
Chemoselective and fast decarboxylative allylation by photoredox catalysis under mild conditions†
Chenchen
Hu
and
Yiyun
Chen
*
State Key Laboratory of Bioorganic and Natural Products Chemistry, Collaborative Innovation Center of Chemistry for Life Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: yiyunchen@sioc.ac.cn
First published on 3rd August 2015
Abstract
Here we report a visible-light-induced decarboxylative allylation to build C(sp3)–allyl bonds. This allylation reaction works for primary, secondary, tertiary, benzyl, and alpha-heteroatom-substituted alkyl N-acyloxyphthalimides and is compatible with many sensitive functional groups. The reaction is complete in minutes at room temperature and can be run in both organic solvents and neutral aqueous solutions. A series of mechanistic experiments have been implemented and a functional-group-rich oligosaccharide modification is demonstrated with the decarboxylative allylation.
Introduction
The allylation is a powerful synthetic method to build carbon–carbon bonds and the resulting alkene is a versatile synthetic building block. Transition-metal-catalyzed allylations are widely-used, while their applications for C(sp3)-allylations are limited to α-activated alkyl groups to minimize β-hydrogen elimination (eqn (1)).1 In contrast, the radical allylations are effective to build C(sp3)–allyl bonds; however, the traditional use of heating or UV light compromises their functional group compatibility and chemoselectivity.2 Recently, visible-light-catalysis has enabled radical initiations under mild photoredox conditions including neutral aqueous solutions.3 Here we report a chemoselective and fast decarboxylative allylation for primary, secondary, tertiary, benzyl, and alpha-heteroatom-substituted alkyl N-acyloxyphthalimides by photoredox catalysis (eqn (2)).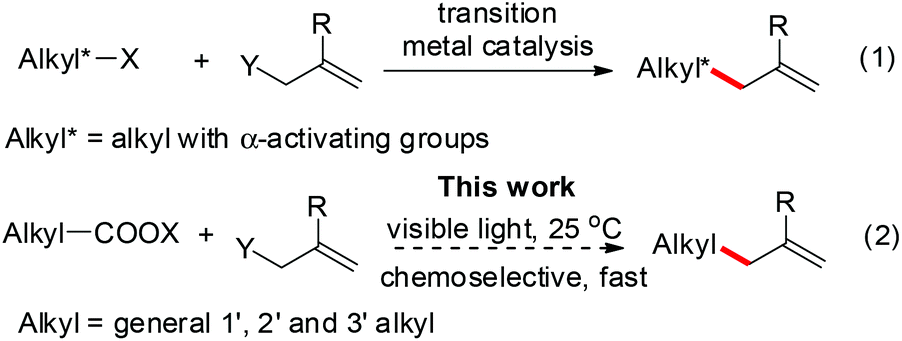
Results and discussion
Using [Ru(bpy)3](PF6)2 as the photocatalyst under blue LED irradiation, we start our investigation with adamantyl N-acyloxyphthalimide 1 and find that the C(sp3)-allylation product 3 is obtained with allyl bromides in 81% yield (entry 1 in Table 1).4 With diisopropylethylamine and Hantzsch ester as reductants, the allyl sulfone is more effective with 86% yield and fewer side products (entry 2).5 This reaction can be run with the addition of formic acids to neutralize the diisopropylethylamine, and the optimal 92% allylation yield is obtained within 30 minutes (90% isolated yield, entry 3). The photocatalyst is necessary to accelerate the reaction where the direct photo-induced reaction without the photocatalyst results in little conversion (entry 4).6 Light irradiation and reductants are both critical for the reaction (entries 5 and 6).|
|
||||
|---|---|---|---|---|
| Entry | Conditionsa | Time | Conversionb | Yieldb |
| a Reaction conditions: 1 (0.10 mmol), 2 (0.15 mmol), Ru(bpy)3(PF6)2 (0.001 mmol), iPr2NEt (0.20 mmol), and Hantzsch ester (HE, 0.15 mmol) in 1.0 mL CH2Cl2 under nitrogen with 468 nm LED irradiation. b Conversions and yields were determined by 1H NMR analysis, isolated yields are in parentheses. c iPr2NEt (0.10 mmol), HCOOH (0.10 mmol), and Hantzsch ester (HE, 0.15 mmol) were used as reductants. X = phthalimide. R = adamantyl. | ||||
| 1 | X′ = Br | 30 min | >95% | 81% |
| 2 | X′ = SO2Ph | 30 min | >95% | 86% |
| 3c | Entry 2, w/HCO2H | 30 min | >95% | 92% (90%) |
| 4 | Entry 3, no [Ru] | 2 h | <5% | <5% |
| 5 | Entry 3, no blue LED | 2 h | <5% | 0% |
| 6 | Entry 3, no reductants | 2 h | <5% | 0% |
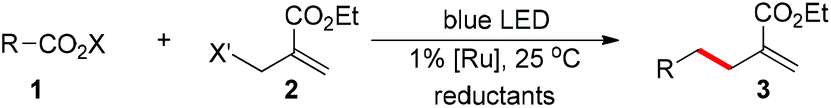
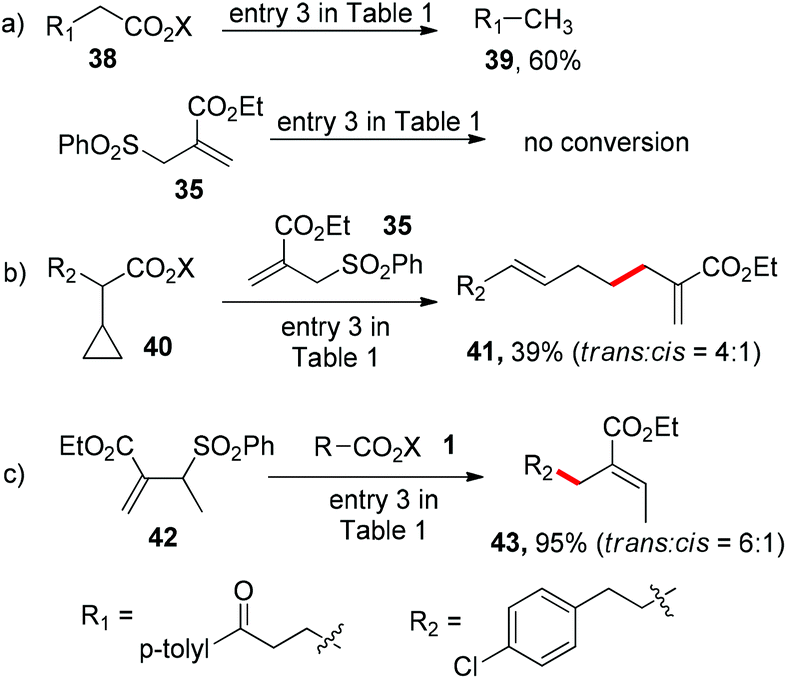
With the mild reaction conditions at room temperature, we next explore the substrate scope of the reaction. Primary, secondary and tertiary alkyl substituted N-acyloxyphthalimides all react well to give the allylation adducts (products 4–7 in Scheme 1). It is worth noting that bulky alpha-quaternary substituted alkyl substituted N-acyloxyphthalimides yield allylation adduct 8 smoothly (trans isomer only). The benzyl allylation product 9 is obtained in 64% yield, and the dimerization side products are not observed. The α-heteroatom substituted alkyl N-acyloxyphthalimides including substitutions with oxygen, nitrogen and sulfur are all tolerated in the reaction (products 10–15). The allyl acceptors are not limited to electron-deficient alkenes with ester groups, and allyl acceptors with phenyl, chloride, or bromide substitution all reacted to give slightly decreased yields (products 12–19).7
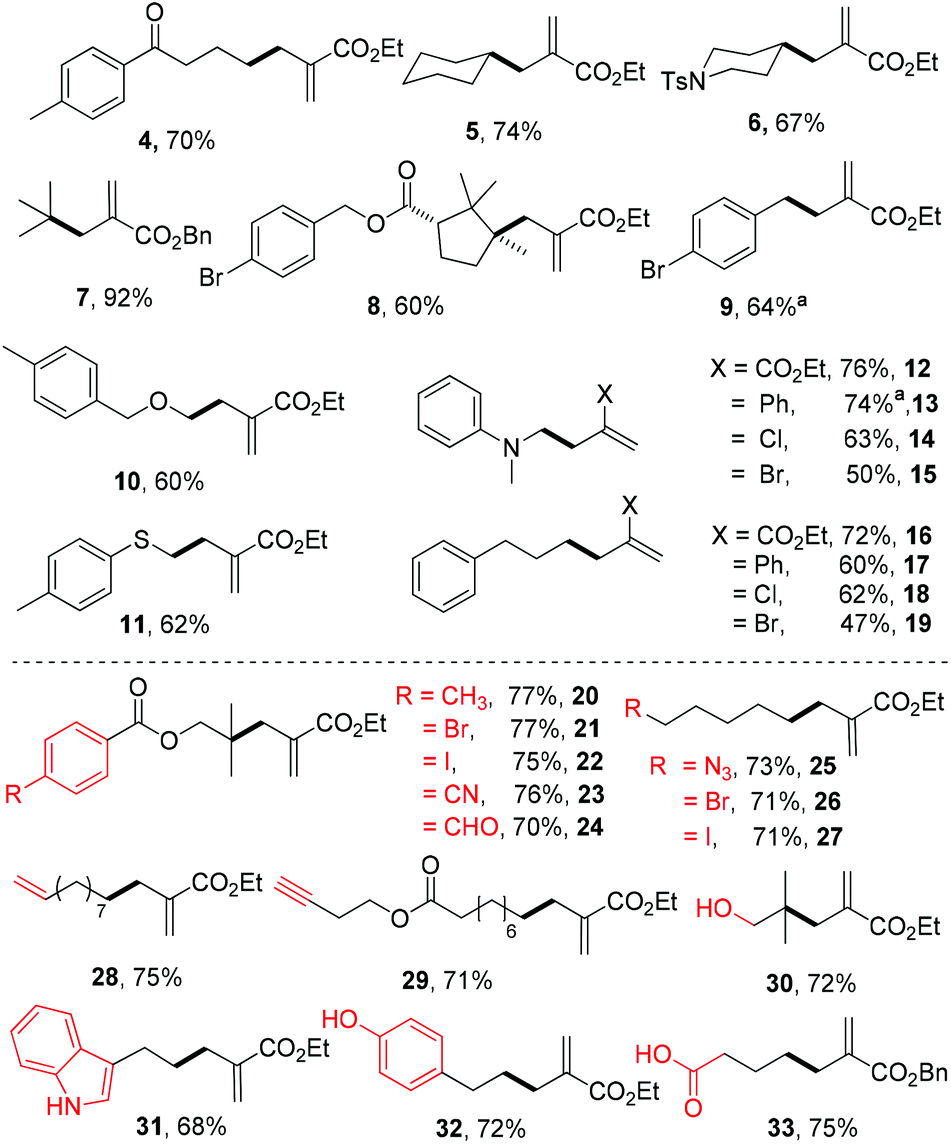 | ||
| Scheme 1 Substrate scope and functional group compatibility of the decarboxylative allylation. Reaction conditions: entry 3 in Table 1. a3 equiv. of allyl sulfone was used. | ||
We further tested the functional group tolerance of the reaction. The transition-metal-sensitive aryl bromides and iodides have no interference with the reaction (products 20–22). The reduction-sensitive aldehydes, nitriles, and alkyl azides are well tolerated (products 23–25). Alkyl bromides and iodides as traditional alkyl radical precursors remain intact under the reaction conditions (products 26 and 27). Both unactivated alkenes and alkynes are tolerated in the allylation reaction (products 28 and 29). Further screenings indicate that typical reactive groups on biomolecules do not interfere with the reaction, including unprotected alcohols, indoles, phenols, and carboxylic acids (products 30–33), which suggest their potential biomolecular applications.
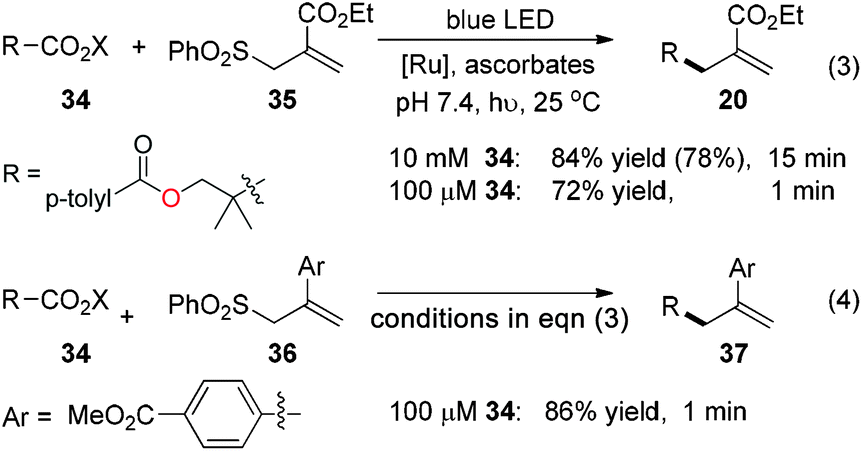
With the excellent chemoselectivity and mild reaction conditions, we envision that this visible-light-induced decarboxylative allylation will be useful for biomolecular studies if neutral aqueous reaction conditions can be achieved.8 The ascorbic acid is a mild water-soluble reductant suitable for biomolecular studies.4g,9 We test the ascorbates in pH 7.4 PBS buffer (phosphate buffered saline) and observe the smooth allylation adduct 20 in 84% yield within 15 minutes (78% isolated yield, eqn (3)). Under aqueous conditions from pH 6 to 10 the allylations are all effective (ESI Table S1†). This allylation can be run very efficiently at as low as 100 μM of N-acyloxyphthalimide 34, in which the allylation product 20 is obtained in 72% yield in less than 1 minute (eqn (3)). The aryl-substituted allyl sulfone 36 also reacts uneventfully to give the allyl product 37 in 86% yield in less than 1 minute at 100 μM concentrations, which is useful for time-sensitive applications where reaction kinetics as fast as minutes are required (eqn (4)).10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 4:1) after cyclopropyl ring opening, which further validates the alkyl radical intermediate (Scheme 2b). We also tested the methyl substituted allyl sulfone 42 with N-acyloxyphthalimide 1 and obtained trisubstituted alkene 43 in 95% yield (trans:cis = 6:1), whose regioselectivity suggests the alkyl radical addition to the alkene followed by sulfonyl radical elimination (Scheme 2c).
:cis = 4:1) after cyclopropyl ring opening, which further validates the alkyl radical intermediate (Scheme 2b). We also tested the methyl substituted allyl sulfone 42 with N-acyloxyphthalimide 1 and obtained trisubstituted alkene 43 in 95% yield (trans:cis = 6:1), whose regioselectivity suggests the alkyl radical addition to the alkene followed by sulfonyl radical elimination (Scheme 2c).
 | ||
| Scheme 2 Mechanistic investigations of the decarboxylative allylation. | ||
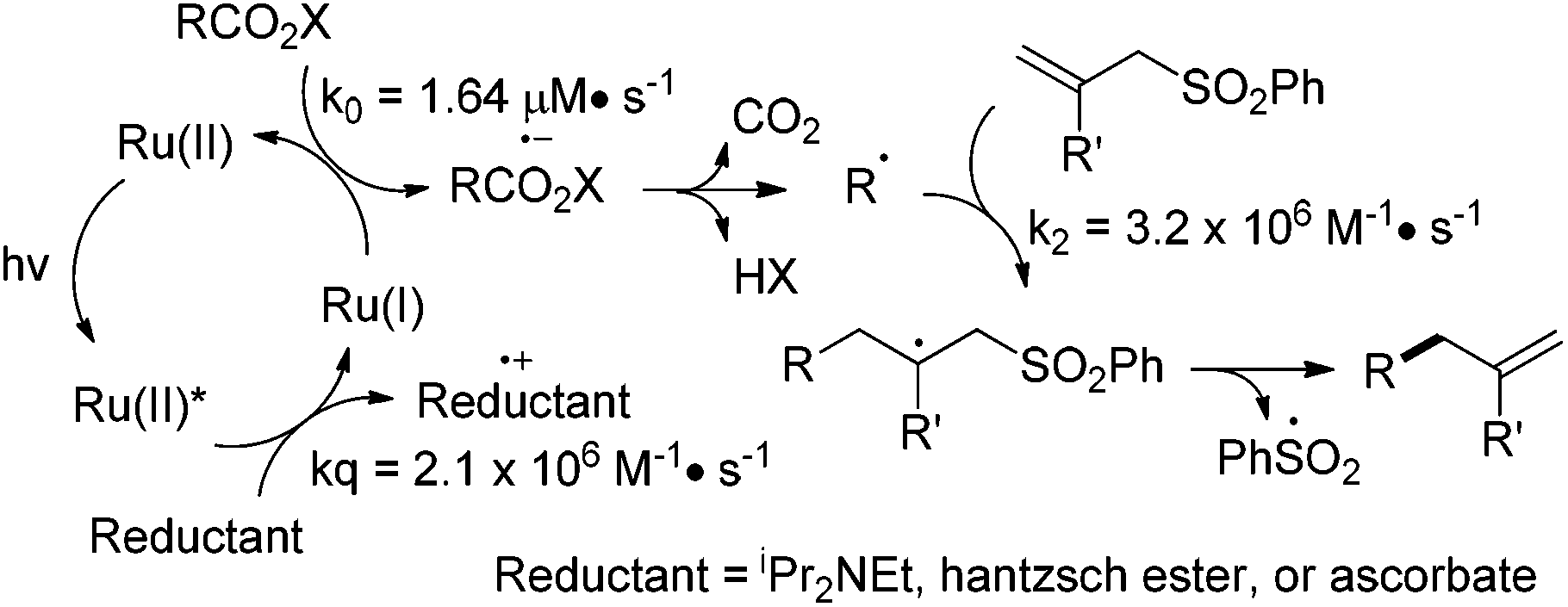
Based on the above mechanistic investigations, we propose that Ru(bpy)32+ is photoexcited to Ru(bpy)32+* and reduced by diisopropylethylamine, Hantzsch ester, or ascorbates to Ru(bpy)3+ (Scheme 3).11 The resulting Ru(bpy)3+ reduces N-acyloxyphthalimide via single electron transfer and generates the alkyl radical after decarboxylation and phthalimide elimination.4 The alkyl radical undergoes addition to the allyl sulfone and yields the allylation adduct after desulfonation.
 | ||
| Scheme 3 Mechanistic proposal of the decarboxylative allylation. | ||
As the alkyl allylation reaction under aqueous conditions is exceptionally fast, we envision that the quantitative study of kinetic parameters is useful. The allylation reaction includes three elementary reaction processes: the single-electron-transfer between the photoexcited Ru(bpy)32+* and the ascorbate (kq), the reductive decarboxylation to generate the alkyl radical (k0), and the carbon–carbon bond-forming allylation (k2) (Scheme 3). The electron transfer rate (kq) from the ascorbate to the photoexcited Ru(bpy)32+* in pH 7.4 Tris buffer is obtained by measuring the fluorescence lifetime quenching at increasing ascorbate concentrations, and is determined by Stern–Volmer analysis to be 2.1 × 106 M−1 s−1 (ESI Scheme S9†).13 The zero-order rate constant k0 of the alkyl radical formation is measured by the consumption of N-acyloxyphthalimide 34 to be 1.64 μM s−1 (ESI Scheme S12†). The second-order carbon–carbon bond-forming rate constant of radical allylation k2 is measured by the competition experiments with thiophenol-induced hydrogenation (kH is 1.3 × 108 M−1 s−1), and is determined to be 3.2 × 106 M−1 s−1 (ESI Scheme S15†).14 The light-induced carbon–carbon bond-forming reactions have been widely used for light-modulated biomolecule manipulations,3d,10c,d which has the advantage of high temporal and spatial precision.15 The use of visible light instead of UV light for this allylation imposes better biomolecular compatibilities and applications.16
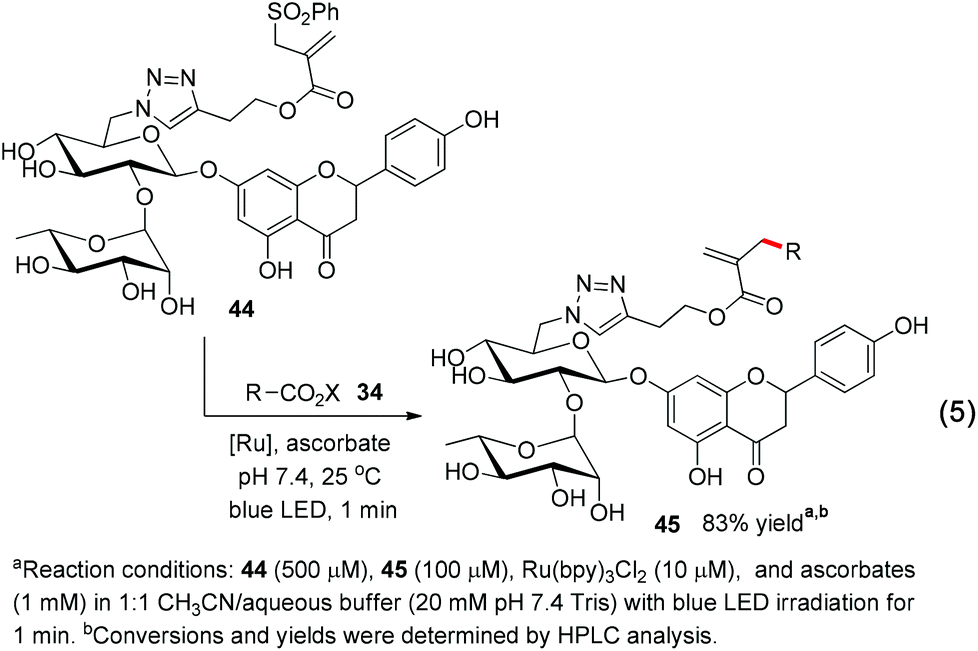
Finally, we tested if this chemoselective and fast decarboxylative allylation can be used for protecting-group-free modification of functional-group-rich complex molecules. We treated the oligosaccharide naringin conjugate 44 under the allylation conditions and gratifyingly obtained the allylation adduct 45 in 83% yield within 1 minute, with no detectable occurrence of a side reaction (eqn (5)).
Conclusions
In conclusion, we have developed a visible-light-induced chemoselective and fast decarboxylative allylation for primary, secondary, tertiary, benzyl, and alpha-heteroatom-substituted C(sp3)-allylations. This reaction is complete in minutes at room temperature and demonstrates excellent chemoselectivity in both organic solvents and neutral aqueous solutions. Further biomolecular application of this decarboxylative allylation is under investigation in our laboratory.Acknowledgements
Financial support was provided by National Basic Research Program of China 2014CB910304, National Natural Science Foundation of China 21272260, 21472230, “Thousand Talents Program” Young Investigator Award, and start up fund from State Key Laboratory of Bioorganic and Natural Products Chemistry, and Chinese Academy of Sciences.Notes and references
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (b) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed.
- (a) D. H. R. Barton and D. Crich, J. Chem. Soc., Perkin Trans. 1, 1986, 1613 RSC; (b) B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 1996, 118, 1209 CrossRef CAS; (c) F. Le Guyader, B. Quiclet-Sire, S. Seguin and S. Z. Zard, J. Am. Chem. Soc., 1997, 119, 7410 CrossRef CAS; (d) B. Quiclet-Sire, S. Seguin and S. Z. Zard, Angew. Chem., Int. Ed., 1998, 37, 2864 CrossRef; (e) S. Kim and C. J. Lim, Angew. Chem., Int. Ed., 2002, 41, 3265 CrossRef CAS; (f) A. P. Schaffner and P. Renaud, Angew. Chem., Int. Ed., 2003, 42, 2658 CrossRef CAS PubMed; (g) S. Lee, C. J. Lim and S. Kim, Bull. Korean Chem. Soc., 2004, 25, 1611 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) C. Hu and Y. Chen, Tetrahedron Lett., 2015, 56, 884 CrossRef CAS PubMed.
- (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401 CrossRef CAS; (b) K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377 CrossRef CAS; (c) K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 2021 CrossRef CAS; (d) M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576 CrossRef CAS PubMed; (e) G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342 CrossRef CAS PubMed; (f) G. Kachkovskyi, C. Faderl and O. Reiser, Adv. Synth. Catal., 2013, 355, 2240 CrossRef CAS PubMed; (g) J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275 RSC; (h) G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025 CrossRef CAS PubMed.
- (a) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef CAS PubMed; (b) M.-H. Larraufie, R. Pellet, L. Fensterbank, J.-P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 50, 4463 CrossRef CAS PubMed; (c) M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Org. Chem. Front., 2014, 1, 551 RSC; (d) S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606 CrossRef CAS PubMed; (e) J. Xuan, T. Zeng, Z. Feng, Q. Deng, J. Chen, L. Lu, W. Xiao and H. Alper, Angew. Chem., Int. Ed., 2015, 54, 1625 CrossRef CAS PubMed.
- The direct photo-induced reaction of N-acyloxyphthalimide 1 without the photocatalyst becomes obvious after extended 12 h light irradiation (12 h, 73% conversion, 65% yield). The direct photo-induced reaction of N-acyloxyphthalimide is also observed in ref. 4a and h.
- The allylation with unsubstituted allyl sulfones (X = H) results in a very little product.
- (a) R. A. Shenvi, D. P. O'Malley and P. S. Baran, Acc. Chem. Res., 2009, 42, 530 CrossRef CAS PubMed; (b) C. J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed; (c) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (d) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- (a) J. B. Borak and D. E. Falvey, J. Org. Chem., 2009, 74, 3894 CrossRef CAS PubMed; (b) Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146 CrossRef CAS PubMed.
- (a) S. S. Gambhir, Nat. Rev. Cancer, 2002, 2, 683 CrossRef CAS PubMed; (b) T. F. Massoud and S. S. Gambhir, Genes Dev., 2003, 17, 545 CrossRef CAS PubMed; (c) G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS PubMed; (d) M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- The incubation of the allyl bromide under the reaction conditions without N-acyloxyphthalimides results in the consumption of the allyl bromide (ESI Scheme S6†), which may suggest the side reactions from allyl bromides.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- J. A. Franz, B. A. Bushaw and M. S. Alnajjar, J. Am. Chem. Soc., 1989, 111, 268 CrossRef CAS.
- Z. Yu and Q. Lin, J. Am. Chem. Soc., 2014, 136, 4153 CrossRef CAS PubMed.
- R. P. Sinha and D. P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00187k |
| This journal is © the Partner Organisations 2015 |