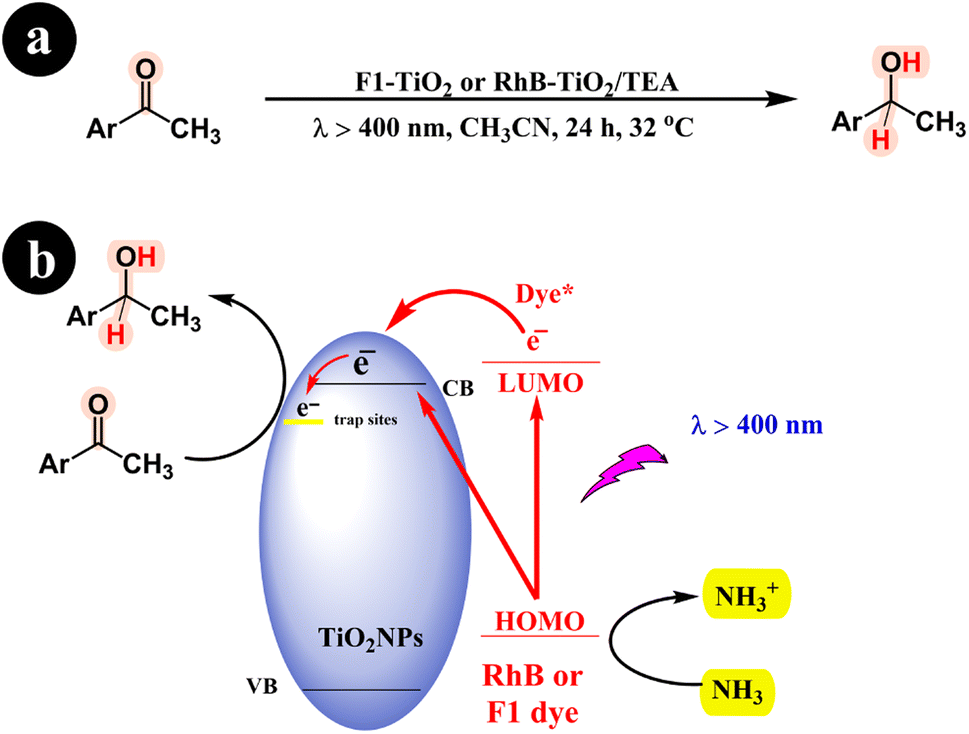
Fluorescein (F1) contains a hydroxyl group, while Rhodamine B features a carboxyl acid group, enabling them to form robust bonds with the surfaces of metal oxides (see Fig. 2 and 3 for reference). Shigeru Kohtani and his team (2014) significantly enhanced the photocatalytic performance of TiO2. Their innovative approach involved harnessing the power of sensitizers, specifically rhodamine B (RhB) and fluorescein (Fl) dyes, which could be activated using visible light. The team successfully fabricated two catalysts by incorporating these sensitizers: RhB–TiO2 and F1–TiO2.65 These catalysts were then utilized to convert aromatic ketones into their respective alcohols, showcasing their remarkable efficiency and potential in catalytic transformations. The study demonstrated the successful catalytic activity of both RhB–TiO2 and F1–TiO2 catalysts in the desired reaction. However, the team found that F1–TiO2 exhibited superior performance compared to RhB–TiO2. For a more comprehensive understanding of the results, refer to Scheme 22 and (Table 11).
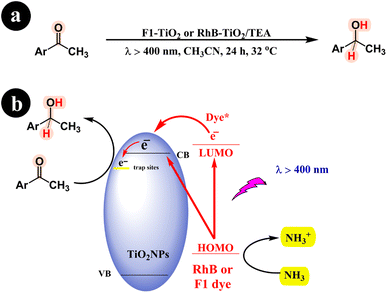 |
| | Scheme 22 The specific reaction conditions (a) and detailed information regarding the conversion of aromatic ketones to alcohols, including the reaction mechanism (b). Reproduced with permission from ref. 65, copyright 2014, Elsevier. | |
Table 11 Application of Rhodamine B (RhB) and Fluorescein (Fl) dyes as photosensitizers for the selective reduction of ketones, conducted under different experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
Fl–TiO2/TEA |
Vis. (λ > 400 nm) |
CH3CN |
24 |
52–78 |
48–74 |
92–100 |
65 |
| 2 |
RhB–TiO2/TEA |
Vis. (λ > 400 nm) |
CH3CN |
24 |
37–78 |
35–73 |
94–97 |
2.6. Coumarin dyes-sensitized metal oxide nanoparticles for the reduction of acetophenone (AP)
Coumarins, a class of organic compounds known for their distinct benzopyrone structure, have found significant utility as photosensitizers. These compounds possess remarkable ability to absorb and transfer light energy. As photosensitizers, coumarins play a crucial role in capturing light energy and transferring it to other molecules or surfaces. Their conjugated molecular structure allows them to efficiently interact with light, making them versatile tools for harnessing and manipulating light-driven reactions in diverse fields.66 Certain coumarin compounds have been employed as photosensitizers, contributing to organic transformations. However, not all coumarins exhibit dye properties. The potential of a specific coumarin to function as a dye or photosensitizer relies on its molecular structure and electronic characteristics.67 A crucial aspect of binding with metal oxides is the presence of carboxylic acid groups.
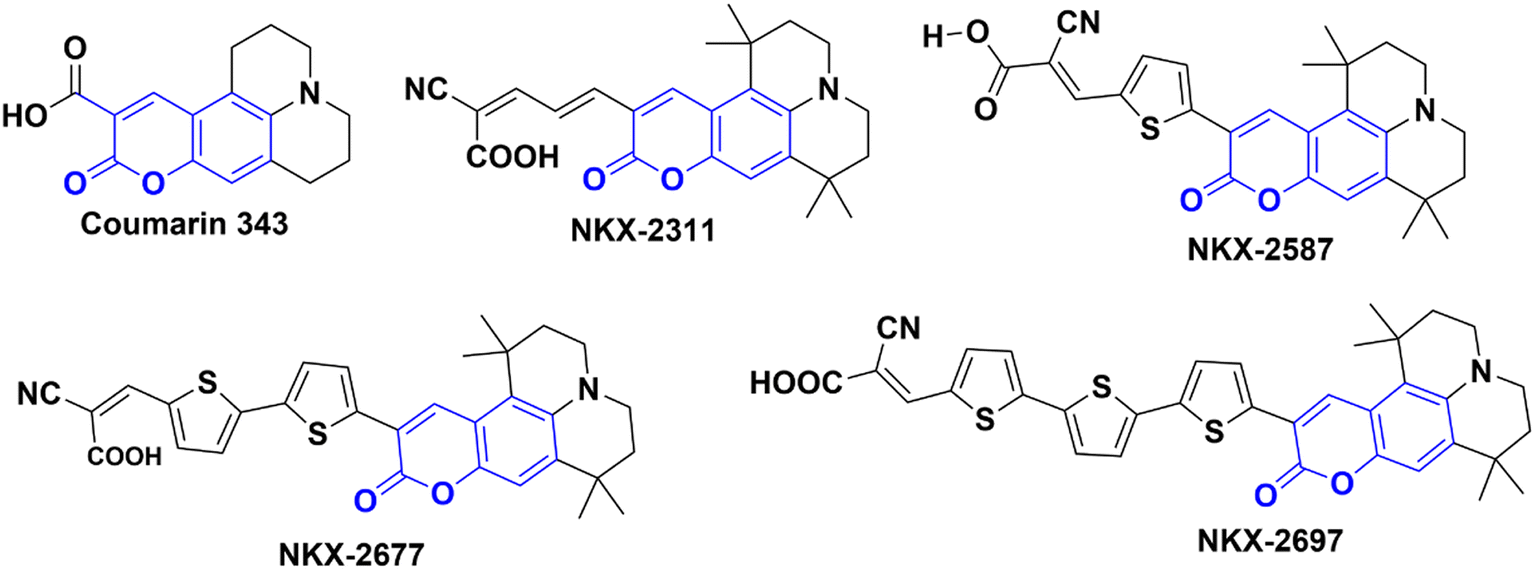
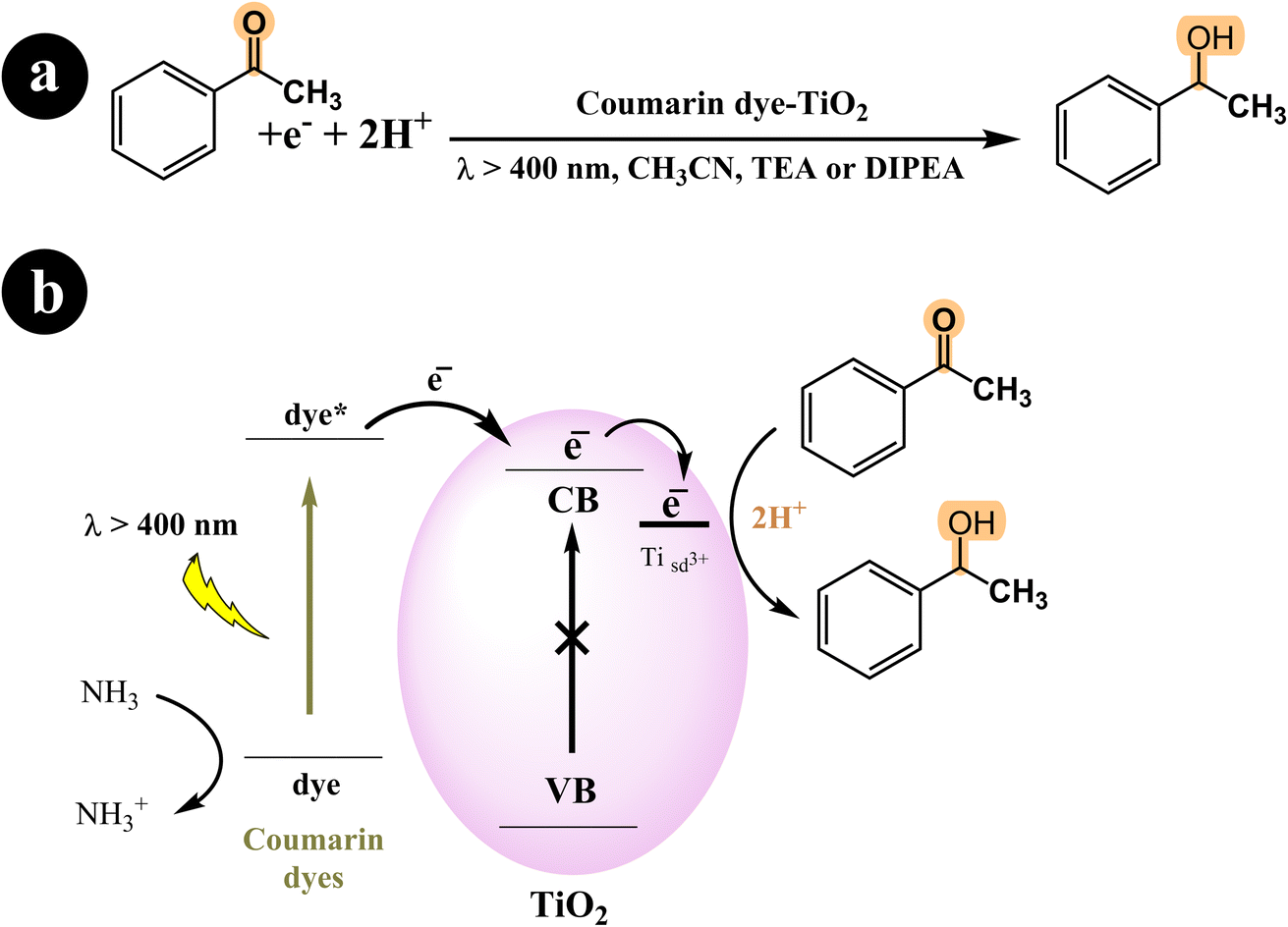
In their study, Shigeru Kohtani and his team (2015) explored the activation of TiO2 nanoparticles using coumarin dyes under visible light. They demonstrated that coumarin dyes adsorbed on titanium dioxide (TiO2, P25) are highly effective in hydrogenating acetophenone, yielding favorable chemical efficiencies. Furthermore, they delved into the influence of sacrificial reagents on this dye-sensitized system and shared their discoveries.68 During their research, the authors utilized a range of coumarin dyes, including Coumarin 343, NKX-2311, NKX-2587, NKX-2677, and NKX-2697 (Fig. 5), to sensitize TiO2 nanoparticles. These combinations of coumarin dye and TiO2 were then employed in the acetophenone hydrogenation process, and after 48 hours, they performed a comparative analysis to identify the combination with the best catalytic activity. The results indicated that C343–TiO2 and NKX2311–TiO2 exhibited the most favorable catalytic activities when triethylamine (TEA) served as the sacrificial electron donor. In addition, they observed that the catalytic activities of NKX2587–TiO2, NKX2677–TiO2, and NKX2697–TiO2 were significantly enhanced when they replaced TEA with DIPEA. For a deeper understanding of the research, including the reaction conditions and the underlying mechanism, please refer to Scheme 23 and (Table 12).
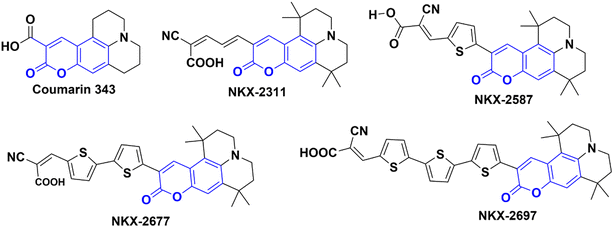 |
| | Fig. 5 The structures of coumarin dyes used by Shigeru Kohtani and his team68 in their work. | |
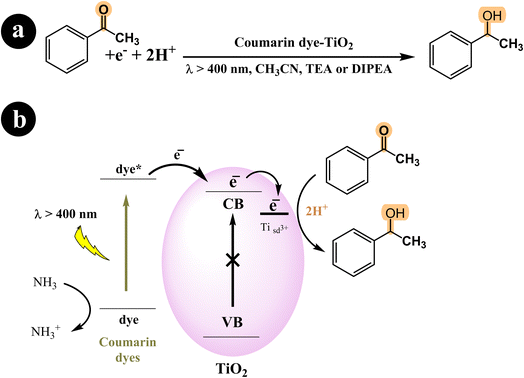 |
| | Scheme 23 (a) The reaction conditions employed for the hydrogenation of acetophenone. (b) The mechanistic pathway of photocatalysis under visible light irradiation using coumarin dyes-TiO2. | |
Table 12 Application of different types of coumarin dyes as photosensitizers for the photohydrogenation of acetophenone, conducted under different experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
C343-TiO2/TEA |
Vis. (λ > 400 nm) |
CH3CN |
48 |
100 |
Not given |
Not given |
68 |
| 2 |
NKX2311-TiO2/TEA |
Vis. (λ > 400 nm) |
CH3CN |
48 |
96 |
Not given |
Not given |
| 3 |
NKX2587-TiO2/DIPEA |
Vis. (λ > 400 nm) |
CH3CN |
48 |
84 |
Not given |
Not given |
| 4 |
NKX2677-TiO2/DIPEA |
Vis. (λ > 400 nm) |
CH3CN |
48 |
75 |
Not given |
Not given |
| 5 |
NKX2697-TiO2/DIPEA |
Vis. (λ > 400 nm) |
CH3CN |
48 |
79 |
Not given |
Not given |
2.7. Erythrosine B dye-sensitized metal oxide nanoparticles
Belonging to the xanthene dye family, Erythrosine B dye stands out prominently. Its unique red coloration characterizes its exceptional capability to absorb light within the visible spectrum (450–550 nm).69 This distinct trait positions it ideally for capturing light energy and driving various photochemical reactions. Erythrosine B dye has proven invaluable across various disciplines, from its role in enhancing dye-sensitized solar cells70 to its application in advanced photodynamic therapy.71 This versatility can be attributed to its proficiency in efficiently channelling absorbed light energy to adjacent molecules or materials. In addition, the dye exhibits the potential to serve as a photosensitizer in heterogeneous catalysts, further broadening its scope of utility.
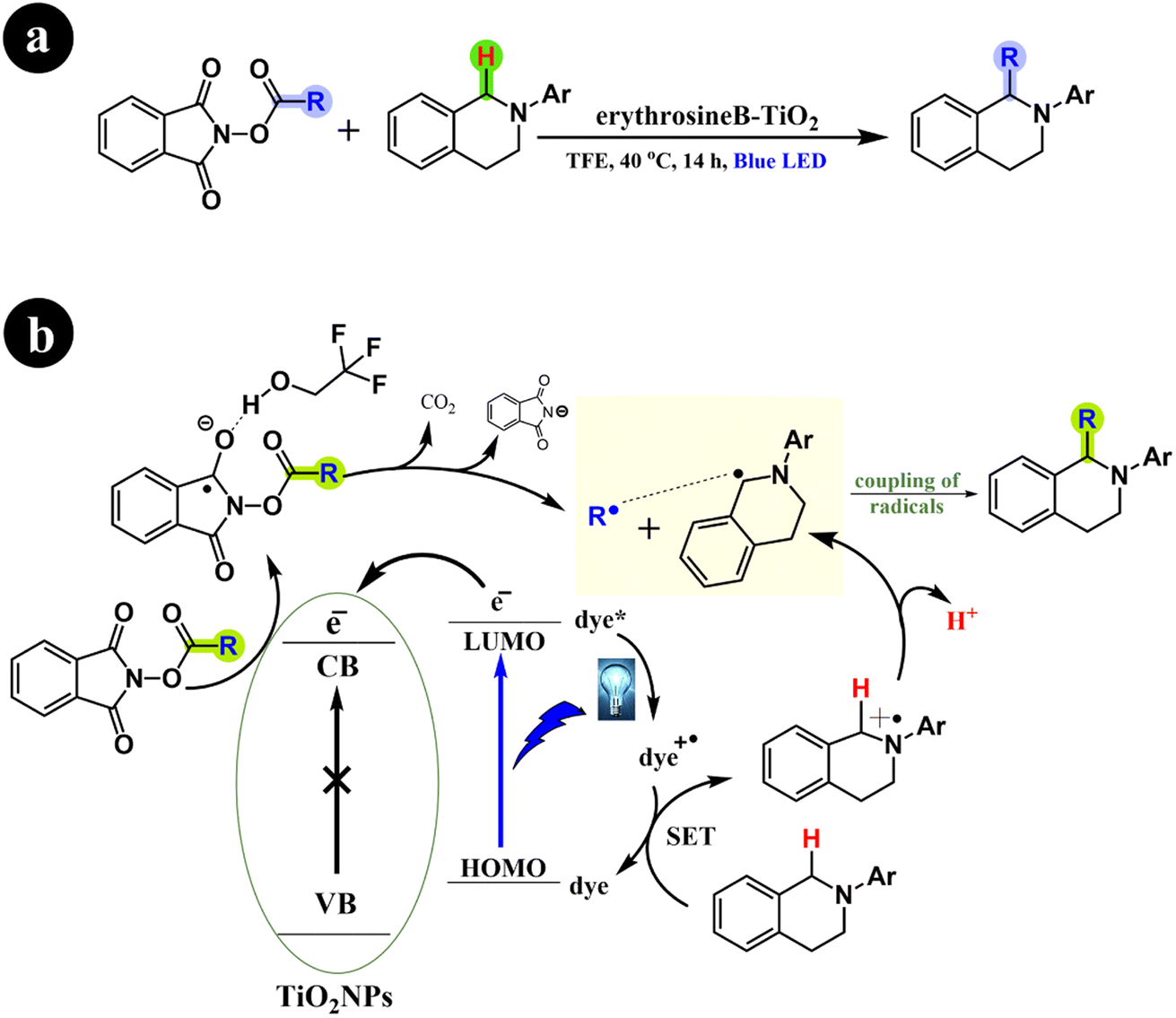
Li Ren and Huan Cong (2018) recently presented their significant findings on the decarboxylative alkylation of tetrahydroisoquinolines. This remarkable advancement involves the utilization of an innovatively engineered catalyst comprising erythrosine B-TiO2, which exhibits excellent performance under visible-light conditions.72 Their findings demonstrated the successful catalytic activity of the erythrosine B-TiO2 catalyst under visible light, affirming its pivotal role in the reaction. In addition, the study conclusively established the indispensability of both the catalyst and visible light (specifically blue light) in conjunction with a protic solvent for the reaction to proceed effectively. Furthermore, the authors elucidated the reaction mechanism, which involves the formation of radical intermediates. For more comprehensive information, refer to Scheme 24.
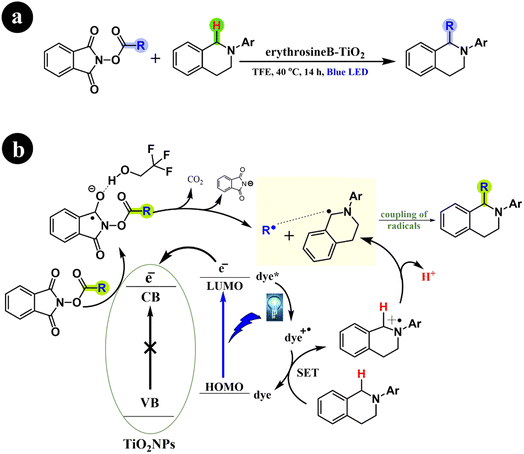 |
| | Scheme 24 (a) Reaction conditions and (b) possible mechanism for carboxyl removal and alkyl addition of tetrahydroisoquinolines. Reproduced with permission from ref. 72. Copyright 2018, American Chemical Society. | |
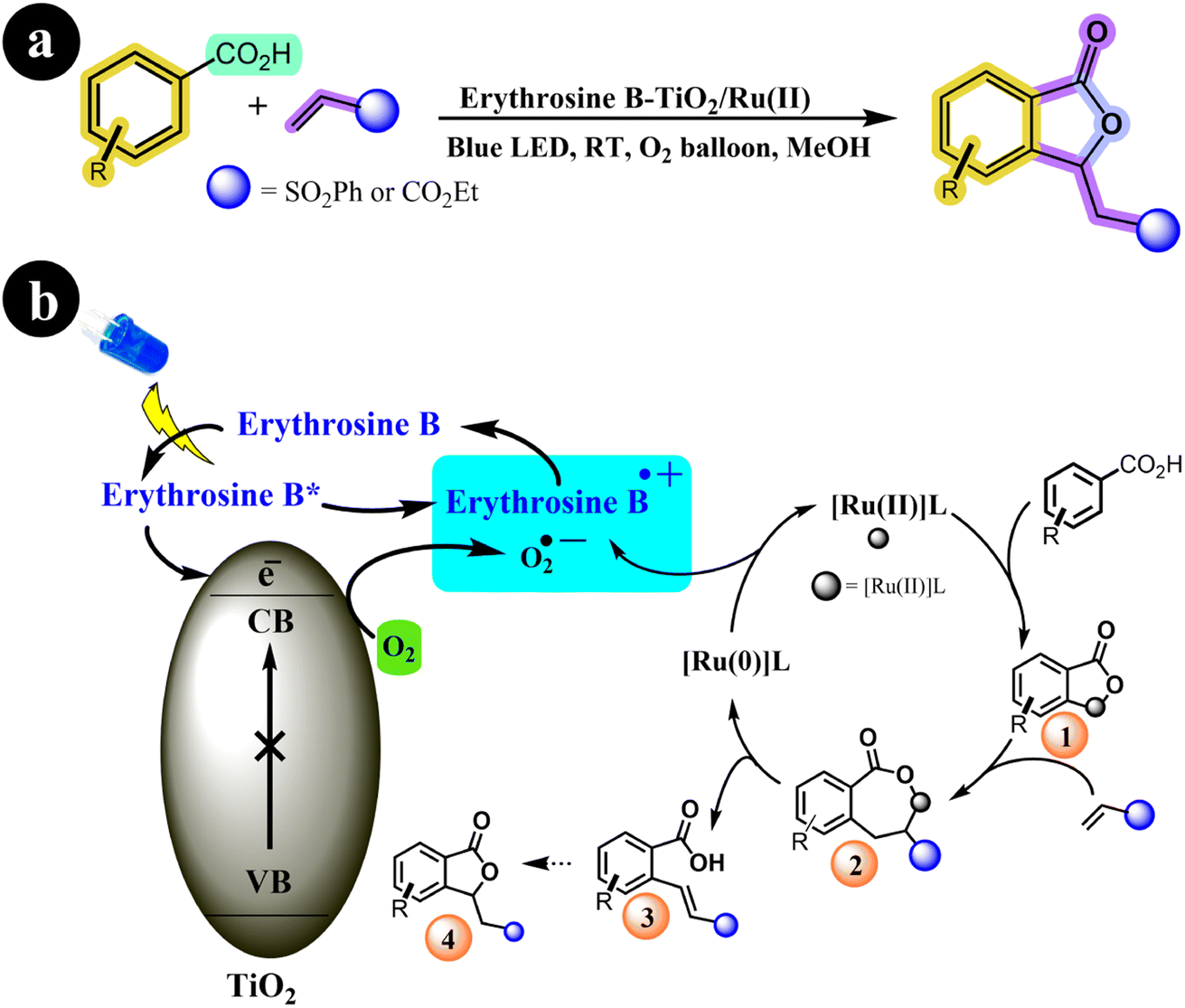
The olefination and annulation processes continue to captivate researchers in the field of organic synthesis, offering a fascinating avenue for creating numerous novel molecules. Through these methods, thousands of previously unexplored compounds can be synthesized, unlocking exciting possibilities for scientific and industrial applications.73,74 In their research, Suman Dana et al. (2020) achieved a notable breakthrough by developing a TiO2 photocatalyst sensitized with Erythrosine B, which was further bolstered by the addition of a Ru(II)-catalysis system as a co-catalyst. Notably, Erythrosine B-TiO2 served as a terminal oxidant in their experimental setup. This pioneering method facilitated the oxidative annulations and olefinations of arene carboxylic acids under gentle reaction conditions. The study showcases the remarkable potential of this approach for synthesizing diverse compounds, thus holding significant implications within the realm of organic chemistry.75 The authors presented a potential mechanistic pathway for the reaction along with the corresponding reaction conditions (refer to Scheme 25). According to their proposal, the process is initiated with the absorption of visible light by Erythrosine B, elevating it to the LUMO energy level. Subsequently, Erythrosine B donates an electron to the conduction band (CB) of TiO2, forming a radical cation of Erythrosine B. At the same time, molecular oxygen is transformed into a superoxide anion through the electron in the CB of TiO2. Concurrently, the arene carboxylic acid undergoes C–H bond activation facilitated by the Ru(II)-catalyst to form (1), followed by an insertion step that generates intermediate (2). This intermediate (2) then proceeds to yield the vinylated product (3), accompanied by the formation of Ru(0) species through β-hydride elimination. Finally, the Ru(0) species regenerated the Ru(II)-catalyst through an electron transfer process with the superoxide anion radical and the cationic dye radical, allowing for the continuation of the catalytic cycle (Table 13).
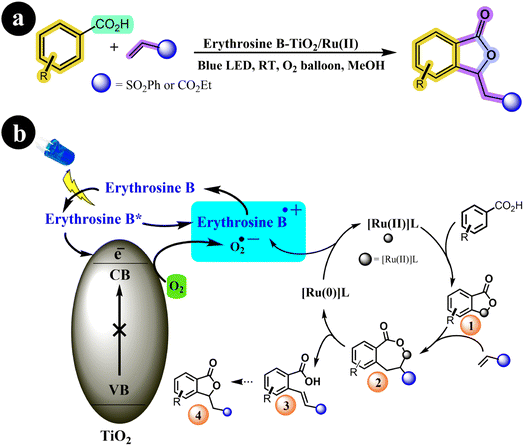 |
| | Scheme 25 (a) Reaction conditions and (b) catalytic activation of C–H bonds facilitated by Ru(II) and photoredox using O2 as an oxidant under blue LED irradiation. Reproduced with permission from ref. 75. Copyright 2020, John Wiley and Sons. | |
Table 13 Application of Erythrosine B dye as a photosensitizer for the decarboxylative alkylation of C–H bond and oxidative C–H olefination of arene carboxylic acids, conducted under different experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
Erythrosine B-TiO2 |
Blue LEDs |
TFE |
14 |
95 |
44–90 |
Not given |
72 |
| 2 |
Erythrosine B-TiO2/Ru(II)/O2 balloon |
Blue LEDs |
MeOH |
24 |
Not given |
31–84 |
Not given |
75 |
2.8. 1,2-Dihydroxyanthraquinone (1,2-DHA)-sensitized metal oxide nanoparticles
1,2-Dihydroxyanthraquinone (1,2-DHA) is derived from ARS and has a structural arrangement with two adjacent hydroxyl groups. This configuration empowers it to exhibit strong adsorption onto metal oxide surfaces, forming a durable dye-MO system akin to the scenario with ARS. Operating as a photosensitizer, 1,2-DHA activates metal oxides within the visible spectrum (400–650 nm).76
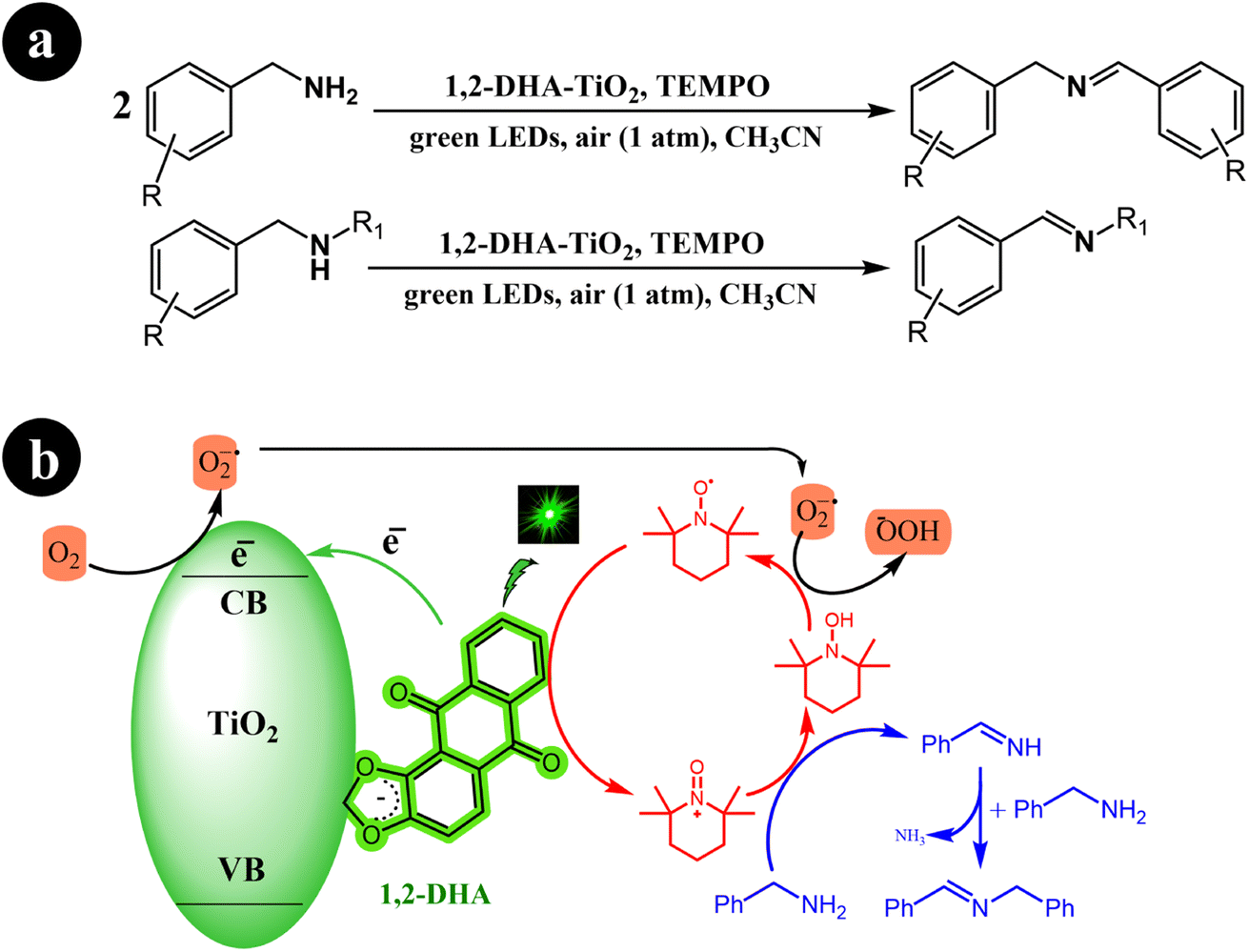
Xia Li et al. (2021) developed a highly efficient photocatalysis system by combining 1,2-dihydroxyanthraquinone (1,2-DHA) with TiO2. They prepared multiple anthraquinones (AQs) derived from ARS and loaded them onto TiO2, subsequently conducting a comparative analysis of the results. Interestingly, they observed that 1,2-DHA exhibited superior photocatalytic performance in selectively converting amines to imines under green light irradiation.76 In their study, the researchers employed TEMPO as a redox mediator. The reaction conditions and the proposed mechanism are depicted in Scheme 26.
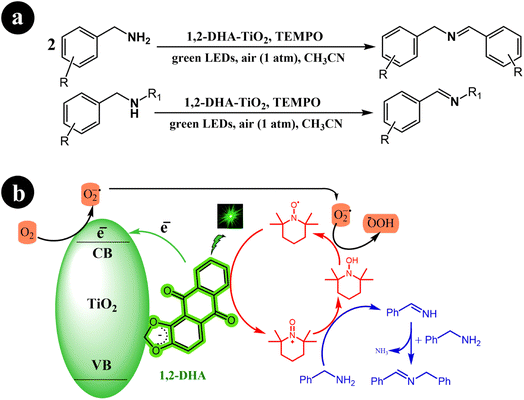 |
| | Scheme 26 (a) 1,2-DHA-TiO2/TEMPO co-catalyst for the green light-induced photocatalytic selective oxidation of primary and secondary amines into imines using aerobic O2 as the oxidizing agent, and (b) the proposed mechanism for the reactions. Reproduced with permission from ref. 76, copyright 2021, Elsevier. | |
The initial step involves the self-assembly of 1,2-DHA on the surface of anatase TiO2 through its catechol group, forming the 1,2-DHA-TiO2 photocatalyst. This unique photocatalyst allows the reaction to take place within the green light spectrum range. Next, the surface charge transfer complex absorbs green light, generating electron–hole pairs (e−/h+). The electrons are then injected from HOMO of 1,2-DHA into the conduction band of TiO2. These electrons subsequently convert O2 to O2˙−. Simultaneously, holes are left at the surface complex 1,2-DHA-TiO2 as radical cations. In the third step, the surface complex radical cation undergoes self-restoration by interacting with TEMPO, while TEMPO itself is transformed into TEMPO+. TEMPO+ then oxidizes benzylamine through a direct two-electron transfer, leading to the formation of benzylideneamine. Finally, benzylideneamine couples with unreacted benzylamine to produce the final product, N-benzylidenebenzylamine. Meanwhile, TEMPO+ is converted to TEMPOH and restored to TEMPO through a reaction with O2˙− (Table 14).
Table 14 Application of 1,2-DHA as a photosensitizer for the selective conversion of amines to imines, conducted under specific experimental conditions
| No. |
Catalyst design |
Light source |
Solvent |
T [h] |
Conv. [%] |
Yield [%] |
Sel. [%] |
Ref. |
| 1 |
1,2-DHA-TiO2/TEMPO/air |
Green LEDs |
CH3CN |
0.6–5 |
49–97 |
Not given |
68–98 |
76 |
3. Conclusion and outlook
Photocatalysis involving dye-synthesized metal oxides is in its initial stages within organic synthesis. Nevertheless, it exhibits the potential to tackle concerns related to catalyst recyclability, byproduct formation, selectivity, and environmental impact. However, several challenges demand attention, encompassing aspects such as scaling up, the stability of the dye-metal oxide catalyst, and limitations associated with substrates. This review article aims to elucidate fundamental aspects crucial for aspiring young researchers intrigued by this field. This objective is achieved by showcasing the synergistic relationship between sensitizers and metal oxides while delving into the intricacies of mechanistic investigations within organic synthesis. This review underscores the vital role of meticulous design and selection of dye sensitizers, which is crucial for achieving efficient and high-performance dye-sensitized systems. Significant strides have been achieved in visible light-induced dye-sensitized heterogeneous photocatalysis, enabling the activation of metal oxide nanoparticles upon exposure to visible light.
Looking forward, the field of dye-sensitized metal oxide photocatalysis holds exceptional promise in driving advancements in green chemistry and organic synthesis. The optimization of dye-metal oxide design and the resolution of challenges such as electron–hole recombination will open doors to more sustainable and cost-effective catalysts. Furthermore, exploring new dye categories and innovative combinations of metal oxides has the potential to unlock cutting-edge and highly-efficient photocatalytic systems tailored for specific organic transformations. The ongoing progression in dye-sensitized metal oxide photocatalysis lays the foundation for a more environmentally conscious and resource-aware future in organic synthesis.
Author contributions
Original draft preparation was done by Dana A. Kader, while the review and editing were conducted by Sewara J. Mohammed and Dana A. Kader. The authors have carefully reviewed and accepted the final published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
References
- A. Zupanc and M. Jereb, Green Chem. Lett. Rev., 2020, 13, 77–84 CrossRef.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- N. Kaur, J. Iran. Chem. Soc., 2015, 12, 9–45 CrossRef CAS.
- R. C. Larock and J. C. Bernhardt, J. Org. Chem., 1977, 42, 1680–1684 CrossRef CAS.
- H. A. A. Jamjoum, K. Umar, R. Adnan, M. R. Razali and M. N. Mohamad Ibrahim, Front. Chem., 2021, 9, 1–24 Search PubMed.
- J. Zhao, C. Chen and W. Ma, Top. Catal., 2005, 35, 269–278 CrossRef CAS.
- B. L. Gadilohar and G. S. Shankarling, J. Mol. Liq., 2017, 227, 234–261 CrossRef CAS.
- M. Watanabe, Sci. Technol. Adv. Mater., 2017, 18, 705–723 CrossRef CAS PubMed.
- M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. Þ. A. C. Pereira and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS PubMed.
- M. Skunik-nuckowska, K. Grzejszczyk, P. J. Kulesza, L. Yang, N. Vlachopoulos, L. Häggman, E. Johansson and A. Hagfeldt, J. Power Sources, 2013, 234, 91–99 CrossRef CAS.
- Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2020, 7, 300–339 RSC.
- D. Kishore Kumar, J. Kříž, N. Bennett, B. Chen, H. Upadhayaya, K. R. Reddy and V. Sadhu, Mater. Sci. Energy Technol., 2020, 3, 472–481 CAS.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- M. Gayathri, P. Senthil Kumar, M. Santhameenakshi and S. Karuthapandian, Sep. Sci. Technol., 2021, 56, 1466–1474 CrossRef CAS.
- S. Venkatesan, I. P. Liu, C. W. Li, C. M. Tseng-Shan and Y. L. Lee, ACS Sustain. Chem. Eng., 2019, 7, 7403–7411 CrossRef CAS.
- L. Alibabaei, H. Luo, R. L. House, P. G. Hoertz, R. Lopez and T. J. Meyer, J. Mater. Chem. A, 2013, 1, 4133–4145 RSC.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- N. Hoffmann, ChemSusChem, 2012, 5, 352–371 CrossRef CAS PubMed.
- X. Yu, L. Wang and S. M. Cohen, CrystEngComm, 2017, 19, 4126–4136 RSC.
- G. Han, G. Li, J. Huang, C. Han, C. Turro and Y. Sun, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- L. M. Peter, Phys. Chem. Chem. Phys., 2007, 9, 2630–2642 RSC.
- M. Grätzel, J. Photochem. Photobiol. C Photochem. Rev., 2003, 4, 145–153 CrossRef.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- J. Moon, C. Y. Yun, K. W. Chung, M. S. Kang and J. Yi, Catal. Today, 2003, 87, 77–86 CrossRef CAS.
- M. Koohgard, Z. Hosseinpour, A. M. Sarvestani and M. Hosseini-Sarvari, Catal. Sci. Technol., 2020, 10, 1401–1407 RSC.
- S. Kaur and V. Singh, Ultrason. Sonochem., 2007, 14, 531–537 CrossRef CAS PubMed.
- D. Chatterjee and A. Mahata, J. Photochem. Photobiol., 2004, 3, 56–58 Search PubMed.
- D. Chatterjee and A. Mahata, Catal. Commun., 2001, 2, 1–3 CrossRef CAS.
- R. Abe, K. Hara, K. Sayama, K. Domen and H. Arakawa, J. Photochem. Photobiol. A Chem., 2000, 137, 63–69 CrossRef CAS.
- C. Zhu, D. Peng, J. Hu, Y. Liu, Y. Liao, Q. Xie and C. Zhong, Monatsh. Chem., 2017, 148, 217–225 CrossRef CAS.
- H. Rahaman and S. K. Ghosh, RSC Adv., 2016, 6, 4531–4539 RSC.
- T. M. Underwood and R. S. Robinson, RSC Adv., 2019, 9, 24259–24266 RSC.
- S. C. Sharma, Optik, 2016, 127, 6498–6512 CrossRef CAS.
- Y. dong Liang, Y. jun He, Y. hang Zhang and Q. qian zhu, J. Environ. Chem. Eng., 2018, 6, 416–425 CrossRef.
- F. Yan, J. Sun, Y. Zang, Z. Sun, H. Zhang, J. Xu and X. Wang, Dyes Pigm., 2021, 195, 109720 CrossRef CAS.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299–301 RSC.
- X. Li, J. L. Shi, H. Hao and X. Lang, Appl. Catal. B Environ., 2018, 232, 260–267 CrossRef CAS.
- S. Liang, S. Shaaban, N. W. Liu, K. Hofman and G. Manolikakes, Recent Advances in the Synthesis of C–S Bonds via Metal-Catalyzed or -Mediated Functionalization of C–H Bonds, Elsevier Inc., 1st edn., 2018, vol. 69 Search PubMed.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- H. Hao, Z. Wang, J. L. Shi, X. Li and X. Lang, ChemCatChem, 2018, 10, 4545–4554 CrossRef CAS.
- W. R. Leow and X. Chen, Bull. Chem. Soc. Jpn., 2019, 92, 505–510 CrossRef CAS.
- F. Huang, H. Hao, W. Sheng and X. Lang, Chem. Eng. J., 2021, 423, 129419 CrossRef CAS.
- F. Huang, H. Hao, W. Sheng, X. Dong and X. Lang, Chem. Eng. J., 2022, 432, 134285 CrossRef CAS.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Müller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- M. Lv, Q. Ma, S. Zhang and H. Xu, Bioorganic Med. Chem. Lett., 2021, 48, 128246 CrossRef CAS PubMed.
- Z. Wang and X. Lang, Appl. Catal. B Environ., 2018, 224, 404–409 CrossRef CAS.
- H. Xu, J. L. Shi, H. Hao, X. Li and X. Lang, Catal. Today, 2019, 335, 128–135 CrossRef CAS.
- J. Zhou, X. Li, X. Ma, W. Sheng and X. Lang, Appl. Catal. B Environ., 2021, 296, 120368 CrossRef CAS.
- X. Ma, X. Li, J. Zhou, Y. Wang and X. Lang, Chem. Eng. J., 2021, 426, 131418 CrossRef CAS.
- K. Portillo-Cortez, A. Martínez, A. Dutt and G. Santana, J. Phys. Chem. A, 2019, 123, 10930–10939 CrossRef CAS PubMed.
- S. Bhar and R. Ananthakrishnan, RSC Adv., 2015, 5, 20704–20711 RSC.
- N. Prabavathy, S. Shalini, R. Balasundaraprabhu, D. Velauthapillai, S. Prasanna and N. Muthukumarasamy, Int. J. Energy Res., 2017, 41, 1372–1396 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS PubMed.
- S. Füldner, R. Mild, H. I. Siegmund, J. A. Schroeder, M. Gruber and B. König, Green Chem., 2010, 12, 400–440 RSC.
- S. Füldner, T. Mitkina, T. Trottmann, A. Frimberger, M. Gruber and B. König, Photochem. Photobiol. Sci., 2011, 10, 623–625 CrossRef PubMed.
- H. S. Yoon, X. H. Ho, J. Jang, H. J. Lee, S. J. Kim and H. Y. Jang, Org. Lett., 2012, 14, 3272–3275 CrossRef CAS PubMed.
- K. Mori, M. Kawashima, M. Che and H. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 8598–8601 CrossRef CAS PubMed.
- L. Zeininger, L. Portilla, M. Halik and A. Hirsch, Chem.–Eur. J., 2016, 22, 13506–13512 CrossRef CAS PubMed.
- L. Ren, M. M. Yang, C. H. Tung, L. Z. Wu and H. Cong, ACS Catal., 2017, 7, 8134–8138 CrossRef CAS.
- Y. Zhang, Z. Wang and X. Lang, Catal. Sci. Technol., 2017, 7, 4955–4963 RSC.
- N. Wang, J. L. Shi, H. Hao, H. Yuan and X. Lang, Sustain. Energy Fuels, 2019, 3, 1701–1712 RSC.
- K. Lu, T. Wang, L. Zhai, W. Wu, S. Dong, S. Gao and L. Mao, J. Colloid Interface Sci., 2019, 539, 553–562 CrossRef CAS PubMed.
- X. Yang, H. Zhao, J. Feng, Y. Chen, S. Gao and R. Cao, J. Catal., 2017, 351, 59–66 CrossRef CAS.
- S. Kohtani, S. Nishioka, E. Yoshioka and H. Miyabe, Catal. Commun., 2014, 43, 61–65 CrossRef CAS.
- A. Gualandi, G. Rodeghiero, E. Della Rocca, F. Bertoni, M. Marchini, R. Perciaccante, T. P. Jansen, P. Ceroni and P. G. Cozzi, Chem. Commun., 2018, 54, 10044–10047 RSC.
- Z. S. Wang, Y. Cui, Y. Dan-Oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17011–17017 CrossRef CAS.
- S. Kohtani, M. Mori, E. Yoshioka and H. Miyabe, Catalysts, 2015, 5, 1417–1424 CrossRef CAS.
- O. Kravchenko, T. C. Sutherland and B. Heyne, Photochem. Photobiol., 2022, 98, 49–56 CrossRef CAS PubMed.
- D. Wang, W. Wei and Y. H. Hu, Ind. Eng. Chem. Res., 2020, 59, 10457–10463 CrossRef CAS.
- A. Claudia Pedrozo da Silva, C. Fabiano de Freitas, C. Aparecida Errerias Fernandes Cardinali, T. Lazarotto Braga, W. Caetano, M. Ida Bonini Ravanelli, N. Hioka and A. Luiz Tessaro, J. Mol. Liq., 2022, 345, 117898 CrossRef CAS.
- L. Ren and H. Cong, Org. Lett., 2018, 20, 3225–3228 CrossRef CAS PubMed.
- J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- J. W. Lehmann, D. J. Blair and M. D. Burke, Nat. Rev. Chem, 2018, 2, 1–20 CrossRef.
- S. Dana, P. Dey, S. A. Patil and M. Baidya, Chem.–Asian J., 2020, 15, 564–567 CrossRef CAS PubMed.
- X. Li, H. Hao and X. Lang, J. Colloid Interface Sci., 2021, 581, 826–835 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Sewara J. Mohammed
bc
*a and
Sewara J. Mohammed
bc