
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted methacrylation of chitosan for 3D printable hydrogels in tissue engineering†
Michael
Zanon
a,
Annalisa
Chiappone
 *ad,
Nadia
Garino
a,
Marta
Canta
b,
Francesca
Frascella
ab,
Minna
Hakkarainen
c,
Candido Fabrizio
Pirri
ab and
Marco
Sangermano
a
*ad,
Nadia
Garino
a,
Marta
Canta
b,
Francesca
Frascella
ab,
Minna
Hakkarainen
c,
Candido Fabrizio
Pirri
ab and
Marco
Sangermano
a
aDipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Turin, Italy. E-mail: annalisa.chiappone@polito.it
bPolitoBIOMed Lab, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Turin, Italy
cDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, 100 44 Stockholm, Sweden
dDipartimento di Scienze Chimiche e Geologiche, Università di Cagliari, Complesso Universitario di Monserrato, S.S. 554 bivio Sestu, 09042 Monserrato, Italy
First published on 16th November 2021
Abstract
Light processable natural polymers are highly attractive for 3D printing of biomedical hydrogels with defined geometries and sizes. However, functionalization with photo-curable groups, such as methacrylate or acrylate groups, is required. Here, we investigated a microwave-assisted process for methacrylation of chitosan to replace conventional methacrylation processes that can be time consuming and tedious. The microwave-assisted methacrylation reaction was optimized by varying the synthesis parameters such as the molar ratio of chitosan to the methacrylic agent, the launch and reaction times and process temperature. The optimized process was fast and efficient and allowed tuning of the degree of substitution and thereby the final hydrogel properties. The successful methacrylation and degree of substitution were verified by 1H NMR and attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR). The influence of the degree of methacrylation on photo-rheology, mechanical stiffness, swelling degree and gel content was evaluated. Furthermore, favourable 3D printability, enzymatic degradability, biocompatibility, cell migration and proliferation were demonstrated giving promise for further applications in tissue engineering.
1. Introduction
In the last few years materials science based on non-toxic natural polymers has gained growing interest due to their peculiar physical–chemical properties and their environmental sustainability. The combination with innovative additive manufacturing techniques, such as 3D printing, is especially attractive.1–6 Of particular interest is the utilization of biopolymers in the biomedical field and the creation of complex structures and scaffolds for tissue engineering applications. A key point that must be considered is that natural polymers generally need to be treated and functionalized before their use and these organic reactions are commonly driven by traditional thermal routes activated by conventional external heat sources.7 Despite their consolidated applications, traditional syntheses present many drawbacks; indeed, the heat transfer is usually relatively slow and often hard to control, leading to an inhomogeneous thermal gradient during the process, incomplete reaction and formation of side products.8–10 Moreover, local overheating may lead to product, substrate, and reagent decomposition, affecting one of the most essential properties in tissue engineering, reproducibility.11,12 To overcome these issues microwave-assisted functionalization reactions could be a novel synthesis approach.Microwave-assisted organic synthesis (MAOS) was first proposed by Gedye et al. in 1986,13 who used a standard kitchen microwave to dramatically decrease the reaction times. The reason behind this behaviour stands in “microwave dielectric heating”. Indeed, the irradiation of the sample causes an alignment of its dipoles or ions following the electric field. As the applied field oscillates, the dipoles or the ions tend to realign themselves and, during the process, lose heat through molecular friction and dielectric loss. The amount of heat generated is directly dependent on the ability of the matrix (or the solvent) to align with the field.14 Since then, the time saving have been clearly evidenced and also the existence of a so-called “specific” or “non-thermal” microwave effect is proposed, able to accelerate the reaction rate and sometimes to alter the product distributions.15,16 In addition to the faster reaction rates and energy saving procedures MAOS represents a green, cheap, and easy chemistry17 with less side reactions or generation of toxic products;18 all features essentials when functionalization reaction are exploited aiming at biomedical applications. Several studies confirmed the MAOS efficacy and convenience, even in the biological field,19,20 but less efforts were put in the synthesis and functionalization of natural polymers21,22 for biomedical applications.23 Chitosan is a natural polysaccharide produced by partial deacetylation of chitin, which is a component of crab/shrimp shells and the second most abundant polysaccharide on the earth, after cellulose. The N-deacetylation of the chitin constitutional repeating unit, N-acetyl-D-glucosamine, creates a random backbone of N-acetyl-D-glucosamine and D-glucosamine, leaving three different types of functional groups, the amino groups and the primary and secondary hydroxyl groups.1,24,25 The amino group is generally protonated at neutral pH and interacts with the other polar group, decreasing the chitosan solubility in water.26 However, chitosan has excellent properties, such as biocompatibility, biodegradability, anti-inflammatory activity, antioxidant effect, and it can diminish foreign body reaction with little or no fibrous encapsulation.27 Chitosan hydrogels are 3D networks able to absorb and retain liquid and swell, maintaining their mechanical integrity3,28,29 and have been used in several biomedical applications as drug carriers, absorbable sutures, injectable and 3D-printable biomaterials, due to the hydrophilic nature, biocompatibility and flexibility.30 Both physical and chemical crosslinking methods are employed to develop chitosan hydrogels. Photo-crosslinking is one of the most promising ways to obtain chemically crosslinked hydrogels in the presence of a photo-initiator, thanks to the fast processing, mild reaction conditions and the ability to tailor the hydrogel geometry. Moreover, light-based 3D printing techniques open new avenues for the preparation of biomedical scaffolds with defined geometries and sizes. The light-responsive precursor is generally functionalized with acrylates or methacrylates using different chemical species, such as glycidyl methacrylate or 2-aminoethyl methacrylate.31,32 Methacrylic anhydride is the best candidate for biomedical applications4,6,33,34 cause of its mild solubility in water.35 Nevertheless, methacrylic anhydride exhibits a certain degree of cytotoxicity.23 For this reason, low usages of the functionalizing agent are always suggested. The application of microwave-assisted processes could offer a convenient, mild and effective route to such functionalization and replace the conventional heating processes that can be time consuming and tedious. A systematic study was therefore performed concerning the influence of different parameters (i.e., reaction temperature, microwave launch time, reaction time and reagents concentration) on degree of substitution during microwave-assisted methacrylation of chitosan. This led to the development of an optimized protocol that allows degree of substitution comparable to the traditional heating bath synthesis with lower amount of methacrylation agent, shorter reaction time and high reproducibility. Furthermore, it was possible to tune the methacrylation degree to design hydrogels with different mechanical properties, thus conceivable for different applications. The hydrogels were characterized for their photo-rheology, mechanical stiffness, swelling degree and gel content. Furthermore, the 3D printability, enzymatic degradability, biocompatibility, cell migration and proliferation were also demonstrated.
2. Materials and methods
2.1. Materials
Chitosan medium molecular weight (CH, Mw = 190–310 KDa, 75–85% degree of N-deacetylation), methacrylic anhydride (MA, 94%), acetic acid (≥99%), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, ≥95%), dialysis sacks (Avg. flat width 35 mm, MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), phosphate-buffered solution (PBS, pH 7.4) and lysozyme from chicken egg white (LYS, protein ≥90%, ≥40000 units per mg protein) were all purchased from Sigma-Aldrich (Milano, Italy) and used as received without further purification.
000 Da), phosphate-buffered solution (PBS, pH 7.4) and lysozyme from chicken egg white (LYS, protein ≥90%, ≥40000 units per mg protein) were all purchased from Sigma-Aldrich (Milano, Italy) and used as received without further purification.
2.2 Synthesis of methacrylated chitosan (CHI-MA)
1.5 wt% of medium molecular weight chitosan was dissolved in a 2 wt% acetic acid–distilled water solution and stirred overnight. Once the solution was homogeneous, the methacrylic anhydride was added. The ratios between the aminoglucose moieties of chitosan and MA were varied from 1:1 to 1:6. For all the samples, the mixtures were transferred in a 100 mL clamped Teflon reactor, equipped with pressure and temperature probes, and placed into a microwave furnace (Milestone STARTSynth, Milestone Inc., Shelton, Connecticut). Besides the ratios of MA and the aminoglucose moieties, different reaction parameters such as temperature, reaction time, and launch time (i.e., the time needed for the system to reach the reaction temperature from room temperature) were investigated. The furnace was irradiated at maximum power of 800 W during the reaction and then cooled back down to ambient temperature. The different synthesis protocols tested are reported in Table 1.
| Synthesis no. | Aminoglucose ring:methacrylic anhydride ratio |
Max power (W) | Synthesis temperature (°C) | Launch time (s) | Synthesis time (min) |
|---|---|---|---|---|---|
| 1 | 1:1 |
800 | 100 | 30 | 5 |
| 2 | 1:2 |
800 | 100 | 30 | 5 |
| 3 | 1:4 |
800 | 100 | 30 | 5 |
| 4 | 1:6 |
800 | 100 | 30 | 5 |
| 5 | 1:1 |
800 | 60 | 30 | 5 |
| 6 | 1:1 |
800 | 80 | 30 | 5 |
| 7 | 1:1 |
800 | 100 | 60 | 5 |
| 8 | 1:1 |
800 | 100 | 120 | 5 |
| 9 | 1:1 |
800 | 100 | 30 | 10 |
| 10 | 1:1 |
800 | 100 | 30 | 15 |
| 11 | 1:1 |
800 | 80 | 60 | 10 |
A selection of the proposed protocols was repeated in triplicate and analyzed aiming to check the repeatability of the process showing good reproducibility with a variability of the results of ±1%. After seven days of dialysis against distilled water, the obtained product was freeze-dried and stored at 4 °C for further use.
2.3 Hydrogel preparation via photo-crosslinking
Freeze-dried CHI-MA (2 and 5 wt%) was dissolved in a 2 wt% of acetic acid–distilled water solution. At the same time, 2 phr of LAP was added and the mixture was stirred until a homogeneous solution was reached. The formulation was poured into PDMS mold and irradiated for 5 min with a visible light Hamamatsu LC8 lamp with a cut-off filter below 400 nm equipped with a light guide (50 mW cm−2).2.4 Characterizations
Attenuated total reflectance-infrared spectroscopy (ATR-FTIR) was used to evaluate the chemical composition of the products. The experiments were conducted on freeze-dried samples utilizing a Thermo Scientific Nicolet iS50 FTIR Spectrometer (Milano, Italy) equipped with a diamond crystal ATR accessory. Thirty-two ATR spectra were collected with a resolution of 4 cm−1 in the range of 4000–600 cm−1 for each sample. The spectrum of neat chitosan was taken as a reference. All 1H NMR spectra were recorded by a Brucker Avance DPX-400 nuclear magnetic resonance spectrometer at 25 °C in D2O with small amount of trifluoroacetic acid. Real-time photorheological measurements were performed using an Anton PAAR Modular Compact Rheometer (Physica MCR 302, Graz, Austria) in parallel-plate mode (25 mm diameter) and the visible-light source was provided by positioning the light guide of the Hamamatsu LC8 lamp under the bottom plate. During the measurements, the gap between the two glass plates was set to 0.2 mm, and the sample was kept under a constant shear frequency of 1 Hz. The irradiating light was switched on after 60 s to allow the system to stabilize before the onset of polymerization. According to preliminary amplitude sweep measurements, all the tests were carried out in the linear viscoelastic region at a strain amplitude of 1% (the test was carried out between 0.1 and 100% of strain, frequency of 1 Hz). The photo-rheology was studied as a function of the changes in the shear modulus (G′) and in the loss modulus (G′′) of the sample versus the exposure time. The crosslinking density was determinated using the formula:28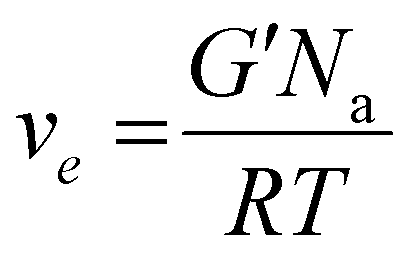 | (1) |
The measurements were repeated three times for each precursor formulation to verify reproducibility. Viscosity was evaluated with continuous flow measurements performed in triplicate with a range of shear rate from 0.01 to 1000 1 s−1, setting a gap between the two plates of 0.5 mm. The compression tests were performed with MTS QTestTM/10 Elite controller using TestWorks® 4 software (MTS Systems Corporation, Edan Prairie, Minnesota, USA). The unconfined uniaxial compression was performed with cell loaded of 10 N and a test speed of 1 mm min−1. The samples tested had cylindrical geometry: D = 8 mm × H = 7 mm. The data acquisition rate was set as 20 Hz. The compressive modulus was calculated by TestWorks® 4 software. The modulus (E) was estimated as the slope of the linear region of the stress–strain curves while the ultimate compression strength (UCS) and the strain at break (εr) were extrapolated from the plots. All the experiments were performed in triplicate. The swelling kinetics was determined by soaking the samples in distilled water. The samples were taken out at different time intervals and weighted once the surface droplets were wiped off with wet paper until constant weight. The swelling ratio (SW%) was calculated as:
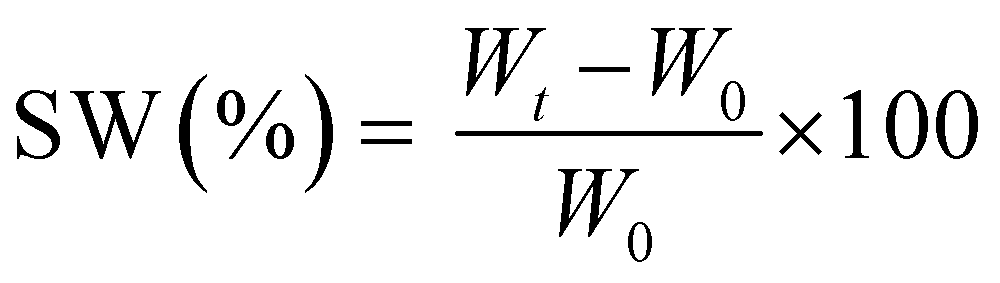 | (2) |
To determine the gel content (GC), previously dried samples were held in a metal net, weighed, and then immersed in water (25 °C) for 24 h to dissolve the uncrosslinked polymer. The samples were then dried for 24 hours (40 °C) in a vacuum oven and weighed again. The gel content was determined as:
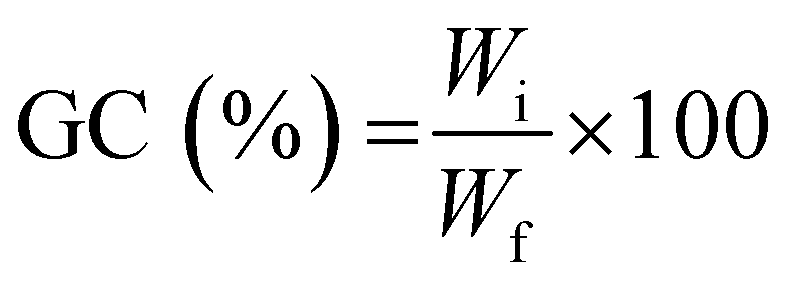 | (3) |
2.5 3D printing
Direct 3D printing of CHI-MA (5 wt%) was conducted on a BioX (CELLINK, Gothenburg, Sweden) using a 0.8 mm nozzle. While the ink temperature was set to 37 °C, the printer bed temperature was 4 °C. The printer pressure and speed were set at 120 kPa and 20 mm s−1, respectively. For each layer, the process was stopped, and the structure irradiated for 2 min at 50 mW cm−2. Ten more minutes of UV-light exposure were carried out at the end of the printing.2.6 Enzymatic biodegradation, cells migration and proliferation assays
Prior to the degradation test, the freeze-dried photo-cured samples were firstly weighted and washed overnight in distilled water to neutralize the pH of each specimen in the enzyme activity range (6.0–9.0). Then the samples were sterilized 2 h in ethanol to remove any trace of bacteria. Two solutions were prepared, one as a control containing PBS (pH 7.4) and one containing PBS (pH 7.4) plus 1.5 mg ml−1 of LYS. The LYS dose was determined based on the human physiological concentration in serum.36 The samples, in a crux form to appreciate the change in geometry caused by degradation, were transferred into an incubator with 5% CO2 at 37 °C for 1, 7, 14 and 21 days (replacing both media after 14 days to prevent the enzyme denaturation). After the extraction, the samples were washed in water to eliminate the enzyme leftovers, freeze-dried, and weighted again. The weight loss percentage (D%) was followed using the equation: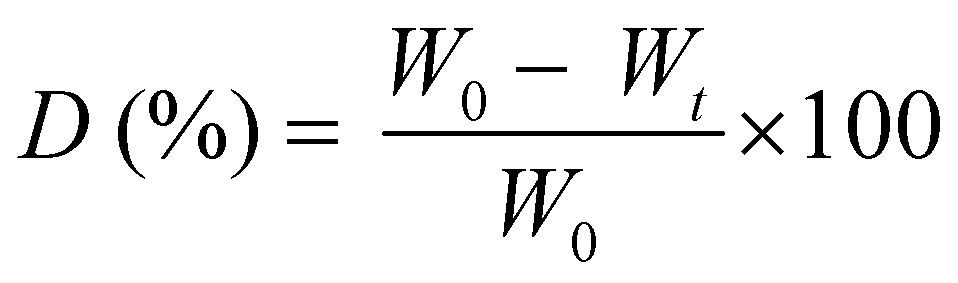 | (4) |
:30 solution of EtOH:DI water followed by 2 h in PBS with 1% penicillin/streptomycin and 1 h (30 min for each side) exposure to UV light. For the migration assay, the scaffolds were positioned inside the 48 well cell culture plate (Greiner bio-one). 4.5 × 105 cells were resuspended in 40 μl of culture medium and seeded on each scaffold basin. Cells were then left for 3 h to get attached to the scaffold, after which the cell culture medium was added to completely cover the samples. After 96 h and 7 days the scaffolds were washed with PBS, fixed with formalin free fixative solution (Sigma-Aldrich), permeabilized with 0.25% Triton x-100 (Sigma-Aldrich) for 30 min at RT and stained with 0.4 mM DAPI (Sigma-Aldrich) for 3 h at RT. Fluorescent images of the cells migrated inside the scaffold were collected using the microscope Eclipse Ti2 Nikon (Tokyo, Japan) equipped with a Crest X-Light spinning disk.
Chitosan-conditioned medium was produced for cell proliferation tests. The scaffolds were soaked in the cell medium (1.5 ml per scaffold) for 48 h in the dark at 37 °C. Then the supernatant was filtered with a 0.22 μM filter and used for cell seeding. 3 × 105 cells per well were seeded on 48 well cell culture plate (Greiner bio one) in 500 μl per well of normal and conditioned medium. After 24 h and 96 h of incubation cell proliferation was determined with Resazurin (Sigma-Aldrich). In particular, 50 μl of Resazurin were added to each well, which were left 3 h in the dark at 37 °C, 5% CO2. The fluorescence signal of the resazurin's reduced form resorufin (exc/em: 530/590) proportional to the number of living cells was detected by the SynergyTM HTX Multi-Mode Microplate Reader (BioTek, Winooski, VM, USA). The signal of the normal and conditioned medium without cells was used as background. The cell proliferation experiments were performed three times. Differences between groups were analyzed by two-way ANOVA using GraphPad PRISM 7.04 (GraphPad Software Inc.).
3. Results
3.1. Synthesis of methacrylated chitosan
Aiming to obtain chitosan-based hydrogels chemically crosslinkable by photopolymerization, the methacrylation reaction of chitosan was performed by utilizing microwave irradiation instead of a traditional heating. The chemical structure of chitosan, showing the different types of functional groups available on the molecule (i.e. the amino groups and the primary and secondary hydroxyl groups) is shown in Fig. 1 followed by methacrylation reaction scheme to produce CHI-MA. Based on previous research with traditional methacrylation, it is expected that the methacrylation can occur either on the hydroxyl or amine groups of chitosan.37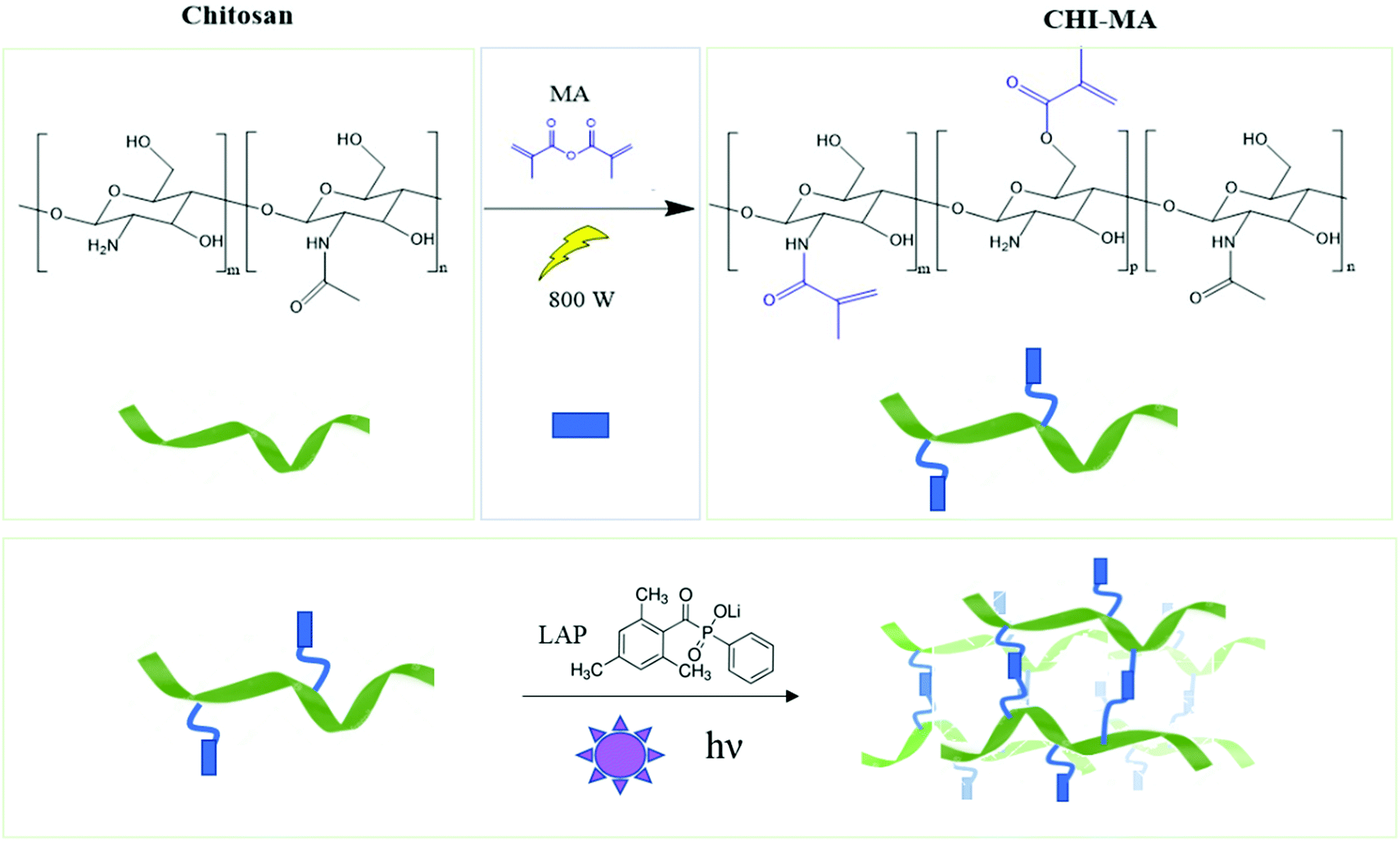 | ||
| Fig. 1 Scheme of microwave-assisted methacrylation of chitosan (upper part) and representation of the light curing of the CHI-MA hydrogel. | ||
Aiming to optimize the innovative MW functionalization process, different synthesis parameters were studied: the ratio of MA and aminoglucose moieties, launch time, reaction time and temperature (Table 1).
At first, different molar ratios between the aminoglucose ring of chitosan and MA were investigated (i.e., 1:1, 1:2, 1:4, 1:6) while the microwave process parameters were set and maintained constant according to similar synthesis reported in literature38,39 (i.e. temperature = 100 °C, launch time = 30 s, reaction time = 5 min, irradiation power = 800 W).
Fig. 2 reports the FTIR spectra collected for raw chitosan and CHI-MA synthesized with different ratios of MA (Fig. 2A, other FT-IR spectra are plotted in Fig. S1, ESI†). A representative 1H-NMR spectrum of CHI-MA is also presented (Fig. 2B, Spectre referred to other synthesis are reported in Fig. S2, ESI†). The successful incorporation of methacrylate groups was confirmed by the presence of FTIR signal at 1620 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)4 and by the shifting of the characteristic peak of amine I stretching at lower wavenumbers (1639 cm−1)37 for all the samples. The coupling of MA with the hydroxyl group of chitosan was demonstrated by the formation of the ester band at 1710 cm−1.40 Interestingly, below a certain amount of MA (ratio 1:4), no ester band was evidenced, suggesting the primary amine as a favourite spot for the reaction.41,42 Moreover, the peak indicating CO–NH groups appeared at 3091 cm−1.431H-NMR spectra further confirmed the incorporation of the methacrylic groups. Three new peaks were noticed in the region 5.4–6 ppm. These peaks can be attributed to the methylene protons of acrylates. In particular, the peaks at 5.4 and 5.6 (yellow and green of Fig. 2B) are assigned to the protons in CCH2 after methacrylation of amine-groups and those at 5.6 and 6 (green and blue on Fig. 2B) indicated same protons after methacrylation of hydroxyl groups.34,37,44 Integrating and normalizing these peaks and those of the potential reactive sites permits the calculation of the degree of substitution (DS) for methacrylation of the NH2 and OH groups; the data collected for the samples prepared with different chitosan:MA ratios are illustrated in Fig. 2C.
C)4 and by the shifting of the characteristic peak of amine I stretching at lower wavenumbers (1639 cm−1)37 for all the samples. The coupling of MA with the hydroxyl group of chitosan was demonstrated by the formation of the ester band at 1710 cm−1.40 Interestingly, below a certain amount of MA (ratio 1:4), no ester band was evidenced, suggesting the primary amine as a favourite spot for the reaction.41,42 Moreover, the peak indicating CO–NH groups appeared at 3091 cm−1.431H-NMR spectra further confirmed the incorporation of the methacrylic groups. Three new peaks were noticed in the region 5.4–6 ppm. These peaks can be attributed to the methylene protons of acrylates. In particular, the peaks at 5.4 and 5.6 (yellow and green of Fig. 2B) are assigned to the protons in CCH2 after methacrylation of amine-groups and those at 5.6 and 6 (green and blue on Fig. 2B) indicated same protons after methacrylation of hydroxyl groups.34,37,44 Integrating and normalizing these peaks and those of the potential reactive sites permits the calculation of the degree of substitution (DS) for methacrylation of the NH2 and OH groups; the data collected for the samples prepared with different chitosan:MA ratios are illustrated in Fig. 2C.
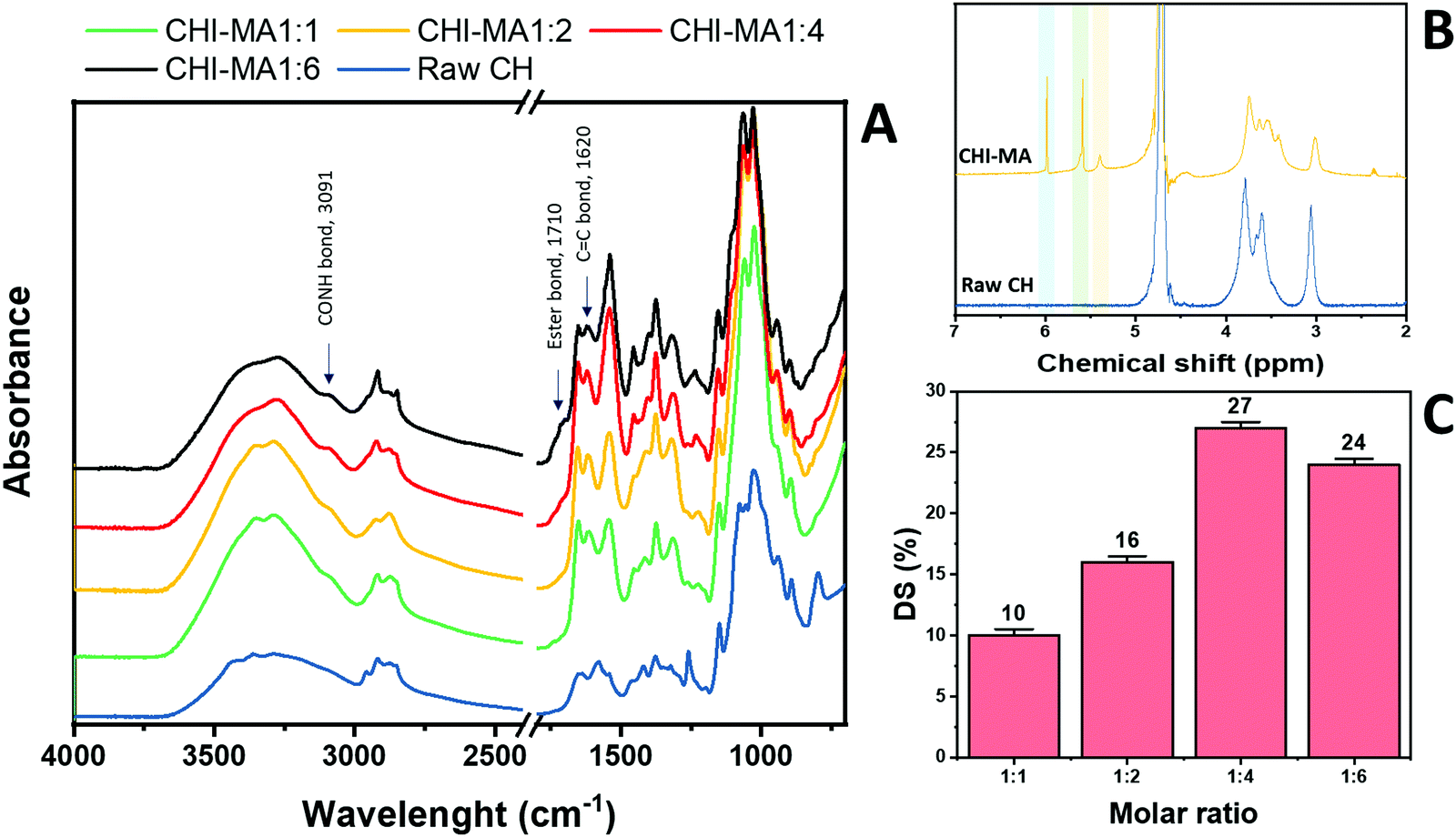 | ||
| Fig. 2 FTIR spectra of raw chitosan and CHI-MA at different ratios of MA (A) and 1H-NMR of CHI-MA (B). Degree of metachrylation of the samples prepared at different ratios of MA (C). | ||
Analysing the data, it can be observed that a higher quantity of methacrylic anhydride resulted in an almost linear increase of %DS on the aminoglucose ring of chitosan, which is explained by the excess of MA during the reaction. The highest DS level is reached at a ratio of 1:4 followed by a sort of plateau. The obtained value is similar to values reported in literature for traditional syntheses,37 while the DS plateau can be attributed to steric hindrance limitations. Once the effect of the molar ratio was evaluated, the investigation moved on the optimization of the microwave parameters. For this a low molar ratio, i.e., 1:1, was chosen aiming to work far from the steric hindrance limit and to isolate the effect of microwave parameters more clearly on the methacrylation of chitosan. The parameters investigated were the reaction temperature (synthesis no. 1, 5, 6 in Table 1), launch time (Synthesis no. 1, 7, 8) and reaction time (Synthesis no. 1, 9,10). The achieved %DS are collected in Fig. 3. Interestingly, non-linear trend was noticed by changing the parameters launch time or temperature but rather two maxima on %DS in correspondence to 80 °C and 60 s, respectively, as visible in Fig. 3. The variation on %DS at different reaction times was also investigated. In this case, longer reaction time resulted in higher substitution levels with a maximum at 10 min, after which further prolonging the reaction time (15 min) leads to gelation. According to the observe trends, low values of %DS at mild conditions (high launch time and low temperature) can be explained by lower efficiency of the reaction,45 while at severe conditions (low launch times and high temperature) might be attributed to a small MA vaporization during the first instants of the synthesis and again to a not complete efficiency at high temperatures.39,45 Considering the calculated values of the degree of substitution it can be observed that, even with a low amount of MA (aminoglucose ring:MA ratio 1:1), modification of microwave parameter can increase the %DS to similar value as obtained with high MA concentrations (1:4 or 1:6). The sample produced just setting a launch time of 60 s, indeed, shows a %DS maximum value of 25%.34,37 Aiming to intersect the best promising parameters and to determine the best protocol one last sample was prepared. The MW parameters that gave the highest %DS among the performed synthesis where chosen (molar ratio 1:1, launch time = 60 s, reaction time = 10 min, temperature = 80 °C, irradiation power = 800 W, Synthesis no. 11 in Table 1). The obtained sample reached a %DS of 24%. This indicates that at low MA concentration, an efficient synthesis can be performed at 80 °C. According to the collected results it can be hypothesized that the extension of the launch time has a higher influence on the efficiency of the reaction than the extension of the reaction time. In fact, also in traditional synthesis the methacrylic agent is often added dropwise to prevent side products and inefficient reactions,3,46,47 so a proper launch time might increase the efficiency.
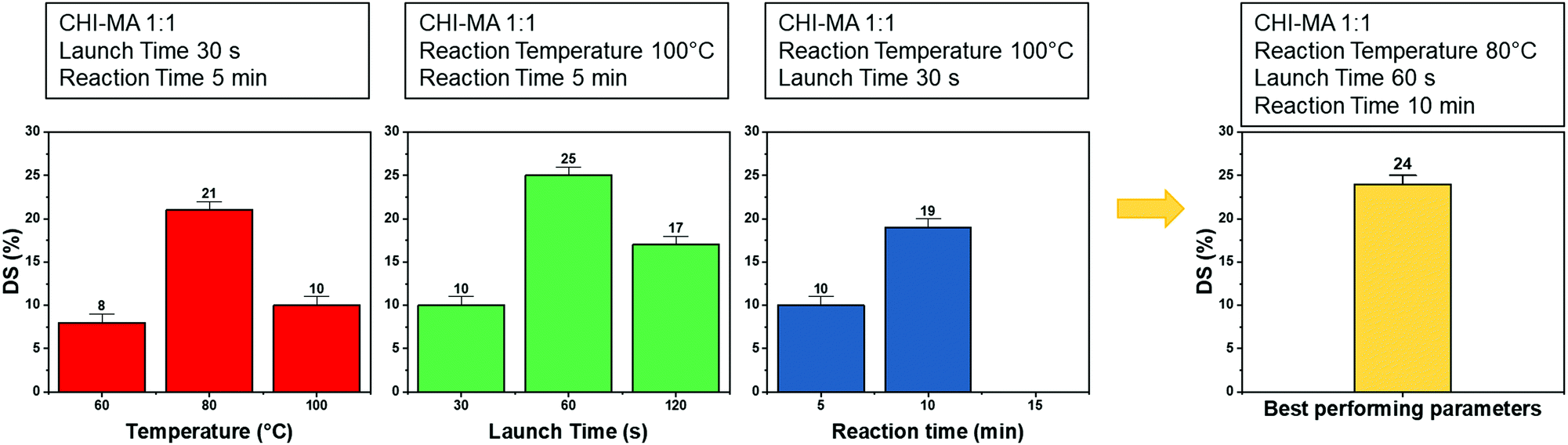 | ||
| Fig. 3 %DS of the different synthesis batch varying the microwave parameters (i.e., reaction temperature, launch time and reaction time). | ||
In conclusion, while traditional heating synthesis is performed carefully adding the methacrylic agent dropwise, at temperature ranges between 50 and 60 °C, from 4 to 6 hours and with high excess of the methacrylic agent (molar ratio CH:MA from 1:4 to 1:6),4,34,37 MAOS drastically reduce the reaction time, while maintaining high efficiency, reproducibility and permitting the reduction of toxic chemicals usage, leading to a greener process. The sample presenting the highest %DS and two other batches of synthesis with intermediate degree of methacrylation (named CHI-MA DS 24, CHI-MA DS 17 and CHI-MA DS 10, where the number indicates the %DS) were selected for preparation of photocured hydrogels with tailored stiffnesses.
3.2. Photopolymerization, mechanical properties and swelling test
Water solutions of CHI-MA DS 24 at different concentrations (2 wt% and 5 wt%) were firstly prepared and evaluated by photo-rheology and viscosity test aiming to identify the best concentration to be used. The obtained data are reported in Fig. S3 (ESI†). According to the photo-rheology test, turning on the lamp after 60 s, the formulation containing the higher content of CHI-MA (5 wt%) shows an instantaneous increase of the storage modulus meaning the fast beginning of the photopolymerization and thus a better reactivity compared with the formulation containing CHI-MA at 2% for which the reaction starts with a delay of 44 s. Envisaging to move toward direct bioprinting, high viscosity η of the formulation with a marked shear-thinning behaviour and high storage modulus G′ of the crosslinked hydrogel are needed, both to grant to the scaffold to withstand before and after the bioprinting and to let the formulation flow through the nozzle (Fig. S3A and B, ESI†). Those are the reasons why a concentration of 5 wt% of CHI-MA was selected for further tests. Since the stiffness and composition of the natural ECM influence cell morphology, cytoskeletal structure, signalling, and function for many normal and cancer cell types,48–50 three different batches with increasing %DS (CHI-MA DS 10, 17, and 24) were selected for an in-deep rheological characterization to explore the scaffold stiffness. Fig. 4A reports photo-rheology data are collected for the three batches (concentration 5%). All the batches showed a high initial reactivity during photo-rheology with the instantaneous increase of G’ when the lamp was turned on? while the storage modulus plateau and the kinetics of crosslinking followed the degree of substitution, with values in according with literature.37 When also observing the variation of G′′ modulus during irradiation (G′/G′′ plots reported in Fig. S4, ESI†) samples CHI-MA 10 and 17 showed gel points (the intersection between the two moduli) after 15 and 7 seconds of irradiation respectively, indicating the fastening of the onset of the reaction with the increase of the methacrylation degree. Interestingly, sample CHI-MA DS 24 was characterized by G′ slightly higher than G′′ even before irradiation, indicating that the uncured hydrogel already presents a more elastic behaviour, nevertheless as the light is turned on, G′ presents a much evident increase than G′′ as expected with the formation of the chemically crosslinked network. The mechanical properties of the photocured hydrogels, were then tested by amplitude sweep (Fig. 4B) and confirmed both the enhanced stiffness and relative brittleness (the yielding point at lower strain) at higher %DS, likely due to a higher degree of crosslinking.51 In particular, before the yielding point, CHI-MA DS 24 presented a storage modulus of ≈ 3 kPa, while CHI-MA DS 17 and 10, respectively showed a G′ of 1 and 0.75 kPa. The evaluation of G’’ modulus during this test (G′/G′′ plots reported in Fig. S4, ESI†) confirms the predominant elastic nature of the networks also demonstrated by the tanδ (G′′/G′) values, decreasing with the increase of the DS%. The obtained crosslinking density (ve) values supports the calculated %DS for the CHI-MA and explains the differences in stiffness. In fact, the measured ve for CHI-MA 10 was 1.75 × 1023 m−3, while the same property was 2.42 × 1023 m−3 and 7.42 × 1023 m−3 for CHI-MA 17 and 24, respectively.
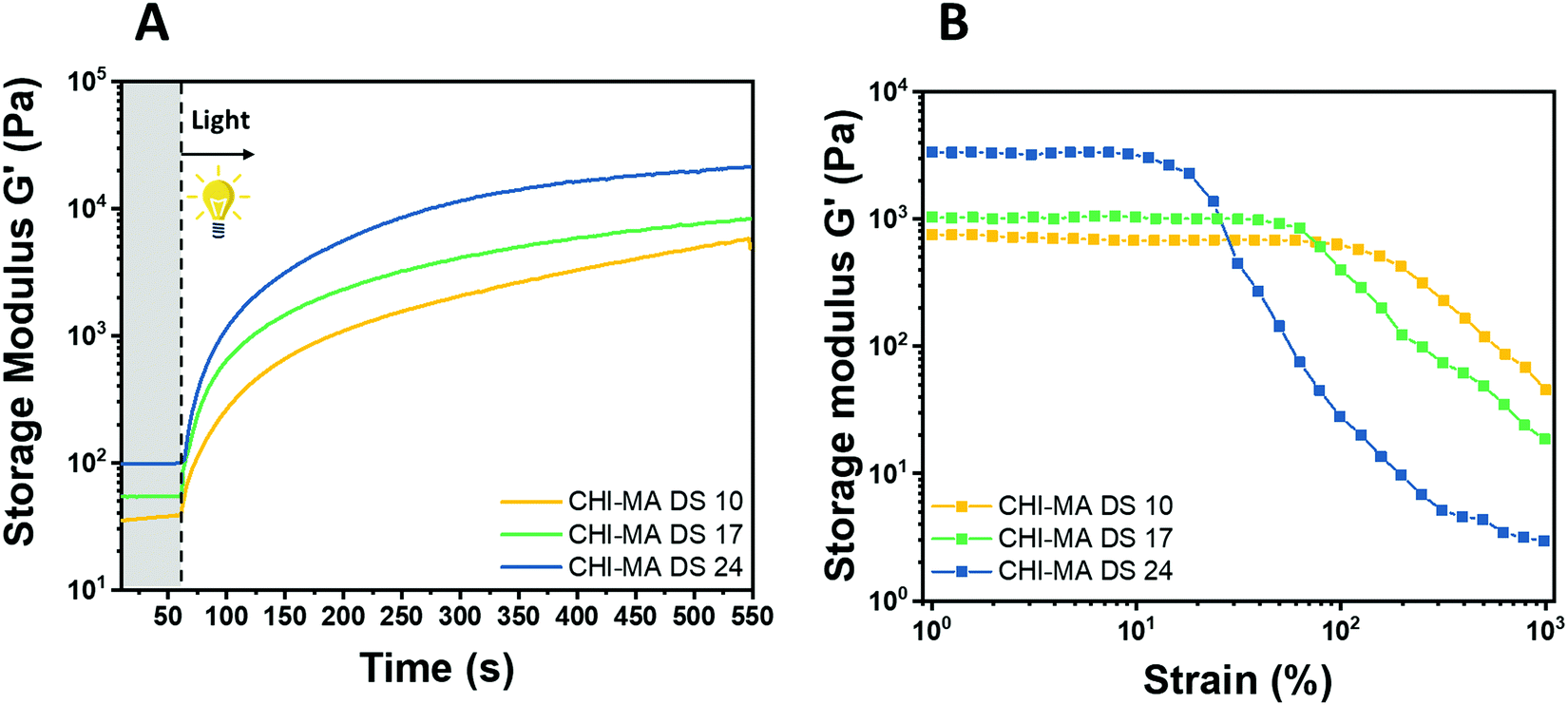 | ||
| Fig. 4 Photo-rheology (A) and amplitude sweep (B) of CHI-MA at different %DS. | ||
The selected formulations at different degree of substitution were also photocured in molds to obtain cylindrical hydrogels. The samples were then subjected to unconfined uniaxial compression to test their mechanical properties. The obtained (Fig. 5A) storage modulus, ultimate compressive strength and compression at break show an increment of the mechanical response with the %DS as summarized in Table 2, confirming the enhanced crosslinking density. With a storage modulus ranging between 20 (DS = 10%) and 39 kPa (DS = 24%), the reported values are comparable with previous investigations52,53 and with suitable stiffnesses for soft tissue applications such as lung, hearth or muscles scaffolds.54–56
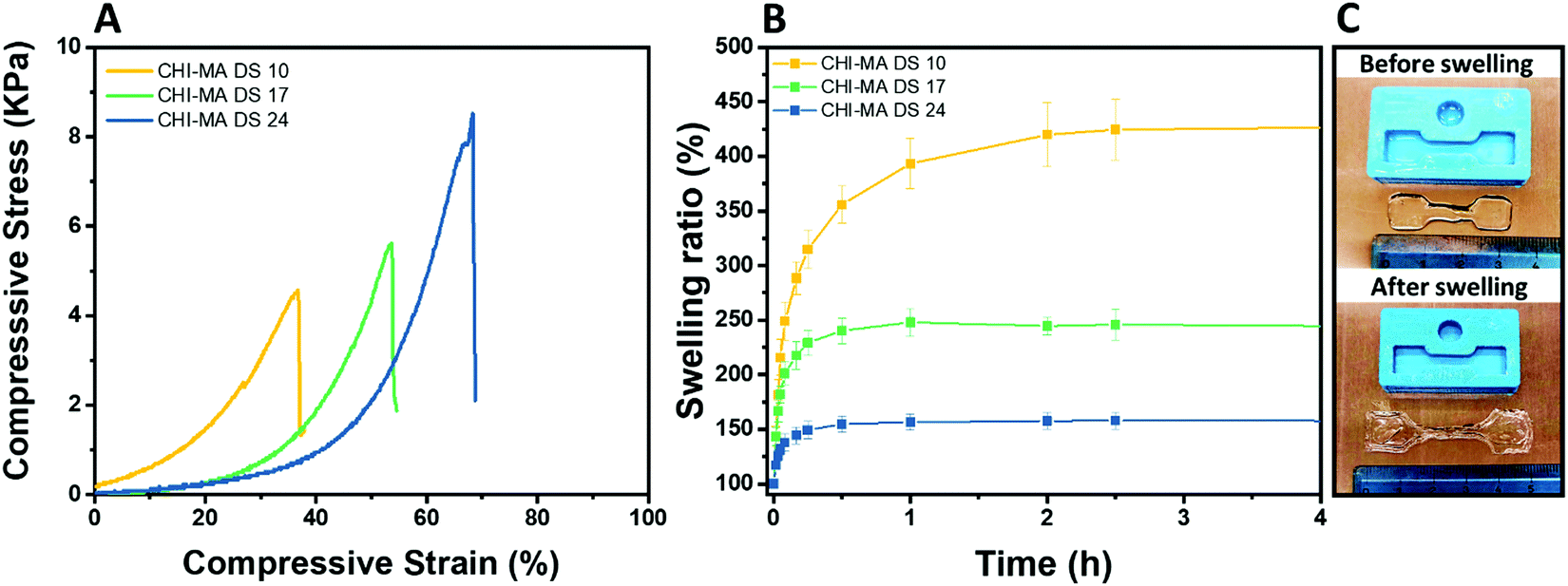 | ||
| Fig. 5 Compression test of the hydrogels prepared from CHI-MA at different DS% (A), swelling kinetics in distilled water at pH 7.3 (B) and images of CHI-MA samples before and after swelling (C). | ||
| Sample | E (kPa) | UCS (kPa) | ε r (%) | Sw eq (%) | GC (%) |
|---|---|---|---|---|---|
| CHI-MA DS 10 | 20 ± 4 | 5.3 ± 0.7 | 46 ± 9 | 441 ± 87 | 69 ± 3 |
| CHI-MA DS 17 | 24 ± 17 | 5.5 ± 3.8 | 51 ± 7 | 245 ± 45 | 79 ± 4 |
| CHI-MA DS 24 | 39 ± 6 | 7.2 ± 0.5 | 59 ± 5 | 157 ± 21 | 87 ± 2 |
The same typology of specimens (in triplicate) was immersed in distilled water and weighted at different time points to evaluate the swelling equilibrium and swelling kinetics (Fig. 5B, extracted values on Table 2); both these properties reflect the %DS of the samples and their degree of crosslinking. Indeed, according with literature,34,57 the swelling equilibrium was 157% after 30 min, 245% after 1 h, and 441% after 2 h for DS 24, 17, and 10, respectively. The observed behaviour can be explained considering that a gel swells when an external pressure (i.e. swelling pressure) is applied by the solvent. At equilibrium, the swelling pressure becomes equal to zero due to a balance between two opposing tendencies: the ordinary gain in entropy resulting from the mixing of the polymer and the solvent, and the decrease in entropy due to distortion (i.e. expansion) of the network.58 It is clear then that the swelling properties strongly depend on the affinity of the polymer with the selected solvent.59–61 In this case the major content of the non-reacted hydrophilic groups at lower %DS corresponds to an increased capacity of the hydrogel to absorb and retain water. Moreover, higher crosslinking densities (at higher %DS) prevent the stretching of the network and its ability to adsorb water. The swelling kinetics of the CHI-MA hydrogel was further evaluated using Fickian diffusion and Shott's second order dynamic model62–65 (Fig. S5 and Table S1, ESI†). The extracted values revealed a swelling mechanism where the influence of the chains’ relaxation is predominant over the solvent diffusion. SEM images collected on swollen samples (see Fig. S6, ESI†) allowed to observe the formation of relatively irregular architectures with larger and connected pores when chitosan with lower degrees of substitution was prepared, while sample CHI_MA DS 24 presented a more close-packed and regular structure, as expected from the swelling test results. In the same way, the gel content (the amount of polymer effectively crosslinked) confirmed the previous tests with 87%, 79%, and 69% gel content for the same samples (Table 2). Considering that tumor tissues are up to ten times stiffer compared to the healthy tissue,66–68 the stiffer hydrogel (CHI-MA DS 24) was selected for the biological investigation.
3.3. 3D printing
The water-based solution of CHI-MA DS 24 (5 wt%) presenting a shear thinning behaviour (see Fig. S3, ESI†), was successfully extruded into continuous and homogeneous stands through a 0.8 mm nozzle. Different geometries were fabricated layer-by-layer. The formulation (at 37 °C) was printed on a platform set at 4 °C to partly freeze the filaments and maintain the filament shape. The pressure and extrusion speed parameters were empirically set to obtain the homogeneous deposition of the hydrogel avoiding the clogging of the syringe. Different photocuring possibilities tests were then evaluated (see Fig. S7, ESI†): the continuous deposition of multiple layers and subsequent irradiation with an external lamp (50 mW cm−2) was considered but, in this case, the bottom layers were deformed during the deposition of the upper ones resulting in a loss of resolution; the instantaneous irradiation of the deposited material exploiting the BIO-X embedded LED (intensity <5 mW cm−2) was then tested, resulting in an insufficient crosslinking of the material due to the low light source intensity. In the end, an external fibre lamp (50 mW cm−2) was connected to the printer, and the printing was developed by subsequently deposing one layer and irradiating it for 2 min (the printing was stopped for this time period after each layer); these steps were reiterated until the completion of the object to permit the correct crosslinking of the structure that allows a good final resolution (Fig. 6). No dissolution was evidenced by immersing the printed grid into an acidic solution (pH ≈ 4) for one day, suggesting a good adhesion between layers and the stability of the hydrogel.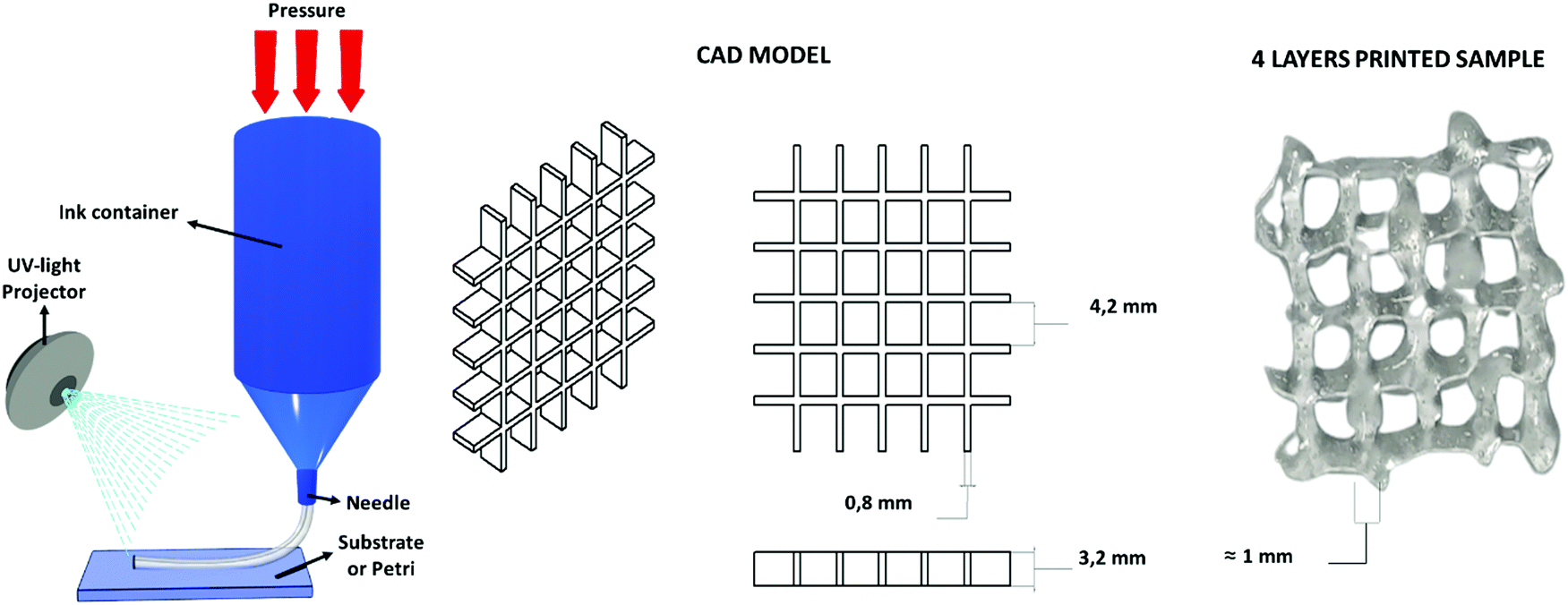 | ||
| Fig. 6 Extrusion-based 3D printing scheme, the used CAD model and the four-layer grid immediately after printing. | ||
3.4 Enzymatic biodegradation
Polysaccharide's degradation into the mammalian body is generally driven by two different routes: hydrolytic and enzymatic degradation.69 In both cases there is a cleavage of the glycosidic bond between the polysaccharide units. The enzymatic degradation rate is, however, faster and the saccharide by-products are incorporated inside of the extracellular matrix (ECM) proteoglycans or metabolized by the body.70 Two solutions were therefore prepared, one PBS solution as a control (to evidence if chemical hydrolysis takes place) and one of PBS solution with lysozyme (to appreciate the enzymatic degradation rate). The crux formed freeze-dried hydrogels were immersed in these solutions. The solution was replaced after 14 days and the pH was controlled at every time point to ensure the enzyme activity (pH ≈ 7 at each measurement). In Fig. 7 the gravimetric curves during degradation are reported as well as images of the samples at the different time points after aging in the two solutions. As expected and in agreement with the literature,69–73 the weight loss was significantly higher when the hydrogels were exposed to lysozyme, leading to a net final weight of 43% after 21 days while the hydrolytic degradation resulted in a net final weight of 92%. Interestingly, the enzymatic mass loss was homogeneously distributed among all the structures, enabling the maintenance of the geometry until 7 days of enzymatic hydrolysis, while the size of the scaffold shrank. On prolonged aging the scaffold started to disintegrate. During purely chemical hydrolysis the geometry of the scaffold was maintained until the end of experiment at 21 days. This result was further demonstrated by a morphological characterization on the degraded samples. Indeed, scaffold morphology is critical to control the cells colonization rates and their organization within the structure so then porosity needs to be monitored during degradation.74–76 In Fig. S7 (ESI†) are shown the FESEM images of the two series of samples, mediated by enzymatic (LYS) and hydrolytic (PBS) degradation at the different time points. In according with the gravimetric results and literature,69 while no major differences in the pores morphology were evidenced in PBS solution, the increment of the pores size was clear from the first day in the presence of lysozyme. In the first time points (from 1 to 7 days) the existing larger pores tended to increase in size while at 14 days numerous smaller pores started to emerge, leading to a complete destruction of the inner structure at 21 days. This could be explained by a retarded penetration of lysozyme inside of the smaller pores.69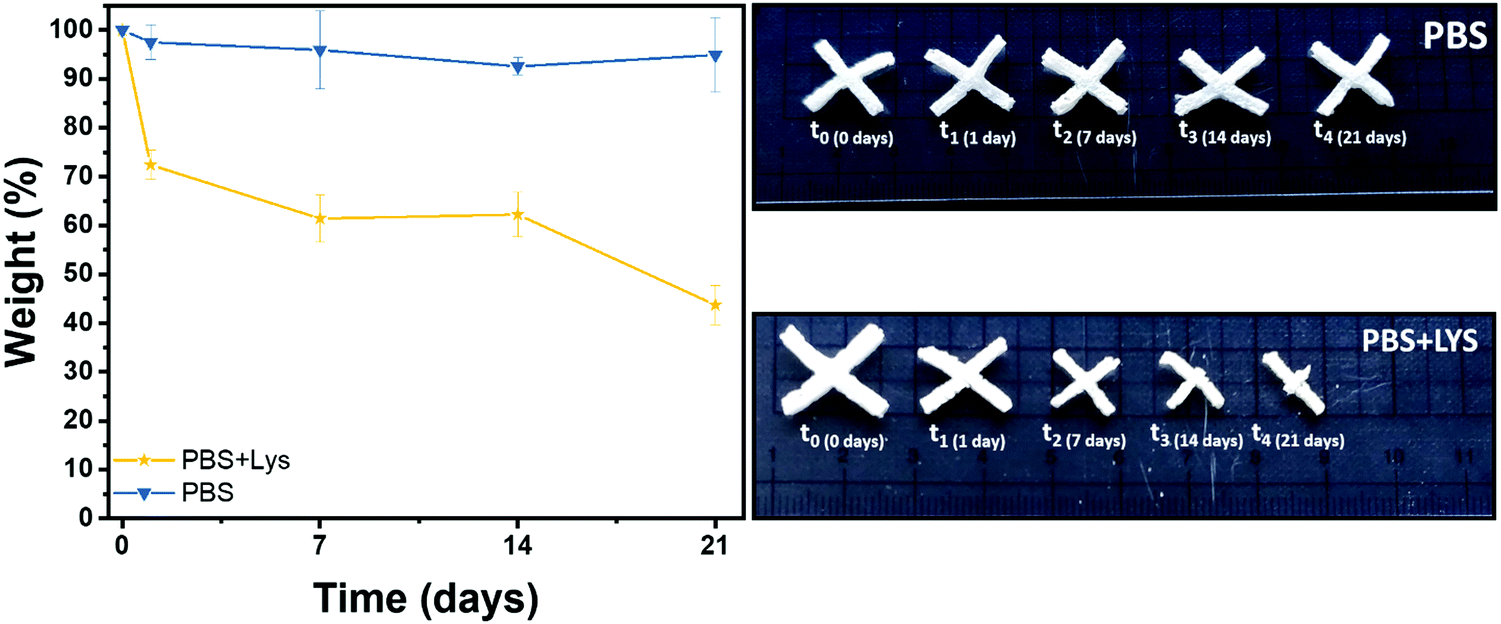 | ||
| Fig. 7 Remaining weight in % after hydrolytic (PBS) and enzymatic (PBS + LYS) degradation and images of the aged samples. | ||
3.5 Cytotoxic effect of chitosan and migration of tumor cell lines
As previously described, chitosan is a biocompatible, low toxic and bioactive polymer, widely investigated in recent years for biological applications.77 In particular, the hydrogel nature of this biopolymer allows the production of printable scaffolds with different shapes and microstructures, adaptable for specific biological needs.78 However, the different modification and manufacturing conditions confer to this material different physicochemical (degree of deacetylation, degree of substitution by e.g. methacrylation, porosity, molecular weight) and biological properties (e.g. adhesive surface, biocompatibility) that must be deeply analyzed for applications in the biological field. Here, in order to assess the biocompatibility of the microwave-assisted methacrylated chitosan and to explore its eventual bioactivity, the effect of chitosan conditioned medium on cell proliferation was explored. Two kinds of tumor cell lines, the H1299 and the A549 GFP-transduced were exposed for 24 h and 96 h to the chitosan conditioned cell culture medium and the extent of proliferation was compared with the growth in normal culture conditions. As clearly visible in Fig. 8, the chitosan conditioned medium does not affect the viability of any of the cell lines even after long time of exposure. Interestingly, no statistically significant differences in the cell proliferation were observed between the treatments, the higher fluorescence values of the cells exposed to the condition medium could suggest a positive bioactive effect of this formulation. These data indicate good biocompatibility for the new formulation, with no release of toxic species or interference with the cell culture conditions. | ||
| Fig. 8 Cell proliferation of H1299 (A) and A549GFP (B) cell lines grown in normal and chitosan conditioned medium for 24 and 96 h. No statistically significant differences in the cell proliferation were observed between the treatments. | ||
Therefore, considering the good biocompatibility and the enzymatic degradability features previously demonstrated, the production of scaffolds to sustain the in vivo cell growth was explored. Scaffolds with a shape that resembles the 48 well cell culture plate and with a basin to host the cells were produced. Both cell lines were seeded into the basin and their growth and migration through the scaffold structure were explored after 4 and 7 days. In particular, the longer time point was added to increase the possibility to observe cellular migration. The fluorescence analysis showed that both cell lines are able to attach to the scaffold surface (Fig. 9) demonstrating that the chitosan formulation enables the cell adhesion, a parameter essential to sustain the cell growth of this kind of cells. However, the two cell lines showed two different migration behaviors trough the scaffold structure. The H1299 cells were detected both at the top/upper level on which they were seeded, but also at the bottom of the scaffold, thus demonstrating migration through the entire scaffold structure. Instead, the A549 GFP were only detected in the upper part of the scaffold, showing a minor ability of these cells to penetrate inside the scaffold architecture.
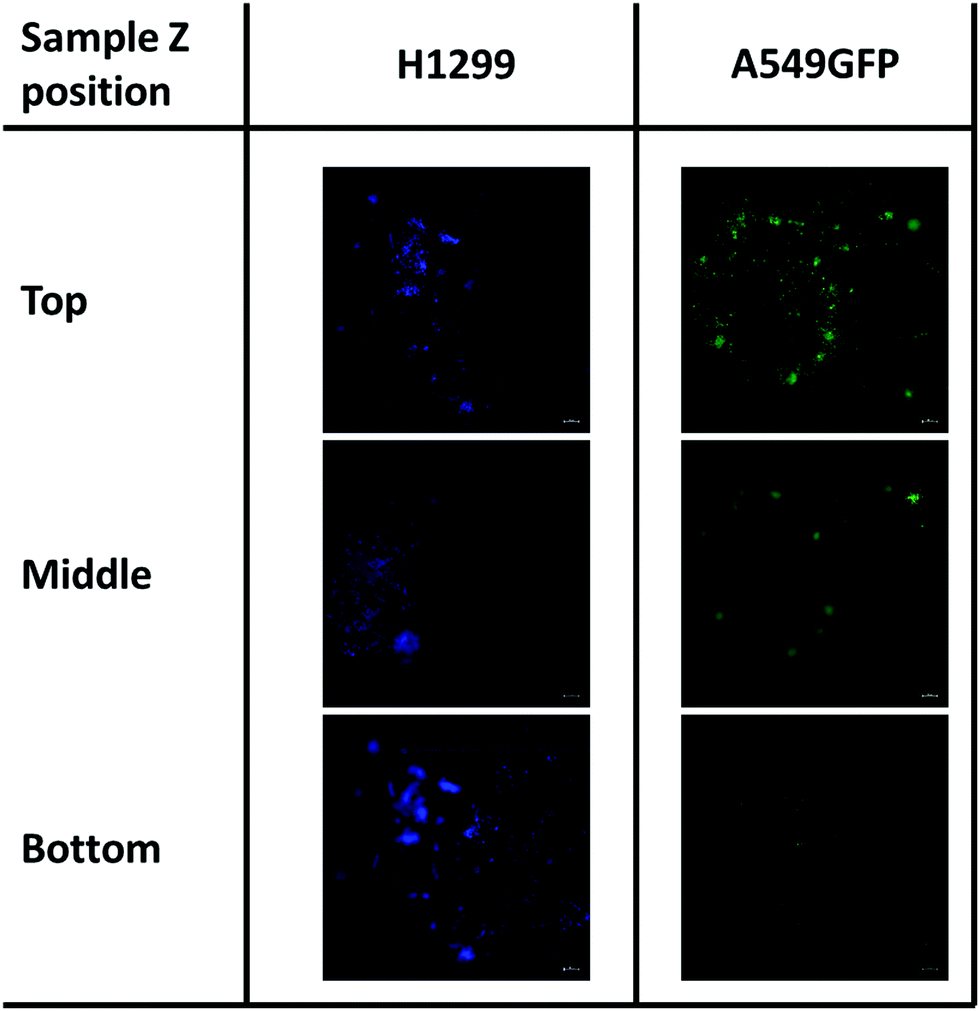 | ||
| Fig. 9 Cell migration of H1299 (DAPI stained) and A549GFP cell lines on chitosan scaffold after 7 days. The scale bar denotes 100 μm for all images. | ||
Although more investigations are needed, this different migration behavior is possibly ascribable to the different source of these tumoral cell lines. Indeed, while the A549 is derived from an adenocarcinoma of the lung, the H1299 is isolated from a metastatic lymphatic site and harbors well documented migration and invasion activities.79 In addition, the presence of both cell lines was detected even after day 7 of cell culture. This demonstrates that the chitosan scaffold could sustain the cell growth also for a longer period, and in some cases allow the cell penetration inside its porous structure.
4. Conclusions
The facile production of methacrylated chitosan by MAOS confirmed that this kind of process can drastically speed up chemical reactions by shortening the reaction time. Here the methacrylation reaction on chitosan was completed in a few minutes, instead of hours under mild conditions in dilute aqueous acetic acid solution. In addition, by optimizing the microwave parameters, it was possible to achieve a high degree of methacrylation in the chitosan backbone, by maintaining the stoichiometric molar ratio of aminoglucose: methacrylate anhydride at 1:1, which lowers the usage of toxic reagents. We also demonstrated the possibility to tune the degree of substitution, and so even the final mechanical properties of the hydrogel, by simply varying the process conditions. Moreover, direct 3D printing of the optimized formulation was performed. The biodegradation and cell tests demonstrated that this microwave-methacrylated chitosan is a promising candidate for the production of scaffolds for biomedical applications. Indeed, this formulation harbours many desirable characteristics, it does not release toxic species or interfere with the cell growth. The biocompatibility and adhesivity ensured the cell growth for long periods of time, which together with the particular porous microstructure allowed cell migration/penetration. We conclude that this rapid and mild functionalization approach is a reliable ad promising alternative to conventional processes taking a step further towards combining green chemistry and tissue engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank Simona Villata for her support in the 3D printing tests. Regione Piemonte is kindly acknowledged for the funding.References
- M. Rajabi, M. Mcconnell, J. Cabral and M. A. Ali, Carbohydr. Polym., 2021, 260, 117768 CrossRef CAS PubMed.
- K. S. Lim, J. H. Galarraga, X. Cui, G. C. J. Lindberg, J. A. Burdick and T. B. F. Woodfield, Chem. Rev., 2020, 120, 10662–10694 CrossRef.
- G. Melilli, I. Carmagnola, C. Tonda-Turo, F. Pirri, G. Ciardelli, M. Sangermano, M. Hakkarainen and A. Chiappone, Polymers, 2020, 12, 1–11 CrossRef CAS.
- C. Tonda-Turo, I. Carmagnola, A. Chiappone, Z. Feng, G. Ciardelli, M. Hakkarainen and M. Sangermano, Bioprinting, 2020, 18, e00082 CrossRef.
- M. Shahbazi and H. Jäger, ACS Appl. Bio Mater., 2021, 4, 325–369 CrossRef CAS.
- Y. He, F. Wang, X. Wang, J. Zhang, D. Wang and X. Huang, Mater. Des., 2021, 202, 109588 CrossRef CAS.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS.
- R. Cerón-Camacho, J. A. Aburto, L. E. Montiel and R. Martínez-Palou, C. R. Chim., 2013, 16, 427–432 CrossRef.
- F. Langa, P. de la Cruz, A. de la Hoz, A. Díaz-Ortiz and E. Díez-Barra, Contemp. Org. Synth., 1997, 4, 373–386 RSC.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- J. P. Tierney and P. Lidström, Microwave Assisted Org. Synth., 2009, 57, 1–280 Search PubMed.
- L. S. Baptista, K. R. Silva, A. Beatrici, G. N. Fontes, J. M. Granjeiro and L. C. Boldrini, J. Sci. Ind. Metrol., 2016, 1 DOI:10.21767/2472-1948.100008.
- R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett., 1986, 27, 279–282 CrossRef CAS.
- G. A. Tompsett, W. C. Conner and K. S. Yngvesson, ChemPhysChem, 2006, 7, 296–319 CrossRef CAS PubMed.
- L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199–9223 CrossRef CAS.
- K. C. Westaway and R. N. Gedye, J. Microwave Power Electromagn. Energy, 1995, 30, 219–230 Search PubMed.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794–806 RSC.
- F. A. Bassyouni, S. M. Abu-Bakr and M. A. Rehim, Res. Chem. Intermed., 2012, 38, 283–322 CrossRef CAS.
- N. Garino, A. Sacco, M. Castellino, J. A. Muñoz-Tabares, A. Chiodoni, V. Agostino, V. Margaria, M. Gerosa, G. Massaglia and M. Quaglio, ACS Appl. Mater. Interfaces, 2016, 8, 4633–4643 CrossRef CAS PubMed.
- N. Garino, T. Limongi, B. Dumontel, M. Canta, L. Racca, M. Laurenti, M. Castellino, A. Casu, A. Falqui and V. Cauda, Nanomaterials, 2019, 9, 1–17 CrossRef.
- J. Cheng, H. Zhu, J. Huang, J. Zhao, B. Yan, S. Ma, H. Zhang and D. Fan, Food Sci. Nutr., 2020, 8, 1987–1994 CrossRef CAS.
- J. Yao, K. Odelius and M. Hakkarainen, ACS Appl. Polym. Mater., 2021, 3(7), 3538–3548, DOI:10.1021/acsapm.1c00438.
- G. Irmak, T. T. Demirtaş and M. Gumusderelioglu, ACS Biomater. Sci. Eng., 2019, 5, 831–845 CrossRef CAS PubMed.
- M. Naveed, L. Phil, M. Sohail, M. Hasnat, M. Muhammad, F. Ashraf, A. Ullah, M. Shumzaid, M. Ullah, T. Mehmood, M. Imtiaz and Q. Zhou, Int. J. Biol. Macromol., 2019, 129, 827–843 CrossRef CAS PubMed.
- C. Choi, J. P. Nam and J. W. Nah, J. Ind. Eng. Chem., 2016, 33, 1–10 CrossRef CAS.
- B. Qu and Y. Luo, Int. J. Biol. Macromol., 2020, 152, 437–448 CrossRef CAS.
- C. Shi, Y. Zhu, X. Ran, M. Wang, Y. Su and T. Cheng, J. Surg. Res., 2006, 133, 185–192 CrossRef CAS.
- C. Noè, C. Tonda-Turo, A. Chiappone, M. Sangermano and M. Hakkarainen, Polymers, 2020, 12, 1359 CrossRef.
- A. Cosola, A. Chiappone, C. Martinengo, H. Grützmacher and M. Sangermano, Polymers, 2019, 11, 1–9 CrossRef.
- M. Rajabi, A. Ali, M. McConnell and J. Cabral, Mater. Sci. Eng., C, 2020, 110, 110612 CrossRef CAS.
- Z. K. Cui, S. Kim, J. J. Baljon, B. M. Wu, T. Aghaloo and M. Lee, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- F. Araiza-Verduzco, E. Rodríguez-Velázquez, H. Cruz, I. A. Rivero, D. R. Acosta-Martínez, G. Pina-Luis and M. Alatorre-Meda, Materials, 2020, 13(3), 34, DOI:10.3390/ma13030534.
- R. Lin, Y. Chen, R. Moreno-luna, A. Khademhosseini and J. M. Melero-martin, Biomaterials, 2013, 34, 6785–6796 CrossRef CAS.
- Y. Shen, H. Tang, X. Huang, R. Hang, X. Zhang, Y. Wang and X. Yao, Carbohydr. Polym., 2020, 235, 115970, DOI:10.1016/j.carbpol.2020.115970.
- J. G. Werner, S. Nawar, A. A. Solovev and D. A. Weitz, Macromolecules, 2018, 51, 5798–5805 CrossRef CAS.
- T. Porstmann, Clin. Biochem., 1989, 22, 349–355 CrossRef.
- Z. Feng, M. Hakkarainen, H. Grützmacher, A. Chiappone and M. Sangermano, Macromol. Chem. Phys., 2019, 220, 1–6 Search PubMed.
- S. H. Kim and C. C. Chu, J. Biomed. Mater. Res., 2000, 49, 517–527 CrossRef CAS.
- L. F. Wang, S. S. Shen and S. C. Lu, Carbohydr. Polym., 2003, 52, 389–396 CrossRef CAS.
- S. Samani, S. Bonakdar, A. Farzin, J. Hadjati and M. Azami, Int. J. Polym. Mater. Polym. Biomater., 2020, 0, 1–12 Search PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- D. J. Trader and E. E. Carlson, Mol. BioSyst., 2012, 8, 2484–2493 RSC.
- S. E. Darmon and K. M. Rudall, Discuss. Faraday Soc., 1950, 9, 251–260 RSC.
- B. G. Amsden, A. Sukarto, D. K. Knight and S. N. Shapka, Biomacromolecules, 2007, 8, 3758–3766 CrossRef CAS.
- S. Hajirahimkhan, P. J. Ragogna and C. (Charles) Xu, Biomass Bioenergy, 2019, 120, 332–338 CrossRef CAS.
- X. Huang, L. Jing, S. V. Kershaw, X. Wei, H. Ning, X. Sun, A. L. Rogach and M. Gao, J. Phys. Chem. C, 2018, 122, 11109–11118 CrossRef CAS.
- S. S. Stalling, S. O. Akintoye and S. B. Nicoll, Acta Biomater., 2009, 5, 1911–1918 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y. L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Cell Motil. Cytoskeleton, 2005, 60, 24–34 CrossRef PubMed.
- K. Xu, K. Ganapathy, T. Andl, Z. Wang, J. A. Copland, R. Chakrabarti and S. J. Florczyk, Biomaterials, 2019, 217, 119311, DOI:10.1016/j.biomaterials.2019.119311.
- D. Holthusen, P. Pértile, J. M. Reichert and R. Horn, Geoderma, 2017, 295, 129–141 CrossRef.
- U. G. T. M. Sampath, Y. C. Ching, C. H. Chuah, R. Singh and P. C. Lin, Cellulose, 2017, 24, 2215–2228 CrossRef CAS.
- A. A. D. N. Pomari, T. L. D. A. Montanheiro, C. P. de Siqueira, R. S. Silva, D. B. Tada and A. P. Lemes, J. Compos. Sci., 2019, 3(3), 84, DOI:10.3390/jcs3030084.
- V. C. Shukla, N. Higuita-Castro, P. Nana-Sinkam and S. N. Ghadiali, J. Biomed. Mater. Res., Part A, 2016, 104, 1182–1193 CrossRef CAS PubMed.
- B. Bhana, R. K. Iyer, W. L. K. Chen, R. Zhao, K. L. Sider, M. Likhitpanichkul, C. A. Simmons and M. Radisic, Biotechnol. Bioeng., 2010, 105, 1148–1160 CAS.
- G. Huang, L. Wang, S. Wang, Y. Han, J. Wu, Q. Zhang, F. Xu and T. J. Lu, Biofabrication, 2012, 4(4), 042001, DOI:10.1088/1758-5082/4/4/042001.
- C. M. Valmikinathan, V. J. Mukhatyar, A. Jain, L. Karumbaiah, M. Dasari and R. V. Bellamkonda, Soft Matter, 2012, 8, 1964–1976 RSC.
- P. J. Flory, Chem. Rev., 1944, 35, 51–75 CrossRef CAS.
- R. K. Farag and R. R. Mohamed, Molecules, 2013, 18, 190–203 CrossRef CAS.
- S. M. Taghizadeh and G. Davari, Carbohydr. Polym., 2006, 64, 9–15 CrossRef CAS.
- M. Davidovich-Pinhas and H. Bianco-Peled, Carbohydr. Polym., 2010, 79, 1020–1027 CrossRef CAS.
- C. Wang, Y. Li and Z. Hu, Macromolecules, 1997, 30, 4727–4732 CrossRef CAS.
- N. Xia, W. Wan, S. Zhu and Q. Liu, Int. J. Biol. Macromol., 2020, 163, 1518–1528 CrossRef CAS.
- L. Masaro and X. X. Zhu, Physical models of diffusion for polymer solutions, gels and solids, 1999, vol. 24 Search PubMed.
- H. Schott, J. Pharm. Sci., 1992, 81, 467–470 CrossRef CAS.
- S. H. Medina, B. Bush, M. Cam, E. Sevcik, F. W. Delrio, K. Nandy and J. P. Schneider, Biomaterials, 2019, 202, 1–11 CrossRef CAS.
- K. R. Levental, H. Yu, L. Kass, J. N. Lakins, M. Egeblad, J. T. Erler, S. F. T. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi and D. L. Gasser, Cell, 2009, 139, 891–906 CrossRef CAS PubMed.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed.
- K. Nawrotek, M. Tylman, A. Adamus-Włodarczyk, K. Rudnicka, J. Gatkowska, M. Wieczorek and R. Wach, Carbohydr. Polym., 2020, 244, 116484 CrossRef CAS PubMed.
- T. Freier, H. S. Koh, K. Kazazian and M. S. Shoichet, Biomaterials, 2005, 26, 5872–5878 CrossRef CAS PubMed.
- S. Kim, Z. K. Cui, B. Koo, J. Zheng, T. Aghaloo and M. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 41138–41145 CrossRef CAS.
- J. Hu, Y. Hou, H. Park, B. Choi, S. Hou, A. Chung and M. Lee, Acta Biomater., 2012, 8, 1730–1738 CrossRef CAS PubMed.
- C. Xu, S. Guan, S. Wang, W. Gong, T. Liu, X. Ma and C. Sun, Mater. Sci. Eng., C, 2018, 84, 32–43 CrossRef CAS.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., Part B, 2010, 16, 371–383 CrossRef CAS.
- S. Van Vlierberghe, V. Cnudde, P. Dubruel, B. Masschaele, A. Cosijns, I. De Paepe, P. J. S. Jacobs, L. Van Hoorebeke, J. P. Remon and E. Schacht, Biomacromolecules, 2007, 8, 331–337 CrossRef CAS.
- S. Gorgieva and V. Kokol, J. Biomed. Mater. Res., Part A, 2012, 100 A, 1655–1667 CrossRef.
- M. M. Islam, M. Shahruzzaman, S. Biswas, M. Nurus Sakib and T. U. Rashid, Bioact. Mater., 2020, 5, 164–183 CrossRef.
- P. Kumar, B. S. Dehiya and A. Sindhu, Int. Nano Lett., 2017, 7, 285–290 CrossRef CAS.
- J. T. Pai, C. Y. Hsu, Y. S. Hsieh, T. Y. Tsai, K. T. Hua and M. S. Weng, Food Sci. Nutr., 2020, 8, 1534–1545 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00765c |
| This journal is © The Royal Society of Chemistry 2022 |