Palladium-catalysed regioselective hydroamination of 1,3-dienes: synthesis of allylic amines†
Debasis
Banerjee
,
Kathrin
Junge
and
Matthias
Beller
*
Leibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de; Web: http://www.catalysis.de Fax: (+49)-381-1281-5000
First published on 18th March 2014
Abstract
Catalytic hydroamination of unactivated 1,3-dienes represents a sustainable and atom-economic C–N bond forming process. Here, we present a novel catalytic system consisting of Pd(cod)Cl2 in combination with DPEphos for the selective 1,4-hydroamination (anti-Markovnikov reaction) of a series of acyclic and cyclic dienes. The reactions proceed in good yields and allow for the exclusive formation of allylic amines with high regioselectivity and do not need any additives.
Introduction
Transition metal catalysed additions of amines to olefins and alkynes, commonly known as hydroamination,1 represent attractive and benign C–N bond forming processes.2 In this respect, the hydroamination reaction of unactivated 1,3-dienes offers a straightforward and atom-economic pathway for the synthesis of allylic amines, which are of importance as pharmaceuticals and intermediates in the fine chemical industry.3 In general, the reaction of butadiene or isoprene with amines using palladium or nickel catalysts leads to the formation of longer chain amines by so-called telomerisation processes.4 However, in some cases minor amounts of allylic amines were also observed as by-products.5To date, the regioselective hydroamination of alkenes has been an important challenge in catalysis development. Particularly, the formation of the so-called anti-Markovnikov addition product is desired, because most of the known methods allow only the synthesis of Markovnikov addition products.6 Hence, the development of novel catalytic processes for the functionalization of alkenes or dienes with amines is of continuing interest in catalysis and synthesis.7 Notable developments from the past decade include the rhodium-catalysed anti-Markovnikov hydroamination of styrene derivatives or olefins,8 and related hydroaminations of 1,3-dienes. In the latter case, Hartwig and co-workers used Pd- or Ni-phosphine complexes as catalysts in the presence of CF3COOH as an additive.9 In addition, Ozawa and co-workers described the well-defined (η3-allyl)palladium complex bearing diphosphinidenecyclobutene ligands for the hydroamination of 1,3-diene. However, the scope of dienes was limited, giving mainly the 1,2-addition products.10 Later on, the group of Yi also employed a cationic ruthenium complex for the coupling of aniline with 1,3-butadiene. Again, the reaction led to the Markovnikov addition products as the major isomer.11 More recently, Schmidt and co-workers have developed a series of well-defined Pd(II) 3-iminophosphine (3IP) complexes for the intermolecular hydroamination of allenes and 2,3-dimethyl-1,3-butadiene with aliphatic amines.12
Here, we report a convenient hydroamination protocol using Pd(cod)Cl2 in combination with DPEphos, which allows for the selective hydroamination of a series of acyclic and cyclic dienes with a variety of aromatic and aliphatic amines without using an additive.13 Notably, the presented catalyst system allows the selective formation of allylic amines with high regioselectivity.
Results and discussion
Based on our previous work on catalytic hydroamination of alkynes with nitrogen nucleophiles,14 and also our interest in the synthesis of allylic amines, we recently reported the Pd-catalysed amination of unactivated allylic alcohols with amines as well as the electron deficient N-heterocycles. Both reactions occurred with high regio- and chemoselectivity.15 However, attempts to expand this chemistry towards hydroamination of 1,3-dienes resulted in no conversion.Hence, we initiated a systematic study on the palladium-catalysed hydroamination of isoprene with 4-methoxyaniline. In order to control the regiochemistry a broad range of phosphine ligands and different metal precursors were tested. Selected results of this study are shown in Scheme 1.
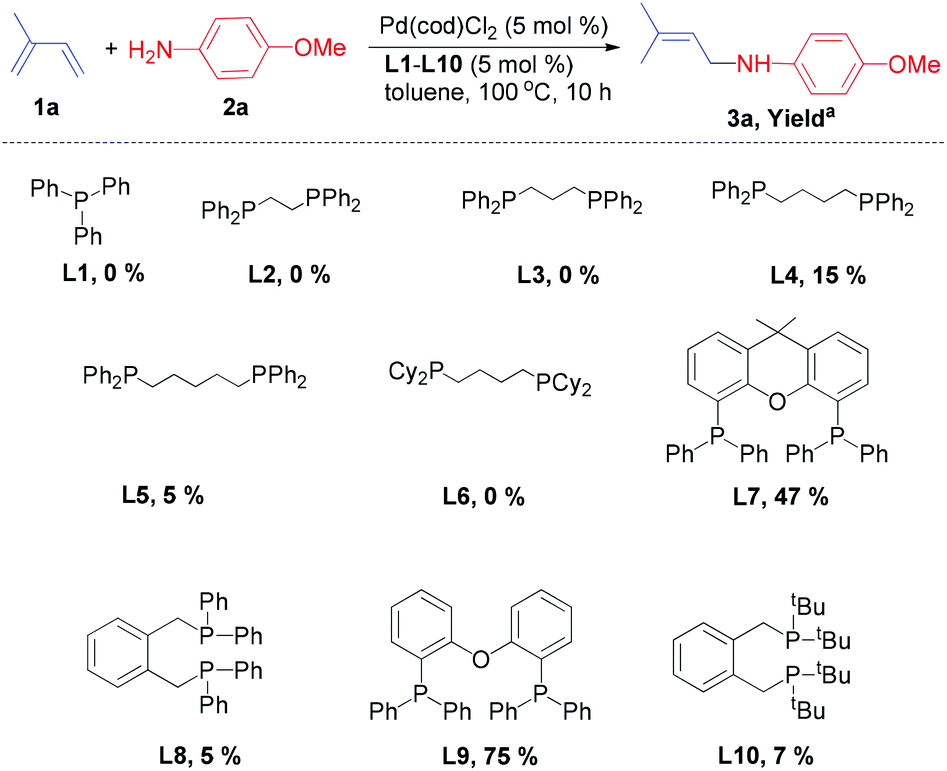 | ||
| Scheme 1 Pd-catalysed hydroamination of isoprene (1a) with 4-methoxyaniline (2a): Influence of ligands. aIsolated product yield. | ||
Typically, we performed the hydroamination of isoprene (1a, 4 equiv.) with 4-methoxyaniline (1 equiv.) in toluene using Pd(cod)Cl2 in the presence of ligands L1–L10. Employing the standard ligand triphenylphosphine (L1) did not result in any desired product 3a. Similarly, the application of bidentate phosphine ligands such as 1,2-bis(diphenylphosphino)ethane (L2), 1,3-bis(diphenylphosphino)propane (L3), and 1,4-bis(dicyclohexylphosphino)butane (L6) was not efficient for this model reaction.
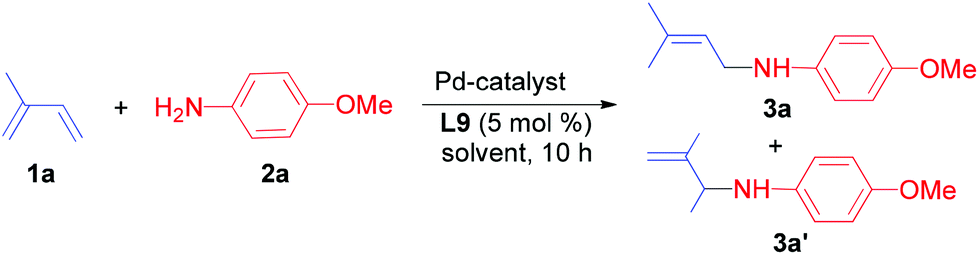
However, the use of 1,4-bis(diphenylphosphino)butane (L4) and 1,5-bis(diphenylphosphino)pentane (L5) resulted in low yields (5–15%) of the desired allylic amine. Hence, we investigated more bidentate phosphines with larger bite angles. Indeed, in the presence of ligands such as Xantphos (L7), 1,2-bis(diphenylphosphinomethyl)benzene (L8) and 1,2-bis(di-tert-butylphosphinomethyl)benzene (L10) the allylic amine 3a was obtained in yields of 5–47%. To our delight, the commercially available DPEphos (L9) was the most promising ligand and afforded 3a in 75% isolated yield.
While in some cases we also observed the formation of minor amounts of the 1,2-addition product (3a′) and the bis-allylamine, the reaction using DPEphos occurred with complete selectivity towards the 1,4-addition product 3a. Interestingly, the chelating phosphine ligand (L9) with a comparably large bite-angle allows selective hydroamination and plays a crucial role in controlling both the regio- and chemoselectivity due to the formation of a stable palladium–phosphine complex.
Next, we studied the influence of different catalyst precursors, solvents, and temperatures for the model reaction using L9 as the ligand of choice (Table 1). Under similar reaction conditions, various palladium-catalysts were found to be inefficient and resulted in no or only small amounts of the desired product (Table 1, entries 2–6). Apparently, Pd(cod)Cl2 is a unique precursor for this hydroamination reaction giving 75% yield of the single regio-isomer 3a (Table 1, entry 1). Apparently, this pre-catalyst allows for straightforward formation of a coordinatively unsaturated Pd-complex, whereas the presence of chloride ligands stabilizes the corresponding Pd–H species. Changing the solvent to 1,4-dioxane or 1,2-dichloroethane (DCE) gave the linear hydroamination product, albeit in lower yields (Table 1, entries 7 and 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | T (°C) | 3a/3a′, Yieldb,c (%) |
| a Reaction conditions: 1a (4 mmol), 2a (1 mmol), Pd-catalyst (1–5 mol%), solvent (3.0 mL). b Isolated yield. c Isomeric ratio was determined by GC-MS analysis. d (2.5 mol%) L9 used. e (1 mol%) L9 used. f Reaction with no ligand. | ||||
| 1 | Pd(cod)Cl2 (5) | Toluene | 100 | 75/2 |
| 2 | Pd(OAc)2 (5) | Toluene | 100 | 5/2 |
| 3 | Pd(OCOCF3)2 (5) | Toluene | 100 | 10/7 |
| 4 | Pd(dba)2 (5) | Toluene | 100 | 0 |
| 5 | {Pd(π-allyl)Cl}2 (2.5) | Toluene | 100 | 10/8 |
| 6 | {Pd(π-cinn)Cl}2 (2.5) | Toluene | 100 | 33/10 |
| 7 | Pd(cod)Cl2 (5) | 1,4-Dioxane | 100 | 62/9 |
| 8 | Pd(cod)Cl2 (5) | 1,2-DCE | 100 | 52/12 |
| 9 | Pd(cod)Cl2 (5) | Toluene | 80 | 60/2 |
| 10 | Pd(cod)Cl2 (5) | Toluene | 60 | 41/4 |
| 11 | Pd(cod)Cl2 (5) | Toluene | 25 | 5/0 |
| 12d | Pd(cod)Cl2 (2.5) | Toluene | 100 | 30/8 |
| 13e | Pd(cod)Cl2 (1) | Toluene | 100 | 5/2 |
| 14f | Pd(cod)Cl2 (5) | Toluene | 100 | 0 |
| 15 | No catalyst | Toluene | 100 | 0 |
Interestingly, it was also possible to run the model reaction at room temperature, though with poor product yield (Table 1, entry 11). Lowering the catalyst concentration revealed an optimal loading of 5 mol% of Pd(cod)Cl2. As expected, we did not observe any product either in the absence of a catalyst or a ligand (Table 1, entries 14 and 15). Notably, in all reactions, where we observed a low yield of 3a, unreacted amine was recovered from the reaction mixture. In some cases, we also observed 5–10% of the bis-allylamine and 2–12% of the 1,2-addition product by GC-MS analysis of the crude reaction mixture. Prolonging the reaction time increased the ratio of bis-allylamine and also the 1,2-addition product.16
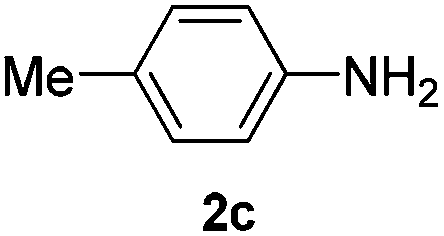
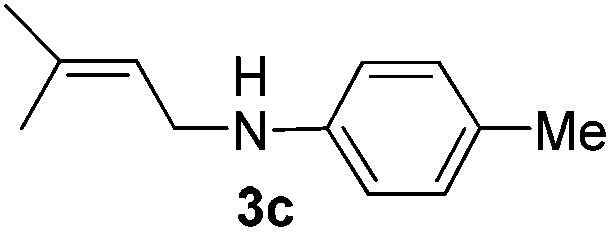
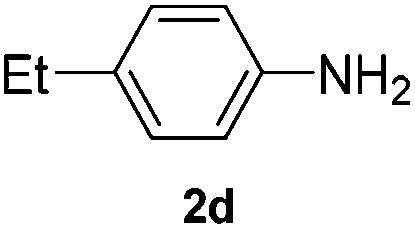
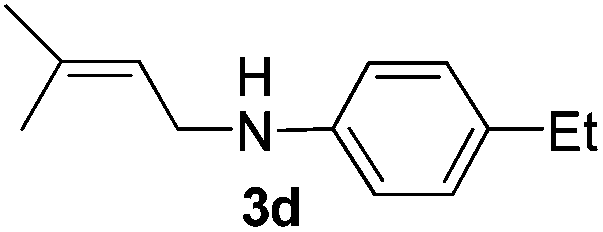
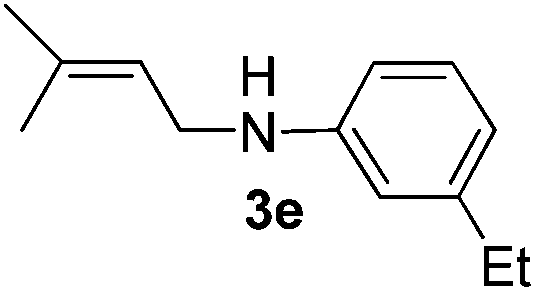
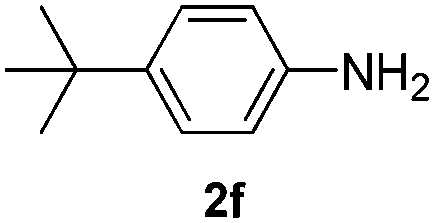
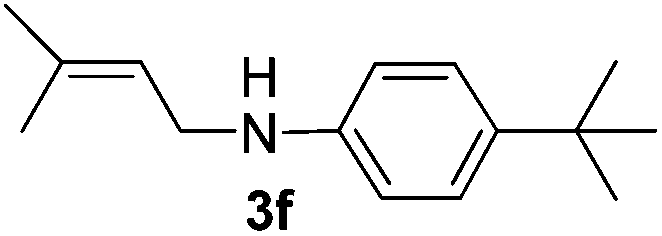
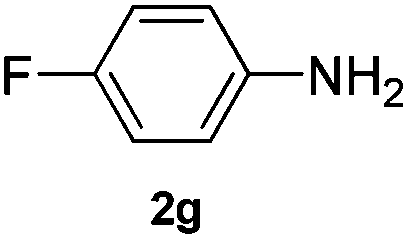
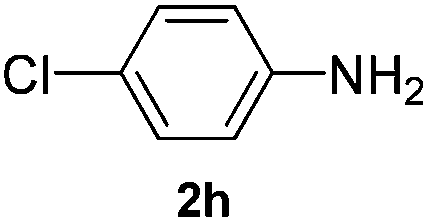
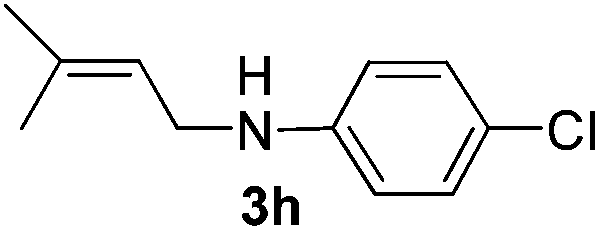
After having found promising results in the model reaction, we explored the general reactivity of our novel catalyst system with a range of anilines and alkylamines. As shown in Table 2 the reaction of isoprene with aniline or electron-rich aniline derivatives proceeded with similar efficiency. Hence, reaction of isoprene (1a) with methyl-, ethyl-, and tert-butyl-substituted aniline resulted in 3c–3f in 69–78% isolated yields. Similar to the model reaction in all cases the 1,4-addition product was isolated as a single regioisomer (Table 2, entries 2–6). Further, reactions of isoprene with 4-fluoroaniline (2g) and 4-chloroaniline (2h) afforded the corresponding allylic amines in 70–83% isolated yield under our optimized reaction conditions (Table 2, entries 7 and 8).
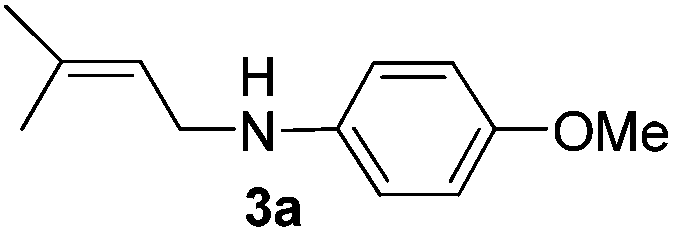
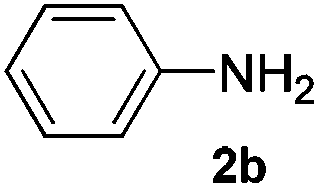
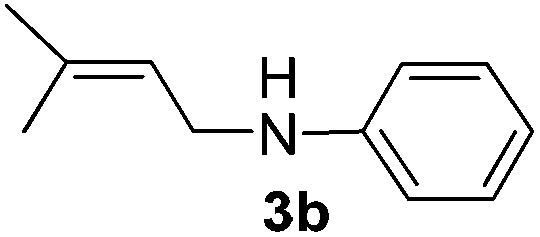
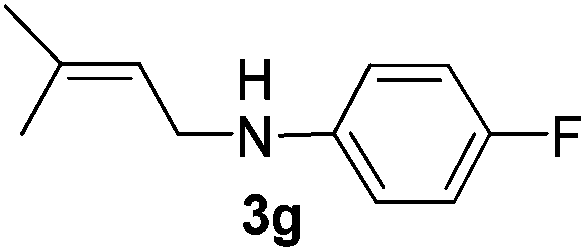
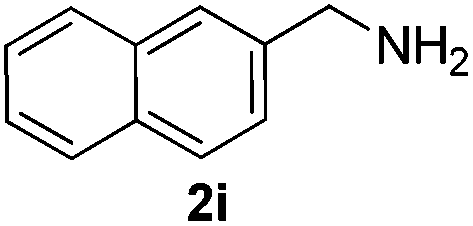
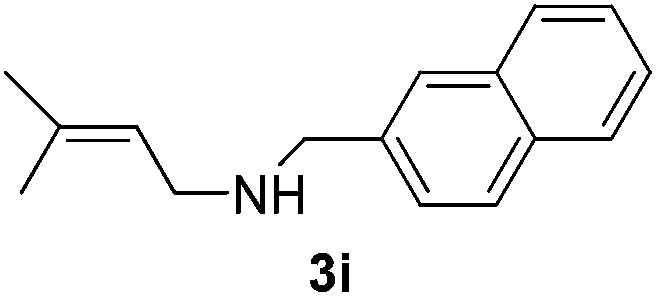
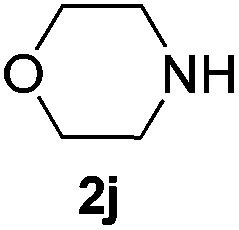
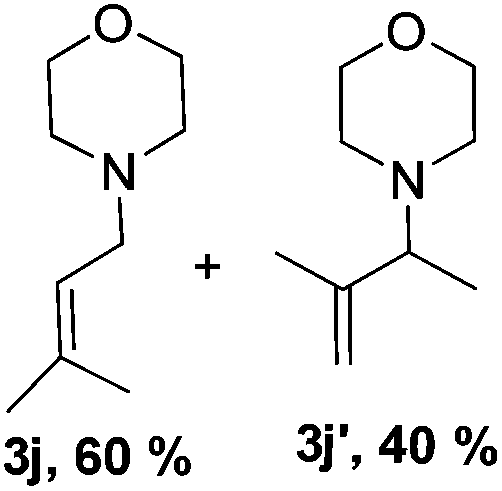
Then, we studied the reaction of 1a with a more challenging primary alkylamine derivative, which is known to be more prone to undergo bis-allylation. Gratifyingly, the reaction of isoprene (1a) with 1-methylnaphthylamine (2i) gave mainly the mono-allylamine derivative in 62% yield (Table 2, entry 9). Nevertheless, in this reaction we also observed a minor amount of another isomer, likely the branched regioisomer. Finally, the reaction of the secondary amine morpholine 2j afforded a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-mixture of the linear and branched allylic amines in 63% yield (Table 2, entry 10).
:1-mixture of the linear and branched allylic amines in 63% yield (Table 2, entry 10).
Next, we became interested in the hydroamination of acyclic and cyclic 1,3-dienes. It should be noted that these substrates are commonly known to be less reactive. Selected reactions with aniline derivatives are summarized in Table 3.
|
|
||||
|---|---|---|---|---|
| Entry | 1,3-Diene, 1 | Amine, 2 | Product, 3 | Yieldb (%) |
| a Reaction conditions: 1 (4 mmol), 2 (1 mmol), Pd(cod)Cl2 (5 mol%), L9 (5 mol%), toluene (3.0 mL). b Isolated yield. c 1c (1 equiv.), 2b (2 equiv.). d Pd(cod)Cl2 (10 mol%), L9 (10 mol%), 120 °C, 20 h. e 2b (4 equiv.). f 1e (1 equiv.), 2b (4 equiv.), 20 h. 2k = 2-Bromoaniline, 2l = 4-cyanoaniline, 2m = 4-trifluoromethylaniline. | ||||
| 1 |
|
2a |
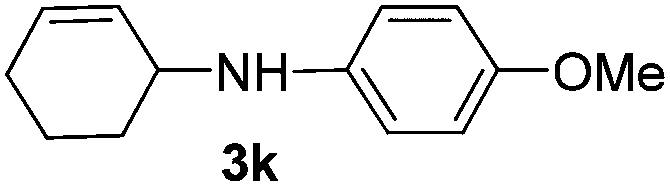
|
94 |
| 2 |
|
2b |
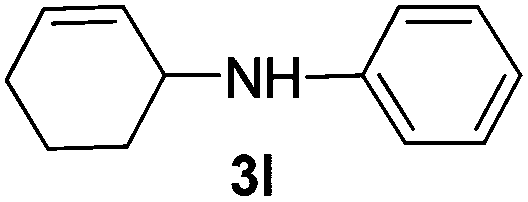
|
95 |
| 3 |
|
2k |
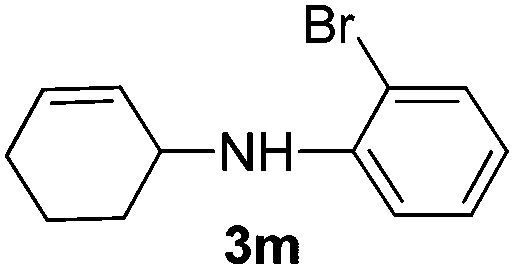
|
40 |
| 4 |
|
2l |
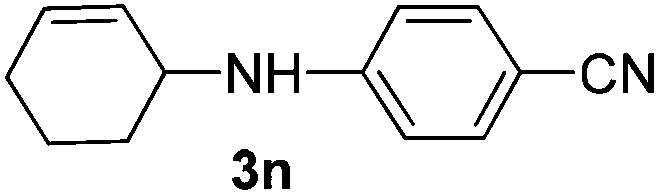
|
49 |
| 5 |
|
2m |
|
77 |
| 6 |
|
2h |
|
82 |
| 7 |
|
2e |
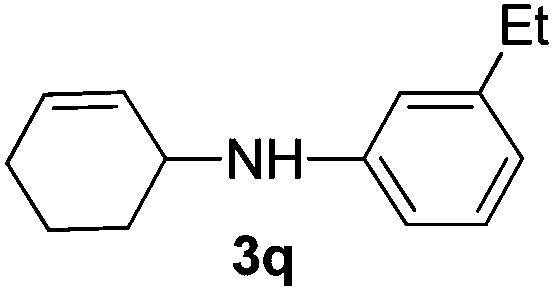
|
78 |
| 8c,d |
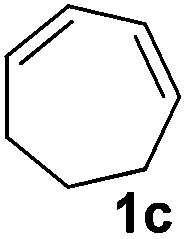
|
2b |
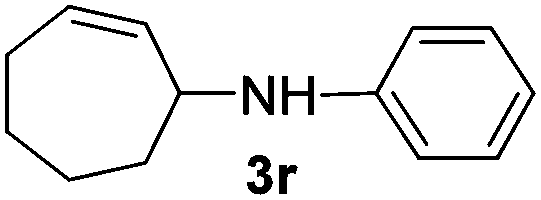
|
53 |
| 9d,e |
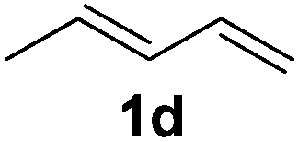
|
2b |
|
49 |
| 10f |
|
2b |
|
55 |
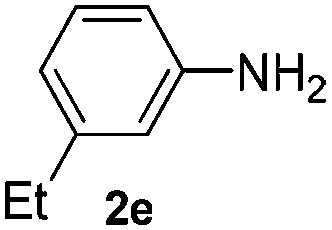
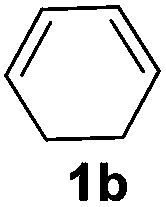
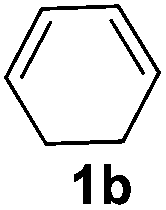
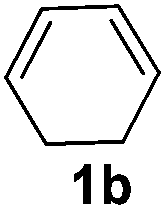
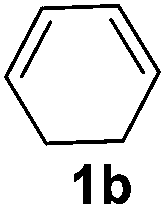
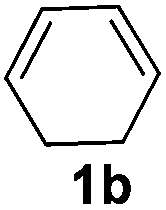
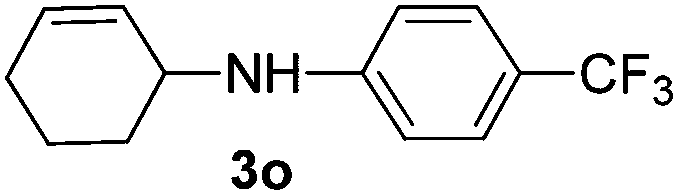
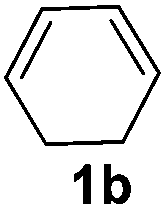
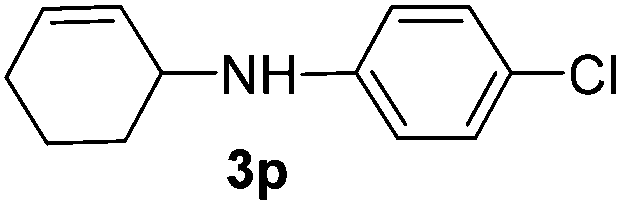
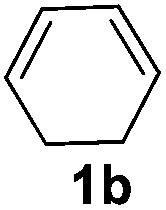
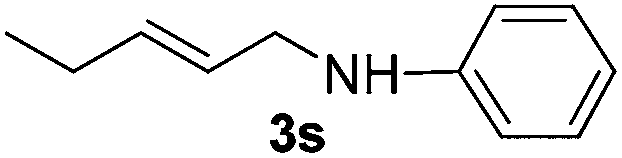
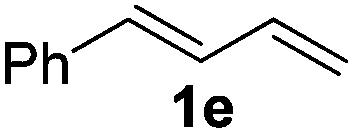
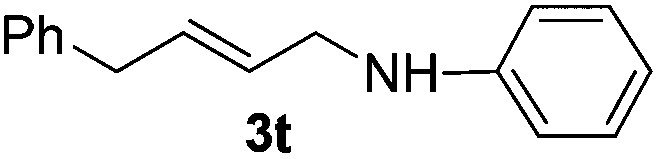
The reaction of 1,3-cyclohexadiene 1b with 4-methoxyaniline 2a and aniline 2b gave almost quantitative yields of 3k and 3l (Table 3, entries 1 and 2). Further reaction with 2-bromoaniline 2k and 4-cyanoaniline 2l resulted in 40–49% yield of 3m and 3n (Table 3, entries 3 and 4). Notably, the reaction of 1,3-cyclohexadiene 1b with 4-trifluoromethyl, 4-chloro and 3-ethyl substituted aniline derivatives afforded 77–82% yields of 3o–3q (Table 3, entries 5–7). A similar reaction with 1,3-cycloheptadiene 1c resulted in only poor yield of the corresponding allylic amine derivative. However, on increasing the catalyst loading and temperature, the reaction resulted in 53% isolated yield of 3r (Table 3, entry 8). Similarly, 4-methyl- and 4-phenyl-substituted 1,3-butadienes, 1d and 1e, also efficiently reacted with aniline 2b, and afforded 49–55% yield of the 1,4-addition product as a single regioisomer (Table 3, entries 9 and 10).
During the reaction with less reactive dienes (Table 3, entries 8–10), we observed that the diene moiety was not fully consumed and 20–30% unreacted dienes were recovered. Importantly, in all these reactions we did not observe any branched isomers or the bis-allylation product of the corresponding amine.
After having observed the intermolecular 1,4-hydroamination of a variety of 1,3-dienes, we became interested in the active Pd-catalyst responsible for high regioselectivity and the probable reaction mechanism. Initially, we prepared the defined (DPEPhos)Pd(π-allyl)Cl complex starting from the Pd(π-allyl)Cl dimer and the DPEPhos ligand. To gain insight into the reaction mechanism, we studied the stoichiometric hydroamination reaction of the defined (DPEPhos)Pd(π-allyl)Cl complex 4 with aniline 2b. The reaction of the π-allyl-Pd-complex resulted in the formation of allylamine 5 in 60% isolated yield (Scheme 2, a). Similarly, reactions of (DPEPhos)Pd(π-allyl)Cl complex 4 with isoprene and aniline 2b resulted in the regioselective formation of 1,4-hydroamination product 3b in 65% yield (Scheme 2, b).
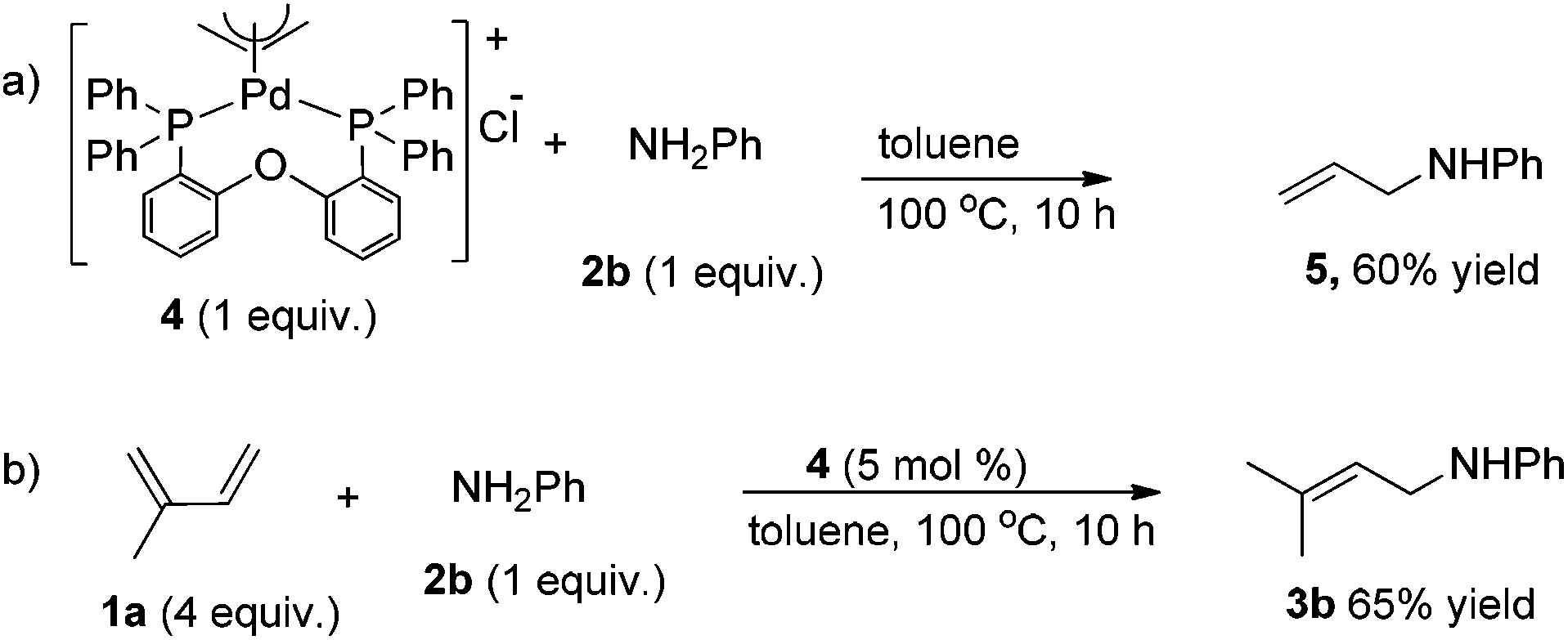 | ||
| Scheme 2 Intermolecular hydroamination of aniline with the defined (DPEPhos)Pd(π-allyl)Cl complex 4. | ||
The above experimental studies allow to understand the highly regioselective 1,4-hydroamination for the formation of allylic amines (Scheme 3). It is proposed that under the reaction conditions, hydrochloric acid is generated from the palladium chloride and aniline. Therefore, the reaction involves the initial formation of a transient Pd–H species A, followed by reaction with the diene that allowed the formation of cationic π-allyl-Pd-complex intermediate B. The regioselective 1,4-hydroamination product 3b resulted from the nucleophilic attack of aniline 2b on the less-substituted carbon of the intermediate species B.
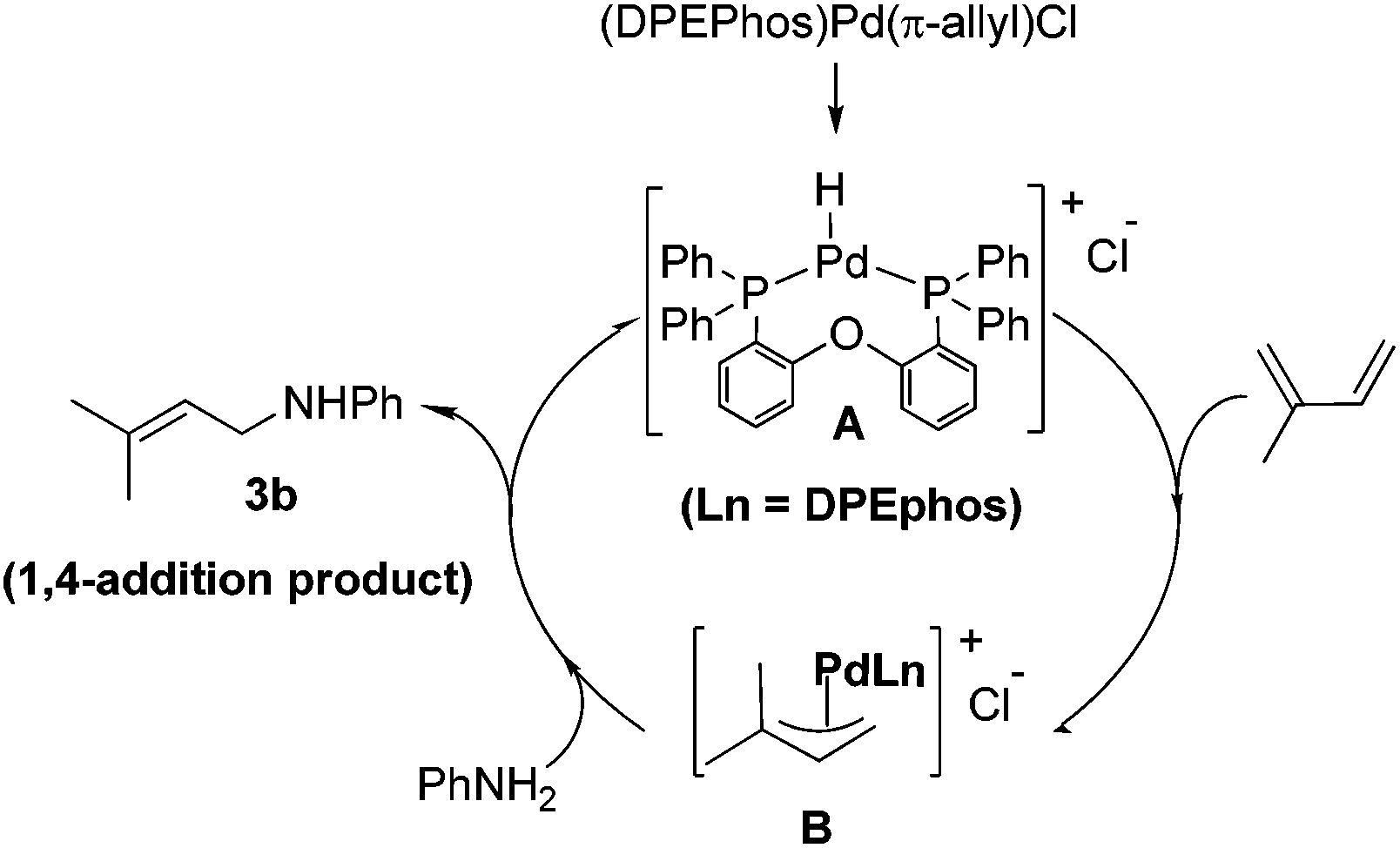 | ||
| Scheme 3 Proposed mechanism for the intermolecular hydroamination of 1,3-dienes with aniline. | ||
Experimental
Under an Ar atmosphere, an oven dried Schlenk tube was charged with 4-methoxyaniline (1 mmol), followed by Pd(cod)Cl2 (5 mol%) and L9 (5 mol%). Toluene (3 mL) and a magnetic stirrer bar were added followed by isoprene (4 equiv.) and the reaction mixture was stirred at 100 °C for the reported time. After completion, the reaction mixture was cooled to rt, diluted with ethyl acetate (10 mL) and dried over anhydrous Na2SO4. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using ethyl acetate–hexane as an eluent to afford the corresponding allylic amine derivative.Conclusions
In summary, we presented a convenient Pd-catalysed hydroamination of a series of acyclic and cyclic 1,3-dienes with a variety of aromatic amines under relatively mild conditions. The available catalytic system consisting of Pd(cod)Cl2 in combination with the DPEphos ligand allows for an exclusive formation of the 1,4-addition product with high regioselectivity. The reaction is atom-economic and does not need any additives, making it a valuable method for the synthesis of allylic amines.Acknowledgements
The research has been funded by the State of Mecklenburg-Western Pomerania and the BMBF. We thank Dr W. Baumann, Dr C. Fischer, S. Buchholz, S. Schareina, A. Koch, and S. Rossmeisl (all at LIKAT) for their excellent technical and analytical support.Notes and references
- For reviews, see: (a) R. Taube, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996, vol. 1, p. 507 Search PubMed; (b) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef PubMed; (c) P. W. Roesky and T. E. Müller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS PubMed; (d) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed; (e) M. Nobis and B. Drießen-Hölscher, Angew. Chem., Int. Ed., 2001, 40, 3983 CrossRef CAS; (f) J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795 CrossRef CAS.
- M. Beller and G. Centi, ChemSusChem, 2009, 2, 459 CrossRef CAS PubMed.
- For reviews on allylic amines, see: (a) R. B. Cheikh, R. Chaabouni, A. Laurent, P. Mison and A. Nafti, Synthesis, 1983, 685 CrossRef PubMed; (b) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (c) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- For selected examples of telomerisation processes see: (a) T. Prinz, W. Keim and B. D. Hölscher, Angew. Chem., Int. Ed., 1996, 35, 1708 CrossRef CAS and references cited therein; (b) A. Behr and M. Urschey, J. Mol. Catal. A: Chem., 2003, 197, 101 CrossRef CAS; (c) J. Mesnager, E. Kuntz and C. Pinel, J. Organomet. Chem., 2009, 695, 2513 CrossRef PubMed; (d) S. Bigot, J. Lai, I. Suisse, M. Sauthier, A. Mortreux and Y. Castanet, Appl. Catal., A, 2010, 382, 181 CrossRef CAS PubMed.
- (a) J. Kiji, E. Sasakawa, K. Yamamoto and J. Furukawa, J. Organomet. Chem., 1974, 77, 125 CrossRef CAS; (b) R. Baker, A. Onions, R. G. Popplestone and T. N. Smith, J. Chem. Soc., Perkin Trans. 2, 1975, 1133 RSC; (c) S. M. Maddock and M. G. Finn, Organometallics, 2000, 19, 2684 CrossRef CAS.
- (a) Catalytic anti-Markovnikov additions of nucleophiles to olefins have been described as one of the “top 10 challenges for catalysis.” See: J. Haggin, Chem. Eng. News, 1993, 71, 23 CrossRef PubMed; (b) For a review see: M. Beller, J. Seavad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed.
- (a) K. Takahashi, A. Miyake and G. Hata, Bull. Chem. Soc. Jpn., 1972, 45, 1183 CrossRef CAS; (b) R. W. Armbruster, M. M. Morgan, J. L. Schmidt, C. M. Lau, R. M. Riley, D. L. Zabrowski and H. A. Dieck, Organometallics, 1986, 5, 234 CrossRef CAS; (c) L. Besson, J. Góre and B. Cazes, Tetrahedron Lett., 1995, 36, 3857 CrossRef CAS; (d) M. Al-Masum, M. Meguro and Y. Yamamoto, Tetrahedron Lett., 1997, 38, 6071 CrossRef CAS; (e) R. Dorta, P. Egli, F. Zurcher and A. Togni, J. Am. Chem. Soc., 1997, 119, 10 857 CrossRef CAS; (f) U. Radhakrishnan, M. Al-Masum and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 1037 CrossRef CAS; (g) M. Beller, H. Trauthwein, M. Eichberger, C. Breindl and T. E. Müller, Eur. J. Inorg. Chem., 1999, 1121 CrossRef CAS; (h) M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller and O. R. Thiel, Chem.–Eur. J., 1999, 5, 1306 CrossRef CAS; (i) K. C. Hultzsch, Adv. Synth. Catal., 2005, 347, 367 CrossRef CAS; (j) A. Hu, M. Ogasawara, T. Sakamoto, A. Okada, K. Nakajima, T. Takahashi and W. Lina, Adv. Synth. Catal., 2006, 348, 2051 CrossRef CAS; (k) N. Nishina and Y. Yamamoto, Angew. Chem., Int. Ed., 2006, 45, 3314 CrossRef CAS PubMed; (l) D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748 CrossRef CAS PubMed; (m) J. Guin, C. Mück-Lichtenfeld, S. Grimme and A. Studer, J. Am. Chem. Soc., 2007, 129, 4498 CrossRef CAS PubMed; (n) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496 CrossRef CAS PubMed; (o) Z. Zhang, S. D. Lee and R. A. Widenhoefer, J. Am. Chem. Soc., 2009, 131, 5372 CrossRef CAS PubMed; (p) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS PubMed; (q) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS PubMed; (r) K. Manna, S. Xu and A. D. Sadow, Angew. Chem., Int. Ed., 2011, 50, 1865 CrossRef CAS PubMed.
- (a) M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608 CrossRef CAS PubMed; (b) M. S. Sanford and J. T. Groves, Angew. Chem., Int. Ed., 2004, 43, 588 CrossRef CAS PubMed.
- (a) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366 CrossRef; (b) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS.
- T. Minami, H. Okamoto, S. Ikeda, R. Tanaka, F. Ozawa and M. Yoshifuji, Angew. Chem., Int. Ed., 2001, 40, 4501 CrossRef CAS.
- C. S. Yi and S. Y. Yun, Org. Lett., 2005, 7, 2181 CrossRef CAS PubMed.
- G. Kuchenbeiser, A. R. Shaffer, N. C. Zingales, J. F. Beck and J. A. R. Schmidt, J. Organomet. Chem., 2011, 696, 179 CrossRef CAS PubMed.
- (a) B. Liu, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943 CrossRef CAS PubMed; (b) Y. Li and T. J. Marks, Organometallics, 1996, 15, 3770 CrossRef CAS.
- (a) A. Tillack, V. Khedkar, H. Jiao and M. Beller, Eur. J. Org. Chem., 2005, 5001 CrossRef CAS; (b) A. Tillack, H. Jiao, I. Garcia Castro, C. G. Hartung and M. Beller, Chem.–Eur. J., 2004, 10, 2409 CrossRef CAS PubMed; (c) V. Khedkar, A. Tillack, M. Michalik and M. Beller, Tetrahedron, 2005, 61, 7622 CrossRef CAS PubMed; (d) V. Khedkar, A. Tillack, M. Michalik and M. Beller, Tetrahedron, 2005, 61, 7622 CrossRef CAS PubMed; (e) V. Khedkar, A. Tillack, M. Michalik and M. Beller, Tetrahedron Lett., 2004, 45, 3123 CrossRef CAS PubMed; (f) A. Tillack, H. Jiao, I. G. Castro, C. G. Hartung and M. Beller, Chem.–Eur. J., 2004, 10, 2409 CrossRef CAS PubMed; (g) K. Alex, A. Tillack, N. Schwarz and M. Beller, ChemSusChem, 2008, 1, 333 CrossRef CAS PubMed.
- (a) D. Banerjee, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, ChemSusChem, 2012, 5, 2039 CrossRef CAS PubMed; (b) D. Banerjee, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 11556 CrossRef CAS PubMed; (c) D. Banerjee, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 1630 CrossRef CAS PubMed.
- When the hydroamination reaction was performed using a 1:1 ratio of isoprene and amine, the reaction gave a poorer yield of the desired product. Using 4 equiv. of isoprene resulted in the complete consumption of amine and a high product yield. It should be noted that in the presence of the Pd-catalyst, isoprene easily undergoes dimerisation as well as oligo- and polymerisation reactions. For example, we observed the dimerisation of isoprene (molecular mass 136) in the GC-MS analysis of some crude reaction mixtures.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00023d |
| This journal is © the Partner Organisations 2014 |