
Back to the oligomeric state: pH-induced dissolution of concanavalin A amyloid-like fibrils into non-native oligomers†
Abstract
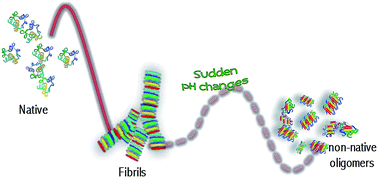
The subtle interplay between long range electrostatic forces, hydrophobic interactions and short range protein–protein interactions regulates the onset/evolution of protein aggregation processes as well as the stability of protein supramolecular structures. Using a combination of FTIR spectroscopy, light scattering and advanced imaging, we present evidence on the main role of electrostatic forces in the formation and stability of amyloid-like fibrils formed from concanavalin A (ConA), a protein showing structural homology with the human serum amyloid protein. At high protein concentration, where protein–protein interactions cannot be neglected, we highlight a thermal-induced aggregation pathway in which amyloid-like aggregates are readily formed. When dissolved in solutions at different pHs, these aggregates show either a reduced β-sheet content keeping the same morphology (3 < pH < 10) or they are promptly dissolved leaving in solution non-native oligomers (pH > 10). The latter result can be ascribed to the change of the charge state for the ConA amino acid side chain with high pKa values. Our results support the idea of fibrils as a reservoir of oligomeric species that can be released if changes or discontinuities in the aggregate microenvironment occur.
 Please wait while we load your content...
Please wait while we load your content...

