
Structural, optical and photo-electrochemical properties of hydrothermally grown ZnO nanorods arrays covered with α-Fe2O3 nanoparticles†
Abstract
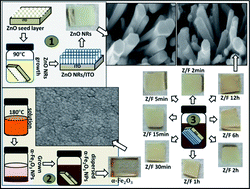
A low cost hydrothermal method and subsequent wet-chemical process has been used for the preparation of a ZnO nanorod (NR) array film grown on tin doped indium oxide (ITO) coated glass substrates, post decorated by α-Fe2O3 nanoparticles (NPs). The effect of the amount of α-Fe2O3 NPs on the properties of ZnO NRs such as electron scattering, surface roughness, photo-charge transport and interface recombination has been investigated. Furthermore, the optimized α-Fe2O3 NPs@ZnO exhibited enhanced light absorption, charge separation and charge transport properties, as characterized by UV-vis absorption spectra and the photo-electrochemical characterization by Linear Sweep Voltammetry (LSV) and Electrochemical Impedance Spectroscopy (EIS). An optimum amount of α-Fe2O3 NPs on the ZnO NRs enhanced the photocurrent density by 163% and the photo-conversion efficiency by more than 2.5 times compared to those of the bare ZnO NRs. These results indicated that the prepared α-Fe2O3 NPs@ZnO NRs could serve as an efficient photo-electrode material for photo-electrochemical cells.
 Please wait while we load your content...
Please wait while we load your content...

