Accelerator mass spectrometry in the attomolar concentration range for 14C-labeled biologically active compounds in complex matrixes
Niklas
Forsgard
,
Mehran
Salehpour
and
Göran
Possnert
Ion Physics, Ångström Laboratory, Department of Engineering Sciences, Uppsala University, Box 534SE-751 21, Uppsala, Sweden
First published on 15th October 2009
Abstract
Accelerator mass spectrometry (AMS) is an ultra-sensitive analytical method suitable for detection of sub-nanomolar concentrations of labeled biological substances such as pharmaceutical drugs in body fluids. A limiting factor in extending the concentration measurements to the sub-picomolar range is the natural 14C content in living tissues. This can be circumvented by separating the labeled drug from the tissue matrix with, for example, liquid chromatography. The analysis of drugs and their metabolites or endogenous compounds in biological fluids by liquid chromatography is usually complicated and the sample preparation step remains the most serious problem both with regard to losses and degradation of the analyte, and also automation of the analysis. In this article a method for detection and quantification of extremely low concentrations of 14C-labeled biomolecules in biological fluids by AMS is described. The use of a column switched chromatographic system incorporating a restricted-access media (RAM) column allowed the direct injection of untreated human plasma samples, which reduces the total time of analysis and makes automation of the sample preparation step possible. As the separated total drug amount is in the attogram to femtogram region, it is not possible to use a standard AMS sample preparation method, where mg sizes are required. We have utilized a sensitive carbon carrier method where a 14C-deficient compound is added to the HPLC fractions and the composite sample is prepared and analysed by AMS. The method shows remarkable sensitivity, low background values and good linearity, allowing the detection and quantification of a pharmaceutical drug in human plasma in the low femtomolar and down to the attomolar concentration range.
Introduction
Organic compounds enriched with 14C in combination with isotopic measurement techniques offer a very sensitive method for detection of low concentrations of molecular markers in different biological samples. 14C can easily be incorporated into most types of biomolecules and has been used within various areas of science including chemistry, biology and medicine.Accelerator mass spectrometry (AMS) was developed in the late 1970s out of the need to radiocarbon date smaller samples than were possible with standard decay counting techniques. Today AMS offers an extensive increase in sensitivity over decay-counting methods that are routinely used in biological studies that involve radioisotopes.1 During the last years the use of AMS has expanded greatly with biomedical applications becoming increasingly important and the availability of AMS and its ease of use have improved considerably during the last decade.2 This is partly a result of the technology having become more mature and thus more reliable, and partly due to the fact that new, more compact and less expensive, accelerators have been introduced to the market. The advancements of the technique have facilitated successful commercialization of AMS into the pharmaceutical and biochemical field. A detailed description of the methodology for quantitation of isotopic molecular labels with accelerator mass spectrometry can be found elsewhere.3 All different experiments that have been performed throughout the years using decay-counting methods can now be performed with AMS with an improvement in sensitivity up to six orders of magnitude.
One exciting application of biological AMS in the pharmaceutical research and development sector is Microdosing which is based on using small doses of drugs (below 100 µg per person) to reduce toxicity and side-effect risk issues associated with therapeutic doses. Consequently, this requires an ultra sensitive method, such as AMS, to determine the drug concentration. Lappin and Garner performed the first AMS Microdosing experiments4 and demonstrated the capability of the method in shortening the drug development time. Microdosing has been endorsed by the FDA5 and also by EMEA.6 Furthermore, other important parameters such as bioavailability, mass-balance studies and metabolic profiling of pharmaceutical drugs are measured routinely with AMS.7
Improvements in the detection sensitivity of the method have obvious advantages. Zeptomole sensitivity for a pharmaceutical drug in human blood in AMS has been demonstrated without the use of a separation method.8 For drug concentration measurements, high performance liquid chromatography (HPLC) is routinely combined with AMS,9–12 facilitating detection down to the 20 femtomolar (fM) region.11 The analysis of drugs and their metabolites by HPLC is both tedious and time consuming. In recent years an increasing number of HPLC methods utilizing solid phase extraction for sample clean-up and pre-concentration by means of column switching have been developed.13 The switching methodology permits the off-line multi-step procedures, often including filtering, protein precipitation and centrifugation, to be performed as a single-step, on-line and directly coupled to the separation step.
In this article we describe a method for detection and quantification of extremely low concentrations of 14C-labeled biomolecules in biological fluids by AMS. A column switched chromatographic system incorporating a restricted-access media (RAM) column allowed the direct injection of untreated human plasma samples to the HPLC. The column switching methodology also provides the possibility to fully automate the sample clean-up, separation and fraction collection steps. The combined method shows remarkable sensitivity, low background values and good linearity, allowing the detection and quantification of pharmaceutical drugs in human plasma in the low femtomolar (amol/mL) down to the attomolar (zmol/mL) concentration range.
Experimental
The different experimental steps for a typical analysis of biological samples are schematically illustrated in figure 1. The sections below describe the experimental steps.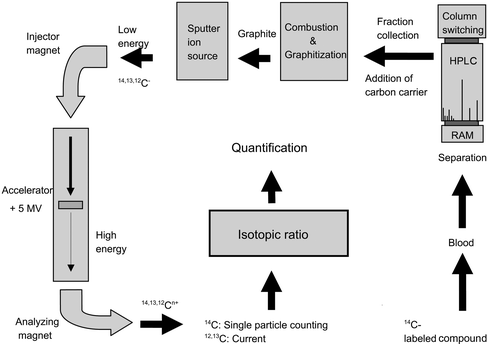 | ||
| Fig. 1 Schematics of a typical biological AMS experiment, illustrating the different steps involved in the analysis of biological samples (see text). | ||
Chemicals
Methanol (LiChrosolv) and formic acid (p.a.) were purchased from Merck (Darmstadt, Germany), ammonium formate (pro analysis) was bought from Fluka (Dorset, U.K.), triethylamine (HiPerSolv Chromanorm) was purchased from VWR BDH Prolabo (Leicestershire, UK). Copper oxide (ACS reagent), iron powder (puriss) and zinc powder (<150 µm, 99.995% trace metals basis) were bought from Sigma-Aldrich (St. Louis, MO). The unlabeled and 14C labeled anti-psychotic drug, remoxipride was supplied by AstraZeneca (Södertälje, Sweden).Plasma samples
A plasma stock solution was prepared by spiking human plasma with 14C-enriched remoxipride (C16H23BrN2O3, Mw = 371 g/mol). The drug has been 14C-marked with a specific activity of 2.035 GBq/mmol, dissolved in water and then in human plasma. Seven samples with a concentration range from approximately 60 fM down to 900 aM of labeled remoxipride were prepared by diluting this stock solution with plasma. To each sample unlabeled remoxipride was added to give a final concentration of 0.5 µM. The samples were vortex mixed and centrifuged at 17000x g for 5 minutes to remove particles. The spiked samples were then directly injected to the chromatographic system without any protein precipitation step.Blank samples were prepared by adding unlabeled remoxipride to plasma (0.5 µM). The blank samples were then prepared exactly like the labeled samples.
Chromatography
A column-switching method was used for on-line clean-up and pre-concentration of the sample prior to separation. A buffer containing 100 mM ammonium formate and 25 mM triethylamine was prepared and adjusted to pH 3.0 with formic acid. Two different mobile phases, A and B, were prepared by mixing the buffer with methanol to 95:5 and 70:30 v/v respectively. Two six port injection valves (Valco Instruments Co. Inc., Huston, TX) and two Jasco-1580 pumps (Jasco, Japan) were used in the experimental setup. In the first valve a 1000 µl injection loop was fitted and in the other valve the extraction column (LiChrosphere® RP-18 ADS 25 µm, 25 × 4 mm, Merck, Darmstadt, Germany) and the separation column (Reprosil.Pur C18-AQ 5 µm, 150 × 2 mm, Dr. Maisch, Ammerbuch, Germany) were fitted. In the loading position the extraction column was pumped with mobile phase A at 500 µl min−1 to load the sample onto the column and wash away the proteins, salts and other matrix components. After 10 minutes the valve was switched to unload the analyte, and both columns were pumped with mobile phase B at 200 µl min−1. The separation was accomplished by isocratic elution with mobile phase B. Detection of the unlabeled remoxipride was accomplished with a Jasco UV-1575 UV detector (Jasco, Japan) and a 5.5 minutes (1.1 mL) fraction, starting at the beginning of the peak, was collected in a quartz vial.The recovery of the column-switching clean-up step was evaluated by injecting four standard solutions of unlabeled remoxipride (ranging from 0.30 to 2.4 µM) dissolved in mobile phase A, directly through both the columns without switching the flow. A standard curve with the integrated areas from remoxipride was created by linear regression. The UV-signals from the unlabeled remoxipride in the samples, measured on the column-switched system, were then compared with the signal from the standards and the recovery was determined.
The chromatographic setup in this report offers several benefits compared to the previous method.9–12 Sample clean-up normally consists of a number of steps such as filtration, protein precipitation and/or centrifugation etc. All of these steps are both time consuming and increase the risk of contamination and low recoveries, especially when working at very low concentrations. In this setup, the protein precipitation step was replaced by an on-line column-switched clean-up step where the analyte was first trapped on a restricted access media column (RAM) and subsequently separated on a C18-material. With this method it was possible to avoid the precipitation step, which might cause large losses due to adsorption to the precipitate. The use of RAM in a column-switching approach has several advantages such as direct injection of untreated biofluids, on-column enrichment, unattended operation, safer handling of hazardous or infectious samples and a decrease in total analysis time. The column-switching approach also made it possible to increase the injection volume from 50 µl up to 1 ml without compromising the chromatographic performance.
AMS sample preparation
The collected fractions from the chromatographic step were evaporated to dryness in a Scanvac Modulespin 40 freeze-drier (Scanlaf A/S, Denmark) for 5 h at 2000 rpm to remove water and the volatile carbon containing components from the mobile phase. The dry fractions from the separation typically contain a few attomoles corresponding to sub-fg in weight for an average 14C-labeled pharmaceutical drug. As AMS requires about 1 mg of carbon, the fractions can not be studied using standard AMS sample preparation. To increase the carbon content 1.5 mg of a petrochemical compound, tributyrin (TRB, C15H26O6), with low 14C-content (0.18 percent Modern Carbon) was added as a carbon carrier to each sample, corresponding to a total carbon amount of about 1 mg. As described by Buchholz et. al.,10 the combination of TRB and the HPLC fraction are then treated as a composite AMS sample and undergo the usual oxidation and reduction process to convert the sample into graphite. The combustion and graphitisation method is described elsewhere11 and will not be described here.Accelerator mass spectrometry
For the AMS studies, the accelerator facility at Uppsala University was used, which is a 5 MV Pelletron tandem accelerator installed in 2001. The setup has been described elsewhere11 and is only outlined here. Two negative ion SIMS (Secondary Ion Mass Spectrometry) sputter ion sources are available for automated, overnight operation.It is standard procedure to have one 14C-free sample (old carbon) and 3 standard reference material samples (Oxalic acid II, obtained from NIST, Boulder, Colorado) in the holder for every AMS experiment. A sample is typically analysed between 3–5 acquisition periods of 5 minutes each and the average value is presented. Isotopic fractionation is compensated for and precision is typically about 0.6%. The 14C/12C ratio, R, is presented in units of Modern or as a percentage of Modern Carbon. 1 Modern is roughly equivalent to 97.8 amole 14C/mg 12C.
Results and discussion
Body fluids and other biological samples taken from living organisms normally contain a natural level of 14C which is about 1 Modern (100 percent Modern Carbon). Consequently, standard AMS measurements suffer from the limitation of not being able to detect concentrations of labeled compounds below this limit. In an earlier publication Salehpour et al. have shown, using the same drug (specific activity of 2035GBq/mmol), that the concentration limit, below which it is not possible to separate the signal from the natural background in blood, is about 700 fM.8In order to measure concentrations below the limit of the natural background, the labeled biomarkers have to be separated from the biological matrix, which might contain numerous carbon-containing compounds such as proteins, peptides, carbohydrates etc.9,10 In a previous article a liquid chromatography separation method was described where a labeled drug was separated and collected in fractions.11 The fractions were treated with the carbon carrier method and analysed with AMS. The method allowed detection of drug concentrations below 20 fM in human plasma. As described earlier, in these experiments a RAM column is used in combination with column switching into a C18-material. Apart from practical improvements, one particular advantage of this approach is that it is possible to increase the injection volume from 50 µl up to 1 ml without compromising the chromatographic performance.
The collected fractions are evaporated to dryness before combustion. Thus, the components of the HPLC mobile phases have to be selected carefully to minimize unwanted carbon addition to the sample as all non-volatile components will be mixed with the sample and cause an increased background. In this study ammonium format and formic acid were used as buffer, with an addition of triethylamine to minimize unwanted interaction with the silanol groups on the columns. These are all volatile components and were straightforwardly removed by evaporation.
Quantification
Seven plasma samples were prepared with a predicted 14C-labeled remoxipride concentration range of 0.9–60 fM. The drug was separated by HPLC and the collected fractions were prepared by the carbon carrier method and analysed by AMS.The concentration, K, of the drug in a carrier sample is given by:3
| K = Ccarrier·(Rsample − Rcarrier)/(L·V) | (1) |
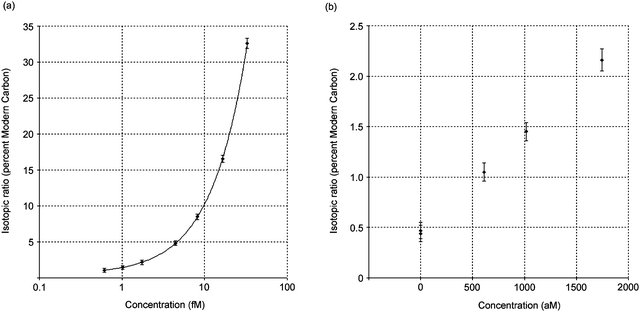 | ||
| Fig. 2 (a) The measured isotopic ratio of the composite sample with carbon carrier and the HPLC fraction in percent Modern carbon plotted as a function of the measured molar concentration of 14C-remoxipride (semi-log plot). (b) The figure is expanded for the low concentration region. The R-values for two blank samples are also included in the figure. | ||
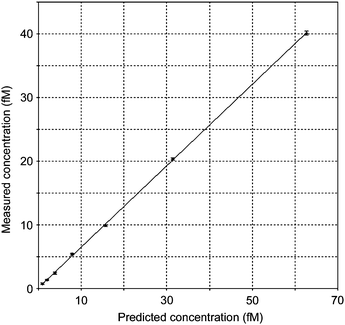 | ||
| Fig. 3 The measured concentrations of 14C-remoxipride versus the predicted concentrations in plasma. | ||
The detection limit for remoxipride in plasma was calculated from the predicted concentrations, defined as the analyte concentration giving a signal equal to the blank signal plus three standard deviations of the blank. The detection limit was calculated to 0.45 fM which is, to our knowledge, the lowest detectable concentration of a drug in plasma ever reported using AMS.
Conclusions
The method presented in this article utilizes online pre-concentration and sample clean-up prior to HPLC separation and AMS analysis. The analyte is readily separated from the carbon containing background and is prepared by a sensitive carbon carrier method which can detect sub-amole amounts of a labeled drug. This methodology provides the possibility to automate the whole separation step, a decrease in total analysis time, lower contamination risk and safer handling of hazardous or contagious samples. It also offers the possibility to detect and quantify extremely low concentrations of, for example, drugs or drug metabolites in complex biological matrixes, such as blood or plasma. A limit of detection at 450 aM was achieved for a 14C-labeled drug in human plasma. The isotopic ratio measurement sensitivity can be improved even more by decreasing the amount of the carbon carrier and by reducing the residual carbon in the blanks, which would facilitate detection limits in the 100 aM region.The extremely low concentration measurements reported here should be beneficial for future drug development processes in the pharmaceutical industry. This may be specifically suitable for an unexplored AMS Microdosing research field involving large molecular drugs such as recombinant proteins and polypeptides. This field, apart from one publication,14 is still uncharted. The larger molecular drugs are inherently more hazardous as they can interact with the human immune system. Therefore, the first-in-man studies would require lower concentrations for safety reasons.
Acknowledgements
Uppsala BIO, Uppsala, Sweden, is gratefully acknowledged for the funding of the project. The staff at Tandemlaboratoriet, Uppsala University are acknowledged for their professional help with the accelerator operation. We would also like to thank Ira Palminge-Hallén from the Swedish Medical Products Agency and Lars Ståhle from AstraZeneca in Södertälje, Sweden, for providing us with the labeled substance and Prof. Jonas Bergquist from Uppsala University for stimulating discussions and creative ideas.References
- K. W. Turteltaub and J. S. Vogel, Curr. Pharm. Des., 2000, 6(10), 991–1007 CrossRef CAS.
- M. Suter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 223, 139–148 CrossRef.
- J. S. Vogel and A. Love, Methods in Enzymology, ed. A. L. Burlingame, Academic Press, New York, 2005, pp. 402–422 Search PubMed.
- G. Lappin and R. C. Garner, Nat. Rev. Drug Discovery, 2003, 2(3), 233–240 CrossRef CAS.
- Guidelines for Industry Investigators and Reviewers: Exploratory IND Studies, Food and Drug Administration, US Department of Health and Human Services, Rockville, MD, 2006 Search PubMed.
- Position Paper on Non-clinical Safety Studies to Support Clinical Trials with a Single Microdose. Position Paper CPMP/SWP/2599, EMEA, London, UK, 2004 Search PubMed.
- G. Lappin, Radiotracers in Drug development, CRC press, Taylor and Francis Group, Florida, 2006 Search PubMed.
- M. Salehpour, G. Possnert and H. Bryhni, Anal. Chem., 2008, 80(10), 3515–3521 CrossRef CAS.
- S. D. Gilman, S. J. Gee, B. D. Hammock, J. S. Vogel, K. Haack, B. A. Buchholz, S. Freeman, R. C. Wester, X. Y. Hui and H. I. Maibach, Anal. Chem., 1998, 70(16), 3463–3469 CrossRef CAS.
- B. A. Buchholz, E. Fultz, K. W. Haack, J. S. Vogel, S. D. Gilman, S. J. Gee, B. D. Hammock, X. Y. Hui, R. C. Wester and H. I. Maibach, Anal. Chem., 1999, 71(16), 3519–3525 CrossRef CAS.
- M. Salehpour, N. Forsgard and G. Possnert, Rapid Commun. Mass Spectrom., 2009, 23(5), 557–563 CrossRef CAS.
- R. C. Garner, I. Goris, A. A. E. Laenen, E. Vanhoutte, W. Meuldermans, S. Gregory, J. V. Garner, D. Leong, M. Whattam, A. Calam and C. A. W. Snel, Drug Metab. Dispos., 2002, 30(7), 823–830 CrossRef CAS.
- P. Sadilek, D. Satinsky and P. Solich, TrAC, Trends Anal. Chem., 2007, 26(5), 375–384 CrossRef CAS.
- G. Lappin, R. C. Garner, T. Meyers, J. Powell and P. Varley, J. Pharm. Biomed. Anal., 2006, 41(4), 1299–1302 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |