
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trifluoromethylation of haloarenes with a new trifluoro-methylating reagent Cu(O2CCF2SO2F)2†
Gang
Zhao‡
,
Hao
Wu‡
,
Zhiwei
Xiao
,
Qing-Yun
Chen
* and
Chao
Liu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: chaoliu@sioc.ac.cn
First published on 5th May 2016
Abstract
A new trifluoromethylating reagent Cu(O2CCF2SO2F)2, which easily decomposes to generate active CuCF3 species in DMF at room temperature, has been conveniently prepared from inexpensive starting materials on a large scale. This new reagent can be applied to efficiently trifluoromethylate a variety of haloarenes under mild conditions, providing good-to-excellent yields of the desired products.
1. Introduction
Trifluoromethyl-containing aromatic compounds are becoming increasingly important as privileged structural motifs in pharmaceuticals, agrochemicals and drug candidates due to the unique properties arising from the electron-withdrawing nature, unique lipophilicity and metabolic stability of the trifluoromethyl groups.1 Hence, considerable effort has been made to synthesise them and considerable progress has been achieved in recent decades.2 Representative aromatic trifluoromethylation during the last few decades includes transition-metal-promoted nucleophilic trifluoromethylation2a–d,g–j as well as radical trifluoromethylation.2e,f,j Among these methodologies, copper-mediated aromatic trifluoromethylation has been most extensively studied and applied2,3 due to its good reactivity, relatively low cost of copper and good regiospecificity.4 Various CuCF3 reagents preformed or generated in situ from a variety of CF3 sources have been developed recently and demonstrated good reactivity towards haloarenes or their analogues.2 For example, Vicic,5 Hartwig,6 Grushin7 and Weng8 successively reported relatively stable and well defined complexes of (L)nCuCF3 based on Me3SiCF3 by treatment with a Cu(I) salt and an appropriate ligand. Grushin's group subsequently developed the first reaction of direct cupration of fluoroform by treatment with CuCl and KOtBu followed by stabilization with Et3N·3HF to generate highly active CuCF3 reagents.9 Hu10 and Mikami11 independently reported the preparation of CuCF3 reagent from cuprate and phenyl trifluoromethyl sulfones and trifluoromethyl ketone derivatives, respectively. Xiao found that difluorocarbene derived from various carbene precursors could be decomposed by DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) to generate a fluoride ion and further converted to CuCF3 in the presence of a Cu(I) salt to efficiently finish trifluoromethylation with iodoarenes.12 Zhang reported a new reagent trimethylsilyl chlorodifluoroacetate for the efficient synthesis of trifluoromethyl-substituted arenes.13 Very recently, Weng reported the synthesis and trifluoromethylation reactions of a decarboxylative trifluoromethylating reagent (phen)Cu(O2CCF3) (phen = 1,10-phenanthroline).14 Despite these notable accomplishments, it is still desirable to develop efficient and convenient reagents to access various trifluoromethylated arenes under mild conditions.Because of our long-standing interest in various trifluoromethylation reactions, we have always been turning our attention and putting effort toward developing new trifluoromethylation methods, particularly by using tetrafluoroethene β-sultone-based fluorinating derivatives as reagents.15 Tetrafluoroethylene β-sultone is a unique cyclic compound and can be conveniently synthesized from the reaction between cheap CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2 and SO3 in nearly quantitative yield.16 Dupont commercialized perfluorinated Nafion polymer (Nafion™), a key source of pendant perfluoroalkylsulfonate groups, which has been widely manufactured and used as a basic and important industrial product.17 Hence, fluorinating reagents derived from it are inexpensive and readily available in large quantities. In 1989, to the best of our knowledge, we developed the first catalytic trifluoromethylation reaction of haloarenes with FSO2CF2COOMe (ref. 17) in DMF at 60–80 °C in the presence of catalytic amounts of CuI, leading to various trifluoromethylated arenes in good yields.3h Later on, several related trifluoromethylating reagents were developed to efficiently furnish a variety of desired trifluoromethylated arenes.15 It has been proposed that the reaction pathway involves the formation of the corresponding Cu(I)(O2CCF2SO2F) as the intermediate, followed by decarboxylation and generation of difluorocarbene, which in combination with a fluoride ion produces a CF3Cu species in the presence of Cu(I) (Scheme 1).3h,15 However, this Cu(I)(O2CCF2SO2F) has not been isolated or detected spectroscopically in the reaction system. We then sought to isolate and characterize the intermediate Cu(I)(O2CCF2SO2F). During the study, it was found that fluorosulfonyldifluoroacetic acid copper(II) salt Cu(II)(FSO2CF2COO)2,19 instead of Cu(I)(O2CCF2SO2F), was easily prepared in a large quantity and could be used as an efficient and mild trifluoromethylating agent. Herein, we present the results.
CF2 and SO3 in nearly quantitative yield.16 Dupont commercialized perfluorinated Nafion polymer (Nafion™), a key source of pendant perfluoroalkylsulfonate groups, which has been widely manufactured and used as a basic and important industrial product.17 Hence, fluorinating reagents derived from it are inexpensive and readily available in large quantities. In 1989, to the best of our knowledge, we developed the first catalytic trifluoromethylation reaction of haloarenes with FSO2CF2COOMe (ref. 17) in DMF at 60–80 °C in the presence of catalytic amounts of CuI, leading to various trifluoromethylated arenes in good yields.3h Later on, several related trifluoromethylating reagents were developed to efficiently furnish a variety of desired trifluoromethylated arenes.15 It has been proposed that the reaction pathway involves the formation of the corresponding Cu(I)(O2CCF2SO2F) as the intermediate, followed by decarboxylation and generation of difluorocarbene, which in combination with a fluoride ion produces a CF3Cu species in the presence of Cu(I) (Scheme 1).3h,15 However, this Cu(I)(O2CCF2SO2F) has not been isolated or detected spectroscopically in the reaction system. We then sought to isolate and characterize the intermediate Cu(I)(O2CCF2SO2F). During the study, it was found that fluorosulfonyldifluoroacetic acid copper(II) salt Cu(II)(FSO2CF2COO)2,19 instead of Cu(I)(O2CCF2SO2F), was easily prepared in a large quantity and could be used as an efficient and mild trifluoromethylating agent. Herein, we present the results.
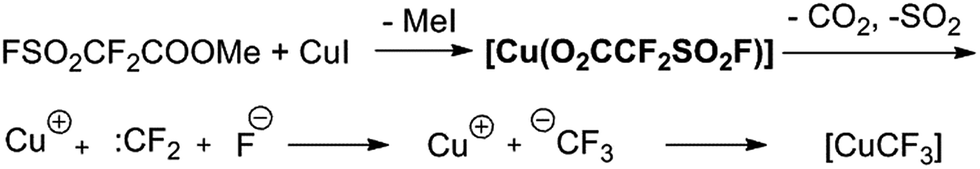 | ||
| Scheme 1 Proposed decomposition mechanism of FSO2CF2COOMe in the presence of CuI in the literature. | ||
2. Results and discussion
At the beginning, we became aware of the potential to synthesize and isolate the key intermediate Cu(I)(O2CCF2SO2F) as a new trifluoromethylating reagent. Attempts were carried out to prepare it by mixing FSO2CF2COOH (ref. 18) with excess amounts of the corresponding Cu2O in Et2O. The color of the contents turned to dark-blue after stirring for 24 hours at room temperature. After filtration and evaporation of the solvent Et2O and water generated during the reaction, a blue powder was obtained (Scheme 2). To our surprise, it was not the desired Cu(I)(O2CCF2SO2F) but Cu(II)(O2CCF2SO2F)2 after thorough characterization by 19F NMR spectroscopy, elemental analysis and X-ray structural analysis. The redox processes involved in the reaction was unclear at this stage. The 19F NMR spectrum shows a typical paramagnetic resonance due to the Cu(II) center. The single crystals obtained by solvent diffusion (Et2O/hexane) were subjected to X-ray structural analysis. The copper(II) center is bound by two fluorosulfonyldifluoroacetic ions and four water molecules as ligands and demonstrates an axially-elongated octahedron configuration. As expected, the reaction of FSO2CF2COOH and a copper(II) salt Cu2(OH)2CO3 under similar conditions afforded the corresponding Cu(O2CCF2SO2F)2 in almost quantitative yield. The reaction was run on a >50 gram scale, and the procedure was very simple. Both starting materials are commercially available and inexpensive. As such, Cu(O2CCF2SO2F)2 is very convenient to prepare on a large scale.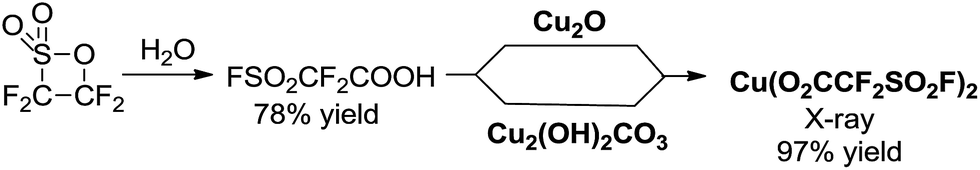 | ||
| Scheme 2 Preparation of Cu(O2CCF2SO2F)2. | ||
Cu(O2CCF2SO2F)2 is a stable and hygroscopic blue powder. No obvious decomposition occurred after storage for more than one month at 4 °C under dry air, and it displayed reactivity similar to that of a freshly prepared material. It is relatively stable in low polarity solvents such as CH2Cl2, acetone, toluene, THF and Et2O at room temperature. However, it decomposes quickly in highly polar solvents such as CH3CN, MeOH, DMF and DMSO at room temperature. Notably, it is highly soluble in Et2O and THF, which is beneficial for the work-up. To our delight, both [Cu(CF3)] (δ = −26.6 ppm) and [Cu(CF3)2−] (δ = −31.3 ppm) species were detected in the reaction solution of Cu(O2CCF2SO2F)2 in DMF according to the 19F NMR spectra, which matched well with the previous reported results.3f,9,20
To assess the potential of Cu(O2CCF2SO2F)2 as a trifluoromethylation reagent, its reactivity with 1-iodonaphthalene (1a) was investigated under various conditions. As shown in Table 1, treatment of 1a with 1.5 equivalent of Cu(O2CCF2SO2F)2 in DMF at room temperature for 3 hours resulted in smooth formation of the desired 1-trifluoromethylnaphthalene (2a) in 50% yield. As the Cu(I)CF3 species is widely considered to be the real active intermediate, we attempted to convert Cu(II) to Cu(I) by adding an equal equivalent of Cu in the reaction system. To our delight, the reaction yield increased significantly to 96% in the presence of an equal equivalent of Cu. A survey of stoichiometry of Cu(O2CCF2SO2F)2/Cu and reaction temperatures showed that 1.5 equivalents and room temperature were the best choices. DMF stood out to be a more effective solvent than DMSO, DMAc, NMP and CH3CN. It should be mentioned that in some cases 1-(pentafluoroethyl)naphthalene was observed as a side-product in trace amounts. The role of some additives was also investigated. KF, NEt3 and DBU had no significant effect on the reaction. In addition, prolonging the reaction time to 12 hours was not beneficial. In brief, a combination of 1.5 equivalents of Cu(O2CCF2SO2F)2 and Cu in DMF at room temperature for 3 hours was established as the optimal reaction conditions for the trifluoromethylation.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Additives | Yieldb (2a) |
| a Reaction conditions: 1-iodonaphthalene (0.2 mmol, 1.0 equiv.), Cu(FSO2CF2COO)2 (0.3 mmol, 1.5 equiv.), Cu (0.3 mmol, 1.5 equiv.) in solvent (2 mL) under a nitrogen atmosphere for 3 hours at the reaction temperatures indicated above. b Yields were determined by 19F NMR with trifluorotoluene as an internal standard. c Without Cu. d No additive was used. e 2.5 equiv. of Cu(O2CCF2SO2F)2/Cu was used. f 1.0 equiv. of Cu(O2CCF2SO2F)2/Cu was used. g Reaction time: 12 hours. | ||||
| 1c | DMF | 25 | —d | 50% |
| 2 | DMF | 25 | —d | 96% |
| 3e | DMF | 25 | —d | 96% |
| 4f | DMF | 25 | —d | 85% |
| 5g | DMF | 25 | —d | 85% |
| 6 | DMF | 0 | —d | 74% |
| 7 | DMF | −5 | —d | 68% |
| 8 | DMF | −10 | —d | 57% |
| 9 | DMF | −20 | —d | 30% |
| 10 | DMSO | 25 | —d | 0 |
| 11 | NMP | 25 | —d | 82% |
| 12 | DMAc | 25 | —d | 83% |
| 13 | CH3CN | 25 | —d | 0 |
| 14 | DMF | 25 | KF | 90% |
| 15 | DMF | 25 | Et3N | 96% |
| 16 | DMF | 25 | DBU | 87% |
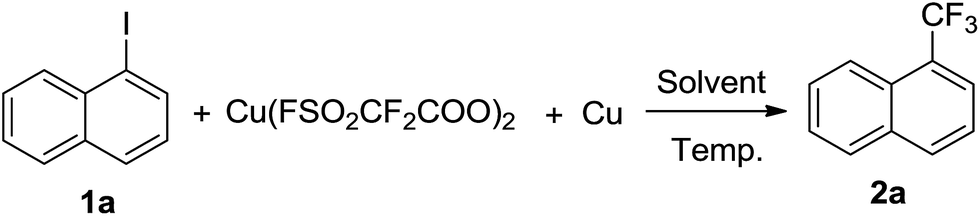
Under these optimal reaction conditions, the scope of iodoarenes was carefully examined. As shown in Scheme 3, a variety of aryl iodides could be subjected to trifluoromethylation under mild conditions, giving the corresponding trifluoromethylated products in good-to-excellent yields. In general, electron-deficient aryl iodides (1h–k, 1n–q) demonstrated better reactivity and yields than electron-rich aryl iodides (1b–g). Remarkably, the enhanced reactivity of ortho-substituted haloarene substrates was observed (1b, 1e, 1l, 1n, 1p), which is consistent with the previously reported ortho-effect.9b,c This ortho-effect is characteristic of copper-mediated aromatic substitution reactions in general. Moreover, ether, nitro, acetyl, halide, ester and nitrile were tolerated under the standard conditions. In addition, we explored the scope of the trifluoromethylation reaction by utilizing heteroaryl iodides. Iodopyridenes, iodopyrimidines, iodoimidazole and iodothiofuran (1r–1x) were all suitable substrates, resulting in the desired trifluoromethylated products in good yields. In addition, it should be noted that in many cases, the isolated yields of the desired trifluoromethylated products were much lower than what could be detected by NMR spectroscopy, which may be attributed to their high volatility or similar polarity to the starting iodoarenes.
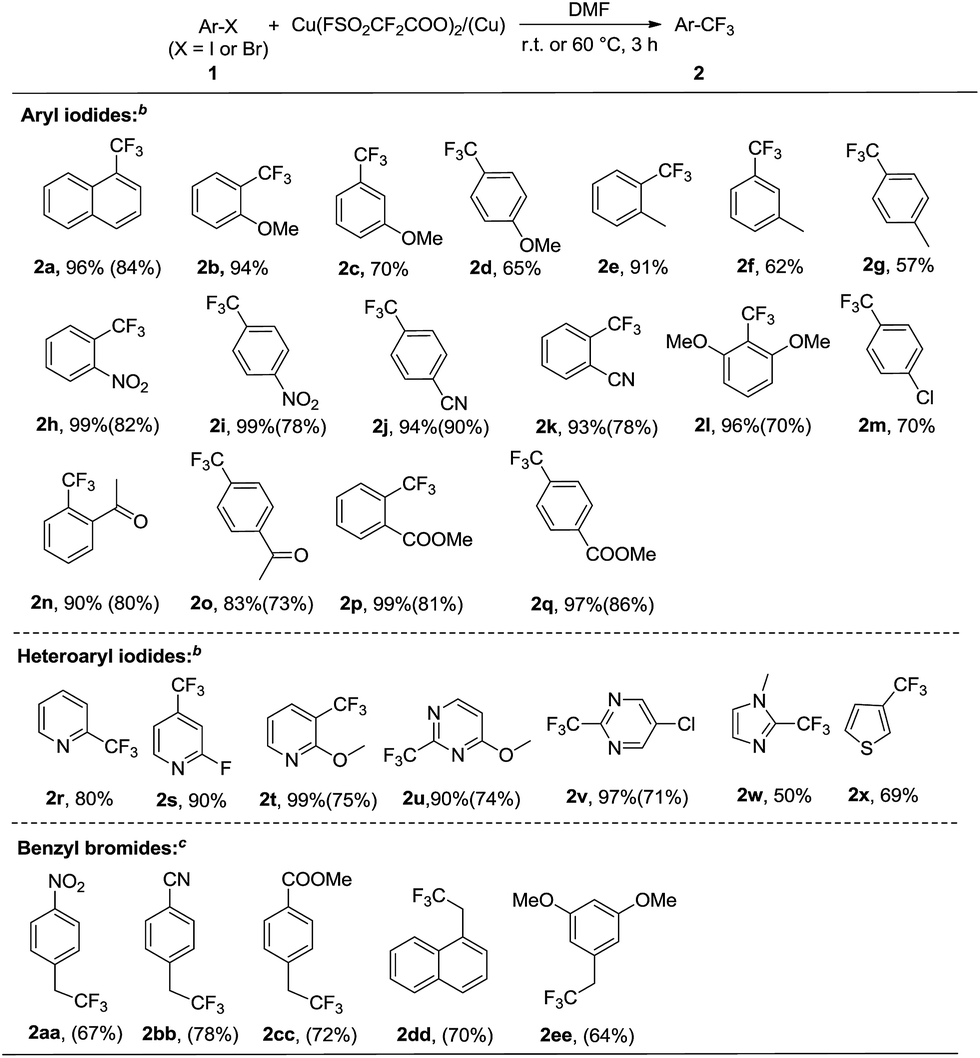 | ||
| Scheme 3 Scope of the trifluoromethylation of various iodoarenesa. aYields were determined by 19F NMR with trifluorotoluene or 4-(trifluoromethoxy)benzotrifluoride as the internal standard. Isolated yields are given in parentheses. bReaction conditions: substrate 1 (0.4 mmol, 1.0 equiv.), Cu(O2CCF2SO2F)2 (0.6 mmol, 1.5 equiv.), Cu (0.6 mmol, 1.5 equiv.) in DMF (4 mL) under a nitrogen atmosphere at room temperature for 3 hours. cReaction conditions: substrate 1 (0.5 mmol, 1.0 equiv.), Cu(O2CCF2SO2F)2 (1.0 mmol, 2 equiv.) in DMF (5 mL) under nitrogen atmosphere at 60 °C for 3 hours. | ||
We next extended trifluoromethylation on various benzyl bromides under the abovementioned standard conditions. Although the reaction proceeded smoothly and successfully produced the desired product, the yield was not good (<30%). After further screening of the reaction conditions, it was found that the trifluoromethylation reactions of benzyl bromides proceeded better without the use of copper and at higher reaction temperatures (60 °C). Various benzyl bromides (Scheme 3, 1aa–ee) were efficiently and conveniently subjected to smooth trifluromethylation under these conditions, providing the desired products in good yields (Scheme 3).
Subsequently, a comparison of the reactivity of the new trifluoromethylating reagent Cu(O2CCF2SO2F)2 with our previously reported trifluoromethylating reagent FSO2CF2COOMe (ref. 3h and 18) with various haloarenes was performed (Scheme 4). The results clearly suggest that Cu(O2CCF2SO2F)2 demonstrates higher reactivity and better yields than FSO2CF2COOMe.
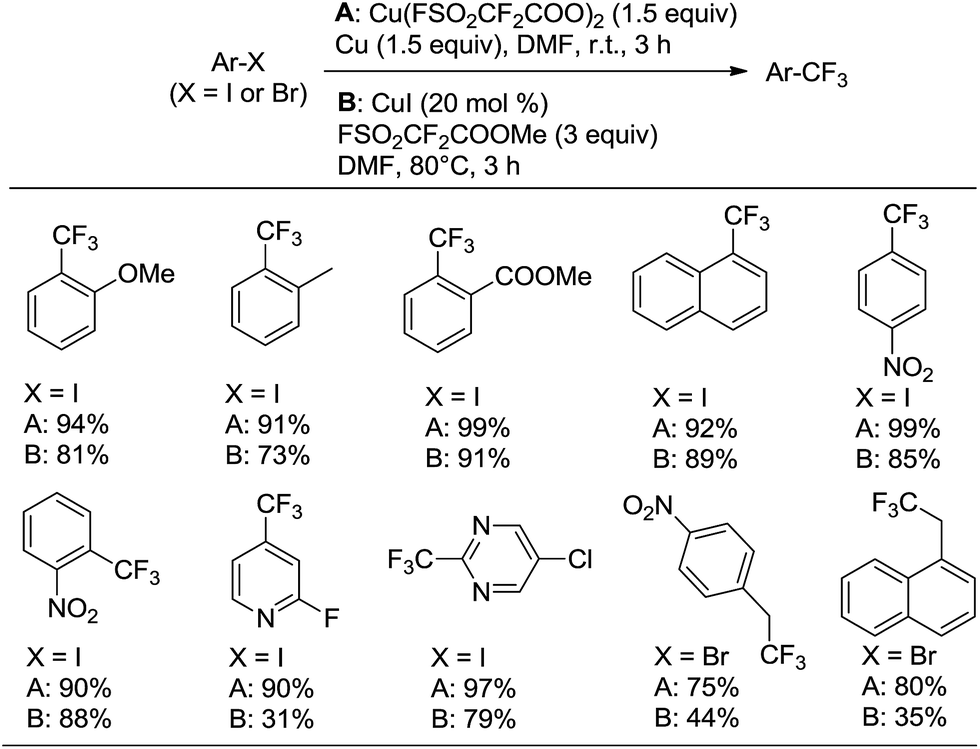 | ||
| Scheme 4 Comparison of trifluoromethylating reagent Cu(O2CCF2SO2F)2 with FSO2CF2COOMe on various haloarenes. Yields were determined by 19F NMR with trifluorotoluene or 4-(trifluoromethoxy)benzotrifluoride as the internal standard. | ||
To determine the mechanism of trifluoromethylation with Cu(O2CCF2SO2F)2, we first carefully examined the decomposition of Cu(O2CCF2SO2F)2 in DMF using 19F NMR analysis (see the ESI†). A singlet peak at −31.3 ppm assigned to [Cu(CF3)2]− was detected in 10 minutes. One hour later, a broad singlet peak at −26.5 ppm, which is considered to be the reactive mono-CuCF3 species according to the literature,3f,9,20 appeared and the intensity of the signal at −31.3 ppm decreased. Two hours later, the singlet at −26.5 ppm further increased, whereas the signal at −31.3 ppm further decreased. Based on these experimental results and the literature, we proposed a plausible mechanism (Scheme 5). First, Cu(O2CCF2SO2F)2 in DMF decomposes into Cu2+, :CF2 and fluoride with the release of SO2 and CO2. Then, difluorocarbene and fluoride combine into a CuCF3 species in the presence of Cu2+ and Cu. CuCF3 generated in situ reacts with the haloarenes to provide the desired trifluoromethylated products. It should be mentioned that CuCF3 generated in our reaction system is a ‘ligandless’ species according to the stoichiometry of the overall process and has excellent reactivity, which is in agreement with the literature.12
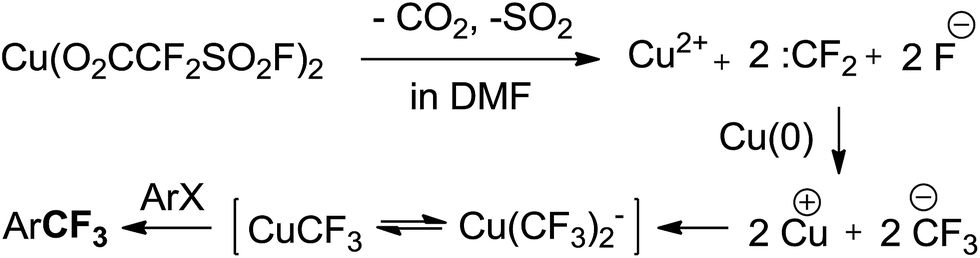 | ||
| Scheme 5 Proposed mechanism of trifluoromethylation reactions with Cu(O2CCF2SO2F)2. | ||
3. Conclusions
In summary, we developed a new trifluoromethylating reagent Cu(O2CCF2SO2F)2, which efficiently trifluoromethylates various haloarenes under mild conditions. It is a blue solid and can be conveniently prepared from inexpensive starting materials on a large scale. Its decomposition in DMF at room temperature readily affords highly active CuCF3 species, and the corresponding trifluoromethylation reactions of various haloarenes proceeded smoothly and efficiently at room temperature in good to excellent yields with good functional group compatibility. Moreover, there is no need for an added ligand, which is in contrast with the vast majority of previously reported copper-mediated trifluoromethylation reactions of haloarenes in which the reaction proceeds efficiently only in the presence of a special ligand. Further investigation on the other fluorosulfonyldifluoroacetic acid metal salts is ongoing in our lab, and the results will be reported in due time.Acknowledgements
The authors gratefully acknowledge the financial support from the National Basic Research Program of China (2012CB821600) and the National Natural Science Foundation of China (21032006, 21302207, 21421002).Notes and references
-
(a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC
; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. D. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed
- Selected recent reviews:
(a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475–4521 CrossRef CAS PubMed
- Selective examples:
(a) V. C. R. McLoughlin and J. Thrower, Tetrahedron, 1969, 25, 5921 CrossRef CAS
- For related reviews, see:
(a) D. J. Burton and Z. Y. Yang, Tetrahedron, 1992, 48, 189 CrossRef CAS
-
(a) G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600–8601 CrossRef CAS PubMed
-
(a) H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793–3798 (
Angew. Chem.
, 2011
, 123
, 3877–3882
) CrossRef CAS PubMed
- O. A. Tomashenko, E. C. Escudero-Adan, M. M. Belmonte and V. V. Grushin, Angew. Chem., Int. Ed., 2011, 50, 7655–7659 (
Angew. Chem.
, 2011
, 123
, 7797–7801
) CrossRef CAS PubMed
- Y. Liu, C. Chen, H. Li, K.-W. Huang, J. Tan and Z. Weng, Organometallics, 2013, 125, 1588–1592 Search PubMed
-
(a) A. Zanardi, M. A. Novikov, E. Martin and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901–20913 CrossRef CAS PubMed
- X. Li, J. Zhao, L. Zhang, M. Hu, L. Wang and J. Hu, Org. Lett., 2015, 17, 298–301 CrossRef CAS PubMed
- H. Serizawa, K. Aikawa and K. Mikami, Chem.–Eur. J., 2013, 19, 17692–17697 CrossRef CAS PubMed
- J. Zheng, J.-H. Lin, X.-Y. Deng and J.-C. Xiao, Org. Lett., 2015, 17, 532–535 CrossRef CAS PubMed
- X. Zhang, J. Wang and Z. Wan, Org. Lett., 2015, 17, 2086–2089 CrossRef CAS PubMed
- X. Lin, C. Hou, H. Li and Z. Weng, Chem.–Eur. J., 2016, 22, 2075–2084 CrossRef CAS PubMed
-
(a) Q.-Y. Chen, J. Fluorine Chem., 1995, 72, 241–246 CrossRef CAS
-
(a) I. L. Knunyants and G. A. Sokolski, Angew. Chem., Int. Ed., 1972, 11, 583–595 CrossRef CAS
-
M. Yamabe and H. Miyake, in Organofluorine Chemistry Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Plenum Press, New York and London, 1994, pp. 403–411 Search PubMed
- FSO2CF2COOMe (CAS No. 680-15-9) and FSO2CF2COOH (CAS No. 1717-59-5) are easily prepared on scale by reaction of tetrafluoroethylene β-sultone with methanol and water, respectively (for detailed preparation procedures, see the ESI†). They are also commercially available, such as from Sigma-Aldrich.
-
Q.-Y. Chen, G. Zhao, C. Liu, Y. Guo, J.-C. Xiao, S. Zhao, W. Wang and D. Jiang, Preparation and application of new trifluoromethylating reagents, CN201310077629.1, March 11 2013
- M. Hu, C. Ni and J. Hu, J. Am. Chem. Soc., 2012, 134, 15257–15260 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1450534. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra09011g |
| ‡ G. Z. and H. W. contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |