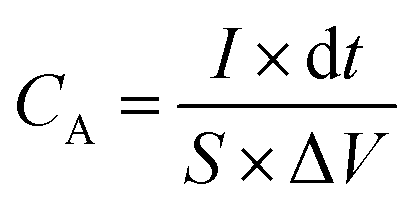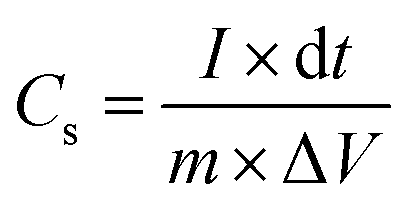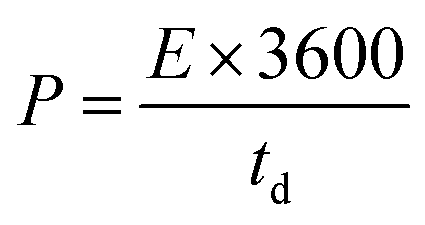DOI:
10.1039/D0NA00670J
(Paper)
Nanoscale Adv., 2020,
2, 4918-4929
Phytic acid controlled in situ synthesis of amorphous cobalt phosphate/carbon composite as anode materials with a high mass loading for symmetrical supercapacitor: amorphization of the electrode to boost the energy density†
Received
13th August 2020
, Accepted 8th September 2020
First published on 9th September 2020
Abstract
Transition metal phosphate (TMPi)-based composites as anode electrode materials in supercapacitor applications are less reported. Herein, we report a phytic acid (PA)-assisted in situ-formed amorphous cobalt phosphate/carbon (CoPi/C) composite grown on a flexible woven carbon cloth (CC) via a simple one-step carbonization approach. The tunable synthesis of amorphous and crystalline composites is shown by simply controlling the concentration of the cobalt salts. The strategy for high mass loading to 12 mg cm−2 is also shown in this report. Importantly, the resulting amorphous electrode materials exhibit electric double-layer capacitance (EDLC) behavior that works over a wide potential range from −1.4 to +0.5 V in an aqueous solution of potassium hydroxide (2 M KOH) and from −1.5 to +1.5 V in sodium sulfate (1 M Na2SO4). The amorphous electrode as an anode is capable of delivering an areal capacitance up to 2.15 F cm−2 at a current density of 4 mA cm−2 (gravimetric capacitance up to 606.1 F g−1 at 1 Ag−1) and has a retention of 94.2% at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The flexible solid-state symmetric device fabricated shows an energy density of approximately 620.0 μW h cm−2 at a power density of 4.7 mW cm−2 (31.1 W h kg−1 at 476.0 W kg−1). This study offers a novel route for the generation of metal phosphate-based anode materials with high capacitance for symmetrical supercapacitor device with high energy density.
000 cycles. The flexible solid-state symmetric device fabricated shows an energy density of approximately 620.0 μW h cm−2 at a power density of 4.7 mW cm−2 (31.1 W h kg−1 at 476.0 W kg−1). This study offers a novel route for the generation of metal phosphate-based anode materials with high capacitance for symmetrical supercapacitor device with high energy density.
1. Introduction
High-performance supercapacitor energy storage devices have been attracting tremendous interest for decades and address the increasing demand for energy storage and supply. At present, carbonaceous materials are still the most commonly used anode materials for supercapacitor devices due to their high power density and conductivity.1–3 Nevertheless the maximum energy density achieved by carbon-based electrode materials that have a high surface area, such as activated carbon, is approximately 12 W h kg−1, which does not satisfy the energy requirement of next-generation electronic devices.4 Research has primarily focused on the fabrication of high-energy-density positive electrode materials, whereas negative electrode materials with a high capacitance and a wide potential window are relatively less explored.
Recently, phosphorous-containing transition metals have attracted the most attention due to their high theoretical capacitance and energy density.5–9 However, reports focusing on the utilization of these materials as anode materials for supercapacitors are limited.5,10–12 On the other hand, they have a tendency to aggregate, undergo quick phase transformation, and deteriorate performance.13 Iron oxides have been broadly studied for use as anode materials with a high specific capacitance and energy density due to having multiple valence states of the metal ions that could enable a fast faradaic redox reaction.3,14 However, the major limitation of iron-based compounds is their relatively poor electrical conductivity and cycle life.15–17 To overcome the drawbacks of transition metal-based electrodes, one strategy is to improve their conductivity and stability by hybridizing them with typical carbon additives, including carbon fibers,18 amorphous carbon19 and graphene oxide.20 On the one hand, the high conductivity of carbon materials renders the composite favorable for electron transfer. On the other hand, carbon materials act as a buffer layer able to prevent the main materials from phase changes due to the stress generated in the electrochemical process, thereby contributing to an improvement in the cycling stability. However, the inherent properties of carbon, such as EDLC charge storage behavior, high conductivity, and long-term stability, are severely hampered by composite formation. To this end, amorphization of the electrode materials can be a way to improve the cycling stability by reducing anisotropic expansion/extraction and eliminating stresses derived from the phase conversion reaction and poor electron conduction.3,21–24 Xia et al.3 found that a crystalline/amorphous interface integrated on graphene delivers a large capacitance of 701 F g−1 at 1 A g−1, which is almost twice the capacitance of conventional iron oxide (Fe2O3δ−) nanorods without an amorphous surface layer on the graphene. Amorphous phases of the materials have several properties that can significantly improve the double-layer capacitance and expand the potential voltages and stability.22,25 Owing to their structural disorder and anisotropic nature, amorphous materials offer fast ion diffusion via an open framework.26 According to the formula [(energy density (E) = 1/2CV2)], the energy density of a supercapacitor device can be improved by maximizing either the capacitance (C) or the cell voltage (V).27–30 Therefore, it can be a good strategy to improve the capacitance and voltage by a rational design of amorphous metal doping in a carbonaceous network. This approach can also support the formulation of stable composites that can work as negative electrode materials. Furthermore, the inclusion of phosphorus into the carbon composite can widen the potential window to increase the energy density of EDLCs.31,32 Therefore, in situ synthesis of amorphous transition metals wrapped within a carbon network with phosphorous-rich compounds can be a potential strategy to achieve a high capacitance and energy density. These strategies can avoid the necessity of using costly and toxic organic and ionic liquid electrolytes, which is the common practice to improve the capacitance and energy density. However, the controlled synthesis of nanomaterials with a well-defined morphology in the amorphous state is challenging because many materials have a high propensity to crystallize during preparation.
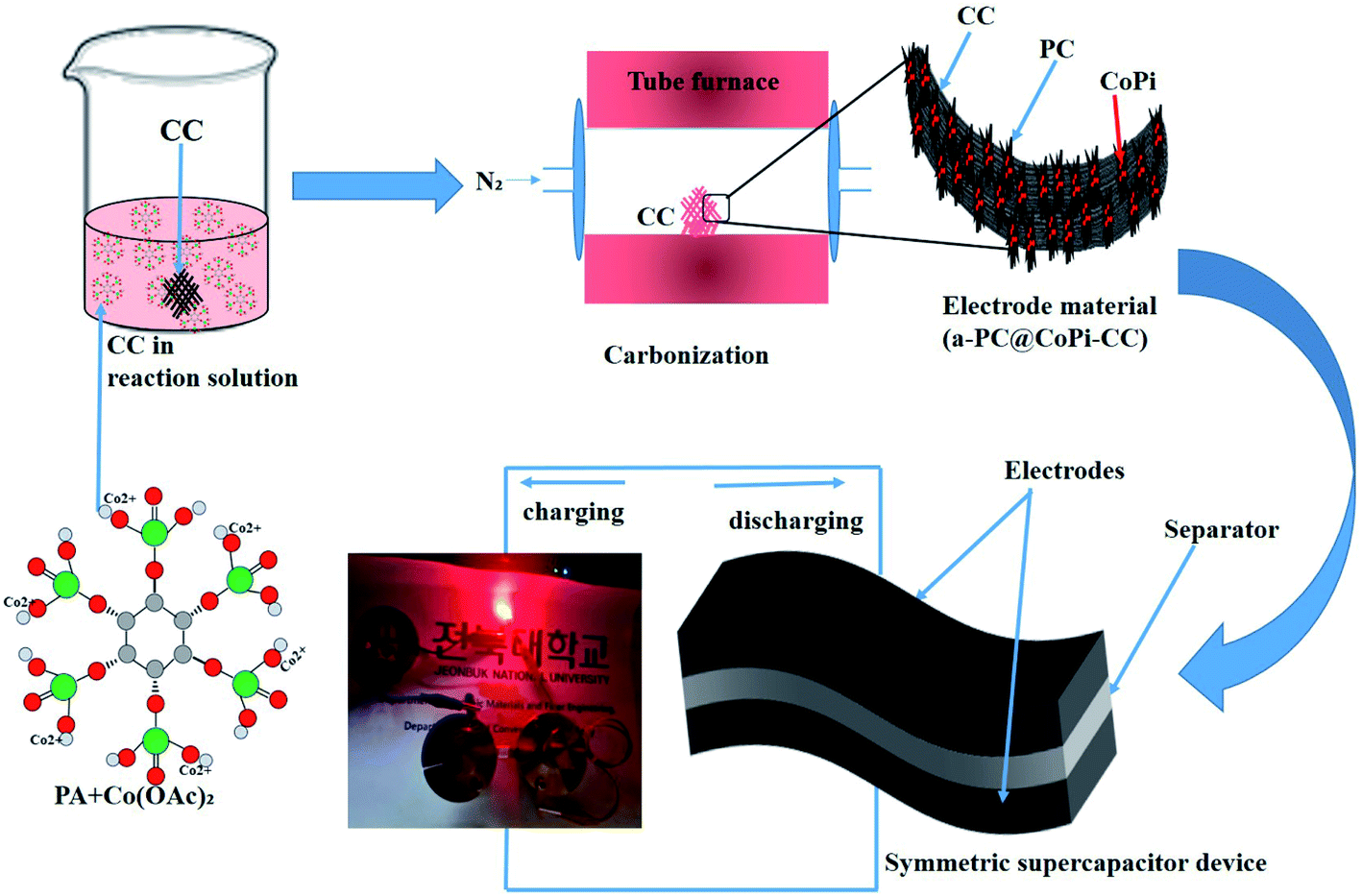
Herein, we used an amorphous composite with a nanostructured transition metal phosphate/carbon in which both components synergistically work together to improve the energy storage performance. Zhang et al.33 prepared transition metal phosphides (TMPs) with an organic phosphorous source, namely, phytic acid (PA). Interestingly, PA-assisted synthesized TMPs have an electrochemical performance that is superior to that of TMPs obtained with the assistance of a common phosphorous source. In another work, Zhao et al.34 showed that the electrochemical performance of PA-assisted synthesized glucose-derived internal carbon/lithium iron phosphate (GC/IC/LFP), such as stability and capacity recovery ability, are better than those of GC/LFP prepared using phosphoric acid as a common phosphorous source; this result was attributed to the higher oxygenic groups provided by PA and their assistance in providing a hierarchical network than that of the latter case. These studies emphasize that PA can be a key component to assist the synthesis of TMP/TMPi, which can possess improved electrochemical properties compared to those of non-PA-assisted synthesis routes for phosphates and phosphides. PA not only assists in the in situ formation of transition metal phosphates/phosphides but also acts as a carbon support for in situ-formed phosphorous-containing TMs.35 Moreover, the use of PA ensures the application of eco-friendly chemicals while avoiding toxic chemicals and gases [e.g., sodium hypophosphite (NaH2PO2), phosphane (PH3), and red phosphorus] as phosphorous sources for the synthesis of TMP/TMPi. Therefore, we motivated synthesizing a PA-derived TMPi as an electrode material via a simple controlled heating approach. PA–metal (PA–M) complexes undergo breakdown at high temperature and reorganize as metal phosphate/phosphide.33,34 In this study, PA-mediated synthesis of amorphous carbon-supported CoPi particles arrayed on a carbon cloth (CC) was proposed for the first time for supercapacitor applications. The synthesis of the electrode material was achieved via a simple pyrolysis process, as shown in a schematic illustration (Scheme 1). The formed electrode material works in both the positive and negative potential ranges, making it possible to assemble a symmetrical supercapacitor (SSC) device. The as-prepared SSC showed high energy density and cyclic stability with super flexibility. This study provides a strategy to further increase the capacitance and energy density of carbon-based materials, making them efficient and cost-effective supercapacitor electrode materials.
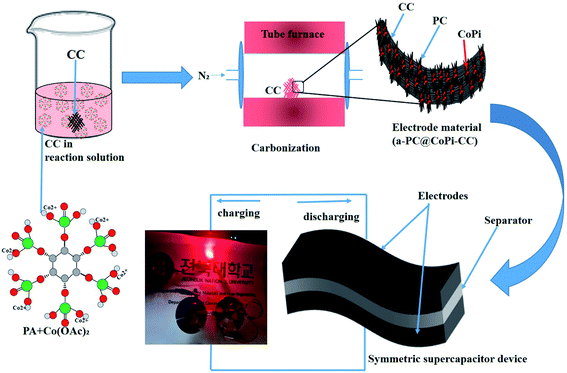 |
| | Scheme 1 Schematic illustration of the fabrication of a flexible and solid-state symmetric supercapacitor (SSC) based on a-PC@CoPi-CC. | |
2. Experimental section
2.1. Materials
A commercial CC with a thickness of 0.55 mm and a density of 120 g m−2 was received from O-Sung Co. Ltd, Korea, and cleaned by acetone, ethanol, and deionized water in sequence before use. Cobalt acetate(II)tetrahydrate [Co(OAc)2·4H2O] and PA (50 wt% aqueous solution) were purchased from Sigma, Korea.
2.2. Electrode preparation
Scheme 1 presents the process for fabricating the electrode material in which a commercial CC was used as the conductive support. In a typical experiment, 44.8 mg cobalt acetate (0.18 mM) was into the 2 mL PA/ethanol (1:1) solution and stirring for 30 minutes until to dissolve. A CC with a size of 1 cm2 was dipped into the PA/ethanol solution containing cobalt salt, dried in an oven at 60 °C for 20 minutes and turned upside down (2–5 times). This process was repeated to increase the loading of the active materials. Later, the electrode material was subjected to carbonization at 750 °C in a nitrogen (N2) atmosphere for 2 h. After cooling, the loading amount of the active material was calculated based on the changes in the weight of the CC before and after deposition of the material. We found that the CC contained the active materials (CoPi/C) in amounts of 2, 5, 8, and 12 mg, which are referred to as a-PC@CoPi-CC2, a-PC@CoPi-CC5, a-PC@CoPi-CC8, and a-PC@CoPi-CC12, respectively. Moreover, the concentration of cobalt acetate was 89.6 mg (0.36 mM) and 179.3 mg (0.72 mM), and the samples were prepared using the same procedure mentioned. Similarly, a sample was prepared without using PA; it used glucose as the carbon source, followed the same protocol, and is referred to as GC@Co-CC. A control sample was also prepared without using cobalt acetate, and is referred to as PC@CC.
2.3. Characterization
Physicochemical characterizations are discussed in ESI.†
2.4. Electrochemical measurements
The electrochemical tests were carried out using electrochemical impedance spectroscopy (EIS) board. An Ag/AgCl reference electrode (Fisher Scientific) and a platinum counter electrode (Fisher Scientific) were used in measurements. The electrochemical properties were measured in a three-electrode configuration in a 2 M KOH electrolyte unless otherwise stated. The cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) curves were recorded within the different potential windows, while EIS was performed at an AC potential amplitude of 5 mV with a variable frequency between 0.01 Hz and 100 kHz. The areal capacitance (CA; F cm−2) and specific capacitance (Cs; F g−1) were calculated from the GCD curves according to eqn (I) and (II), respectively.| | 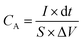 | (I) |
| | 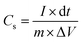 | (II) |
where I (A) is the charge–discharge current at the discharge process; and dt, S, ΔV and m are the discharge time (s), area of the active materials (cm2), potential window (V), and the mass of the active electrode materials (g), respectively.
2.5. Assembly of the SSC device
The SSC was assembled using the a-PC@CoPi-CC5 material as the positive and the negative electrode with a size of 1 cm2 each. The electrochemical performance of the fabricated SSC device was evaluated using a two-electrode configuration with a polyvinyl alcohol/potassium hydroxide (PVA–KOH) gel as a solid electrolyte. The PVA/KOH was prepared by adding 6 g of KOH and 6 g of PVA to 60 mL of water under vigorous stirring at 80 °C for 2 h. The filter paper separator was immersed in the gel electrolyte for 10 minutes. To assemble the SSC, two identical electrodes and a separator were sandwiched together. Total thickness of device was 1.5 mm. The areal capacitance (CA; F cm−2), areal energy density (E; W h cm−2), and power density (P; W cm−2) were evaluated according to eqn (I), (III), and (IV), respectively.| | 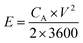 | (III) |
| | 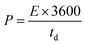 | (IV) |
where V is the potential input (V) and ‘td’ is the discharging time (s). To calculate the volumetric capacitance, energy density and power density, the total volume of the symmetric cell was estimated about 0.15 cm3, including the two symmetrical electrodes and separator.
3. Results and discussion
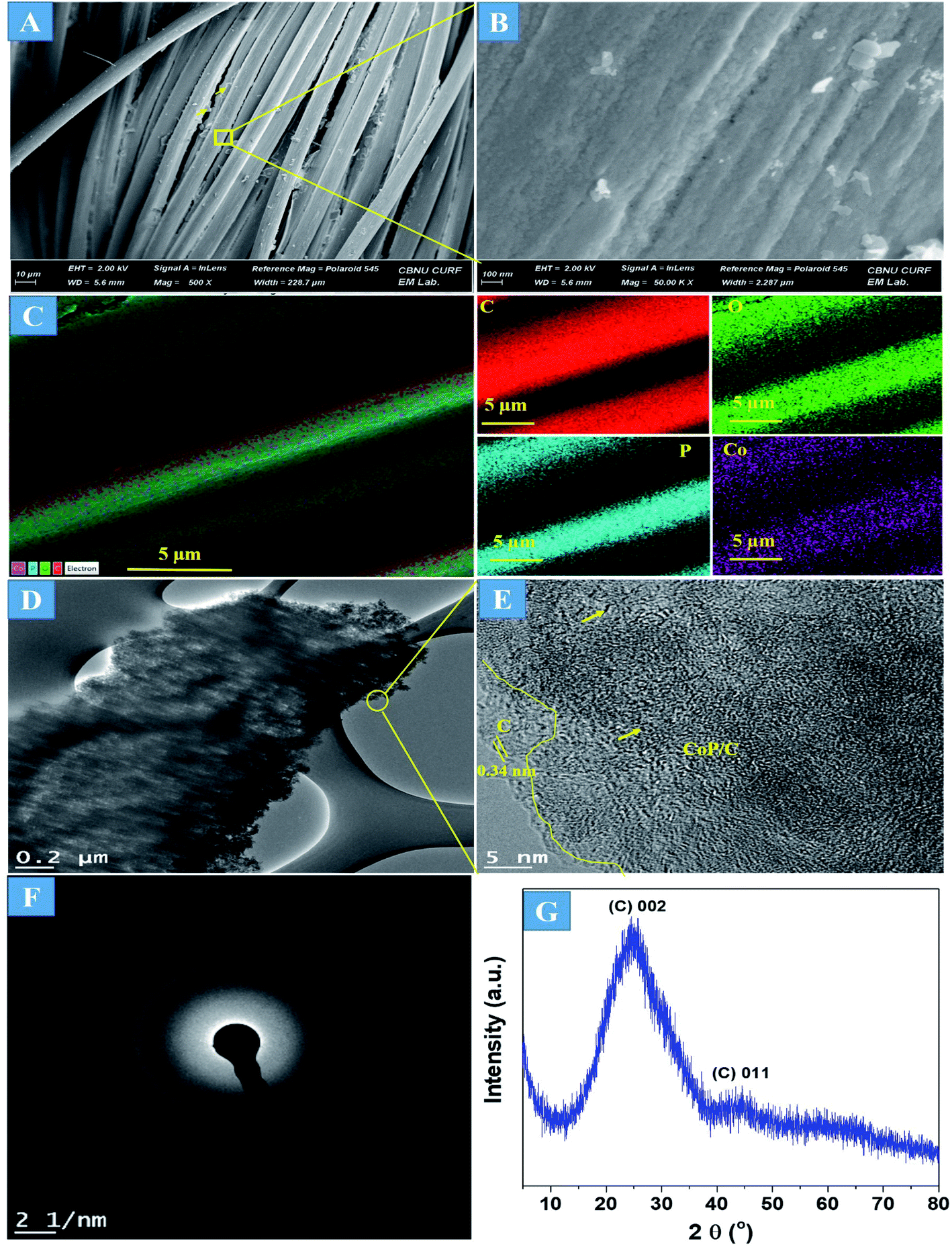
The preparation process of the CoPi/C anchored carbon fiber was carried out successfully by a single-step pyrolysis method as shown in scheme (Scheme 1). First, the CC was saturated with the mixture of PA and cobalt salt followed by carbonization at 750 °C in a N2 atmosphere. The PA bonded to the Co2+ with its negatively charged groups, thus making a metal phytate complex. At subsequent heating at a high temperature, the metal phytates decomposed into the respective metal phosphate along with freshly synthesized PA-derived carbons.33,36 Typical SEM images of the a-PC@CoPi-CC8 show that the sample consisted of a PA-derived sheet-like structure (arrows) along with a layer of nanoparticles attached to the surface of the fiber (Fig. 1A and B). Elemental mapping images show the homogeneous distribution of all expected elements, namely, C, O, P, and Co (Fig. 1C).
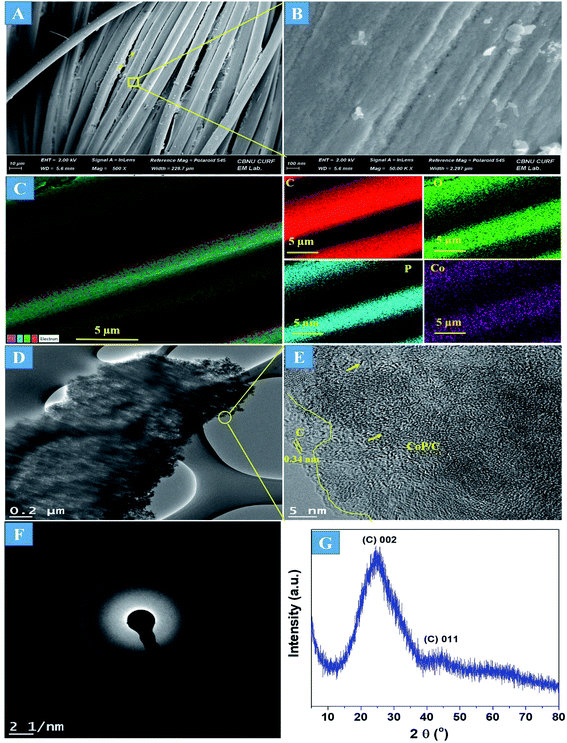 |
| | Fig. 1 FE-SEM images (A and B), elemental mapping images (C), HRTEM images (D and E), SAED pattern (F), and (G) XRD spectrum of the a-PC@CoPi-CC8. | |
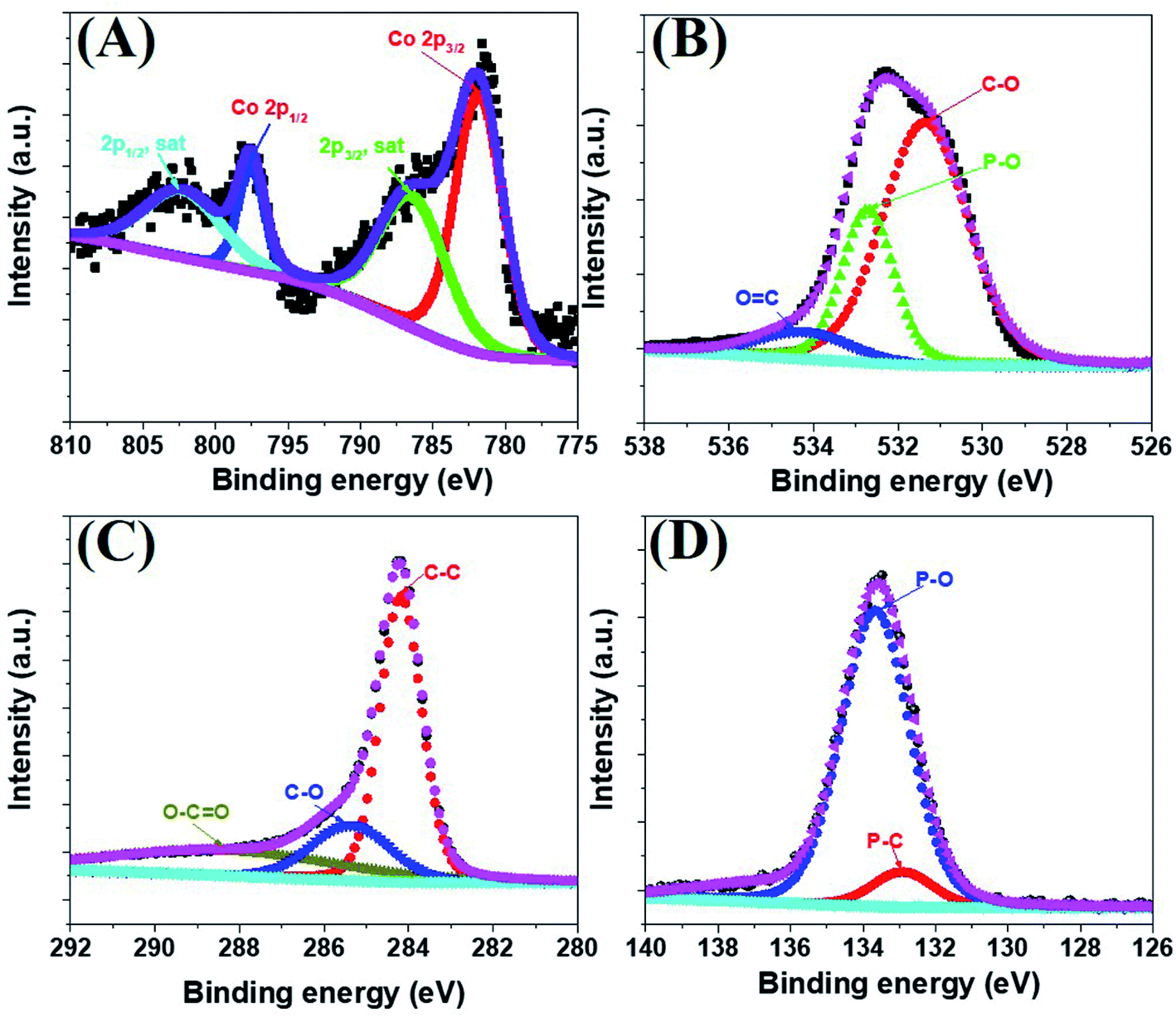
The morphology of the a-PC@CoPi-CC8 was further examined by HRTEM analysis. The TEM image (Fig. 1C) reveals that the apparent coating layer consisted of a layer of Co/C-based particles, which agrees with the SEM (Fig. S1†) and atomic force microscopy results (Fig. S2†). The HRTEM image reveals that the coating layer consisted of many defects (Fig. 1E, yellow arrows), as indicated by the black dots or canals and short-range disorder throughout. The absence of a clear lattice in the TEM image suggests the presence of an amorphous material and is consistent with the diffuse selected area electron diffraction (SAED) pattern from the corresponding image (Fig. 1F). The presence of carbon in the boundary region with a thickness of up to 5 nm can be explained by the presence of a lattice with a parameter of 0.34 nm that does not have long-range order (Fig. 1E). This indicates that the carbon layer shielded the inner structure. The uniform distribution of the elements, as shown in the STEM elemental mapping images (Fig. S3†), highlights the intercalation of all major components throughout. The C, O, P and Co contents were 76.6, 12.8, 7.6, and 3.0%, respectively. The XRD pattern (Fig. 1G) for the a-PC@CoPi-CC8 contains broad reflection peaks located at approximately 25° and 45° that corresponds to carbon, further confirming the amorphous nature of the composite. XPS was conducted for the a-PC@CoPi-CC8 as a representative amorphous structured composite to understand the chemical bonding and elemental composition. Fig. 2A, B, C and D show the Co 2p, O 1s, C 1s, and P 2p high-resolution emission spectra, respectively. In the Co 2p spectra, the peak at 781.9 eV and its satellite peak at 786.4 eV from Co 2p3/2 electrons are ascribed to Co species in the CoPi.37 Besides, the peaks at 797.4 eV and 802.5 eV are attributed to Co 2p1/2 and its shakeup excitation, respectively.38 The binding energies at 531.3 and 534.2 eV are ascribed to C–O and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding, respectively, while the peak at 532.7 eV can be explained by P–O bonding.39 The C 1s XPS spectrum possesses three main peaks at 284.1, 285.3 and 288.2 eV corresponding to functional groups C–C, C–O, and CO, respectively. For the wide-range P 2p spectrum, the binding energy of 132.9 eV corresponds to P–C bonding, suggesting the doping of additional P into the network.40 On the other hand, the peak at 133.6 eV is assigned to the metal phosphate (Co-POx).41 These results confirm the inclusion of the CoPi/C composite into the CC.
C bonding, respectively, while the peak at 532.7 eV can be explained by P–O bonding.39 The C 1s XPS spectrum possesses three main peaks at 284.1, 285.3 and 288.2 eV corresponding to functional groups C–C, C–O, and CO, respectively. For the wide-range P 2p spectrum, the binding energy of 132.9 eV corresponds to P–C bonding, suggesting the doping of additional P into the network.40 On the other hand, the peak at 133.6 eV is assigned to the metal phosphate (Co-POx).41 These results confirm the inclusion of the CoPi/C composite into the CC.
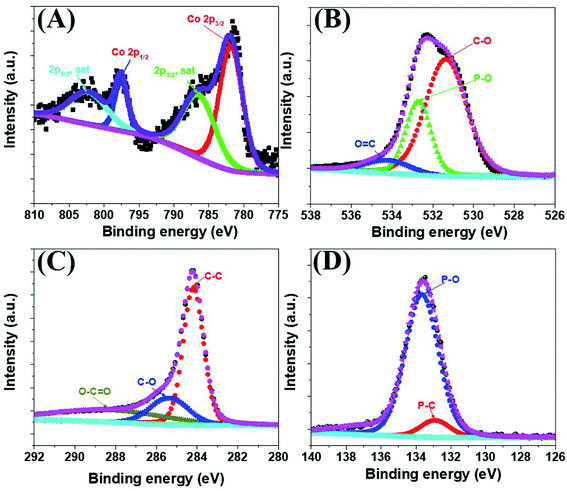 |
| | Fig. 2 Co 2p (A), O 1s (B), C 1s (C), and P 2p (D) high-resolution XPS survey spectra for the a-PC@CoPi-CC8. | |
Supercapacitor electrodes with high mass loading are crucial for obtaining favorable electrochemical performance in real-world applications that require a loading of approximately 10 mg cm−2. Therefore, we loaded different amounts of active materials in the substrate. As the first step of the evaluation, the surface morphology and material phase were studied. Increasing the active materials from 2 mg to 12 mg did not show any morphological differences in terms of the fiber surface (Fig. S4†). However, the fiber junctions were likely to appear filled with active materials in the case of higher mass loaded samples, as shown in the FESEM images (Fig. S4C and D arrows†). The amorphous nature of the sample was observed independent of the loading amount, as indicated by the SAED pattern of the a-PC@CoPi-CC12 (Fig. S4D inset†). However, by increasing the concentration of the cobalt salt to 0.72 mM in the reaction solution, noticeable particle aggregates were observed to be randomly attached to the CC (Fig. 3A and B). Elemental mapping images show that C, O, P, and Co were present (Fig. 3C). The crystallinity was evaluated by XRD. As shown in Fig. 3D, XRD peaks at 2θ values of 14.4, 20.9, 26.1, 28.0, 29.7, 31.2, 34.4, 37.7, 43.2, 53.9, 56.9, and 60.4° correspond to the (011), (−112), (121), (310), (−222), (013), (400), (−323), (−233), (−622), (−440) and (−633) planes, respectively, for CoPi. All the peaks agreed well with the standard pattern for CoPi (JCPDS 00-027-1120). As the results indicated, the sample obtained from the 0.72 mM cobalt salt was crystalline, and the sample is referred to as c-PC@CoPi-CC. The loading amount of the active material was estimated to be 8 mg cm−2. The sample prepared from a cobalt salt concentration of 0.36 mM was also crystalline, as indicated by XRD pattern (Fig. S5†). But only a few peaks with lower intensities at 2θ = 26.1, 29.7, 43.2, and 53.7° were observed in this condition. This may be due to less abundance of the cobalt salt with respect to PA used.
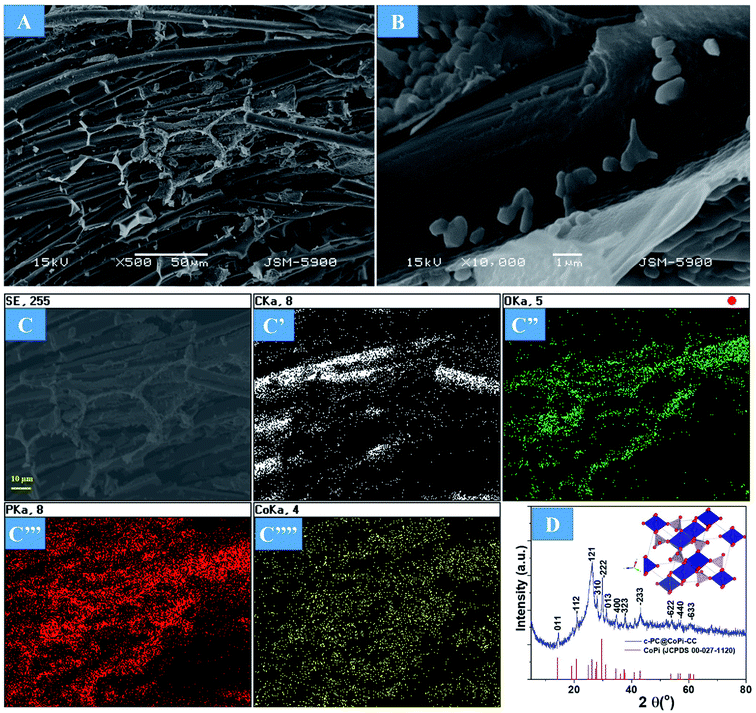 |
| | Fig. 3 FE-SEM images (A and B), corresponding elemental mapping images (C) and XRD of the c-PC@CoPi-CC (D). The inset in (D) shows the schematic crystal structure of the CoPi based on the mp-541790 data. | |
The substantial phase variation of the a-PC@CoPi-CC and c-PC@CoPi-CC hints that the formation of amorphous CoPi or crystalline CoPi was ultimately mediated by the concentration of Co monomers (Fig. S6†). For the a-PC@CoPi-CC, PA sufficiently bound the cobalt ions when there was a low cobalt salt concentration, thereby reducing the chance for mutual interaction of the cobalt ions. This process could prevent crystallization of the in situ formed CoPi during pyrolysis. This was unlikely to occur for the c-PC@CoPi-CC, where the formation of large crystallites occurred, which can be attributed to an insufficient amount of PA with respect to the high cobalt salt concentration.
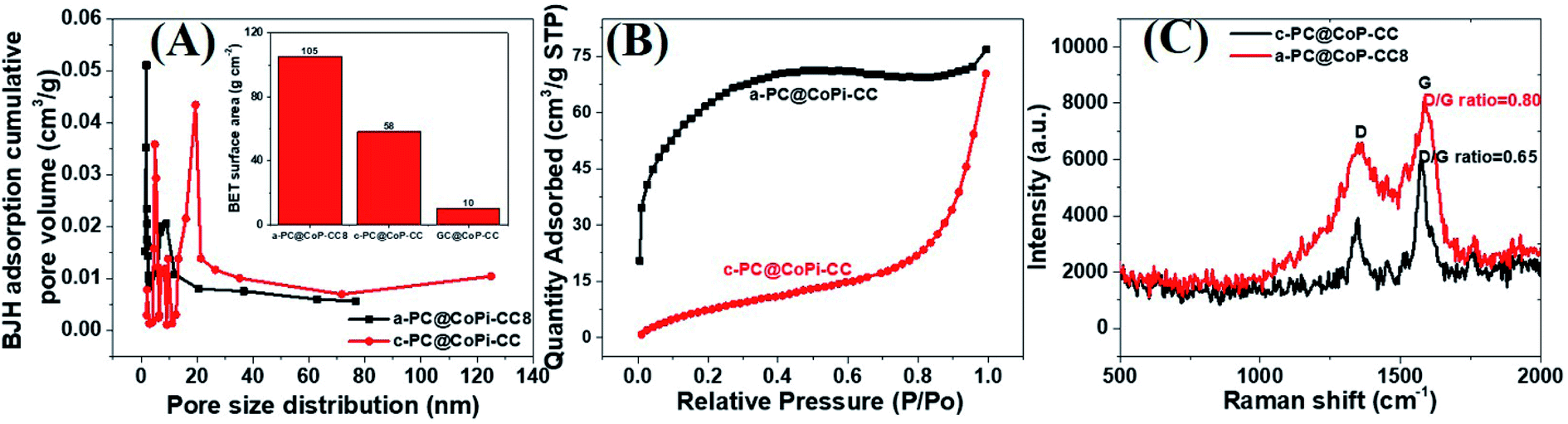
The morphology of the GC@Co-CC as the carbon source was studied by electron microscopy. Results show the pure Co particles were attached on the fiber surface. Details are presented in ESI (Fig. S7†). Upon exposure to Co salt at a high temperature, Co ions are readily reduced in situ to metallic Co,42 thereby forming the glucose-derived carbon/Co composite. The PA/ethanol mixture was evenly distributed on the CC surface, in contrast to the low absorption of the water/ethanol mixture (Fig. S8†). Therefore, all PA-assisted amorphous samples (a-PC@CoPi-CCs) prepared in PA/ethanol showed a uniform particle distribution on the substrate, unlike the samples without PA (GC@Co-CC/GC@CoPi-CC). A glucose-assisted CoPi/C composite on the CC was also prepared for comparative study using the commonly used simple hydrothermal process followed by phosphorization. The sample is referred to as GC@CoPi-CC. CoPi particles with a size from 20–50 nm were found anchored on the carbon fiber surface. Some particle agglomerations were also observed randomly on the fiber surface (Fig. S9A and B†). The XPS results confirmed that the particles were CoPi (Fig. S9D and E†). An overall comparison of the morphologies of the PA-assisted synthesized samples and non-PA synthesized samples showed that the prior sample had a uniform coating layer consisting of small particles attached to the substrate (Fig. 1A and B); this is in contrast to the particle aggregates randomly found in later cases (Fig. S7 and S9†), suggesting that PA acted as a structural directing agent in addition to acting as a precursor for carbon and phosphate. The BET surface area and pore size of the samples were evaluated by N2 adsorption–desorption methods, and the results are summarized in Fig. 4 and Table 1. The a-PC@CoPi-CC8 sample exhibited a higher BET surface area of up to 105.4 m2 g−1 compared to the significantly lower BET surface areas of 58.1 and 10.1 m2 g−1 for the c-@PC-CoPi-CC and GC@CoPi-CC samples, respectively. Additionally, the amorphous sample had a higher pore volume (Table 1). After sufficient PA induction, sufficient porosity developed in contrast to other samples that show large particles embedded into the CC. The PA released carbon dioxide during carbonization, leading to the formation of highly microporous materials on the carbon support. The pore distribution data supported that the a-PC@CoPi-CC8 possessed micropores (<2 nm) (Fig. 4A) followed by mesopores (4–50 nm) whereas the c-PC@CoPi-CC and GC@CoPi-CC had average pore sizes of 9.8 and 139.5 nm, respectively (Table 1). The N2 sorption data in Fig. 4B show that the a-PC@CoPi-CC8 exhibited significant N2 uptake at a relative pressure (P/P0) below 0.01, which was attributed to the presence of micropores.43 The continuous N2 uptake at P/P0 between 0.05 and 0.3 was attributed to N2 adsorption in the mesopores, suggesting the presence of micro- and mesopores in the a-PC@CoPi-CC8 sample, which is in good agreement with the pore distribution data. This can result in improving electrochemical performance. Raman spectra of the amorphous and crystalline samples exhibited typical D and G bands at 1353 and 1596 cm−1, respectively, representing the disordered (D) and graphitic (G) phases in the carbon, respectively (Fig. 4C). However, the ratio of D/G, which reveals the level of ordering and defects in the carbon structure, indicates that the a-PC@CoPi-CC showed a higher D/G ratio (0.80) than the c-PC@CoPi-CC (0.65). This indicates that many defects or disordered sites were present in the a-PC@CoPi-CC8, which agrees well with the HRTEM results. Additionally, this result can be related to the increased porosity of the amorphous sample.
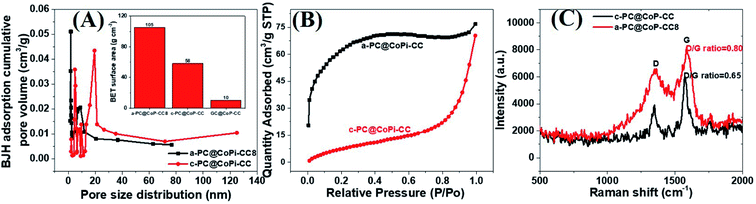 |
| | Fig. 4 BJH pore distribution (A), where the inset shows the BET surface area of the different samples. N2 adsorption isotherms (B) and Raman spectra (C) of the a-PC@CoPi-CC8 and c-PC@COPi-CC. | |
Table 1 The detailed BET surface area, average pore size and BJH pore volume of the as-prepared samples
| Sample |
Surface area (m2 g−1) |
Average pore size (4V/A by BET) |
Total pore volume (cm3 g−1) |
| a-PC@CoPi-CC2 |
93.2 |
1.9 nm |
0.11 |
| a-PC@CoPi-CC5 |
95.3 |
1.9 nm |
0.12 |
| a-PC@CoPi-CC8 |
105.4 |
1.8 nm |
0.14 |
| c-PC@CoPi-CC |
58.1 |
9.8 nm |
0.10 |
| GC@CoPi-CC |
10.1 |
139.5 nm |
0.09 |
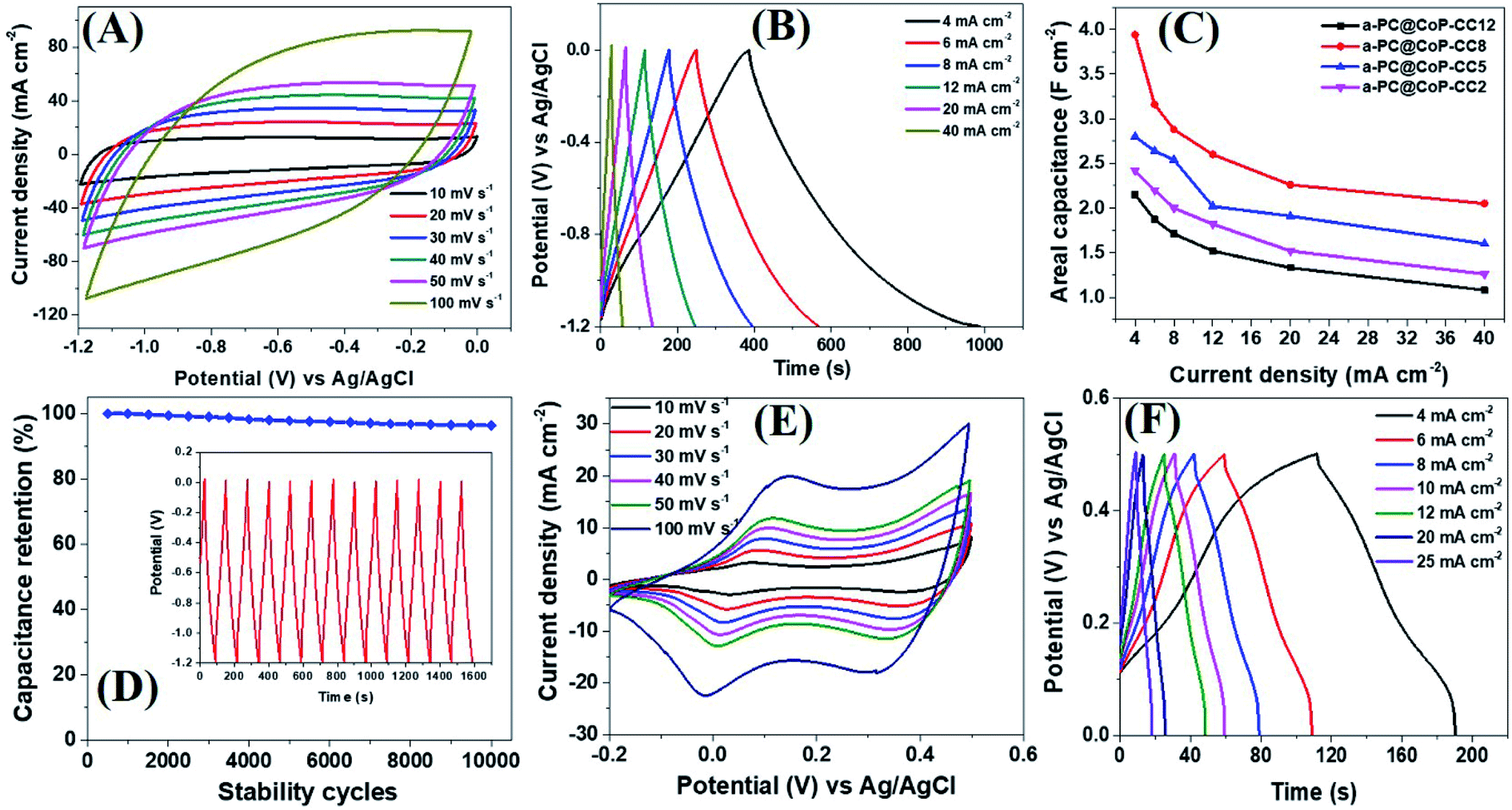
The electrochemical performance of the a-PC@CoPi-CC electrodes with different mass loading capacities (2, 5, 8, and 12 mg) was evaluated by cyclic voltammetry. The obtained results were compared with those from the control samples, such as c-PC@CoPi-CC, PC@CC, GC@CoPi-CC, and GC@Co-CC. The CV results show that all the amorphous samples (a-PC@CoPi-CCs) exhibited an EDLC charge storage behavior over a wide range of negative potentials from −1.2 to 0.0 (Fig. 5A and S10–S12†). The corresponding GCD profiles also showed a linear charge–discharge pattern consistent with that for the EDLC storage-type materials. Fig. 5A and B shows the CV and GCD data, respectively, of the a-PC@CoPi-CC8. The areal capacitance estimated from the GCD curves was 1.99, 1.58, 1.44, 1.30, 1.13, and 1.05 F cm−2 at current densities of 4, 6, 8, 12, 20, and 40 mA cm−2, respectively. Fig. 5C shows that the areal capacitances were a function of the mass loading of the active materials. For instance, a-PC@CoPi-CC12 and a-PC@CoPi-CC8 showed much higher areal capacitances of approximately 2.15 F cm−2 and 1.99 F cm−2 at a current density of 4 mA cm−2, respectively, compared to areal capacitances of 1.60 F cm−2 and 1.21 F cm−2 for a-PC@CoPi-CC5 and a-PC@CoPi-CC2, respectively (Table S1†). However, the specific capacitances were found to have an opposite trend to that of the areal capacitances, i.e., a low mass loading electrode (a-PC@CoPi-CC2) had the highest capacitance of 606.1 F g−1 compared to lower capacitances of 322.0, 248.7 and 179.5 F g−1 for the a-PC@CoPi-CC5, a-PC@CoPi-CC8 and a-PC@CoPi-CC12 at a 1 A g−1 current density, respectively (Table S1†). These values are superior to most reported values for anode materials (Table S2†).44 High mass loading per unit area gives the high current than for the low mass loading which is a general notion. According to the eqn (I), areal capacitance is directly proportional to the current (area of electrode kept constant i.e. 1 cm2). Hence, it was justifying that upon increasing the mass loading from 2 to 12 mg cm−2, the areal capacitance linearly increased. However, the current may not increase with the same proportion to the mass loading because high mass loading may cause lower electrical conductivity, limited accessing of electrolyte ions, and higher series resistance due to longer transport paths for the diffusion of protons.45 Moreover, high mass loading in the same area can lead to the increased thickness of the electrode material and more chances of agglomeration may occur. These also affect negatively on the conductivity of the electrodes and eventual performance. According to eqn (II), the specific capacitance is indirectly proportional to the mass of the material. Therefore high mass loading electrode had lower specific capacitance than for the low mass loading electrodes. All amorphous samples retained 50–60% of the capacitance of the initial value even at a high current density of 40 mA cm−2 and retained about 95% of the capacitance following the stability tests (Fig. 5C, D and Table S1†). In practical applications, the importance of a high mass loading of active materials for the fabrication of supercapacitors has been highlighted.46 High mass loaded electrodes can offer high charge storage, which is critical to increasing device capacity. The high mass loading as reported in this study is attributed to the metal chelating effect of PA. The repeated dipping–drying process of the CC in a solution containing PA/metal salt led to the higher mass loading due to the PA-assisted gelation effect. The GC@Co-CC and GC@CoPi-CC samples prepared without PA had a lower mass loading of only 2 mg cm−2, which supports that claim. Interestingly, amorphous electrode (a-PC@CoPi-CC8) can also work in a positive potential window (Fig. S13A†). Fig. S13B† presents the representative GCD curves for the a-PC@CoPi-CC8 that shows it can work over a wide potential range from −1.4 to 0.5 V under a high current density of more than 10 mA cm−2. Moreover, the a-PC@CoPi-CC8 was efficient in the neutral electrolyte (1 M aqueous Na2SO4) as well (Fig. S14A and B†). For example, the a-PC@CoPi-CC8 exhibits a capacitance of 1.63 F cm−2 at a current density of 4 mA cm−2 (204.5 F g−1 at 1 A g−1 current density). However, according to the CV data shown in Fig. S14C,† the a-PC@CoPi-CC8 can work over a wider potential window of 3.0 V (−1.5 to +1.5 V) when the process was conducted in Na2SO4. This finding can be possibly the highest working potential range to date reported in aqueous electrolytes.47 The widening of the cell voltage has a great advantage in enhancing the energy density of the supercapacitor as it is proportional to the square of the cell voltage. Correspondingly, the widening of the operative voltage for amorphous samples can be ascribed to the presence of phosphate groups as these block the active oxidation at a positive potential and the adsorption of hydrogen at a negative potential.48 This result suggests that the electrode proposed in this work can be used as both an anode and a cathode material.
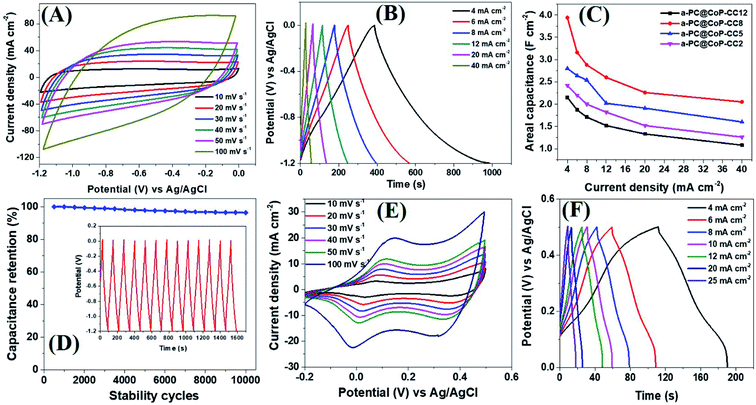 |
| | Fig. 5 CV (A) and GCD (B) curves of the a-PC@CoPi-CC8. A comparative view of the capacitance of the given electrodes (C) and capacitance retention as a function of stability cycles (D). The inset in (D) shows the GCD pattern for 10 cycles after the stability test (10000 cycles at 20 mA cm−2). CV (E) and GCD (F) curves of the c-PC@CoPi-CC. | |
In the absence of CoPi, the sample (PC@CC) had an areal capacitance of only 0.64 F cm−2 at 4 mA cm−2 (Fig. S15 and Table S1†), whereas the sample prepared in the absence of PA and with a glucose replacement as the carbon source (GC@Co-CC) had the lowest areal capacitance among all samples at approximately 0.10 F cm−2 (Fig. S16 and Table S1†). Pristine CC had negligible capacitive storage capability (data not shown). These values are significantly lower than the amorphous (CoPi/C) composites highlighting the synergistic effect of the PA and CoPi for improving the capacitive performance. Interestingly, the c-PC@CoPi-CC, which was prepared by the same method as for the a-PC@CoPi-CC, except for the use of a higher concentration of Co salt, showed pseudocapacitive behavior with clear redox peaks (Fig. 5E and F), unlike the other amorphous samples. The GCD data exhibited an areal capacitance of approximately 0.54 F cm−2 at a current density of 4 mA cm−2 with a stability of ∼76% (Fig. 5F and Table S1†). These values are much lower than those of the corresponding amorphous samples (Table S1†). Similarly, a CoPi/C composite on the CC (GC@CoPi-CC) was prepared using the common phosphorization exhibited clear redox peaks (Fig. S17†) close to the CV/GCD patterns of the c-PC@CoPi-CC. The intense anodic and cathodic peaks are ascribed to the conversion reaction of metal phosphates to metal hydroxides and oxides.49 The capacitance was reported to be 0.25 F cm−2 at a current density of 4 mA cm−2 (Fig. S18†), which is 50% lower than the capacitance obtained for the c-PC@CoPi-CC and 4–8 times lower than that for the a-PC@CoPi-CC samples (Table S1†). This result could be attributed to the lower BET surface area, and randomly found bigger particles on the fiber surface of GC@CoPi-CC in contrast to the PA-derived samples which had higher surface area and PA-induced internal carbon wrapped uniform CoPi particles. Importantly, the a-PC@CoPi-CCs exhibit an EDLC-type energy storage pattern that is different than the pseudocapacitance from the c-PC@CoPi-CC/GC@CoPi-CC. This result can be ascribed to the amorphous phase along with the influence of the phosphorous with short-range disorder and defects.31,50 The PA assisted the formation of an amorphous sample and generated a microporous carbon support with abundant phosphorus content that synergistically contributed to forming the electric double layer. Wang et al. demonstrated that amorphous microporous carbonaceous samples with heteroatom doping can contribute to the electric double-layer capacitance property.31 The crystallized samples show a typical pseudocapacitive behavior that is closely related to that in other works.49,51 The electrochemical stability of the c-PC@CoPi-CC sample exhibited a ∼76% capacitance retention, which is significantly lower than the ∼95% achieved for the a-PC@CoPi-CC, indicating an obvious kinetic limitation of the crystallized sample. It has been reported that crystalline samples undergo faster structural deformation during the charge/discharge process, leading to poor stability. However, transformation into the amorphous form of the samples resulted in a significantly higher retention. Postcycling analysis of the a-PC@CoPi-CC electrode material was done by SEM and TEM, as presented in ESI (Fig. S18A and B†) and showed no apparent changes in the physical morphology, and the particle layer remained intact close to the initial sample. The sample remained amorphous (Fig. S18C inset†) during the round-trip charge and discharge cycles. The a-PC@CoPi-CC electrode contained many micropores that resisted the volume change and structural pulverization.22,25 Moreover, the carbon layer that coated the CoPi/C protected it from overexposure into the electrolyte solution, which enhanced the stability of the material. Moreover, the a-PC@CoPi-CC8 retained its morphological integrity and configuration even after continuous sonication (power 99%) for 30 minutes (Fig. S19†), confirming that the sample was mechanically strong enough to bear a high shear stress and agitation.
The excellent supercapacitive performance of the a-PC@CoPi-CC electrode can be attributed to several factors. First, the preferable synergistic promotion of the PA-derived porous carbon together with the homogenously distributed amorphous CoPi improved the capacitive properties. The proposed mechanism behind the enhanced capacitance can be explained by the uniqueness of the amorphous structured composite which had a higher degree of the disorder as indicated by the TEM image (Fig. 1E) and Raman spectra analysis (Fig. 4C). Based on these observations the mechanism of ion storage and electron transfer was proposed and illustrated in Fig. S20 (left).† The abundance defects and disorders enabled the hydroxide ions to access very deep active sites to a large extent and fast electron transfer using multiple diffusion paths as shown in the illustration (Fig. S20†). Second, the significantly higher surface area of the a-PC@CoPi-CC than its corresponding crystalline counterparts provided more effective ion adsorption/desorption. A third reason could be due to the higher conductivity of the amorphous samples than their crystalline counterparts. Herein, the conductivity of the samples was evaluated by Nyquist plots (Fig. S21†). The semicircular diameter in the high-frequency region corresponds to the charge transfer (Rct) process occurring at the electrode/electrolyte interface and double-layer capacitance.52 It was observed that the c-PC@CoPi-CC had a larger diameter for the semicircle in the high-frequency region than that of other amorphous samples. The Rct values were 0.28, 0.001, 0.16, and 0.23 ohms for the a-PC@CoPi-CC2, a-PC@CoPi-CC5, a-PC@CoPi-CC8, and a-PC@CoPi-CC12, respectively, compared to higher Rct values of 0.7 and 0.4 ohms for the c-PC@CoPi-CC and GC@CoPi-CC, respectively (Table S3†). This suggests that electron transfer was more efficient in the amorphous samples than in their crystalline counterparts. This can explain why a lower capacitance was obtained for the c-PC@CoPi-CC electrode. The linear region parallel to the imaginary axis (Zim) in the low-frequency region was attributed to Warburg impedance (Zw, diffusion of ions in the electrode). The linear region at low frequencies for the PA-assisted electrodes came closer to an ideal straight line along the imaginary axis than the line for the c-PC@CoPi-CC, suggesting higher ion diffusion kinetics through the amorphous electrode samples. The a-PC@CoPi-CC exhibited a more uniform and porous nanostructure that can offer a more efficient exposure of accessible active surfaces for faster electrolyte penetration. The homogeneous carbon content efficiently increased the electron transport for the amorphous sample, which consequently improved the electroconductive performance as shown in Fig. S20.† On the other hand, the crystalline samples demonstrated that the presence of aggregated particles without a sufficient carbon support resulted in an increased resistance. Furthermore, the iR drop evaluation of the a-PC@CoPi-CC8 showed that there was no significant iR drop observed at the beginning of the constant current discharge, indicative of the low contact resistance (Fig. S22†).
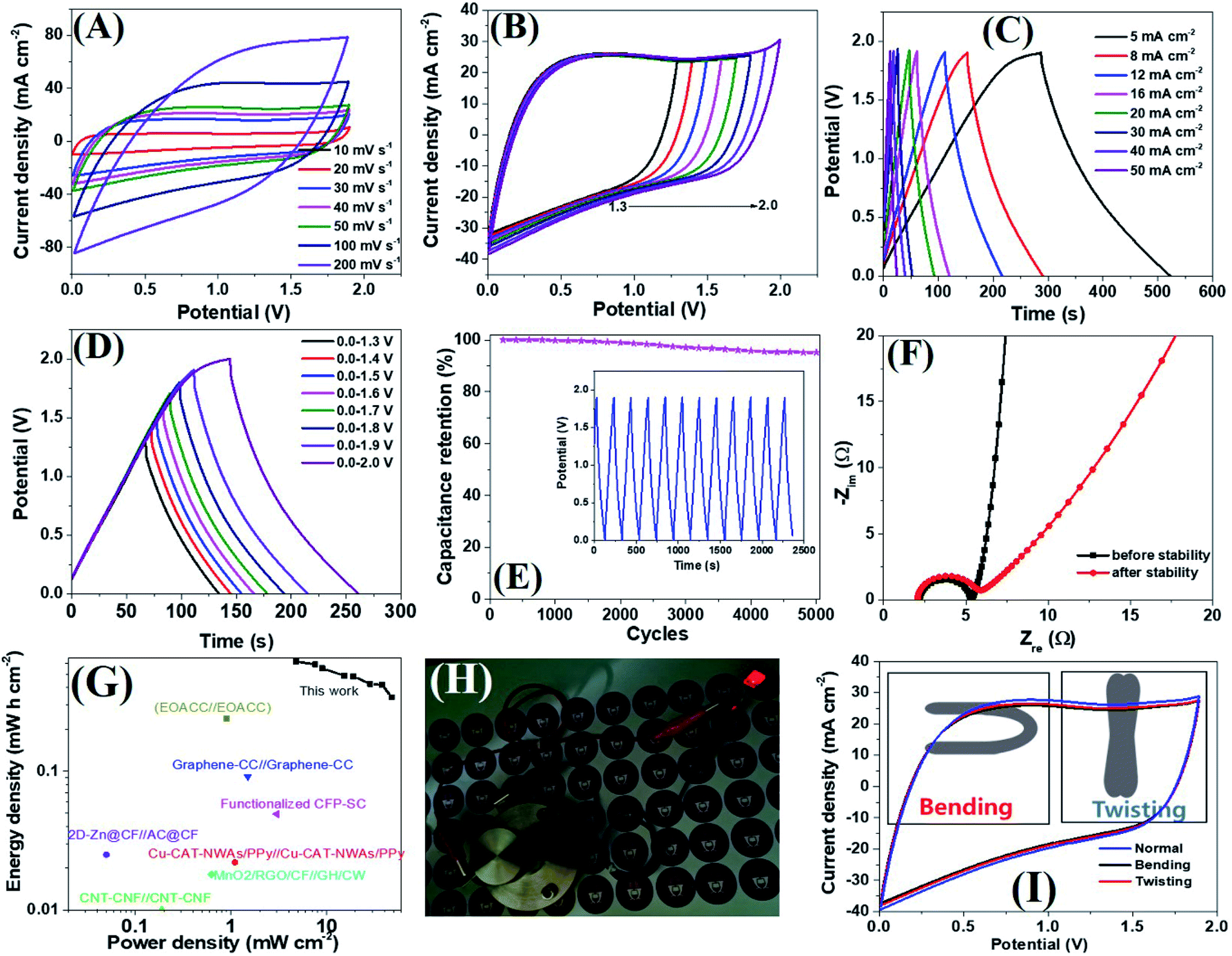
Encouraged by the excellent supercapacitive performance, a symmetric all-solid-state flexible supercapacitor device was prepared by taking an equal weight of amorphous electrodes. Due to the having superior conductivity of a-PC@CoPi-CC5 compared to other amorphous samples as indicated by the lowest Rs and Rct value (Table S3†), we have chosen this electrode to assemble a SSC device. The CV curves of the SSC are shown in Fig. 6A at different scanning rates at voltages ranging from 0.0 to 1.9. Similarly, the SSC is shown in Fig. 6B with operation voltages at a sweep rate of 50 mV s−1. The GCD profiles of the device are presented in Fig. 6C and D, which show a linear GCD curve compatible with the corresponding CV curves. The areal capacitance was calculated from the GCD curves (Fig. 6C) to be 620.0, 589.4, 552.0, 488.1, 484.0, 426.0, and 342.0 mF cm−2 at current densities of 5, 8, 10, 16, 20, 30, 40 and 50 mA cm−2, respectively. In the gravimetric data, the SSC showed specific capacitances of 62.8, 59.2, 55.2, 52.2, 49.4, 42.6, 42.1 and 34.2 F g−1 at 0.5, 0.8, 1.0, 1.6, 2.0, 3.0, and 5.0 A g−1 current density, respectively. Additionally, the device showed an excellent stability with a 95.2% capacitive retention after 5000 cycles at a current density of 10 mA cm−2 (Fig. 6E). Capacitance retention was still achieved 78.6% of initial capacitance when the current density increased from 5 to 20 mA cm−2 which later reached to 55.1% at 50 mA cm−2 while the coulombic efficiency of SSC was 95% or more above 10 mA cm−2 current density (Fig. S23A†). Based on the Nyquist plot (Fig. 6F and Table S3†) the Rs and Rct were similar for SSC before and after the stability test, suggesting the electrodes are capable of fast electron transport for an extended period. The Ragone plot correlating the areal energy densities (E) with areal power densities (P) indicated the highest power density of 47.4 mW cm−2 and a maximum energy density of 620.0 μW h cm−2 (gravimetric E = 31.1 W h kg−1 at P = 476.0 W kg−1). This output surpasses many relevant supercapacitor devices (Fig. 6G).2,18,53–61 The highest volumetric energy density was achieved 2.0 mW h cm−3 at a 31.65 mW cm−3 power density (Fig. S23B†). Importantly as-assembled SSC device alone lit the red LED bulb for about 1 min in 40 seconds charge at 10 mA cm−2 (Fig. 6H). This observation further confirms the wider working potential window up to 1.9 of SSC as claimed by CV/GCD. Further the two SSC devices were assembled to make double SSC (DSSC) devices in series (Fig. S24†). The CV and GCD data show the DSSC works up to 3.8 V. The result showed a red LED which remained lit for 4 min by DSSC with more brightness. The study demonstrated that the as-assembled supercapacitors could store high energy which implies their application in several types of electronic devices. The mechanical stability of the as-prepared flexible supercapacitor was evaluated by monitoring the capacitive behaviors during bending and twisting. As seen in Fig. 6I, the CV curves are identical in conditions including normal, twisting, and bending states, demonstrating the excellent mechanical flexibility and stability of the device. The extended durability of the device could be attributed to the high conductivity and flexibility of carbon fibers.
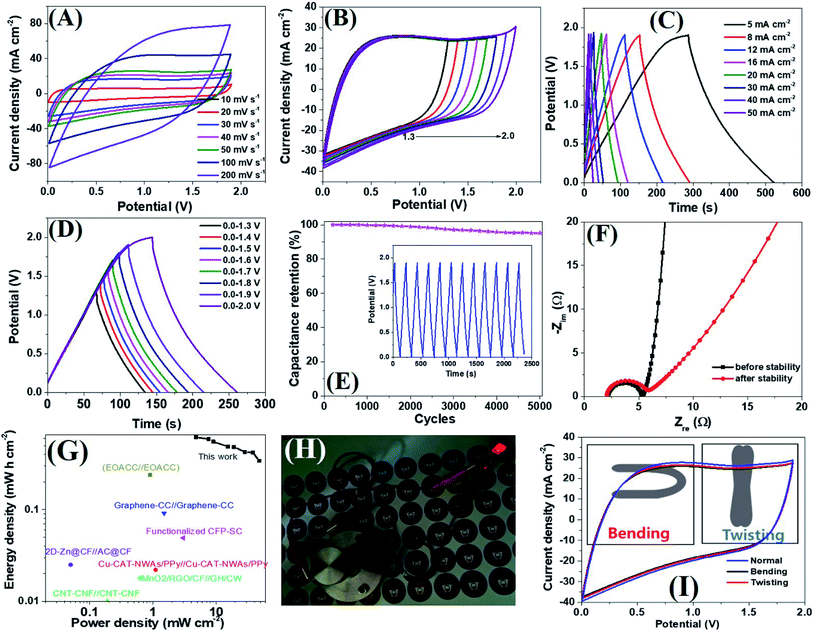 |
| | Fig. 6 CV curves of the SSC (a-PC@CoPi-CC5//a-PC-CoPi-CC5) at different scan rates in two-electrode systems (A), CV curves of the SSC with different potential windows at 50 mV s−1 (B), GCD curves at different current densities (C), GCD curves at different potential windows at 10 mA cm−2 current density (D), capacitance retention as a function of stability cycles (E). The inset in (E) shows the GCD pattern for 10 cycles after the stability test (5000 cycles at 10 mA cm−2). Nyquist plots of a device before and after the stability test (F). Ragone plot of SSC (G). A red LED powered by the SSC device (H) and comparing the CV curve of SSC in the normal state with bending and twisting (I). | |
4. Conclusions
In summary, PA-facilitated controlled synthesis of amorphous CoPi/C composites on CCs was successful with the use of a carbonization process. The PA contributed multidimensionally to the composite synthesis. It acted as a structure-directing agent and carbon precursor, and it assisted with the synthesis of the metal phosphate with the reaction of a metal salt. Additionally, it provided a platform for additional phosphorous doping in carbon. The amorphous samples delivered areal capacitances of up to 2.15 F cm−2 at a current density of 4 mA cm−2 and exhibited an ∼95% capacitance retention after 10000 charge and discharge cycles at 20 mA cm−2. These values were found to be significantly superior to their corresponding crystalline counterpart, which had a capacitance of 0.54 F cm−2 and ∼76% cyclic stability. Increased microporosity, a hierarchically porous carbon network containing short-range disorders or defects, and increased conductivity synergistically contributed to amorphous-based CoPi/C composite samples with a high capacitance. Interestingly, the amorphous composite electrode worked over a broad potential window up to 1.9 V in 2 M KOH and up to 3 V aqueous 1 M Na2SO4. The SSC device (a-PC@CoPi-CC5//a-PC@CoPi-CC5) showed an energy density of 620.0 μW h cm−2 at a power density of 4.7 mW cm−2 (31.1 W h kg−1 at 476.0 W kg−1), demonstrating that a-PC@CoPi-CC is an outstanding candidate for energy storage. Overall, the results support the design and use of amorphous metal phosphate-based carbon composites as anode materials for high-energy storage symmetrical supercapacitor devices.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2019R1A5A8080326). This research was supported by the program for fostering next-generation researchers in engineering of National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (No. 2017H1D8A2030449). We are also thankful to the Korean Basic Science Institute, Jeonju and CURF (JBNU) for providing TEM, FE-SEM analysis.
References
- G. Xu, B. Ding, J. Pan, J. Han, P. Nie, Y. Zhu, Q. Sheng and H. Dou, J. Mater. Chem. A, 2015, 3, 23268–23273 Search PubMed.
- V. T. Le, H. Kim, A. Ghosh, J. Kim, J. Chang, Q. A. Vu, D. T. Pham, J.-H. Lee, S.-W. Kim and Y. H. Lee, ACS Nano, 2013, 7, 5940–5947 Search PubMed.
- S. Sun, T. Zhai, C. Liang, S. V. Savilov and H. Xia, Nano Energy, 2018, 45, 390–397 Search PubMed.
-
P. Simon and Y. Gogotsi, in Nanoscience and technology: a collection of reviews from Nature journals, World Scientific, 2010, pp. 320–329 Search PubMed.
- M. Liu, J. Li, W. Han and L. Kang, J. Energy Chem., 2016, 25, 601–608 Search PubMed.
- C. Chen, N. Zhang, Y. He, B. Liang, R. Ma and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 23114–23121 Search PubMed.
- H. Pang, Y.-Z. Zhang, Z. Run, W.-Y. Lai and W. Huang, Nano Energy, 2015, 17, 339–347 Search PubMed.
- B. Li, P. Gu, Y. Feng, G. Zhang, K. Huang, H. Xue and H. Pang, Adv. Funct. Mater., 2017, 27, 1605784 Search PubMed.
- H. Shao, N. Padmanathan, D. McNulty, C. O'Dwyer and K. M. Razeeb, ACS Appl. Energy Mater., 2019, 2, 569–578 Search PubMed.
- Z. Zheng, M. Retana, X. Hu, R. Luna, Y. H. Ikuhara and W. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 16986–16994 Search PubMed.
- M. Yu, Z. Wang, Y. Han, Y. Tong, X. Lu and S. Yang, J. Mater. Chem. A, 2016, 4, 4634–4658 Search PubMed.
- X. Lei, S. Ge, Y. Tan, Z. Wang, J. Li, X. Li, G. Hu, X. Zhu, M. Huang, Y. Zhu and B. Xiang, ACS Appl. Mater. Interfaces, 2020, 12, 9158–9168 Search PubMed.
- A. M. Elshahawy, C. Guan, X. Li, H. Zhang, Y. Hu, H. Wu, S. J. Pennycook and J. Wang, Nano Energy, 2017, 39, 162–171 Search PubMed.
- Q. Tang, W. Wang and G. Wang, J. Mater. Chem. A, 2015, 3, 6662–6670 Search PubMed.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M.-S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 Search PubMed.
- J. Liu, M. Zheng, X. Shi, H. Zeng and H. Xia, Adv. Funct. Mater., 2016, 26, 919–930 Search PubMed.
- G. Zhu, X. Zhang, Y. Li, G. Zhao, H. Xu and Z. Jin, Nanoscale Adv., 2020 10.1039/D0NA00372G.
- Z. Zhang, F. Xiao and S. Wang, J. Mater. Chem. A, 2015, 3, 11215–11223 Search PubMed.
- X. Zhang, S. Hou, Z. Ding, G. Zhu, H. Tang, Y. Hou, T. Lu and L. Pan, J. Alloys Compd., 2020, 822, 153578 Search PubMed.
- T. Zhou, Y. Du, S. Yin, X. Tian, H. Yang, X. Wang, B. Liu, H. Zheng, S. Qiao and R. Xu, Energy Environ. Sci., 2016, 9, 2563–2570 Search PubMed.
- S. Liu, Z. Tong, J. Zhao, X. Liu, J. Wang, X. Ma, C. Chi, Y. Yang, X. Liu and Y. Li, Phys. Chem. Chem. Phys., 2016, 18, 25645–25654 Search PubMed.
- H. Li, Y. Gao, C. Wang and G. Yang, Adv. Energy Mater., 2015, 5, 1401767 Search PubMed.
- K. A. Owusu, L. Qu, J. Li, Z. Wang, K. Zhao, C. Yang, K. M. Hercule, C. Lin, C. Shi, Q. Wei, L. Zhou and L. Mai, Nat. Commun., 2017, 8, 14264 Search PubMed.
- Y. Xi, B. Dong, Y. Dong, N. Mao, L. Ding, L. Shi, R. Gao, W. Liu, G. Su and L. Cao, Chem. Mater., 2016, 28, 1355–1362 Search PubMed.
- Y. Dong, S. Zhang, X. Du, S. Hong, S. Zhao, Y. Chen, X. Chen and H. Song, Adv. Funct. Mater., 2019, 29, 1901127 Search PubMed.
- J. Chen, J. Xu, S. Zhou, N. Zhao and C.-P. Wong, Nano Energy, 2016, 21, 145–153 Search PubMed.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 Search PubMed.
- B. Dahal, T. Mukhiya, G. P. Ojha, K. Chhetri, A. P. Tiwari, A. Muthurasu, M. Lee, S.-H. Chae, T. Kim, D. C. Chung and H. Y. Kim, Chem. Eng. J., 2020, 387, 124028 Search PubMed.
- G. Prasad Ojha, A. Muthurasu, A. Prasad Tiwari, B. Pant, K. Chhetri, T. Mukhiya, B. Dahal, M. Lee, M. Park and H.-Y. Kim, Chem. Eng. J., 2020, 399, 125532 Search PubMed.
- K. Chhetri, A. P. Tiwari, B. Dahal, G. P. Ojha, T. Mukhiya, M. Lee, T. Kim, S.-H. Chae, A. Muthurasu and H. Y. Kim, J. Electroanal. Chem., 2020, 856, 113670 Search PubMed.
- C. Wang, Y. Zhou, L. Sun, P. Wan, X. Zhang and J. Qiu, J. Power Sources, 2013, 239, 81–88 Search PubMed.
- J. Patiño, N. López-Salas, M. C. Gutiérrez, D. Carriazo, M. L. Ferrer and F. d. Monte, J. Mater. Chem. A, 2016, 4, 1251–1263 Search PubMed.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 Search PubMed.
- Q. Zhao, Y. Zhang, Y. Meng, Y. Wang, J. Ou, Y. Guo and D. Xiao, Nano Energy, 2017, 34, 408–420 Search PubMed.
- X. Wang, Z. Na, D. Yin, C. Wang, Y. Wu, G. Huang and L. Wang, ACS Nano, 2018, 12, 12238–12246 Search PubMed.
- J. Yang, D. Guo, S. Zhao, Y. Lin, R. Yang, D. Xu, N. Shi, X. Zhang, L. Lu, Y.-Q. Lan, J. Bao and M. Han, Small, 2019, 15, 1804546 Search PubMed.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 Search PubMed.
- L. Chen, Y. Zhang, H. Wang, Y. Wang, D. Li and C. Duan, Nanoscale, 2018, 10, 21019–21024 Search PubMed.
- R. Li, Z. Wei, X. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 Search PubMed.
- R. Zhang, C. Zhang and W. Chen, J. Mater. Chem. A, 2016, 4, 18723–18729 Search PubMed.
- P. Li, Z. Jin, J. Yang, Y. Jin and D. Xiao, Chem. Mater., 2016, 28, 153–161 Search PubMed.
- A. P. Tiwari, S.-H. Chae, G. P. Ojha, B. Dahal, T. Mukhiya, M. Lee, K. Chhetri, T. Kim and H.-Y. Kim, J. Colloid Interface Sci., 2019, 553, 622–630 Search PubMed.
- J. W. F. To, Z. Chen, H. Yao, J. He, K. Kim, H.-H. Chou, L. Pan, J. Wilcox, Y. Cui and Z. Bao, ACS Cent. Sci., 2015, 1, 68–76 Search PubMed.
- H. Wang, J. Deng, C. Xu, Y. Chen, F. Xu, J. Wang and Y. Wang, Energy Storage Mater., 2017, 7, 216–221 Search PubMed.
- J.-G. Wang, Y. Yang, Z.-H. Huang and F. Kang, Electrochim. Acta, 2011, 56, 9240–9247 Search PubMed.
- S.-Y. Lin and X. Zhang, J. Power Sources, 2015, 294, 354–359 Search PubMed.
- Y. Zhang, S. Duan, Y. Li, S. Zhang, Y. Wu, M. Ma, C. Tao, Z. Zhang, D. Qin and E. Xie, Dalton Trans., 2020, 49, 1785–1793 Search PubMed.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 Search PubMed.
- X. Chen, M. Cheng, D. Chen and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 3892–3900 Search PubMed.
- Y.-N. Liu, J.-N. Zhang, H.-T. Wang, X.-H. Kang and S.-W. Bian, Mater. Chem. Front., 2019, 3, 25–31 Search PubMed.
- H. C. Chen, S. Jiang, B. Xu, C. Huang, Y. Hu, Y. Qin, M. He and H. Cao, J. Mater. Chem. A, 2019, 7, 6241–6249 Search PubMed.
- W. Pell, B. Conway and N. Marincic, J. Electroanal. Chem., 2000, 491, 9–21 Search PubMed.
- R. Hou, M. Miao, Q. Wang, T. Yue, H. Liu, H. S. Park, K. Qi and B. Y. Xia, Adv. Energy Mater., 2020, 10, 1901892 Search PubMed.
- Z. Dou, Z. Qin, Y. Shen, S. Hu, N. Liu and Y. Zhang, Carbon, 2019, 153, 617–624 Search PubMed.
- P. Suktha, P. Chiochan, P. Iamprasertkun, J. Wutthiprom, N. Phattharasupakun, M. Suksomboon, T. Kaewsongpol, P. Sirisinudomkit, T. Pettong and M. Sawangphruk, Electrochim. Acta, 2015, 176, 504–513 Search PubMed.
- Y. Zheng, X. Wang, W. Zhao, X. Cao and J. Liu, Chem. Eng. J., 2018, 333, 603–612 Search PubMed.
- G.-H. An, J. Hong, S. Pak, Y. Cho, S. Lee, B. Hou and S. Cha, Adv. Energy Mater., 2020, 10, 1902981 Search PubMed.
- J. Li, X. Yun, Z. Hu, L. Xi, N. Li, H. Tang, P. Lu and Y. Zhu, J. Mater. Chem. A, 2019, 7, 26311–26325 Search PubMed.
- K. V. Sankar, S. C. Lee, Y. Seo, C. Ray, S. Liu, A. Kundu and S. C. Jun, J. Power Sources, 2018, 373, 211–219 Search PubMed.
- P. K. Katkar, S. J. Marje, S. S. Pujari, S. A. Khalate, A. C. Lokhande and U. M. Patil, ACS Sustainable Chem. Eng., 2019, 7, 11205–11218 Search PubMed.
- X. Jiang, W. Lu, X. Yu, S. Song and Y. Xing, Nanoscale Adv., 2020, 2, 3865–3871 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00670j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Kisan
Chhetri
a,
Gunendra Prasad
Ojha
a,
Hyoju
Kim
a,
Su-Hyeong
Chae
a,
Bipeen
Dahal
a,
Byoung Min
Lee
c,
Tanka
Mukhiya
a and
Hak Yong
Kim
*ab,
Kisan
Chhetri
a,
Gunendra Prasad
Ojha
a,
Hyoju
Kim
a,
Su-Hyeong
Chae
a,
Bipeen
Dahal
a,
Byoung Min
Lee
c,
Tanka
Mukhiya
a and
Hak Yong
Kim