
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,8-Di(piperidinyl)-naphthalene – rationally designed MAILD/MALDI matrix for metabolomics and imaging mass spectrometry†
Jerrit
Weißflog
and
Aleš
Svatoš
*
Mass Spectrometry/Proteomics Research Group Max Planck Institute for Chemical Ecology, Hans-Knoell-Strasse 8, 07745 Jena, Germany. E-mail: svatos@ice.mpg.de; Tel: +49 3641 57 1700
First published on 29th July 2016
Abstract
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of small molecules requires special matrices, which do not generate interfering signals below m/z 500. Here, we present 1,8-di(piperidinyl)naphthalene (DPN), a new matrix that is suitable for analytes of low molecular weight in negative ion mode and that does not generate such signals. This compound was rationally designed from parent 1,8-bis(dimethylamino)naphthalene (DMAN) by increasing the distance among the di-nitrogen-center-chelated proton and a deprotonated anion and by increasing a steric repulsion. This matrix features superior ionization properties, as its superbasic nature causes analyte molecules to be more effectively deprotonated prior to laser desorption according to the MAILD-principle (Matrix-Assisted-Ionisation/Laser-Desorption). As a result, for a wide range of analytes with varying degrees of acidity, higher signal intensity can be achieved with DPN compared to conventional “classic” matrices as well as already known MAILD-matrices. To demonstrate the applicability of DPN for metabolomical studies, we tested its performance as a matrix on biologically relevant analytes and compared the results with those obtained using established matrices. We also show the advantages of DPN for MS-imaging techniques and MALDI at atmospheric pressure (AP-MALDI). This new matrix reported here represents a considerable improvement on established MAILD-matrices in terms of sensitivity and range of potential analytes.
Introduction
Matrix-Assisted Laser Desorption/Ionisation (MALDI), considered to be a soft way to obtain ions, has proven to be a reliable and powerful technique allowing the analysis of large and fragile biomolecules like peptides, proteins and polysaccharides as well as synthetic organic macromolecules like polymers.1,2 The analysis of low-molecular-weight molecules with this technique is more challenging, because the employed, conventionally used matrices (e.g. α-cyanohydroxycinnamic acids and 2,5-dihydroxybenzoic acid) create a broad background of signals below m/z 500, which interfere with those of the analytes and thus complicate identification.3 However, there are several approaches that aim to address these problems. One simply avoids such classic matrices, relying instead on various synthetic materials (e.g. modified silica and modified graphite)4,5 or even polymeric biomaterials6 to exploit the surface effects of the matrices; these surface effects are beneficial to the laser desorption and ionization. Even so, the fact that most of these materials are insoluble in the usual solvents makes their application to the sample target a cumbersome step in the process of sample preparation. MALDI-MS-imaging in particular relies heavily on a homogeneous distribution of matrix on the sample, and such a surface homogeneity can be achieved by either spraying a solution of matrix or sublimating the matrix onto the target. This can be achieved more easily with soluble low-molecular-weight-matrices, which is why such matrices are still used in MS-imaging experiments despite of the drawbacks. Ionic-liquids have also been reported to be efficient matrices generating homogeneous sample spots.7Nevertheless, various low-molecular-weight matrices have been developed, which produce either only few distinct matrix signals, such as 9-aminoacridine (9-AA),8 or no signals at all (1,8-bis(dimethylamino)naphthalene) (DMAN, 2).9 The superbasic nature of DMAN results in the deprotonation of the acidic analyte in solution during sample preparation, according to the acid–base theory developed by Brønsted–Lowry. The ion pair crystals that formed, which consist of deprotonated analyte ions and protonated DMAN, are desorbed by the UV-Laser. As DMAN itself does not undergo gas-phase deprotonation, no interfering matrix signals are generated. This ionisation principle, in which a preformed matrix/analyte salt is desorbed from the target plate, was coined matrix assisted ionisation/desorption (MAILD).10 This concept was further expanded to superbasic azahelicenes that work effectively for diverse acids.11 DMAN was later found to suffer from higher volatility which led to contaminations on some parts of the ion optics in MALDI-TOF/MS instruments and is therefore prevented it from being suitable for longer measurements.12 Recently series of bisphosphazene superbases have been introduced capable of deprotonation even secondary alcohols like cholesterol. However, this class of sponges produce matrix-related signals.13
In this study, we searched for new superbasic ion-less matrices with a low vapour pressure to address these shortcomings. We also focused on increasing the sensitivity of the analysis.
Experimental section
General
DPN (1) was synthesized from 1,8-diaminonaphthalene (Sigma-Aldrich) and 1,5-dibromopentane (Sigma-Aldrich) as described in literature,14 although the purification steps were modified as follows: after cooling, the reaction mixture was treated with 1 M NaOH (2 ml per mmol of starting compound) and extracted repeatedly with n-hexane. The solvent of the extract was removed via evaporation and the obtained residue subjected to column chromatography on alumina pretreated with triethylamine (1% in n-hexane) using n-hexane as eluent. The obtained material was recrystallized twice from n-hexane to obtain pure DPN as colourless, needle-shaped crystals. Compounds 4–7 (Fig. 1) were synthesized as described in literature.14 9-Aminoacridine was prepared from its hydrochloride (Sigma-Aldrich) by treatment with sodium hydroxide and subsequent washing with water. All matrices were recrystallized prior to use (Fig. 2).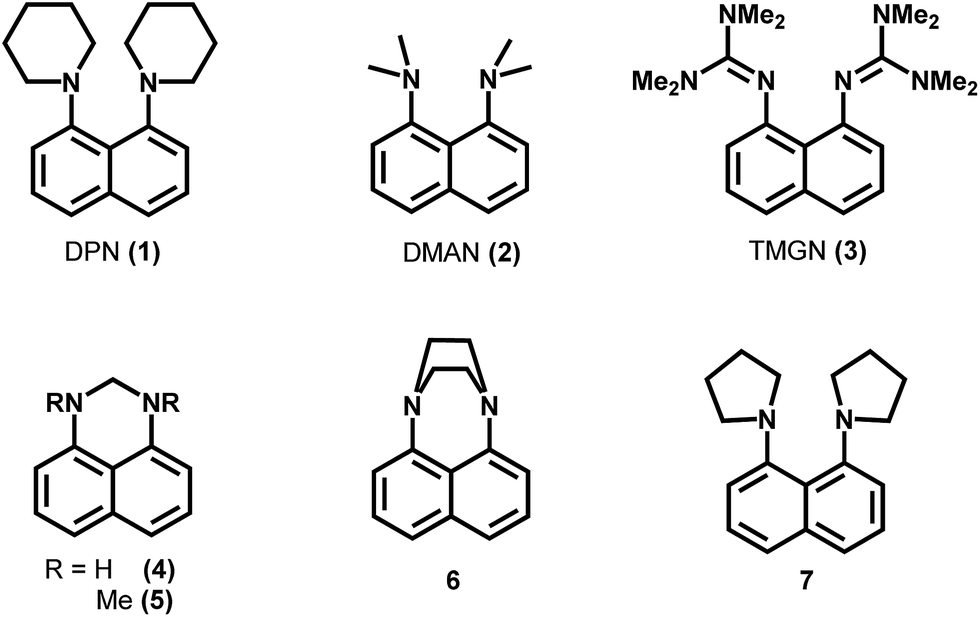 | ||
| Fig. 1 Examples of basic naphthalenes, which were tested for the use as matrices for negative mode MALDI-MS. | ||
 | ||
| Fig. 2 MALDI-ToF mass spectra of model analytes recorded by using DMAN and DPN as matrices. Top left: Mixture of palmitic, stearic and arachidic acid (50 pmol on plate each), top right: oleic acid (125 pmol on plate), middle left: cysteine (250 pmol on plate, contains cystine as an impurity), middle right: Phe-Gly dipeptide (250 pmol on plate), bottom: ascorbic acid (175 pmol, on plate). For all measurements an equimolar ratio of matrix to analyte was used. Each mass spectrum represents a combination of 5 scans after background subtraction. S/N ratios were estimated by comparing the mass peak intensity with the noise intensity in a range next to the peak. | ||
MALDI-MS experiments were performed on a MALDI micro MX mass spectrometer (Waters/Micromass Manchester UK) equipped with a N2-laser (λ = 337 nm, 4 ns pulse duration up to 20 Hz repetition rate and max 320 μJ per pulse) in negative ion mode for data acquisition. The instrument operated at a pressure of 5 × 10−7 bar with a plate voltage of 5.2 kV, an acceleration voltage of −12 kV, a pulse voltage of 1.95 kVA and a detector voltage of 2.25 kV. The laser frequency was set to 10 Hz and laser energy was optimized prior to each measurement (typically 160–190 μJ per pulse) unless stated otherwise. The extraction delay was set to 155 ns. Polyethyleneglycol 600 sulfate (PEG sulfate 600, Sigma-Aldrich) was used for calibration in the range of m/z 100–1000 prior to each set of measurements. A 96-well MALDI-target plate was used featuring circular target spots of 2.5 mm diameter. Mass Lynx v.4.1 (Waters) software was used for data acquisition.
AP-MALDI-MS experiments were performed on either a Q Exactive Plus (Thermo Fisher, San Jose, CA) or an LTQ XL (Thermo) orbitrap mass spectrometer coupled with an AP/MALDI PDF+ (Pulsed Dynamic Focusing) Ion Source (MassTech, Columbia, MD) equipped with an Nd-YaG-laser. The spectra were recorded in negative ion mode using a plate voltage of 3.5 kV, a PDF delay setting of 80 and an injection time of 1000 ms. The laser focus was set to move in a spiral motion over the whole spot with a velocity of 4 mm min−1 and a firing rate of 20 Hz. Xcalibur v.3.0 (Thermo) software was used for data acquisition. A 96-well AP-MALDI-target plate was used featuring square target spots of 4 × 4 mm dimensions.
UV-Vis measurements were performed on a UV-2501PC spectrophotometer (Shimadzu) using quartz cuvettes for the sample and reference.
Quantum chemical calculations were performed by using the TURBOMOLE 6.4 program and the RI-PBE+D3/def2-TZVP//RI-PBE+D3/def2-SVP model chemistry system and are described in literature.11
Comparison of DMAN and DPN
The solutions of model analytes were mixed with matrix solutions of the same molar concentration (DMAN and DPN in EtOH), and these mixtures were applied onto a MALDI-target plate (1 μl per circular target spot). The fatty acid solutions were prepared from 10 nanomolar stock solutions in EtOH and diluted with EtOH to the final concentration immediately prior to use (100 pmol μl−1 each for the mixture of saturated FA's and 250 pmol μl−1 for oleic acid). Cysteine, Phe-Gly-hydrate and the sodium salt of ascorbic acid were dissolved in 10% acetic acid at a concentration of 10 nmol μl−1 and diluted with Milli-Q water to the final concentration (250 pmol μl−1 for cysteine and Phe-Gly hydrate and 350 pmol μl−1 for ascorbic acid). The TOF response curve of stearic acid (ESI Fig. S4†) was created by mixing (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) DPN solution (1.2 nmol μl−1 in EtOH) with stearic acid solutions of varying concentrations (600, 400, 200, 100, 50, 25, 10, 2 and 1 pmol μl−1). These mixtures were applied onto a MALDI target plate (1 μl per spot, 4 spots per concentration) and recorded in negative ion mode. Each data point was created from 4 measurements (i.e. from 4 different spots of the same concentration).
:1 v/v) DPN solution (1.2 nmol μl−1 in EtOH) with stearic acid solutions of varying concentrations (600, 400, 200, 100, 50, 25, 10, 2 and 1 pmol μl−1). These mixtures were applied onto a MALDI target plate (1 μl per spot, 4 spots per concentration) and recorded in negative ion mode. Each data point was created from 4 measurements (i.e. from 4 different spots of the same concentration).
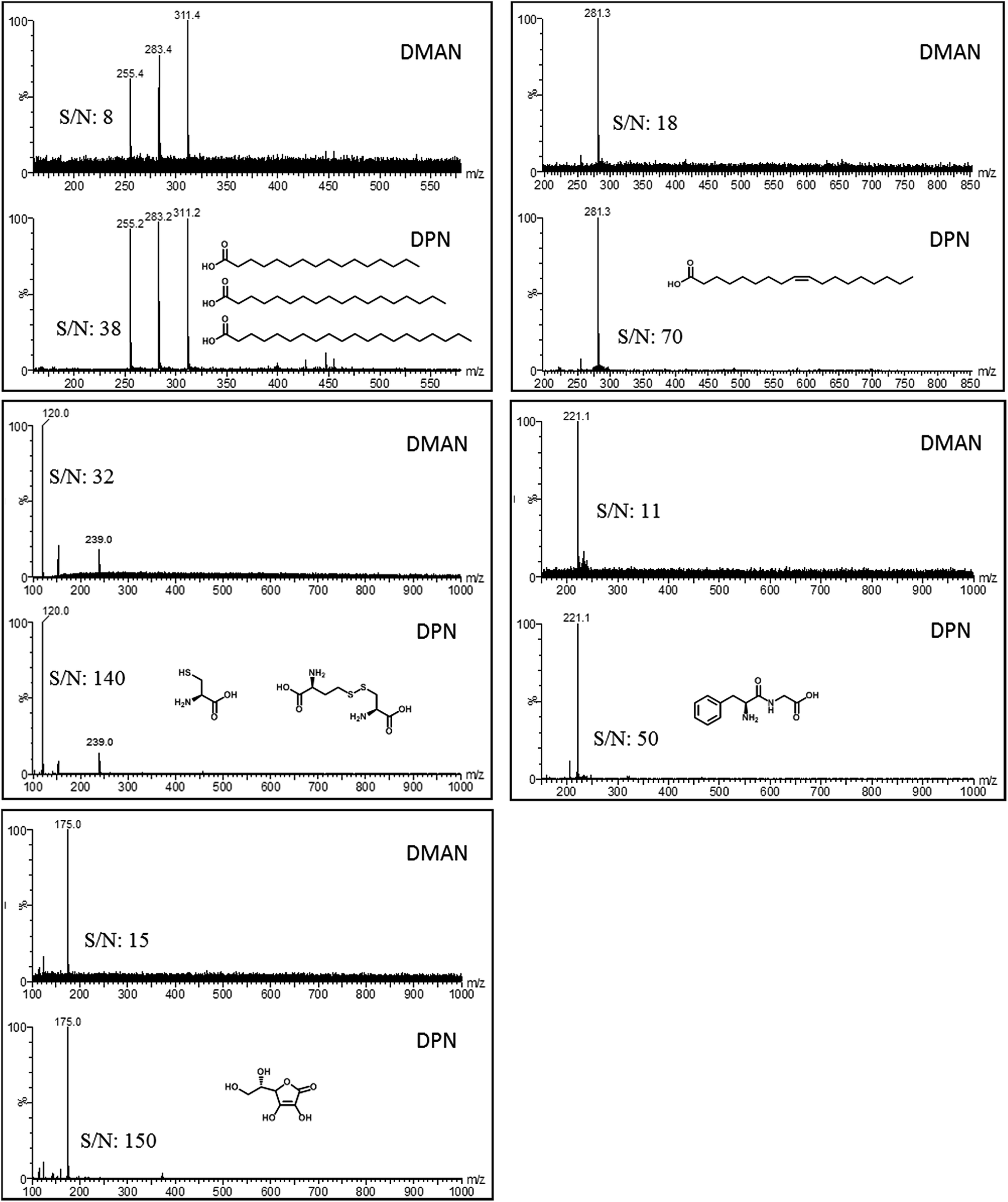
Signal intensity vs. analyte/matrix ratio
A stock solution of DPN in chloroform was sequentially diluted with ethanol to obtain matrix solutions having the following concentrations 16, 8, 2, 1, 0.5, 0.25, 0.1, 0.05 and 0.01 nmol μl−1. Each of these solutions was mixed with a solution of stearic acid (500 pmol μl−1 in EtOH) and applied to a MALDI target plate (1 μl per spot). Every target spot was measured, 20 scans combined, the background subtracted, smoothed and centered, before the intensity of the peak at 281 m/z was read out. 4 replicates were performed for each data point in Fig. 3.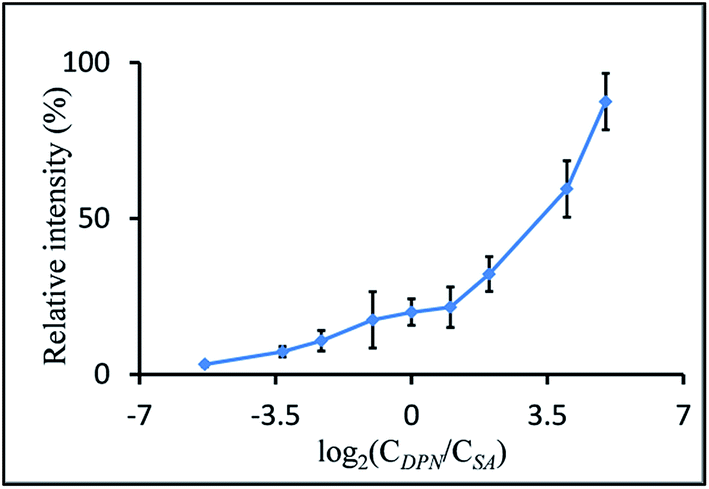 | ||
| Fig. 3 Plot of relative intensity of the mass signal of the stearate anion versus molar ratios of DPN to stearic acid used in the measurement. Plotted on a logarithmic scale for clarity. The amount of analytes was kept constant at 250 pmol, and the matrix concentrations were increased to have the following matrix-to-analyte molar ratios: 0.025:1, 0.1:1, 0.2:1, 0.5:1, 1:1, 2:1, 4:1, 16:1 and 32:1 (100% equals to an intensity of 22500 a.u.). | ||
Dependency of signal intensity on laser energy
A PEG-sulfate solution (500 pmol μl−1 in EtOH) was mixed with a matrix solution (DMAN or DPN, 500 pmol μl−1 in EtOH), and 1 μl of these mixtures was applied to a MALDI-target plate. The mass spectra of these spots were recorded at defined laser energy settings ranging from 200 to 300. Fifteen scans of each measurement were combined.UV-Vis spectra of matrices
A droplet (5 μl) of the respective matrix solution (DMAN or DPN, 50 nmol μl−1 in EtOH each) was carefully placed on the inside of a quartz cuvette and gently dried in an argon stream. The cuvette was then placed into the UV-Vis spectrophotometer and the absorption recorded in the range of 250 to 500 nm. An empty cuvette was used as the reference.Lipid MALDI-imaging
A little finger of one of the authors (JW) was placed on a cleaned MALDI-plate and the obtained print was further imaged. One 1 μl of PEG sulfate 600-solution (1 nmol μl−1 in EtOH) was added on a different part of the plate for mass calibration. DPN was sublimated onto the plate at a pressure of 0.015 mbar and a temperature of 130 °C for 2 minutes. After calibration the area of the fingerprint (14 mm × 12 mm) was imaged at a laser energy setting of 300, a firing rate of 5 Hz and a step size of 0.2 mm. 30 shots were recorded per spot resulting in a total measuring time of 7 h. After the measurement, the MALDI-plate was washed with 3 ml pure n-hexane and the collected solvent was treated with an etheric solution of diazomethane. The mixture was dried in a nitrogen stream, re-dissolved in 1 ml n-hexane and analyzed via GC/MS to confirm the identity of the observed fatty acids.Lipid extraction of chicken egg
The whole content of a chicken egg (yolk + egg white), purchased from a local supermarket, was homogenized. 400 μl of this mixture were transferred into a 4 ml vial; next, 1.5 ml of a 2:1 (v/v) solution of methanol and chloroform was added and the vial was vigorously shaken for 5 min. 500 μl water and 500 μl chloroform were added, and the vial was again shaken for 5 min before it was centrifuged at 7000 rpm for 5 min. The aqueous phase was removed and the organic phase concentrated to dryness in a stream of nitrogen. The obtained material was dissolved in 2 ml of a 1:1 mixture of methanol and chloroform (v/v).
AP-MALDI-MS analysis of hen egg extract
10 μl of this extract was mixed with 10 μl of matrix solution (DMAN [10 mg ml−1, 1 mg ml−1 and 0.1 mg ml−1 in EtOH], 9-AA [10 mg ml−1 in 3:2 isopropanol/acetonitrile] and DPN [10 mg ml−1 in 2:1 chloroform/MeOH]) and this mixture was applied onto an AP-MALDI-target plate (1 μl per target spot). The plate was dried in a gentle N2-stream, placed into the AP-MALDI source and the MS spectra recorded in the range of m/z 70 to 1000.
AP-MALDI-MS analysis of whole blood
Using a sterile needle and syringe, ca. 1 ml of blood was taken from the median cubital vein of one of the authors and immediately diluted with deionized water. A small portion of this diluted blood was mixed 1:1 with the respective matrix solution in ethanol (DMAN, 9-AA and DPN; 10 nmol μl−1 each) and applied to an AP-MALDI target plate (2 μl per target spot). The plate was dried in a gentle N2-stream applied to the AP-MALDI source, and the MS spectra were recorded in the range of m/z 100 to 1000.
Results and discussion
Screening of basic naphthalenes for potential matrices
Using the structure of DMAN as a starting lead structure, several superbasic compounds were designed and/or synthesized and subsequently screened for their potential as a MAILD-matrix (some examples are shown in Fig. 1). All of the tested compounds contained a naphthalene ring system and nitrogen-substitution in positions 1 and 8. Our aim was to improve the performance of DMAN as matrix. We pursued two hypotheses: (1) an increase in basicity would improve the deprotonation of the analyte and thus increase the sensitivity of analysis. (2) Expanding the hydrophobic area around the nitrogen centers would improve the ion-pair charge separation, causing the ion pairs to separate more easily and increasing their signal intensity. Additionally, we were aiming for compounds with less matrix volatility than DMAN.We tested the candidate compounds on several model analytes, and compared the results with those obtained with DMAN. The analytes used in these experiments ranged from weakly to medium acidic and were chosen to represent compound classes of biological relevance i.e. 3 saturated fatty acids, oleic acid, cysteine, Phe-Gly dipeptide and ascorbic acid. We tried to keep the experimental conditions the same for DMAN and the matrix candidates to make the results comparable.
We realized early in our studies that just using a matrix with a high basicity, for example 1,8-bis(tetramethylguanidino)naphthalene (TMGN)15 with pKA (MeCN) of 25.1 (exp.), does not lead to an improvement in signal intensity (see ESI Fig. S1†) rather produce lot of fragments (loss of methyl groups) and does not fulfill our requirement for designing ion-less matrices. Likewise if the two nitrogen atoms were bridged, as in compounds 4–6,14 the experimental results could not compete with those obtained from DMAN (ESI Fig. S1†), as the bridge prevents the proton from being bound by both nitrogen atoms, and so reducing its basicity. When aliphatic rings were used as substituents, as seen in compounds 6 and 1,8-di(piperidinyl)naphthalene (DPN, 1), we observed better ionization than what is obtained when DMAN is used. However, more noisy spectra were measured with matrix compound 6 (ESI S1†), and DPN was selected for further experiments.
Of all tested compounds, DPN features the highest potential as a matrix for negative ion mode MALDI-MS, and it reaches higher signal intensities for all model analytes than obtained with DMAN (Fig. 1). It should be noted that DPN, like DMAN produces no interfering matrix signals of DPN. To see how DPN competes with established classic MALDI-matrices, we compared the results of the measurements using DPN as a matrix with those obtained when we used 9-AA (see ESI Fig. S3†). We observed a significant increase in sensitivity with saturated and unsaturated fatty acids as model analytes. DPN showed a high linearity of response (r2 = 0.998) over more than 2 orders of magnitude like its predecessor DMAN,16 but the limit of detection improved by a factor of 30 (see ESI Fig. S4†).
With a pKA (EtOH) of 9.6 (calculated), the basicity of DPN is roughly the same as that of DMAN (pKA (EtOH) = 9.5). We therefore assume that the reason for DPN's superior ionization properties is not only its basicity but also its kinetic effects. In the protonated state, both piperidinyl rings shield the proton and so reduce the rate at which the proton is exchanged back to the analyte ion in the gas phase after desorption. Additionally, quantum calculations of matrix/acetate ion pairs in ethanol reveal, that the O–H distance is increased from 0.982 Å in free acetic acid to 1.030 Å in DMAN·HOAc and to 1.049 in DPN·HOAc. In ionization processes involving MAILD-matrices, the ion pairs formed in the solution are desorbed during the laser pulse and dissociate further in the gas phase. The longer O–H distance in DPN·HOAc than in DMAN·HOAc facilitates the dissociation of the ion pair and so increase signal intensities. This dissociation energy was calculated for DMAN and DPN matrix acetates as ΔGdiss = −405 kJ mol−1 and ΔGdiss = −374 kJ mol−1, respectively. The weaker ion pairing is manifested as ΔΔGdiss = −32 kJ mol−1 energy gain for DPN.
It is worth noting that DPN is capable of ionizing some compounds which lack an acidic proton. For example glucose can be detected via its [M − H]−-ion using DPN as matrix (see ESI, Fig. S5†), however with very low sensitivity. However cholesterol was not deprotonated in contrast to published bisphosphazenes.13
Effect of matrix/analyte ratio
Reportedly, optimal signal intensities can be achieved only if an equimolar ratio of DMAN to analyte is used in the sample preparation.10 The reason for these intensities is that DMAN forms a salt with the analyte on the target plate, and the process works best when both compounds are present in the same concentration. As similar effects have also been reported for other recently introduced superbasic matrices,11 we deemed it appropriate to use an equimolar ratio for our initial experiments with DPN; this ratio also made our results more comparable with results from experiments in which DMAN was used. But as the superior properties of DPN became apparent, we wanted to investigate whether such effects also apply to DPN. Therefore mass spectra of a model analyte (stearic acid) were recorded using different amounts of DPN while keeping the analyte concentration the same. We were surprised to see, that DPN can be safely used in higher molar ratios and that this even leads to a further increase in signal intensity (Fig. 3).Low dependence of ion intensity on the matrix/analyte ratios is beneficial when mixtures of compounds in a wide range of different concentrations are analyzed. Using an excess of DPN would ensure that all analytes are detected optimally.‡ Based on this property it would appear that DPN is not a typical MAILD matrix and acts instead like a classic MALDI-matrix. However, we found that the UV-absorbance of unprotonated DPN in solid state is slightly higher than that of DPN co-crystallized with acetic acid (see ESI†). It can be assumed that the excess of free base is assisting in the desorption process, as the energy of the laser is more efficiently absorbed by the excessive matrix molecules.
Dependency of signal intensity on laser energy
To investigate the influence of the laser energy applied with each pulse, we recorded the mass spectra of PEG-sulfate at different laser energy settings while employing DMAN, 9-AA and DPN as matrix, and compared the results (Fig. 4). | ||
| Fig. 4 Stacked overlay of mass spectra of PEG-sulfate (250 pmol on plate) recorded at different settings of laser energy using DMAN (top), 9-AA (middle) and DPN (middle) as matrix (each spectrum was created from 10 combined scans). Bottom: Overlay of UV-Vis spectra of DMAN (red), 9-AA (blue) and DPN (green) recorded in solid state. | ||
For DMAN and 9-AA we observed a drastic drop in signal intensity when the laser energy was reduced from 192 μJ per laser pulse to 160 μJ. No analyte signal could be detected at settings below 160 μJ. Aside from the overall higher signal intensity, DPN facilitated excellent ionization even at the lowest setting resulting in high quality mass spectra. Additionally, we recorded the UV-Vis spectra of all 3 matrices in solid states (by creating thin crystal layers of the matrices inside the quartz cuvettes using the dried-droplet method), simulating the conditions on a MALDI-plate (Fig. 4, bottom). These data show that DPN has a high absorption at the excitation wavelength of the N2-laser, which may explain the observed ionization behavior. Using low laser energy for ionization reduces the risk of unwanted fragmentation of analyte molecules/ions, as little energy is introduced into the process. Therefore it is possible to use DPN for fragile analytes and still obtain clean spectra.
Lipid MALDI-imaging
In the literature DMAN is said to desorb from the target plate over time in the high-vacuum conditions of mass spectrometer, resulting in reduced signal intensity in prolonged experiments as well as the formation of deposits contaminating the ion source.17 We assumed that DPN has a higher vapor pressure than DMAN as a result of its higher molecular weight and, therefore, that is was more stable than DMAN under vacuum conditions. To verify these assumptions, we performed an MS-imaging experiment of a fingerprint, which required 7 hours to complete (Fig. 5). We observed mass signals which corresponded with those of several lipids and free fatty acids, all of which are present in the oil on skin. Their identity was confirmed by dissolving the material on the MALDI-target in n-hexane, converting the fatty acids to their respective methyl esters via diazomethane treatment and subsequently analyzing the samples with GC-MS.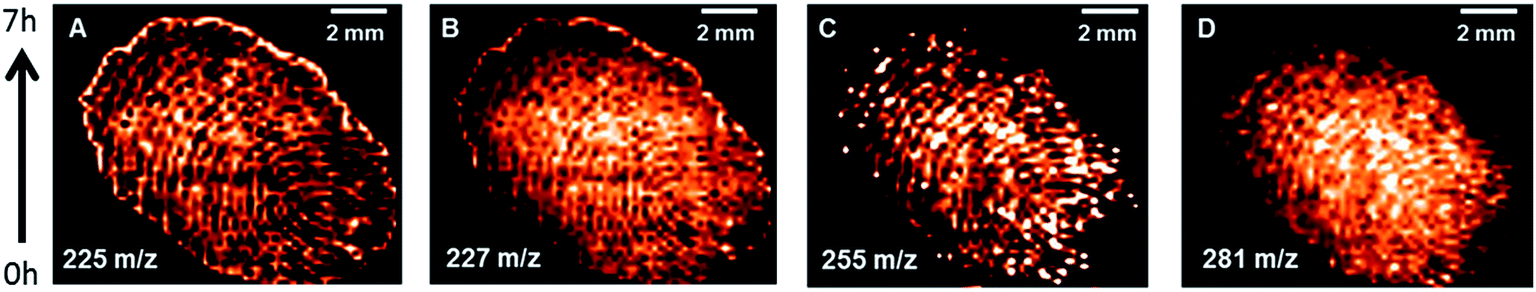 | ||
| Fig. 5 Selected ion maps of a fingerprint (applied directly on the MALDI-plate) acquired via neg. ion mode MALDI-MS using DPN as matrix (applied by sublimation), (A) myristoleic acid ([M − H]−m/z 225), (B) myristic acid ([M − H]−m/z 227), (C) palmitic acid ([M − H]−m/z 255), (D) oleic acid ([M − H]−m/z 281). | ||
As can be deduced from the ion maps of the observed fatty acids, no loss of signal intensity occurred during the time of the experiment. This result not only supports our claim that DPN is particularly stable in a high vacuum but also illustrates DPN's potential for MS-imaging in negative ion mode.
Lipid extract analysis using AP-MALDI-FTMS
To further explore the potential of DPN as MALDI-matrix, we decided to test DPN's ionization performance with more complex samples. Additionally, we decided to examine the samples under ambient conditions, as these are more relevant to the biological samples we are typically investigating at our institute. We chose a lipid extract of a chicken egg as a sufficiently complex analyte and compared the results of DPN with those of DMAN and 9-AA, as the latter is considered the matrix of choice for the analysis of such lipids in negative ion mode.18 It is reported that the efficiency of 9-AA as a matrix depends significantly on the used solvent system.19 Therefore, we took care to use the same conditions as reported for experiments with this matrix (e.g. isopropanol/methanol as solvent), to get optimal results. We observed that matrix concentrations in the range of 10–50 nmol μl−1 were necessary for efficient ionization in AP-MALDI-FTMS when using DPN and 9-AA. DMAN failed to produce detectable analyte ions at these concentrations, because the optimal 1:1 matrix/analyte ratio was exceeded by far. Lower concentrations of matrix however did not improve the results. DPN and 9-AA yielded rich spectra containing mass signals of singly charged phospholipid (PL) anions and, in the case of DPN, several anions of free fatty acids (Fig. 6). The lipids were identified based on accurate masses only. As a result of the higher sensitivity some phospholipid species could only be detected with DPN as can be seen in Table 1. Therefore, DPN represents a suitable matrix to analyze PLs producing negatively charged ions of a given sample. It should be noted that DPN, renders phosphatidylcholines (PC) to some extent detectable in negative ion mode, by inducing the loss of a methyl group and/or trimethylamine to generate negatively charged ions (see ESI Fig. S7†). Similar behavior was already observed for 9-AA.20 We assume this occurs in solution during the sample preparation as a result of the strong basic nature of the matrix. ESI-MS experiments have confirmed the presence of such ions in solution, when DPN is mixed with PC 34:1 (see ESI Fig. S8†). We have not observed such fragmentation for other classes of PLs. Although the intensity of these ions is an order of magnitude lower than those of the isobaric PEs, the analytical information can become distorted if large amounts of PCs are present in the sample.
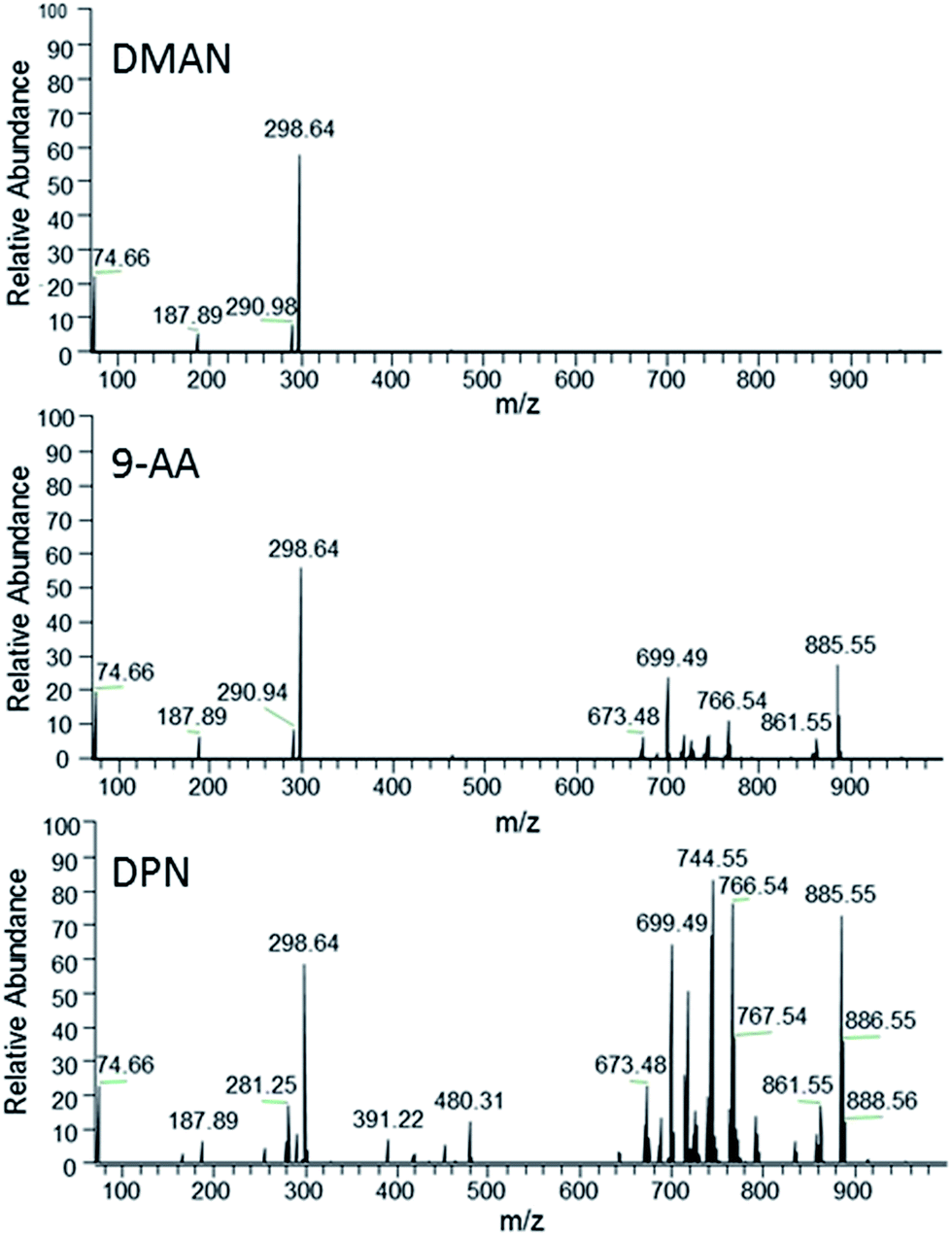 | ||
| Fig. 6 FT-MS spectra of a hen egg extract obtained via AP-MALDI using DMAN (top), 9-AA (middle) and DPN (bottom) as matrix (averaged over 40 scans, normalized to same intensity, 298.64 is generated via a background frequency of the mass spectrometer). | ||
:1) might contribute to these peaks to some degree)
| m/z | Assignment | Detected by | |
|---|---|---|---|
| 9-AA | DPN | ||
| 279.232 | C18H31O2 linolate | − | + |
| 281.248 | C18H33O2 oleate | − | ++ |
| 283.263 | C18H35O2 stearate | − | + |
| 303.231 | C20H31O2 arachidonate | − | + |
| 671.463 | C37H68O8P PA (34:2)–H+ |
− | ++ |
| 673.480* | C37H70O8P PA (34:1)–H+ |
+ | +++ |
| 685.480 | C38H70O8P PA (35:2)–H+ |
− | + |
| 687.496* | C38H72O8P PA (35:1)–H+ |
+ | ++ |
| 697.480 | C39H70O8P PA (36:3)–H+ |
++ | +++ |
| 699.496* | C39H72O8P PA (36:2)–H+ |
+++ | ++++ |
| 714.507 | C39H73O8NP PE (34:2)–H+ |
+ | +++ |
| 716.522 | C39H75O8NP PE (34:1)–H+ |
++ | ++++ |
| 738.507 | C41H73O8NP PE (36:4)–H+ |
− | +++ |
| 742.538 | C41H77O8NP PE (36:2)–H+ |
++ | ++++ |
| 744.554* | C41H79O8NP PE (36:1)–H+ |
++ | ++++ |
| 762.507 | C43H73O8NP PE (38:6)–H+ |
+ | ++ |
| 764.522 | C43H75O8NP PE (38:5)–H+ |
− | + |
| 766.538 | C43H77O8NP PE (38:4)–H+ |
++ | ++++ |
| 768.569 | C43H79O8NP PE (38:3)–H+ |
− | +++ |
| 770.585 | C43H81O8NP PE (38:2)–H+ |
− | ++ |
| 772.589 | C43H83O8NP PE (38:1)–H+ |
− | + |
| 790.537 | C45H77O8NP PE (40:6)–H+ |
− | ++ |
| 792.552 | C45H79O8NP PE (40:5)–H+ |
− | ++ |
| 794.571 | C45H81O8NP PE (40:4)–H+ |
− | + |
| 833.515 | C43H78O13P PI (34:2)–Na+ |
+ | ++ |
| 835.530 | C43H80O13P PI (34:1)–Na+ |
− | + |
| 857.516 | C45H78O13P PI (34:4)–Na+ |
+ | ++ |
| 859.530 | C45H80O13P PI (34:3)–Na+ |
+ | + |
| 861.547 | C45H82O13P PI (34:2)–Na+ |
++ | +++ |
| 885.546 | C47H82O13P PI (38:4)–Na+ |
+++ | ++++ |
| 887.560 | C47H84O13P PI (38:3)–Na+ |
++ | ++++ |
| 911.562 | C49H84O13P PI (40:4)–Na+ |
− | + |
| 913.576 | C49H86O13P PI (40:3)–Na+ |
− | + |
Whole blood analysis via AP-MALDI-FT-MS
Realizing the superior ionization properties of DPN with peptides and lipids, we wanted to see whether DPN could be applied effectively to metabolomic studies. We chose fresh human blood as a test sample and analyzed it via AP-MALDI-FT-MS without any sample preparation except dilution with water, using DMAN, 9-AA and DPN as matrices. For DMAN several analyte/matrix ratios were tested, to give optimal results. In contrast to the lipid experiments, a DMAN concentration of 10 nmol μl−1 could be successfully employed here, as the high amount of analytes in the blood generated a favorable analyte/matrix ratio. A similar MALDI-MS-TOF based experiment is already described in literature using undiluted blood with DMAN.10 In our experiments diluting the blood by a factor of 10, improved the homogeneity of the sample spots and the quality of the spectra. The measurements were performed within minutes after taking the blood sample to cause minimal degradation to as many metabolites as possible. We observed a rapid change in color when the diluted blood was mixed with a solution of 9-AA, and no ions could be generated from sample spots prepared from this mixture. It appeared that the oxidizing properties of fresh blood changed the chemical nature of 9-AA and thus prevented any desorption and ionization. The presence of DMAN and DPN, however, provided rich spectra showing signals derived from amino acids, fatty acids, various small peptides, various forms of heme B as well as lysed and intact phospholipids (see Fig. 7). The majority of these metabolites were detected in their deprotonated form, which simplified their identification. Only few cases of chloride adducts (e.g. glucose) and sodiated anions were observed, despite of the abundance of these electrolytes in the sample. Almost 200 metabolite species were detected with DPN, but fewer than half of them could be detected with DMAN, because of its lower sensitivity (see Table 2S in ESI† for a full list of detected compounds). In fact, most of the peptides and phospholipids could only be detected with DPN.§ These experiments corroborate the effectiveness of DPN as matrix and illustrate its potential for metabolomic studies. When used in tandem with classic matrices in positive ion mode, our method could help to provide complete and fast information about the metabolic composition of various biological fluids without any need for complicated sample preparation and/or extraction procedures.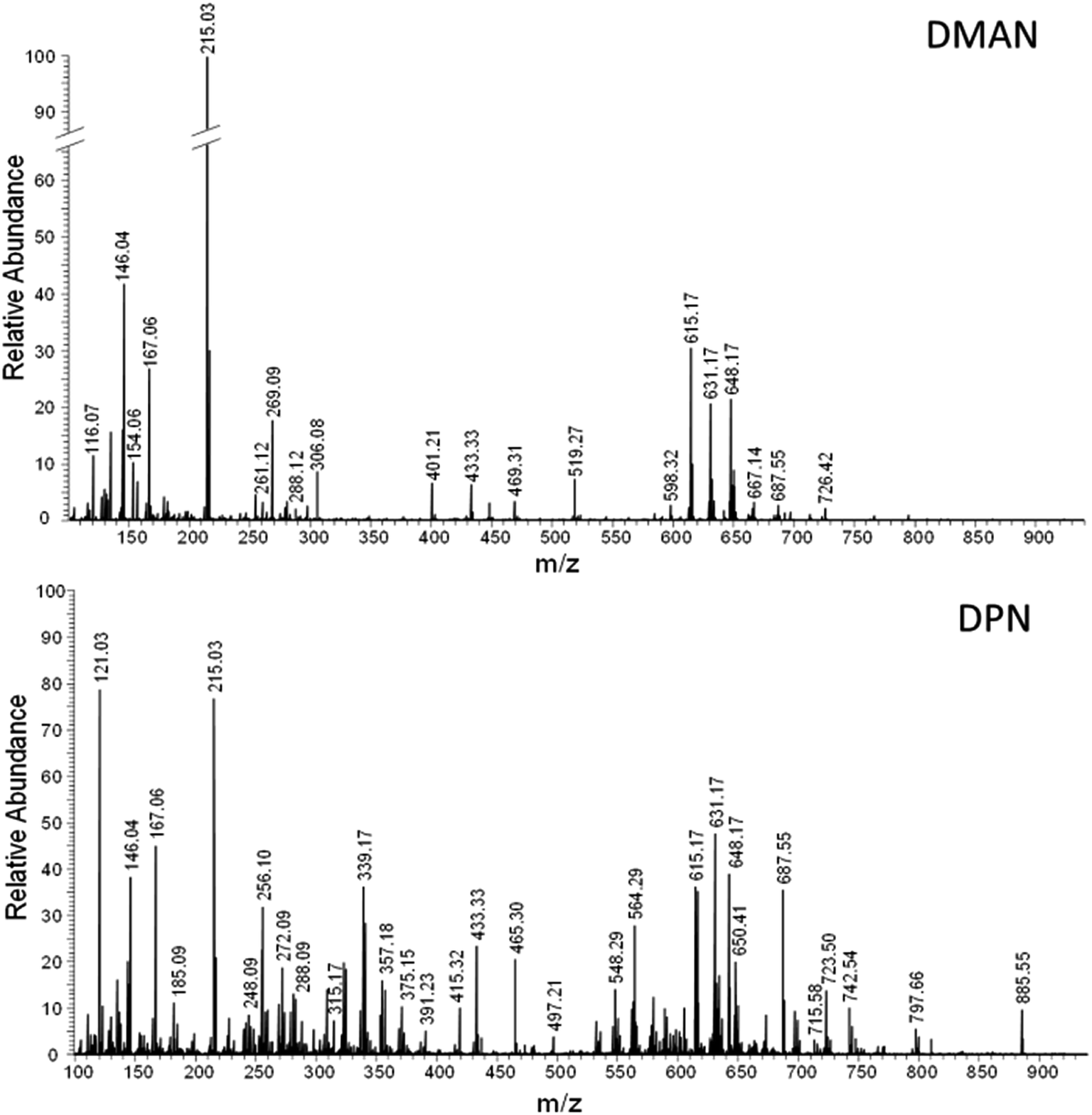 | ||
| Fig. 7 FT-MS spectra of a fresh blood (1:10 diluted) obtained via AP-MALDI using DMAN (top) and DPN (bottom) as matrix (averaged over 40 scans). | ||
Blood was sampled after informed consent was signed and performed according to §6 and §7 of the “Transfusionsgesetz (TFG)”. The blood sample was handled according to the “Schutzmaßnahmen für Tätigkeiten mit biologischen Arbeitsstoffen in Laboratorien (TRBA 100)” regulations. The experiments were approved by ethical committee of Max Planck Institute for Chemical Ecology, Jena, Germany prior the experiments.
Conclusions
In this paper we demonstrated that DPN represents an efficient matrix for negative ion mode MALDI-MS. DMAN was used as a template and ion-pair separation was increased by creating a hydrophobic and bulky shield around the 1,8-dinitrogen center. The charges of the dinitrogen-chelated proton and the resulting anion oxygen are separated by 1.049 Å, for 0.019 Å longer then in DMAN·HOAc. The distance difference corresponds to ca. ΔG = 32 kJ mol−1 gain for DPN. Rationally designed DPN matrix clearly outperforms its “predecessor” DMAN in terms of sensitivity and is even able to compete with established matrices like 9-AA. Efficient UV-absorption and an increased stability in high vacuum conditions further enlarge its applicability. The superior ionization qualities of DPN, especially in the analysis of free fatty acids, lipids and peptides have been proven, confirming its great potential for metabolomic studies in negative ion mode.Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We thank PD Dr Jürgen Schiller (Leipzig University) for providing samples of pure lipids to Lubomir Rulisek from IOCB Prague for quantum calculations, and Max-Planck Society for funding. Also anonymous referees are acknowledged for valuable comments.References
- M. Karas and F. Hillenkamp, Anal. Chem., 1988, 60, 2299–2301 CrossRef CAS PubMed.
- K. Tanaka, H. Waki, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida and T. Matsuo, Rapid Commun. Mass Spectrom., 1988, 2, 151–153 CrossRef CAS.
- L. H. Cohen and A. I. Gusev, Anal. Bioanal. Chem., 2002, 373, 571–586 CrossRef CAS PubMed.
- D. S. Peterson, Mass Spectrom. Rev., 2007, 26, 19–34 CrossRef CAS PubMed.
- M. Rainer, M. N. Qureshi and G. K. Bonn, Anal. Bioanal. Chem., 2011, 400, 2281–2288 CrossRef CAS PubMed.
- T. Jaschinski, K. Thume, M. Klein, P. Richter, J. Popp, A. Svatos and G. Pohnert, Rapid Commun. Mass Spectrom., 2014, 28, 1521–1529 CrossRef CAS PubMed.
- Y. L. Li, M. L. Gross and F. F. Hsu, J. Am. Soc. Mass Spectrom., 2005, 16, 679–682 CrossRef CAS PubMed.
- D. Miura, Y. Fujimura, M. Yamato, F. Hyodo, H. Utsumi, H. Tachibana and H. Wariishi, Anal. Chem., 2010, 82, 9789–9796 CrossRef CAS PubMed.
- R. Shroff and A. Svatos, Rapid Commun. Mass Spectrom., 2009, 23, 2380–2382 CrossRef CAS PubMed.
- R. Shroff, L. Rulisek, J. Doubsky and A. Svatos, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10092–10096 CrossRef PubMed.
- M. Napagoda, L. Rulisek, A. Jancarik, J. Klivar, M. Samal, I. G. Stara, I. Stary, V. Solinova, V. Kasicka and A. Svatos, ChemPlusChem, 2013, 78, 937–942 CrossRef CAS.
- A. Thomas, J. L. Charbonneau, E. Fournaise and P. Chaurand, Anal. Chem., 2012, 84, 2048–2054 CrossRef CAS PubMed.
- C. D. Calvano, T. R. Cataldi, J. F. Kögel, A. Monopoli, F. Palmisano and J. Sundermeyer, Rapid Commun. Mass Spectrom., 2016, 30, 1680–1686 CAS.
- R. W. Alder, M. R. Bryce, N. C. Goode, N. Miller and J. Owen, J. Chem. Soc., Perkin Trans. 1, 1981, 2840–2847 RSC.
- V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, Chem.–Eur. J., 2002, 8, 1682–1693 CrossRef CAS.
- R. Shroff and A. Svatos, Anal. Chem., 2009, 81, 7954–7959 CrossRef CAS PubMed.
- M. Eibisch, R. Suss and J. Schiller, Rapid Commun. Mass Spectrom., 2012, 26, 1573–1576 CrossRef CAS PubMed.
- K. Teuber, J. Schiller, B. Fuchs, M. Karas and T. W. Jaskolla, Chem. Phys. Lipids, 2010, 163, 552–560 CrossRef CAS PubMed.
- G. Sun, K. Yang, Z. Zhao, S. Guan, X. Han and R. W. Gross, Anal. Chem., 2008, 80, 7576–7585 CrossRef CAS PubMed.
- B. Fuchs, A. Bischoff, R. Suss, K. Teuber, M. Schurenberg, D. Suckau and J. Schiller, Anal. Bioanal. Chem., 2009, 395, 2479–2487 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Contains additional mass spectra of fatty acids recorded with TMGN, compounds 4, 6, 7 and 9-AA as matrices (Fig. S1 and S3), LDI-spectra of DPN (Fig. S2), a response curve for stearic acid recorded with DPN (Fig. S4) and a mass spectrum of glucose recorded with DPN (Fig. S5). Furthermore, it contains additional UV-Vis-spectra of pure DPN and DPN cocrystallized with acetic acid (Fig. S6), AP-MALDI and ESI-FTMS spectra of PE 36:1 and PC 34:1 and a complete list of assigned peaks of Fig. S7 (Table S1). See DOI: 10.1039/c6ra17237g |
| ‡ DPN is best used in concentrations between 100 and 2000 pmol μl−1 for MALDI-TOF-MS. Although higher concentrations yield improved signal intensities of the analytes, additional signals were occasionally observed in the spectra which were identified as oxidation products of DPN, that is, these signals represent impurities in the matrix. |
| § Several oxidization products of DPN were detected in the range of 300 to 380 m/z, yet all of them could be easily identified via their accurate mass. We assume that they result from chemical reaction with oxygenated heme. |
| This journal is © The Royal Society of Chemistry 2016 |