
Highly efficient and practical hydrogenation of olefins catalyzed by in situ generated iron complex catalysts†
Na
Guo
,
Meng-Yang
Hu
,
Ye
Feng
and
Shou-Fei
Zhu
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China. E-mail: sfzhu@nankai.edu.cn
First published on 13th April 2015
Abstract
A new method was developed for in situ generation of active Fe catalysts for the hydrogenation of olefins from bench-stable Fe(II) complexes and easily accessible LiAlH4. This method makes the hydrogenation very easy to handle and enables the development of several new Fe catalysts for olefin hydrogenation through practical ligand evaluation. One of the Fe catalysts derived from a Fe complex of a phosphine-bipyridine ligand exhibited unprecedented activity for the hydrogenation of olefins, with turnover numbers up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and turnover frequencies up to 37740 h−1. The NMR studies of the active Fe catalyst showed that a Fe-hydride species stabilized by Al might be a real catalyst.
000 and turnover frequencies up to 37740 h−1. The NMR studies of the active Fe catalyst showed that a Fe-hydride species stabilized by Al might be a real catalyst.
Introduction
Transition-metal-catalyzed hydrogenation of unsaturated compounds has been widely used for the production of value-added bulk and fine chemicals.1 Homogeneous hydrogenation is generally carried out with catalysts derived from precious metals, such as Rh, Ru, Ir, and Os, some of which are highly toxic. The high cost, toxicity, and potential depletion of precious metals have forced chemists to develop green, sustainable catalysts for hydrogenation. Because Fe is abundant, cheap, and environmentally benign, Fe-catalyzed hydrogenation has recently drawn much attention.2Substantial progress has been made in the Fe-catalyzed hydrogenation of highly polarized carbonyl compounds.3 However, progress in the Fe-catalyzed hydrogenation of olefins has been limited,2 even though Fe carbonyl complexes were used to catalyze the hydrogenation of olefins as early as 1960s.4 Because replacing the CO ligand of Fe carbonyl complexes with other ligands is difficult, recent work on the Fe-catalyzed hydrogenation of olefins has focused on the development of Fe complexes with phosphine5 or nitrogen ligands,6 which can be used to tune catalyst reactivity and selectivity. A significant breakthrough was achieved in 2004 by Chirik and co-workers,6 who reported that Fe(N2)2-pyridinediimine complexes are active catalysts for the hydrogenation of olefins. In some cases, the turnover frequencies (TOFs) achieved with Chirik's Fe catalysts surpass those of well-established olefin hydrogenation catalysts, such as Wilkinson's catalyst and Pd/C.6a
However, pre-prepared Fe complexes are extremely sensitive to both oxygen and moisture, which complicates their preparation and use in most synthetic chemistry laboratories. Moreover, the difficulties in preparing these highly sensitive catalysts have limited the development of new Fe catalysts by means of ligand evaluation, which is a widely used strategy for finding efficient catalysts. Several attempts to use Grignard reagents or organolithium reagents to reduce bench-stable Fe complexes to active Fe catalysts for olefin hydrogenation have been reported;7 unfortunately, Fe catalysts generated in situ are much less active than pre-prepared Fe catalysts.
We report here a new method for in situ generation of active Fe catalysts for the hydrogenation of olefins by the reduction of bench-stable Fe(II) complexes with LiAlH4, which is inexpensive and readily available. This method makes the hydrogenation very easy to handle and enables us to develop several new Fe catalysts for olefin hydrogenation through practical ligand evaluation. One of the Fe catalysts derived from a Fe complex of a phosphine-bipyridine ligand (Fe-L8) exhibited unprecedented activity for the hydrogenation of olefins, with turnover numbers (TONs = moles of product per mole of catalyst) up to 10000 and TOFs (TONs per hour) up to 37740 h−1. The preliminary investigations of the structure of the active catalyst were also performed.
Results and discussion
Initially, the hydrogenation was performed with styrene as a substrate under 30 atm of H2 at ambient temperature in THF with a Fe catalyst generated in situ from a FeCl2-L1 complex8 and various reductants (Table 1). Among the tested reductants, NaBEt3H,9 LiAlH4,10 and activated Mg11 afforded complete conversion of the substrate (entries 1, 4, and 8). LiAlH4 and activated Mg accomplished the hydrogenation with a high average TOF (6000 h−1), which surpassed the TOFs obtained with the corresponding pre-prepared Fe complexes.6a The hydrogenation was also promoted by iPrMgCl, but the conversion was only 20% (entry 6). Considering its ready availability and ease of use, LiAlH4 was chosen as the best reductant for in situ generation of Fe catalysts for olefin hydrogenation. LiAlH4 cannot catalyze the hydrogenation in the absence of a Fe complex under the standard reaction conditions, which implies that the Fe is the real catalyst.|
|
|||
|---|---|---|---|
| Entry | Reductant | Time | Conv.b (%) |
|
a Reaction conditions: styrene/FeCl2-L1/reductant = 5:0.005:0.025 (mmol), in 1 mL THF, 30 atm H2, rt.
b Determined by GC using an interCap-1 column.
|
|||
| 1 | NaBEt3H | 12 h | 100 |
| 2 | NaBH4 | Overnight | Trace |
| 3 | NaH | Overnight | Trace |
| 4 | LiAlH4 | 10 min | 100 |
| 5 | BH3·THF | Overnight | Trace |
| 6 | iPrMgCl | Overnight | 20% |
| 7 | NH2NH2·H2O | Overnight | Trace |
| 8 | Mg | 10 min | 100 |
| 9 | iBu3Al | Overnight | Trace |
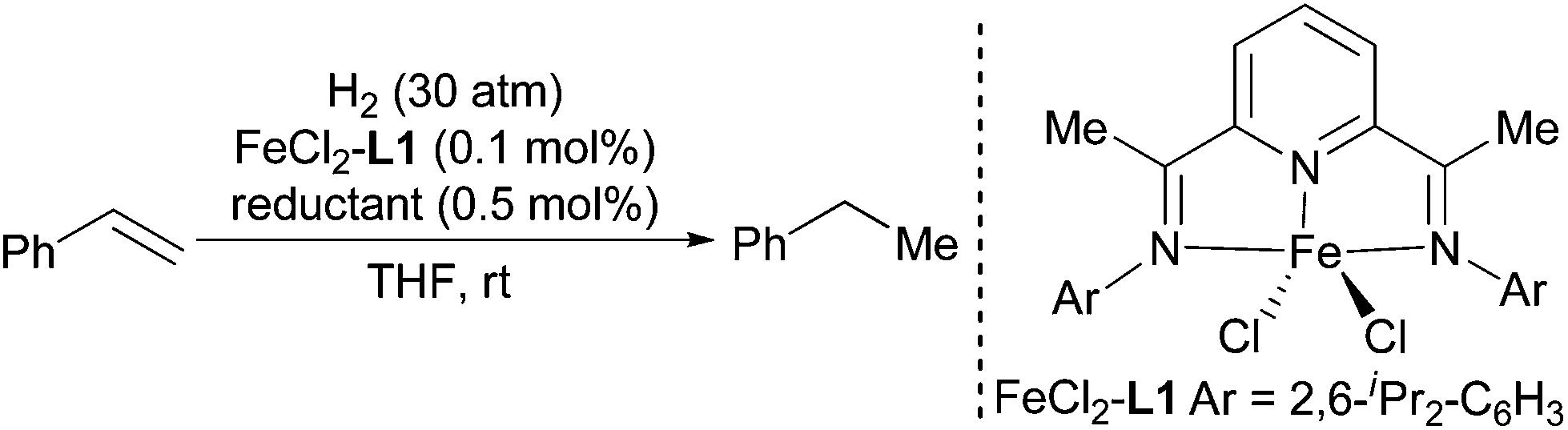
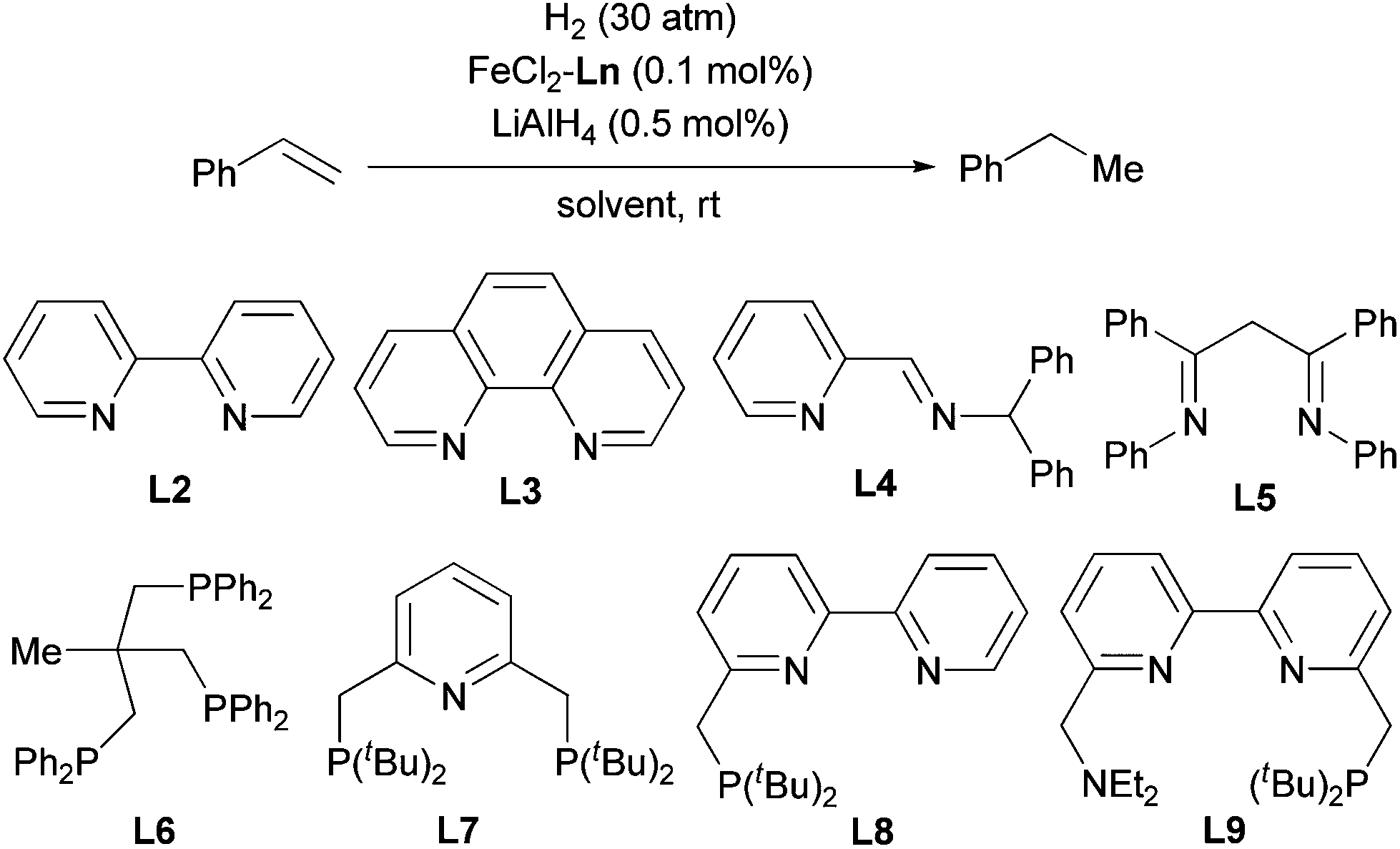
The above-described procedure for in situ generation of active Fe catalysts allows for ligand evaluation, and we tested various stable Fe complexes as catalyst precursors for the hydrogenation of olefins (Table 2). Fe complexes with bidentate sp2 N ligands, such as bipyridine (L2), phenanthroline (L3), pyridine-imine (L4), and biimine (L5) exhibited excellent catalytic activity (entries 2–5). In contrast, N,N,N′,N′-tetramethylethylenediamine, which has sp3 N atoms as coordinating atoms, showed extremely low reactivity (entry 6). Phosphine ligands were generally less active than ligands with sp2 N atoms, and Fe particles were generated after the reaction (entries 7–9). However, a tridentate phosphine ligand, L6, accomplished the reaction within 2 h (entry 10). Several pincer ligands containing pyridine and bipyridine moieties (L7–L9) were also evaluated (entries 11–13). Among these ligands, the PNN ligand (L8)12 exhibited extremely high activity: the hydrogenation was completed within 5 min when 0.1 mol% catalyst was used (entry 12) and within 3 h when 0.01 mol% catalyst was used (entry 14). Furthermore, the Fe-L8 complex catalyzed the hydrogenation at relatively low hydrogen pressure (10 atm, entry 16). The hydrogenation reaction could also be carried out in other ether solvents, such as Et2O, DME (1,2-dimethoxyethane), and 1,4-dioxane (entries 17–19). The TOFs at various conversion states were also determined (Table S1†). An even higher TOF (37740) was observed at the beginning of the hydrogenation, which represents the highest TOF for Fe-catalyzed olefin hydrogenation to the best of our knowledge.
|
|
||||
|---|---|---|---|---|
| Entry | Ln | Solvent | Time | Conv. (%) |
|
a The reaction conditions and analysis are the same as those in Table 1, entry 4 unless otherwise noted. The dppe is 1,2-bis(diphenylphosphino)ethane; the dppp is 1,3-bis(diphenylphosphino)propane.
b PPh3/Fe = 2:1.
c 0.01 mol% catalyst was used.
d 0.25 mol% LiAlH4 was used.
e The reaction was performed at 10 atm H2.
|
||||
| 1 | L1 | THF | 10 min | 100 |
| 2 | L2 | THF | 30 min | 100 |
| 3 | L3 | THF | 20 min | 100 |
| 4 | L4 | THF | 40 min | 100 |
| 5 | L5 | THF | 1 h | 100 |
| 6 | TMEDA | THF | 12 h | 3 |
| 7b | PPh3 | THF | 24 h | 36 |
| 8 | dppe | THF | 12 h | 100 |
| 9 | dppp | THF | 12 h | 83 |
| 10 | L6 | THF | 2 h | 100 |
| 11 | L7 | THF | 1 h | 100 |
| 12 | L8 | THF | 5 min | 100 |
| 13 | L9 | THF | 1 h | 100 |
| 14c | L8 | THF | 3 h | 100 |
| 15d | L8 | THF | 10 min | 100 |
| 16e | L8 | THF | 10 min | 100 |
| 17 | L8 | Et2O | 5 min | 100 |
| 18 | L8 | DME | 5 min | 100 |
| 19 | L8 | 1,4-Dioxane | 10 min | 100 |
Using the optimized reaction conditions, we investigated the substrate scope of the Fe-L8-catalyzed hydrogenation of olefins (Table 3). The steric bulk of the phenyl ring of the olefin substrate had little effect on the hydrogenation reaction (entries 2–5). However, the electronic nature of the substituents on the ring strongly influenced the reaction, with an electron-donating methoxy group giving a faster reaction rate than the electron-withdrawing Cl atom (compare entries 6 and 7). Notably, the TOF of 20000 h−1 was achieved in the hydrogenation of para-methoxy styrene (entry 6). In addition to styrenes, aliphatic terminal olefins could also be hydrogenated with high reaction rates (entries 8–11). However, substrates with a disubstituted olefin moiety or a heteroatom required higher reaction temperatures and longer reaction times, and the TOFs were lower (entries 12–17).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Time | Conv. (%) | TOF (h−1) |
|
a The reaction conditions and analysis are the same as those in Table 2, entry 12 unless otherwise noted.
b The data in parenthesis are the yield determined by the GC method with dodecane as an internal standard.
c Reaction was performed at 50 °C.
d
Z/E = 1:4.
|
||||
| 1 |
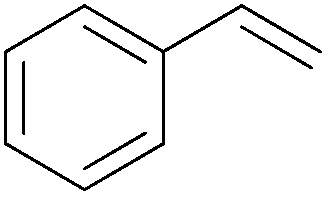
|
5 min | 100 (>99)b | 12000 |
| 2 |
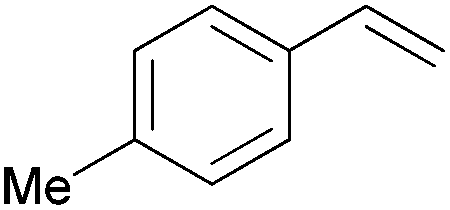
|
10 min | 100 | 6000 |
| 3 |
|
15 min | 100 | 4000 |
| 4 |
|
15 min | 100 | 4000 |
| 5 |
|
5 min | 100 | 12000 |
| 6 |
|
3 min | 100 | 20000 |
| 7 |
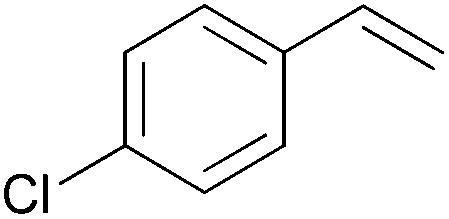
|
6 h | 98 | 163 |
| 8 |
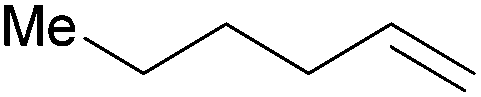
|
20 min | 100 | 3000 |
| 9 |
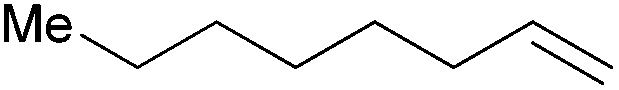
|
30 min | 95.8 | 1916 |
| 10 |
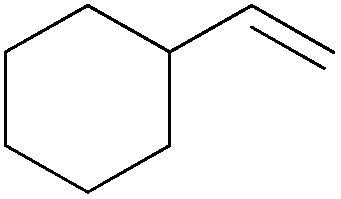
|
20 min | 100 | 3000 |
| 11 |
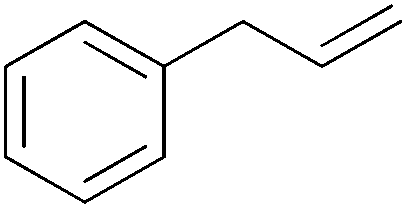
|
15 min | 100 | 4000 |
| 12c |
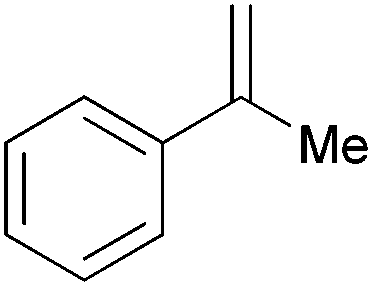
|
1 h | 100 | 1000 |
| 13 |
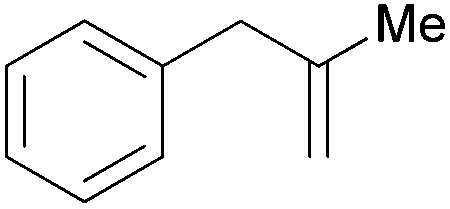
|
1.5 h | 100 | 667 |
| 14c |
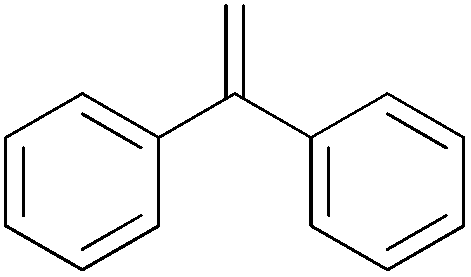
|
2 h | 78.1 | 391 |
| 15c |
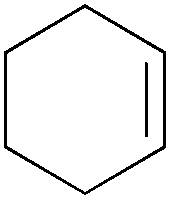
|
1 h | 40 | 400 |
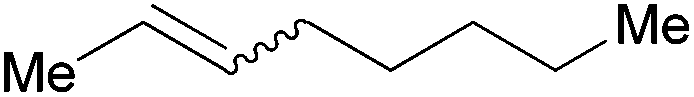
| 16c,d |
|
5 h | 80 | 160 |
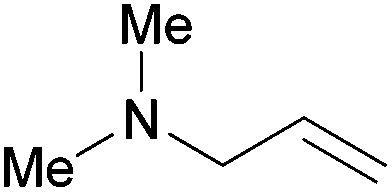
| 17 |
|
6 h | 100 | 167 |
We investigated the in situ generated active Fe species by means of NMR spectroscopy (Fig. 1 and ESI†). A mixture of FeCl2-L8 (0.005 mmol) and LiAlH4 (0.025 mmol) in 1 mL THF-d8 was stirred for 1 min, and the 1H NMR spectrum of the resulting dark red solution was measured. We attribute the signal at −18.92 ppm (d, J = 20 Hz, 2H) to an Fe-hydride, on the basis of spectra in the literature.6e,10 The other signals in the spectrum matched with those of the Fe(CO)2-L8 complex.13 The hydride signal disappeared when the sample was exposed to D2, which implies that a H–D exchange occurs. No hydride signals at < 0 ppm for LiAlH4 were observed under identical conditions. The 31P NMR spectrum contained only one signal at 134.05 ppm. A broad Al signal at 99.3 ppm was also observed in 27Al NMR. The NMR data clearly showed that an L8-Fe-(H)2 species was generated when FeCl2-L8 and LiAlH4 were mixed and the Al may coordinate with the hydride in some way,10c,d which stabilizes the Fe–H catalysts. The studies of the structure of the active Fe catalysts and the mechanism of the hydrogenation are still undergoing in our laboratory.
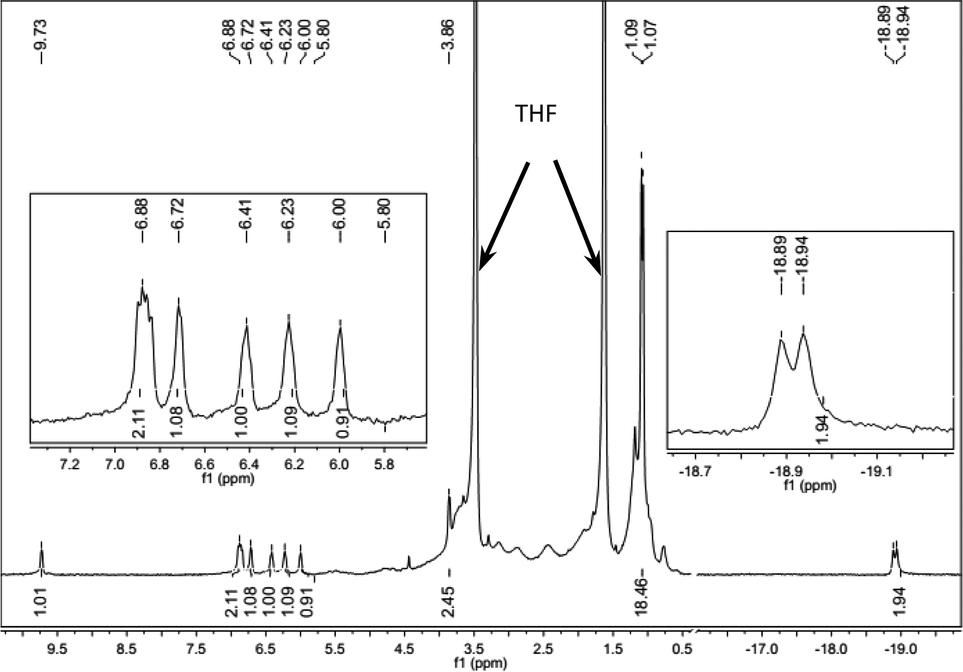 | ||
| Fig. 1 The 1H NMR studies of the in situ generated Fe-L8 catalyst. | ||
The deuterium-labelling experiment clearly implies a significant redistribution of the hydrogen at C1 and C2 of the product during the hydrogenation (Scheme 1).
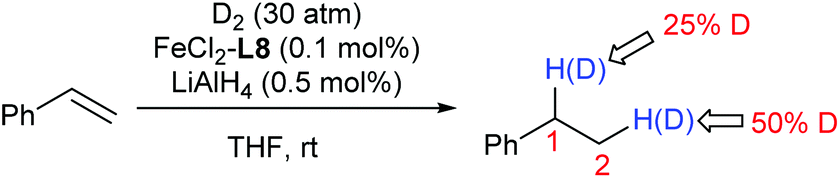 | ||
| Scheme 1 A deuterium-labelling study. | ||
Two catalytic cycles (a–b–c and a–d–e) of the hydrogenation were proposed based on the above observations and the literature (Scheme 2).5,6 The reversible migratory insertion and β-H elimination (step a) might account for the redistribution of the hydrogen of the product.
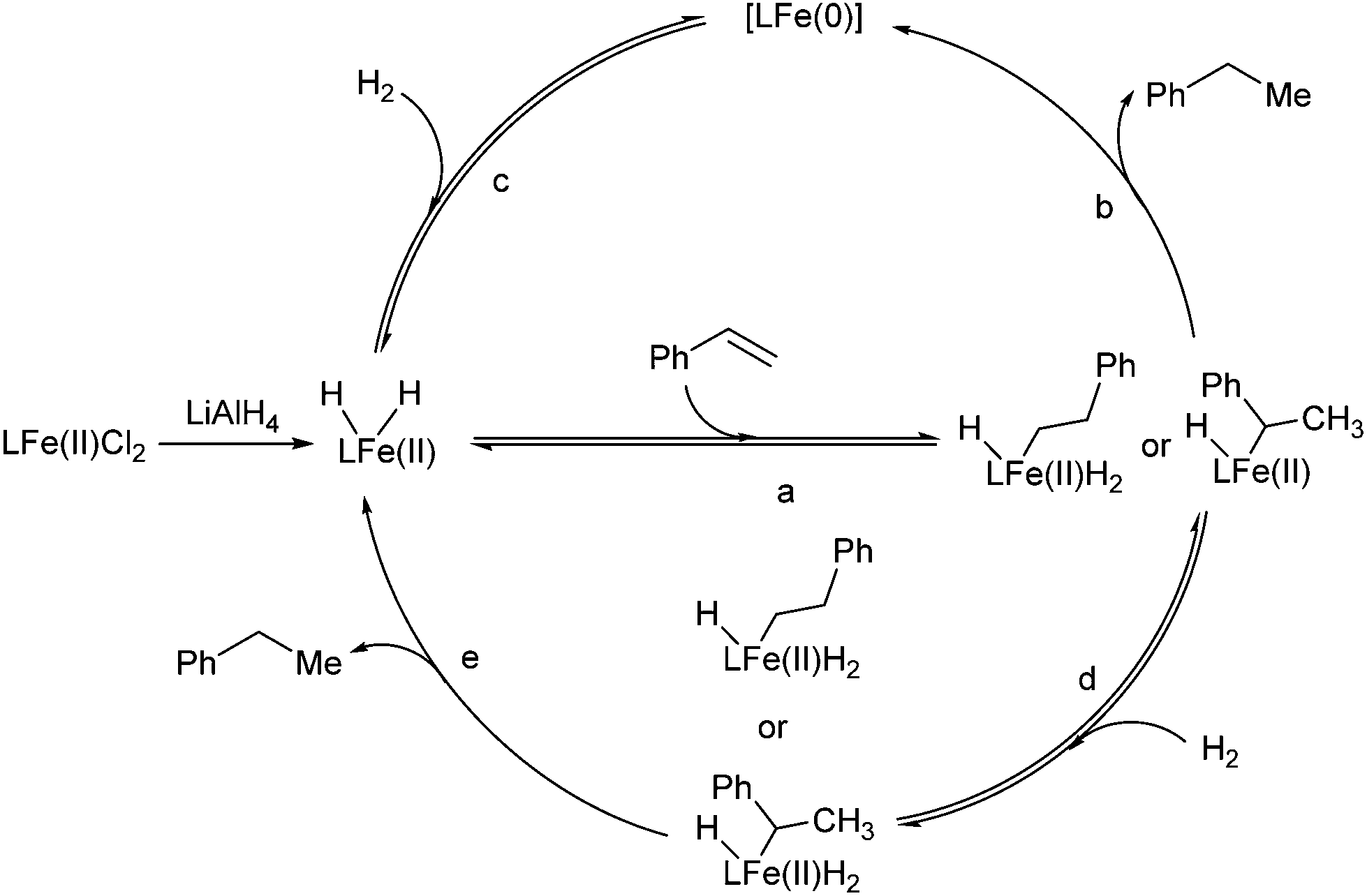 | ||
| Scheme 2 Proposed mechanism of the hydrogenation. | ||
Conclusions
In summary, we have developed a new method for the in situ generation of an active Fe catalyst for the hydrogenation of olefins from bench-stable Fe(II) complexes and LiAlH4, a readily available reductant. This strategy will undoubtedly be useful for developing suitable ligands for Fe-catalyzed olefin hydrogenation and other reactions. We are in the process of developing Fe-catalyzed asymmetric hydrogenation of olefins by using this new method in our laboratory.Acknowledgements
We thank the National Natural Science Foundation of China, the National Basic Research Program of China (2011CB808600), the “111” project (B06005) of the Ministry of Education of China, and the National Program for Support of Top-notch Young Professionals for financial support. We thank Prof. Xun-Cheng Su for his help on NMR analysis.Notes and references
- (a) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (c) D. H. Woodmansee and A. Pfaltz, Chem. Commun., 2011, 47, 7912 RSC; (d) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427 CrossRef CAS (in Chinese); (e) B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed; (f) X.-H. Yang, J.-H. Xie and Q.-L. Zhou, Org. Chem. Front., 2014, 1, 190 RSC.
- (a) S. Enthaler, K. Junge and M. Beller, in Iron Catalysis in Organic Chemistry Reactions and Applications, Vol. 4, ed. B. Plietker, Wiley, Weinheim, 2008 Search PubMed; (b) P. J. Chirik, in Catalysis without Precious Metals, ed. R. M. Bullock, Wiley-VCH, Weinheim, 2010, ch. 4 Search PubMed; (c) H. Nakazawa and M. Itazaki, Top. Organomet. Chem., 2011, 33, 27 CrossRef CAS; (d) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (e) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (f) N. Guo and S.-F. Zhu, Chin. J. Org. Chem., 2015 DOI:10.6023/cjoc201502032.
- For reviews, see: (a) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (b) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed. For selected examples, see: (c) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816 CrossRef CAS PubMed; (d) R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120 CrossRef CAS PubMed; (e) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120 CrossRef CAS PubMed; (f) S. Fleischer, S. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120 CrossRef CAS PubMed; (g) A. Quintard, T. Constantieux and J. Rodriguez, Angew. Chem., Int. Ed., 2013, 52, 12883 CrossRef CAS PubMed; (h) P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wang, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367 CrossRef CAS PubMed; (i) Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong and J. Gao, J. Am. Chem. Soc., 2014, 136, 4031 CrossRef CAS PubMed; (j) T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685 CrossRef CAS PubMed; (k) S. Chakraborty, H.-G. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H.-R. Guan, J. Am. Chem. Soc., 2014, 136, 7869 CrossRef CAS PubMed; (l) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722 CrossRef CAS PubMed.
- E. N. Frankel, E. A. Emken, H. M. Peters, V. L. Davison and R. O. Butterfield, J. Org. Chem., 1964, 29, 3292 CrossRef CAS.
- (a) C. Bianchini, A. Meli, M. Peruzzini, F. Vizza and F. Zanobini, Organometallics, 1989, 8, 2080 CrossRef CAS; (b) E. J. Daida and J. C. Peters, Inorg. Chem., 2004, 43, 7474 CrossRef CAS PubMed; (c) H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053 CrossRef CAS PubMed; (d) D. Srimani, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 14131 CrossRef CAS PubMed.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed; (b) S. C. Bart, K. Chlopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901 CrossRef CAS PubMed; (c) R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 27, 1470 CrossRef CAS; (d) S. C. Bart, E. J. Hawrelak, E. Lobkovsky and P. J. Chirik, Organometallics, 2005, 24, 5518 CrossRef CAS; (e) R. J. Trovitch, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2006, 45, 7252 CrossRef CAS PubMed; (f) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760 CrossRef CAS. For a review, see: (g) P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed.
- (a) Y. Tajima and E. Kunioka, J. Org. Chem., 1968, 33, 1689 CrossRef CAS; (b) R. E. Harmon, S. K. Gupta and D. J. Brown, Chem. Rev., 1973, 73, 21 CrossRef CAS; (c) H. Inoue and M. Suzuki, J. Chem. Soc., Chem. Commun., 1980, 817 RSC; (d) D. J. Frank, L. Guiet, A. Käslin, E. Murphy and S. P. Thomas, RSC Adv., 2013, 3, 25698 RSC; (e) J. M. Hoyt, M. Shevlin, G. W. Margulieux, S. W. Krska, M. T. Tudge and P. J. Chirik, Organometallics, 2014, 33, 5781 CrossRef CAS.
- The complex was prepared according to Brookhart's procedures, see: B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS.
- Lin and co-workers used NaBEt3H as a reductant to generate an active solid catalyst from Fe- or Co-functionalized metal–organic frameworks for olefin hydrogenation, see: K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182 CrossRef CAS PubMed.
- For the preparation of Fe–H species by using LiAlH4 as a reductant, see: (a) G. Argouarch, P. Hamon, L. Toupet, J.-R. Hamon and C. Lapinte, Organometallics, 2002, 21, 1341 CrossRef CAS; (b) T. Liu, S. Chen, M. J. O'Hagan, M. R. DuBois, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 134, 6257 CrossRef CAS PubMed; (c) M. Oishi, T. Endo, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 5100 CrossRef CAS PubMed and ref. 3h. During the preparation of this manuscript, von Wangelin reported hydrogenation of olefins catalyzed by using a FeCl3–LiAlH4 catalyst: (d) T. N. Gieshoff, M. Villa, A. Welther, M. Plois, U. Chakraborty, R. Wolf and A. J. von Wangelin, Green Chem., 2015, 17, 1408 RSC.
- Ritter and co-workers used active Mg to reduce Fe(II) to Fe(0) in 1,4-hydroboration of 1,3-dienes, see: J. Y. Wu, B. Moreau and T. Ritter, J. Am. Chem. Soc., 2009, 131, 12915 CrossRef CAS PubMed.
- For the preparation and selected applications of L8, see: (a) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756 CrossRef CAS PubMed; (b) E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 11702 CrossRef CAS PubMed; (c) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609 CrossRef CAS PubMed; (d) E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122 CrossRef CAS PubMed; (e) L. Zhang, D. Peng, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2013, 52, 3676 CrossRef CAS PubMed.
- T. Zell, P. Milko, K. L. Fillman, Y. Diskin-Posner, T. Bendikov, M. A. Iron, G. Leitus, Y. Ben-David, M. L. Neidig and D. Milstein, Chem. – Eur. J., 2014, 20, 4403 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR characterization and GC analysis data of all hydrogenation products. See DOI: 10.1039/c5qo00064e |
| This journal is © the Partner Organisations 2015 |