
Synthesis, photophysics, and reverse saturable absorption of 7-(benzothiazol-2-yl)-9,9-di(2-ethylhexyl)-9H-fluoren-2-yl tethered [Ir(bpy)(ppy)2]PF6 and Ir(ppy)3 complexes (bpy = 2,2′-bipyridine, ppy = 2-phenylpyridine)†
Zhongjing Li‡
,
Hui Li‡,
Brendan J. Gifford,
Wadumesthrige D. N. Peiris,
Svetlana Kilina and
Wenfang Sun*
Department of Chemistry and Biochemistry, North Dakota State University, Fargo, ND 58108-6050, USA. E-mail: wenfang.sun@ndsu.edu; Tel: +1-701-231-6254
First published on 18th April 2016
Abstract
We report the synthesis, photophysics, and reverse saturable absorption of five iridium(III) complexes 1–5 with 7-(benzothiazol-2-yl)-9,9-di(2-ethylhexyl)-9H-fluoren-2-yl (BTF) pendant attached to the 2-phenylpyridine ligand (1: [Ir(bpy)(BTF-ppy)2]PF6; 2: [Ir(bpy)(BTF–![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) –ppy)2]PF6; 3: Ir(ppy)2(BTF-ppy); 4: Ir(ppy)(BTF-ppy)2; 5: (BTF-ppy)3, where bpy = 2,2′-bipyridine and ppy = 2-phenylpyridine). The effects of the extended π-conjugation of the ppy ligand and the increased number of BTF-ppy ligand, as well as the effects of neutral complex vs. cationic complex were evaluated. All complexes exhibit predominantly ligand-localized 1π,π* transitions below 430 nm and charge-transfer transitions between 430 and 550 nm. They all emit at room temperature and at 77 K, mainly from the metal-to-ligand charge transfer (3MLCT)/ligand-to-ligand charge transfer (3LLCT) states for 1 and 2, and from the BTF-ppy ligand-centered 3π,π* excited states with significant contributions from the 3MLCT states for 3–5. The triplet excited states of 1–5 also manifest broad transient absorption (TA) in the visible to the near-IR spectral region. The electronic absorption, emission, and ns transient absorption are all red-shifted by extending the π-conjugation of the ppy ligand or increasing the number of BTF-ppy ligand. The energies of the lowest singlet and triplet excited states of the neutral complex 4 are lowered compared to those of its cationic counterpart 1; while the transient absorbing triplet excited state of 4 is much longer lived than that of 1. These complexes all exhibit strong reverse saturable absorption (RSA) for ns laser pulses at 532 nm, with a trend of 5 < 4 < 1 ≈ 3 < 2. This trend is primarily determined by the ratio of the excited-state absorption cross section to that of the ground state (σex/σ0) at 532 nm with the triplet quantum yield also playing a role for complexes 3–5. It appears that the increased number of BTF-ppy ligand reduces the RSA of the neutral complexes while the increased π-conjugation of the ppy ligand in the cationic complexes improves the RSA. However, the neutral complex 4 exhibits a weaker RSA at 532 nm than its cationic counterpart 1.
–ppy)2]PF6; 3: Ir(ppy)2(BTF-ppy); 4: Ir(ppy)(BTF-ppy)2; 5: (BTF-ppy)3, where bpy = 2,2′-bipyridine and ppy = 2-phenylpyridine). The effects of the extended π-conjugation of the ppy ligand and the increased number of BTF-ppy ligand, as well as the effects of neutral complex vs. cationic complex were evaluated. All complexes exhibit predominantly ligand-localized 1π,π* transitions below 430 nm and charge-transfer transitions between 430 and 550 nm. They all emit at room temperature and at 77 K, mainly from the metal-to-ligand charge transfer (3MLCT)/ligand-to-ligand charge transfer (3LLCT) states for 1 and 2, and from the BTF-ppy ligand-centered 3π,π* excited states with significant contributions from the 3MLCT states for 3–5. The triplet excited states of 1–5 also manifest broad transient absorption (TA) in the visible to the near-IR spectral region. The electronic absorption, emission, and ns transient absorption are all red-shifted by extending the π-conjugation of the ppy ligand or increasing the number of BTF-ppy ligand. The energies of the lowest singlet and triplet excited states of the neutral complex 4 are lowered compared to those of its cationic counterpart 1; while the transient absorbing triplet excited state of 4 is much longer lived than that of 1. These complexes all exhibit strong reverse saturable absorption (RSA) for ns laser pulses at 532 nm, with a trend of 5 < 4 < 1 ≈ 3 < 2. This trend is primarily determined by the ratio of the excited-state absorption cross section to that of the ground state (σex/σ0) at 532 nm with the triplet quantum yield also playing a role for complexes 3–5. It appears that the increased number of BTF-ppy ligand reduces the RSA of the neutral complexes while the increased π-conjugation of the ppy ligand in the cationic complexes improves the RSA. However, the neutral complex 4 exhibits a weaker RSA at 532 nm than its cationic counterpart 1.
Introduction
The spin–orbit coupling constant of iridium is among the largest in transition metals,1 which leads to fast intersystem crossing to the triplet excited state and high phosphorescence efficiency for iridium(III) complexes. Extensive research has been carried out in utilizing the photoluminescence properties of iridium(III) complexes as dopant for organic light-emitting diodes (OLEDs)2–4 and light-emitting electrochemical cells (LEECs).5–10 Other applications of Ir(III) complexes, including therapeutic and bioimaging,11,12 oxygen sensing,13 photo catalytic water splitting,14 photoredox catalysis,15 and solution electroluminescence16 are also explored.It is well known that the electronic structure of Ir(III) complexes can be efficiently controlled by deliberate structural modifications.14,17 By using different types of diimine or cyclometalating ligands,18 or varying substituent on the ligands,19 the nature of the lowest-energy singlet and triplet excited states can be significantly adjusted. For instance, density functional theory (DFT) calculations and experimental studies carried out by Hay20 revealed that the contribution of Ir(III) d orbital to the low-lying metal-to-ligand charge transfer (MLCT) state varied from 45% to 60% in replacing one 2-phenylpyridine (ppy) ligand in Ir(ppy)3 with acetylacetonate or benzoylacetonate. In another work reported by Watts and Houten, the energy level and lifetime of the emitting 3d,d excited state of the iridium 1,10-phenathroline complexes were found to be readily tuned through modification with different substituents, such as electron withdrawing groups (–NO2 and –Cl) or electron donating group (–CH3) on the 4, 5, 6, or 7 position of the 1,10-phenathroline ligand.21
Further structure–property correlation studies were conducted in more “bulky” molecules with molecular weight up to 3000. In recent reports by Zhao and co-workers, the photophysics of tris-cyclometalated iridium complexes with varied positions or repeating units of the alkylated fluorenyl oligomer on the 2-phenylpyridine ligand was studied.22,23 Spectroscopic studies and DFT calculations revealed that the triplet lifetime increased but the phosphorescence quantum yield decreased with increased chain length of the oligofluorene, due to localization of the triplet excitons. Meanwhile, the contribution of the MLCT state to the emissive excited state is significantly affected by the attachment position of the oligofluorene. Particularly, attaching the trifluorene at the para-position of the cyclometalating carbon of the phenyl ring produced a ligand-centered triplet excited state with long lifetime and moderate quantum yield.
Unlike the extensive study on the light-emitting applications, research on nonlinear absorption (which has potential applications in all-optical switching, laser beam stabilization or compression, optical signal rectification, photodynamic therapy, bioimaging, optical data storage, etc.) of Ir(III) complexes is quite limited. Schanze and co-workers first reported the dual mechanism nonlinear absorption of an Ir(III) complex bearing two 2-phenylpyridine ligands and one 4,4′-(2,2′-bipyridine-5,5′-diylbis(ethyne-2,1-diyl))bis(N,N-dihexylaniline) ligand.24 Strong excited-state absorption in ca. 560–1500 nm region was observed. Meanwhile, the absorbing excited state can be populated by 5 ns, 1064 nm laser via two-photon absorption. Therefore, both one-photon process and two-photon absorption can induce nonlinear transmission by this complex. Our group also reported the photophysics and reverse saturable absorptions of heteroleptic cationic Ir(III) complexes with extended π-conjugation on the diimine and/or the phenylpyridine ligands.25–27 It was found that extending the π-conjugation of the diimine ligand via attaching the BTF motifs can significantly increase the triplet lifetime by admixing the substituted bipyridine-localized 3π,π character into the lowest triplet excited state; while the extended π-conjugation of the phenylpyridine ligand via attaching the BTF motif decreased the triplet lifetime due to predominantly 3MLCT/3LLCT characters in the lowest triplet excited state.25,26 All complexes exhibited very strong RSA at 532 nm for nanosecond laser pulses. These results are quite promising. However, all reported nonlinear absorption work are on the cationic heteroleptic Ir(III) complexes. To date, there has been only one report on the nonlinear absorption of the neutral tris-cyclometalating Ir(III) complexes to the best of our knowledge.28
In this paper, five cationic or neutral Ir(III) complexes 1–5 were synthesized and studied. The structures of these complexes are shown in Chart 1, which feature BTF pendant at the para-position of the cyclometalating carbon of the phenyl ring. The aim of our research is to illustrate the structure–property correlations in neutral and cationic Ir(III) complexes for rational design of nonlinear absorbing materials. Complexes 1 and 2 contain one bipyridine ligand and two BTF substituted 2-phenylpyridine ligands and thus are cationic, while complexes 3–5 bear three 2-phenylpyridine ligands and are neutral. Instead of single bond connection in 1, the BTF unit is connected to the 2-phenylpyridine via a triple bond in 2. In this way, the effect of extended π-conjugation length on the cyclometalating ligands can be evaluated. From 3 to 5, the number of the BTF pendant increases. This variation possibly allows us to evaluate the interplay or mixing between the ligand-centered 3π,π* state predominantly from the BTF substituted cyclometalating ligand and the 3MLCT/3LLCT excited states related to the coordination core. Comparison of the cationic complexes vs. neutral complexes can be realized by assessing the photophysics of 1 and 4, which contain the same cyclometalating ligands but 1 has bipyridine as the 3rd ligand, while 4 has 2-phenylpyridine as the 3rd ligand. The much stronger ligand field of 2-phenylpyridine can induce different emission and absorption characteristics. It should be noted that although complexes 1–5 all contain the BTF substituted 2-phenylpyridine ligands, which appears to be similar to some of the cationic Ir(III) complexes previously reported by our group,25–27 either the diimine ligand is different from the reported complexes (such as bipyridine in complexes 1 and 2 vs. phenanthroline or the substituted bipyridine or phenanthroline in ref. 25–27) or the triplet bond position is different in the BTF substituted 2-phenylpyridine ligand (such as the case in complex 2) or the formal charges of the complexes are different (i.e. 0 charge in complexes 3–5 vs. +1 charge in the cationic Ir(III) complexes reported in ref. 25–27). Finally, the reverse saturable absorption of these complexes is investigated at 532 nm using ns laser pulses, aiming to evaluate the effect of structural variation on the nonlinear absorption of the Ir(III) complexes. The nature of transitions is characterized computationally using time-dependent density functional theory (TDDFT), thereby supporting the aforementioned effects on the optical properties of the Ir(III) complexes.
 | ||
| Chart 1 Structures of complexes 1–5. | ||
Experimental section
Synthesis and characterization
The synthetic routes for 1–5 are illustrated in Scheme 1. Br-F8-BTZ was synthesized according to literature procedure.29 The synthetic details and characterization data for Br-ppy, B-ppy, 1-L, 2-L, and 1–5 are reported herewith. All the chemicals and solvents were purchased from Aldrich Chemical Co. or Alfa Aesar and used as received. Silica gel for chromatography was purchased from Sorbent Technology (60 Å, 230–400 mesh, 500–600 m2 g−1, pH: 6.5–7.5). Complexes 1–5 and ligands 1-L and 2-L were characterized by 1H NMR, electrospray ionization high-resolution mass spectrometry (ESI-HRMS), and elemental analyses. Other intermediates were characterized by 1H NMR. | ||
| Scheme 1 Synthetic routes for complexes 1–5. | ||
1H NMR was obtained on Varian Oxford-VNMR spectrometers (400 or 500 MHz). ESI-HRMS analyses were performed on a Bruker BioTOF III mass spectrometer. Elemental analyses were conducted by NuMega Resonance Laboratories, Inc. in San Diego, California.
The dried oily product from the previous step, 1,3-dibromobenzene (2.5 g, 10.5 mmol), Pd(PPh3)4 (30 mg), and toluene (30 mL) were added together. The reaction mixture was degassed with argon and heated to reflux for 24 hours. After reaction, the precipitate was removed by filtration. The solvent of the filtrate was then removed by distillation. The resultant dark oil was purified by a silica gel column eluted with hexane and ethyl acetate (v/v = 10/1). 1.7 g yellow oil was collected as the final product (yield: 61% from 2-bromopyridine). 1H NMR (CDCl3, 500 MHz): 8.69–8.70 (m, 1H), 8.19 (s, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.73–7.77 (m, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.32–7.35 (m, 1H), 7.24–7.28 (m, 1H).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CH. The mixture of Br-F8-BTZ (3.2 g, 5.3 mmol), (trimethylsilyl)acetylene (1 mL, 7.1 mmol), Pd(PPh3)2Cl2 (10 mg, 0.014 mmol), PPh3 (20 mg, 0.076 mmol), CuI (2 mg, 0.01 mmol), and Et3N (50 mL) was heated to reflux under argon for 24 hours. After reaction, the precipitate was removed by filtration. After being confirmed by HRMS, the yellow oil obtained after distillation of Et3N was used directly for the next step without further purification.
CH. The mixture of Br-F8-BTZ (3.2 g, 5.3 mmol), (trimethylsilyl)acetylene (1 mL, 7.1 mmol), Pd(PPh3)2Cl2 (10 mg, 0.014 mmol), PPh3 (20 mg, 0.076 mmol), CuI (2 mg, 0.01 mmol), and Et3N (50 mL) was heated to reflux under argon for 24 hours. After reaction, the precipitate was removed by filtration. After being confirmed by HRMS, the yellow oil obtained after distillation of Et3N was used directly for the next step without further purification.The yellow oil collected from the previous step, combined with K2CO3 (3.7 g, 27 mmol) and MeOH (50 mL), were stirred at room temperature for 12 hours. After reaction, water (100 mL) was added and the mixture was extracted with hexane (50 mL × 3). The combined hexane solution was then dried over MgSO4. After the solvent was removed, the red oil was purified by running a silica gel column eluted first with hexane and then with the mixed solvent of hexane and CH2Cl2 (v/v = 3/1). 2.65 g yellow oil was collected as the final product (yield: 91% from Br-F8-BTZ). 1H NMR (CDCl3, 400 MHz): 8.06–8.13 (m, 3H), 7.88–7.91 (m, 1H), 7.76–7.78 (m, 1H), 7.67–7.69 (m, 1H), 7.46–7.54 (m, 3H), 7.35–7.39 (m, 1H), 3.13 (t, J = 2.4 Hz, 1H), 1.97–2.12 (m, 4H), 0.48–0.96 (m, 30H).
CH (1.4 g, 2.6 mmol), Pd(PPh3)2Cl2 (20 mg, 0.028 mmol), PPh3 (50 mg, 0.19 mmol), CuI (5 mg, 0.025), and Et3N (30 mL) was heated to reflux under argon for 24 hours. After reaction, the precipitate was removed by filtration. The solvent was removed by distillation and the resultant brown oil was purified by running a silica gel column. The column was first eluted with CH2Cl2/hexane (v/v = 1/1) then with ethyl acetate/hexane (v/v = 1/3). 0.95 g yellow oil was collected as the final product (yield: 52%). 1H NMR (CDCl3, 400 MHz): 8.70–8.72 (m, 1H), 8.22–8.23 (m, 1H), 8.07–8.14 (m, 3H), 7.95–7.98 (m, 1H), 7.88–7.91 (m, 1H), 7.71–7.79 (m, 4H), 7.54–7.62 (m, 3H), 7.44–7.50 (m, 2H), 7.34–7.38 (m, 1H), 7.22–7.25 (m, 1H), 2.03–2.16 (m, 4H), 0.50–0.96 (m, 30H). HRMS: m/z calc. for [C49H52N2S + H]+: 701.3924; found: 701.3924. Anal. calc. for C49H52N2S·0.4H2O·0.8C6H14: C, 83.15; H, 8.30; N, 3.60. Found: C, 82.79; H, 8.76; N, 4.06.The yellow powder collected, 2,2′-bipyridine (16 mg, 0.1 mmol), AgCF3SO3 (26 mg, 0.1 mmol) and 2-ethoxyethanol (10 mL) were heated to reflux under argon for 48 hours. After the mixture was cooled to room temperature, NH4PF6 (17 mg, 0.1 mmol) was added and the mixture was stirred at room temperature for 3 hours. After the reaction was finished, 50 mL ethyl acetate was added. The combined organic solution was washed with brine (50 mL × 3). After drying over MgSO4 and filtered, the solvent was removed by distillation. Purification was conducted by a flash silica gel column eluted with dichloromethane followed by recrystallization from dichloromethane and hexane. 69 mg orange powder was collected as the final product (yield: 37% for two steps). 1H NMR (CDCl3, 500 MHz): 8.70–8.71 (m, 2H), 8.19–8.23 (m, 2H), 8.07–8.16 (m, 10H), 7.99–8.00 (m, 2H), 7.79–7.93 (m, 8H), 7.62–7.65 (m, 6H), 7.48–7.52 (m, 4H), 7.39 (t, J = 7.5 Hz, 2H), 7.30–7.33 (m, 2H), 7.17 (t, J = 6.5 Hz, 2H), 6.50–6.53 (m, 2H), 2.07–2.20 (m, 8H), 0.51–0.99 (m, 60H). HRMS: m/z calc. for [C104H110N6S2Ir]+: 1699.7866; found: 1699.7893. m/z calc. for [C104H110N6S2Ir + H]2+: 850.3969; found: 850.3998. Anal. calc. for C104H110N6S2IrPF6: C, 67.69; H, 6.01; N, 4.55; S, 3.48. Found: C, 67.63; H, 6.32; N, 4.62; S, 3.80.
It is worth mentioning that during the column purification of complexes 4 and 5, the mer-isomers of 4 (Rf = 0.17 eluted with CH2Cl2) and 5 (Rf = 0.27 eluted with CH2Cl2) that have different 1H-NMR spectra and much shorter emission lifetimes were also obtained. However, the mer-isomer of complex 3 was unable to be obtained under the aforementioned experimental conditions. Because the mer-isomers are not the focus of this work, their characterization data and photophysical properties are not included in this paper and will be reported later.
Photophysical and nonlinear transmission measurements
The solvents used for photophysical experiments were spectroscopic grade, and were purchased from VWR International and used as is without further purification. UV-vis absorption spectra were recorded on a Shimadzu UV-2501 spectrophotometer. A FluoMax-4 fluorometer/phosphorometer was used to measure the steady-state emission spectra in different solvents. The emission quantum yields were determined by the relative actinometry30 in degassed solutions, in which the degassed acetonitrile solution of [Ru(bpy)3]Cl2 (Φem = 0.097, λex = 436 nm)31 was used as the reference for complexes 1–5, and a degassed 1 N sulfuric acid solution of quinine bisulfate (Φem = 0.546, λex = 347.5 nm)32 was used as the reference for ligands 1-L and 2-L.The nanosecond transient difference absorption (TA) spectra and decays were measured in degassed toluene solutions on an Edinburgh LP920 laser flash photolysis spectrometer. The third harmonic output (355 nm) of a Nd:YAG laser (Quantel Brilliant, pulse width: 4.1 ns, repetition rate was set to 1 Hz) was used as the excitation source. Each sample was purged with argon for 30 min prior to measurement. The triplet excited-state absorption coefficient (εT) at the TA band maximum was determined by the singlet depletion method.33 After obtaining the εT value, the ΦT could be determined by the relative actinometry using SiNc in benzene as the reference (ε590 = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1, ΦT = 0.20).34
000 M−1 cm−1, ΦT = 0.20).34
The reverse saturable absorption of complexes 1–5 was characterized by nonlinear transmission experiment at 532 nm using a Quantel Brilliant laser as the light source. The pulse width of the laser was 4.1 ns and the repetition rate was set to 10 Hz. The complexes were dissolved in CH2Cl2. The concentration of the sample solutions was adjusted to obtain a linear transmission of 90% at 532 nm in a 2 mm-thick cuvette. The experimental setup and details are similar to that reported previously.35 A 40 cm plano-convex lens was used to focus the beam to the center of the 2 mm thick sample cuvette. The radius of the beam at the focal plane was approximately 96 μm.
Computational methodology
All calculations were performed using Gaussian-09 software package.36 Density functional theory (DFT) was utilized to determine the optimized singlet ground state geometry of each complex. These calculations were performed using the PBE1PBE37,38 functional with LANL2DZ39,40 and 6-31G(d)41–43 basis set for the metal and ligand atoms respectively. The Conductor Polarized Continuum Model (CPCM)44,45 was utilized to simulate the solvent toluene. Time-dependent density functional theory (TDDFT) calculations were performed from the optimized geometries using the same functional, basis set, and CPCM solvent model. Our previous studies have demonstrated that this methodology reasonably reproduced the absorption and emission spectra of the heteroleptic cationic Ir(III) complexes with extended π-conjugation on the diimine and/or the phenylpyridine ligands.25–27 As such, it is expected to be suitable for complexes 1–5. For calculations of absorption spectra, 70 excited states were calculated. Spectra were generated by broadening the resultant optical transitions with a Gaussian function with line-width of 0.1 eV to fit the experimental spectra. Natural transition orbitals (NTOs) were generated to characterize the nature of the optical transitions in two different energetic regions: 413–600 nm and 250–413 nm.Calculations of the emission energies were carried out using analytical TDDFT gradients,46,47 by optimizing either the lowest singlet (fluorescence) or triplet (phosphorescence) state as implemented in the Gaussian-09 software. The same functional, basis set, and solvent model used for the ground state TDDFT calculations were used for the emission calculations as well. For optimization of the excited state, we started with several different initial guesses for the wavefunction choosing either the very lowest triplet state (root = 1) or the second lowest triplet state (root = 2). Both guessing states were initially taken at the ground state geometry with the triplet spin configuration, where their electronic wavefunctions were calculated by regular TDDFT procedure. By starting with different initial wavefunctions, we were able to converge to different triplet states having similar 3π,π* character but very different energies for complex 3. In contrast, other complexes did not recover different states, resulting in the same type and nearly the same energy of the lowest triplet state, independent on the initial wavefunction configuration.
Results and discussion
Geometries of complexes 1–5
It has been widely reported in the literature that the generally formed configuration for the heteroleptic cationic Ir(III) complexes Ir(ppy)2(bpy)+ is the one with the two cyclometalating carbon atoms cis to each other and lying in the same plane with the two nitrogen atoms from the bpy ligand.48–52 Therefore, we believe our two cationic complexes 1 and 2 also adopt this configuration (see the configuration drawn for complexes 1 and 2 in Chart 1). For tris-cyclometalating Ir(C^N)3 complexes, fac and mer isomers are possible.49,53,54 The fac isomers have much longer emission lifetimes and higher emission quantum yields in comparison to their corresponding mer isomers.53,55 It has been reported that when the reaction temperature to convert the Cl-bridged Ir dimmer into the mononuclear Ir complex is below 160 °C, the mer isomer is the dominant product.53,54 However, fac isomers can be the major product when AgCF3SO3 or AgCF3CO2 is used as the catalyst and the reaction temperature is below 160 °C.55,56 Because our reactions for synthesis of complexes 3–5 were carried out at ∼130 °C in the presence of AgCF3SO3, and the emission lifetimes of complexes 3–5 are all on the order of tens of μs (will be discussed in the photoluminescence section), it is reasonable to believe that these complexes adopt the fac geometry although all of our attempts to obtain single crystals to confirm the geometry of these complexes failed due to the presence of branched long alkyl chains in their ligands. The fac geometry is used in all of our DFT calculations for the electronic absorption and emission. It should also be noted that although the C^N ligand contains two cyclometalating units, only the 2-phenylpyridine unit coordinates with the Ir(III) ion.Electronic absorption
The electronic absorption of 1-L, 2-L, and 1–5 in toluene obey Beer's law in the concentration range of 5 × 10−6 to 1 × 10−4 mol L−1, suggesting the absence of ground-state aggregation in the concentration range used. The electronic absorption spectra of 1-L, 2-L, and 1–5 in toluene are shown in Fig. 1, and their absorption band maxima and molar extinction coefficients are presented in Table 1.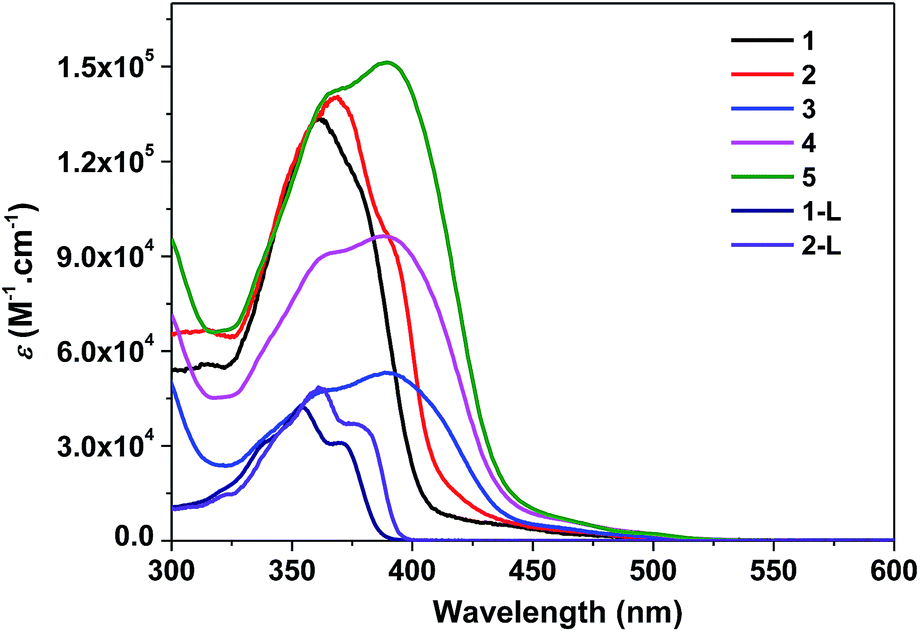 | ||
| Fig. 1 Electronic absorption spectra of 1-L, 2-L, and 1–5 in toluene at room temperature. | ||
| λabsa/nm (logε/L mol−1 cm−1) |
λem/nm (τ/μs); Φema, R.T. | λemb/nm (τ/μs), 77 K | λT1–Tna/nm (τT/μs; logεT1–Tn/L mol−1 cm−1; ΦT) |
|
|---|---|---|---|---|
| a In toluene.b In butyronitrile. | ||||
| 1 | 350 (sh, 5.06), 361 (5.13), 380 (sh, 5.03), 420 (3.85) | 583 (6.8), 633 (7.6); 0.095 | 546 (1447), 594 (966) | 510 (0.06; 5.14; 0.042) |
| 2 | 350 (sh, 5.07), 369 (5.15), 392 (sh, 4.97), 425 (4.03) | 592 (8.5), 644 (6.0); 0.13 | 558 (933), 598 (—) | 568 (7.9; 5.04; 0.063) |
| 3 | 363 (sh, 4.68), 390 (4.73), 460 (3.66), 484 (3.34) | 518 (29.0 (83%), 1.15 (17%)), 556 (27.9 (89%), 1.17 (11%)), 604 (25.6 (85%), 1.53 (15%)); 0.22 | 548 (857), 592 (622) | 524 (27.5; 4.73; 0.28) |
| 4 | 365 (sh, 4.96), 388 (4.98), 464 (4.81), 495 (3.40) | 520 (36.0 (92%), 1.25 (8%)), 555 (34.1 (96%), 1.31 (4%)), 595 (34.2 (78%), 2.40 (22%)), 648 (24.3 (56%), 1.69 (44%)); 0.38 | 552 (426), 590 (—) | 532 (35.3; 5.03; 0.14) |
| 5 | 366 (sh, 5.15), 389 (5.18), 465 (4.81), 498 (3.43) | 519 (35.1), 555 (33.1), 594 (33.1 (85%), 2.9 (15%)), 648 (34.8 (68%), 2.6 (32%)); 0.45 | 556 (502), 586 (411) | 534 (37.7; 5.14; 0.11) |
| 1-L | 353 (4.63), 371 (4.49) | 389 (—), 409 (—), 429 (—); 0.76 | 580 (67.7; —; —) | |
| 2-L | 362 (4.68), 377 (4.57) | 392 (—), 415 (—), 438 (—); 0.67 | 610 (7.1; —; —) | |
For ligands 1-L and 2-L, well-resolved vibronic structures are observed in the 300–400 nm region. Considering the large molar extinction coefficients (on the order of 104 L mol−1 cm−1) and the well-resolved vibronic structures, these absorption bands are attributed to the 1π,π* transitions. The absorption bands of 2-L are red-shifted and have larger extinction coefficients compared to those of 1-L, which is in line with the extended π-conjugation via incorporating the triple bond in 2-L.
Compared to the absorption spectra of ligands 1-L and 2-L, the absorption spectra of 1–5 are broader and red-shifted, and the molar extinction coefficients of the major absorption bands are much larger than their corresponding C^N ligands. The bathochromic shift indicates an electron delocalization induced by the interactions with the metal center. The broadening and red-shift of the ligand centered 1π,π* transitions are more significant for the neutral complexes 3–5 compared to the cationic complexes 1 and 2, suggesting a stronger interaction between the ligand and the metal center in the neutral complexes than in the cationic complexes. This can be explained by the stronger ligand field strength of the 2-phenylpyridine ligand than the bipyridine ligand. It is worth noting that complexes 3–5 exhibit the similar shape and energy of the major absorption bands, and the molar extinction coefficients of the band at ca. 390 nm are roughly proportional to the number of BTF-ppy ligands. This confirms that the major absorption bands should arise mainly from the 1π,π* transitions localized on the BTF-ppy ligand.
In addition to the ligand centered 1π,π* transitions, lower energy absorption bands are observed for all of the five complexes above 430 nm, which can be tentatively attributed to the charge transfer transitions including 1,3MLCT, 1,3LLCT or 1,3ILCT. Similar to the trend observed from the 1π,π* transitions, the charge-transfer absorption bands in the neutral complexes 3–5 are more red-shifted and stronger in comparison to the cationic complexes 1 and 2. With the increased number of the BTF-ppy ligands, the charge-transfer bands become stronger.
Comparison of the spectra of complexes 1 and 4 reveals that the neutral Ir(III) complex 4 exhibits weaker but much broader major absorption bands, while its charge transfer bands are much broader and stronger.
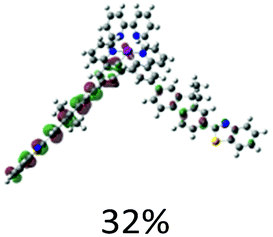
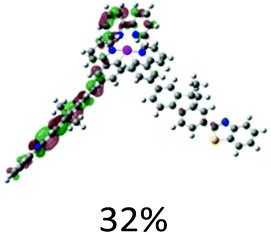
The aforementioned assignments for each of the absorption band are supported by the TDDFT calculation results (Fig. 2). Table 2 displays the NTOs for the major transitions contributing to the absorption bands of 325–600 nm. The major absorption bands in the range of 325–425 nm predominantly arise from the ligand-localized 1π,π* transitions, with minor contributions from the 1MLCT/1LLCT transitions. In complexes 3–5, contributions from the 1ILCT transition are also evident. For the two cationic complexes 1 and 2, the lowest-energy transition in the energy range of 425–600 nm is 1MLCT/1LLCT in nature; and it separates well from the major absorption bands. However, for the neutral complexes 3–5, the low-energy transitions essentially have the 1π,π* character, with some contributions from the 1MLCT/1ILCT transitions, which makes the transitions in the 400 nm region having much higher intensity.
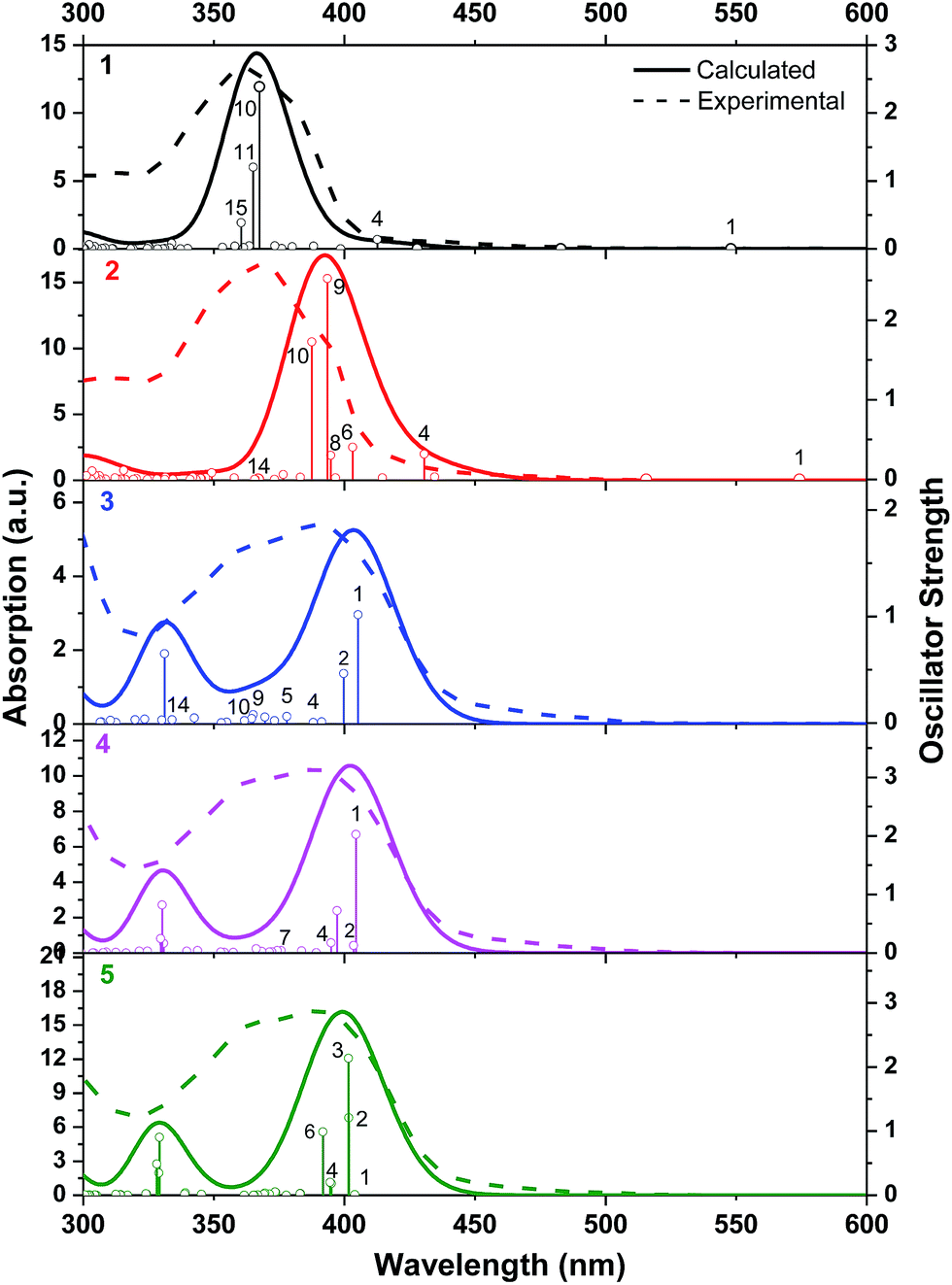 | ||
| Fig. 2 Computed transitions and associated absorption spectra in toluene for complexes 1–5. | ||
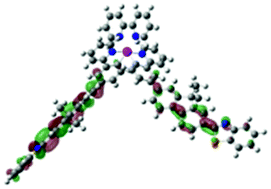
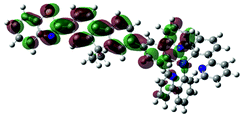
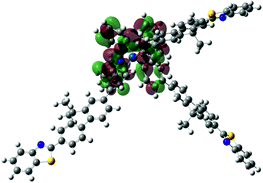
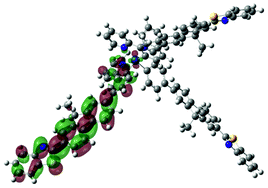
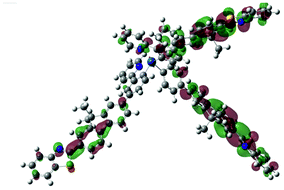
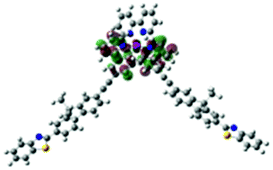
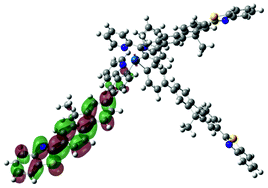
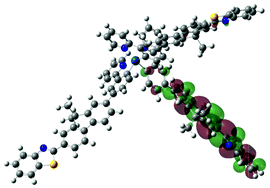
| Excited state and properties | Hole | Electron | Excited state and properties | Hole | Electron | ||
|---|---|---|---|---|---|---|---|
| 1 | S1, 548 nm, f = 0.0004 |  |
 |
2 | S10, 388 nm, f = 1.726 |  |
 |
| S4, 413 nm, f = 0.138 |  |
 |
 |
 |
|||
| S10, 368 nm, f = 2.385 | 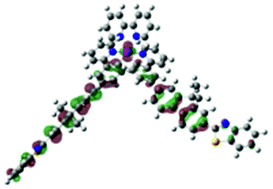 |
 |
3 | S1, 405 nm, f = 1.016 | 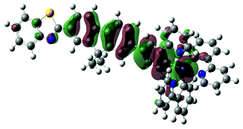 |
 |
|
| S11, 365 nm, f = 1.203 | 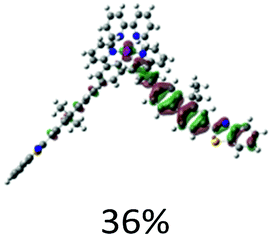 |
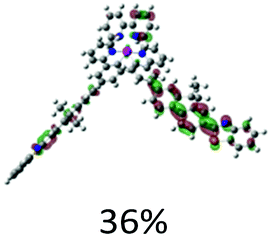 |
S2, 400 nm, f = 0.465 | 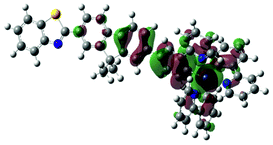 |
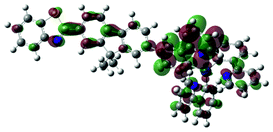 |
||
 |
 |
S15, 331 nm, f = 0.650 | 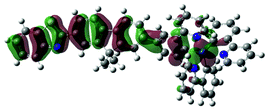 |
 |
|||
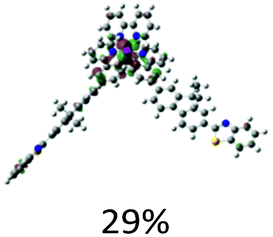 |
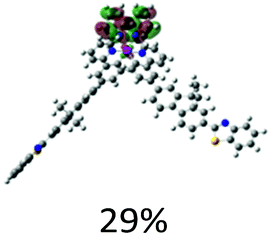 |
4 | S1, 404 nm, f = 2.024 | 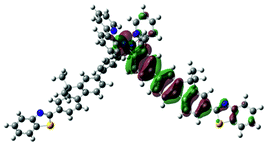 |
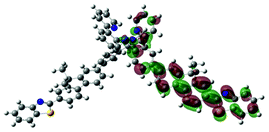 |
||
| S15, 361 nm, f = 0.383 | 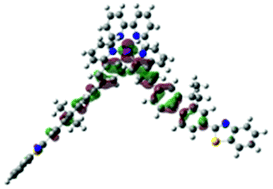 |
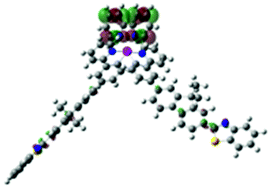 |
S4, 395 nm, f = 0.176 | 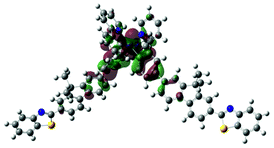 |
 |
||
| S19, 330 nm, f = 0.820 | 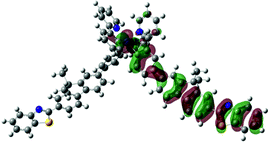 |
 |
|||||
| 2 | S1, 574 nm, f = 0.0008 |  |
 |
5 | S1, 404 nm, f = 0.010 | 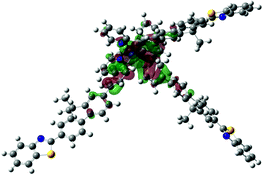 |
 |
| S4, 431 nm, f = 0.316 | 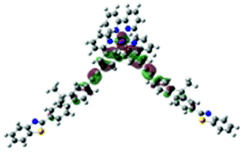 |
 |
S3, 402 nm, f = 2.136 | 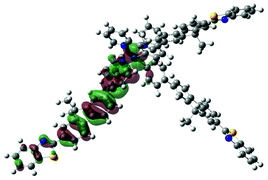 |
 |
||
| S6, 403 nm, f = 0.404 | 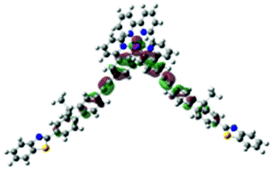 |
 |
S6, 392 nm, f = 0.989 | 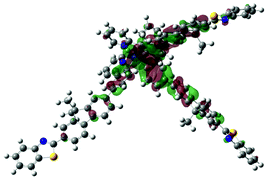 |
 |
||
| S8, 395 nm, f = 0.303 |  |
 |
S22, 329 nm, f = 0.904 | 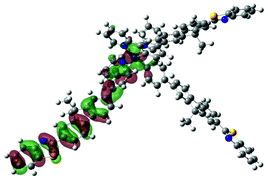 |
 |
||
| S9, 394 nm, f = 2.525 | 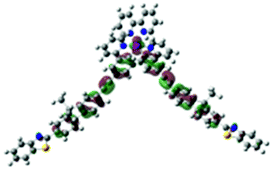 |
 |
S24, 328 nm, f = 0.488 | 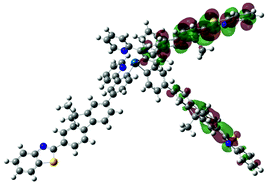 |
 |
The ascription of the absorption bands in complexes 1–5 is further supported by the solvent-dependent study. As illustrated in ESI Fig. S2–S6,† these complexes display a minor solvatochromic effect for both the major absorption bands and the low-energy absorption tails. This supports the assignment of the predominant 1π,π* transitions for the major absorption bands. The insensitivity of the low-energy transitions to the polarity of solvent could be explained by the shielding of the central metal from the solvent due to the octahedral geometry of the Ir(III) complexes. Alternatively, the different directions of the various charge transfer transitions could partially cancel out the dipole moment of the lowest-energy transition, which in turn reduces the solvent influence. Moreover, mixing of the charge transfer transitions with the 1π,π* transitions could also play a role.
Photoluminescence
Complexes 1–5 all exhibit strong emission with relatively long lifetimes (in the scale of several to tens of microseconds) both in room temperature solution and in butyronitrile glassy matrix at 77 K. The emission spectra of 1–5 at room temperature are shown in Fig. 3(a), and the emission spectra of 1–5 at 77 K are shown in Fig. 3(b). The emission spectra of 1-L and 2-L are included in Fig. 3(a) for comparison purpose. The emission band maxima, emission lifetimes, and quantum yields are summarized in Table 1.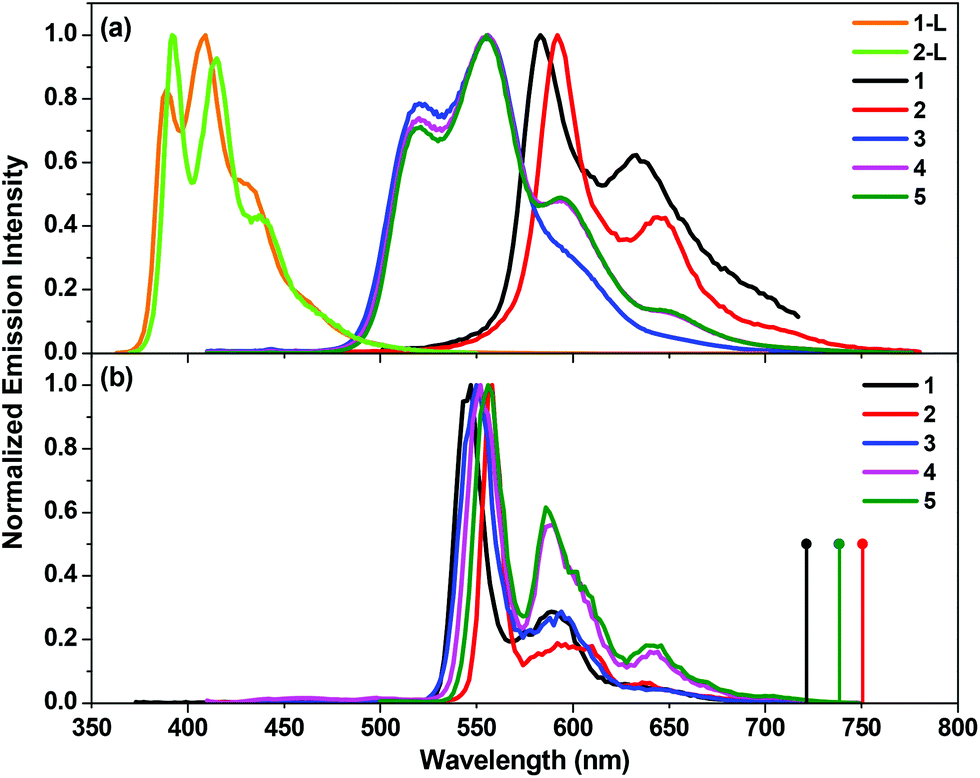 | ||
| Fig. 3 (a) Normalized emission spectra of complexes 1–5 and ligands 1-L and 2-L at room temperature in toluene at the concentration of 1 × 10−5 mol L−1 (λex: 353 nm for 1-L, 360 nm for 2-L, 363 nm for 1, 400 nm for 2, and 390 nm for 3–5). (b) Normalized emission spectra of 1–5 at 77 K in butyronitrile glassy matrix at the concentration of 1 × 10−5 mol L−1 (λex: 363 nm for 1, and 400 nm for 2–5). The calculated triplet emission energies corresponding to the optimized excited states at T = 0 K are shown as droplines in panel (b). | ||
The ligands 1-L and 2-L show strong emission with well-resolved vibronic structures and short lifetimes (<5 ns, too short to be measured on our spectrometer), which can be assigned to the 1π,π* fluorescence. Similar to the trend observed from their electronic absorption spectra, the fluorescence spectrum of 2-L is red-shifted compared to that of 1-L, indicating the electron delocalization induced by the more extended π-conjugation in 2-L with the CC bond.
The emission spectra of complexes 1–5 are drastically red-shifted in comparison to their corresponding ligands. They all possess well-resolved vibronic structures, with the lifetimes on the order of several to tens of microseconds. They are all readily quenched by oxygen as well. Considering these facts, we assign the observed emission for these complexes to phosphorescence. However, the emission characteristics (such as the lifetime and temperature dependence) of the cationic complexes 1 and 2 and the neutral complexes 3–5 appear to be quite different, implying a different nature of the emitting triplet excited state for the cationic and neutral complexes.
For the cationic complexes 1 and 2, the room temperature emission lifetimes are shorter than 10 μs, and the emission quantum yields are ca. 10%. The solvent-dependency study (see ESI Figs. S7 and S8†) reveals a minor negative solvatochromic effect. These features suggest a predominant charge transfer nature of the emitting states. In addition, as shown in Fig. 3(b), the emission spectra of complexes 1 and 2 in butyronitrile matrix at 77 K exhibit well-resolved peaks that are pronouncedly blue-shifted and narrower compared to the room temperature emission spectra due to rigidochromic effect in the solid matrix.57 The thermally induced shifts are on the order of ∼1200 cm−1, which is a characteristics of the charge transfer emitting state.
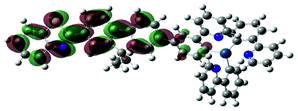
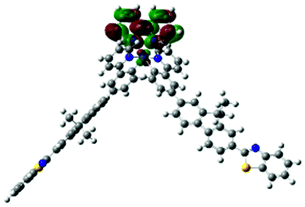
The charge transfer nature of the emitting states for complexes 1 and 2 is further supported by the TDDFT calculation results. As shown in Table 3, the holes of complexes 1 and 2 are located on the cyclometalating C^N ligand and the d-orbitals of the Ir(III) ion; while the electrons are exclusively on the bipyridine ligand. Therefore, the emitting triplet excited states (T1) are the 3MLCT/3LLCT states for these two complexes. The NTOs in Table 3 also show that the calculated singlet states responsible for fluorescence (S1) have predominate 1MLCT/1LLCT character for these two complexes and the similar energies to their corresponding triplet excited states. It was previously shown that the stronger the MLCT character involved in both the singlet and triplet states, the stronger the spin–orbit coupling (SOC) governing the rate of intersystem crossing (ISC) is.58 However, identical orbitals with different spin configurations result in negligible SOC and slow ISC, which is referred to as the “organometallics version of El-Sayed's rule” in ref. 58. Because the singlet states (S1) are similar to the triplet (T1) states in nature, involving a high degree of MLCT and identical character of d-orbitals, the SOC in complexes 1 and 2 should be negligible. Therefore, we conclude that the observed emission for these two complexes is governed by the 3MLCT/3LLCT states.
| S1 (nm) | Hole | Electron | T1 (nm) | Hole | Electron | |
|---|---|---|---|---|---|---|
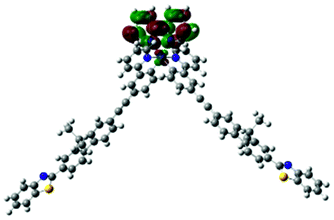
| 1 | 685, f = 0.001 |  |
 |
691 |  |
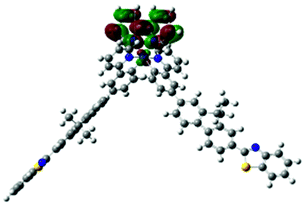 |
| 2 | 720, f = 0.001 |  |
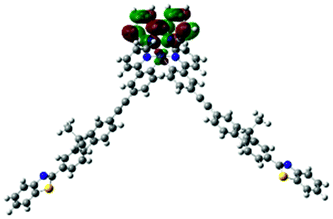 |
725 | 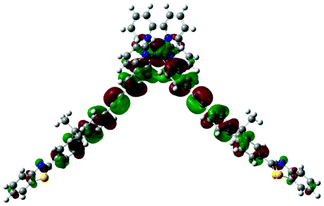 |
 |
| 3 | 472, f = 1.831 | 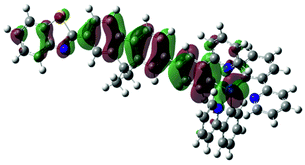 |
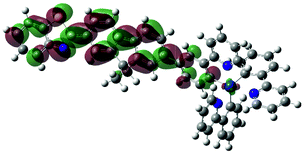 |
738 | 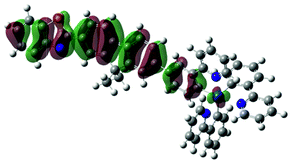 |
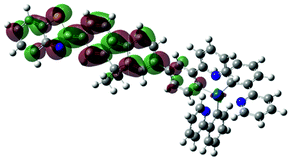 |
| 4 | 470, f = 1.992 |  |
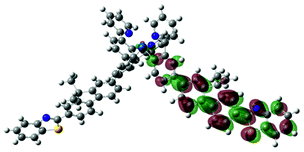 |
738 | 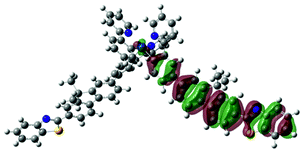 |
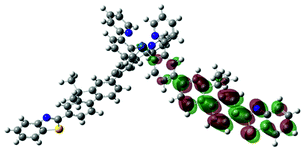 |
| 5 | 482, f = 0.045 | 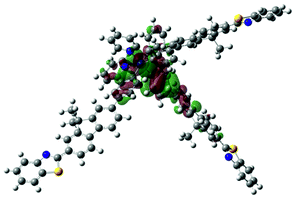 |
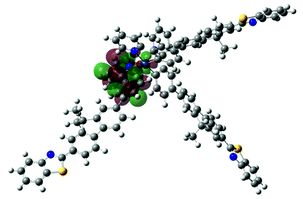 |
738 | 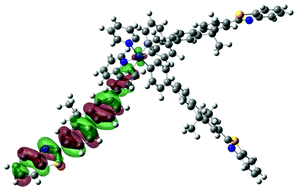 |
 |
The emission energy of 2 is red-shifted compared to that of 1 both at room temperature and 77 K, manifesting the extended electron delocalization due to the triple bond in the cyclometalating ligand in 2. The same trend is reproduced by TDDFT calculations, although the calculated energies of triplet emission are noticeably underestimated compared to the experimental data. This is a typical error for such calculations due to the higher inaccuracy in the performance of the functional in an excited state optimization restricted to triplet spin configuration and a lack of consideration of the SOC, which is important for Ir(III) complexes. Nonetheless, the qualitative trend is reproduced by our calculations and agrees with experimental emission spectra (see Fig. 3).
In contrast to the emission of the two cationic complexes 1 and 2, the emission of these fac-isomers of the neutral complexes 3–5 is drastically blue-shifted and more structured. Most importantly, they exhibit dual emission (see the biexponential decay lifetimes in Table 1), with a long-lived emission of 25–35 μs and a short-lived one of 1–2 μs. Although the dual emission is rarely encountered in the Ir(III) complexes, it has been reported in the literature for both the neutral and cationic Ir(III) complexes.59–61 It was generally regarded that the long-lived high-energy emission emanates from an excited state enriched with 3π,π* character, while the short-lived low-energy emitting state has more charge transfer (3CT) character.
The presence of dual emission is partially supported by the different excitation spectra when monitored at the high-energy and low-energy emission bands. As exemplified in ESI Fig. S11† for complex 5, the excitation spectra monitored at the low-energy emission bands of 595 and 647 nm clearly show the charge-transfer absorption bands at 435–475 nm and 475–550 nm, which are absent in the excitation spectra monitored at the high-energy emission bands of 519 and 555 nm. Another piece of evidence confirming the different natures of the high-energy and low-energy emission bands is the different sensitivities of these bands to oxygen quenching. As shown in Fig. 4 and ESI Fig. S12† for complex 5 in toluene, the relative intensities of the high-energy emission bands increase more pronouncedly than the low-energy emission bands upon purging nitrogen for longer period of time, indicating the higher sensitivity of the high-energy emission bands to oxygen than the low-energy bands due to the different lifetimes of the excited states predominantly contributing to these bands. This phenomenon is consistent with that reported in the literature for a neutral heteroleptic Ir(III) complex exhibiting high-energy 3π,π* and low-energy 3CT dual emission.60 The different natures of the emission bands are also reflected by the relative intensity of these bands in different solvents. As shown in ESI Fig. S13–S15† for complexes 3–5, the relative intensity of the 519 nm band gradually decreases when the polarity of the solvent increases, while the relative intensities of the 595 and 640 nm bands increase with increased solvent polarity. This trend coincides with the proposed 3π,π* and 3CT nature for the respective high-energy and low-energy bands, in which the polar solvents favor the charge transfer emission. TDDFT optimization of the lowest triplet exited state also points to the involvement of 3MLCT transition in complexes 3–5 (see NTOs presented in Table 3) in the lowest triplet excited state.
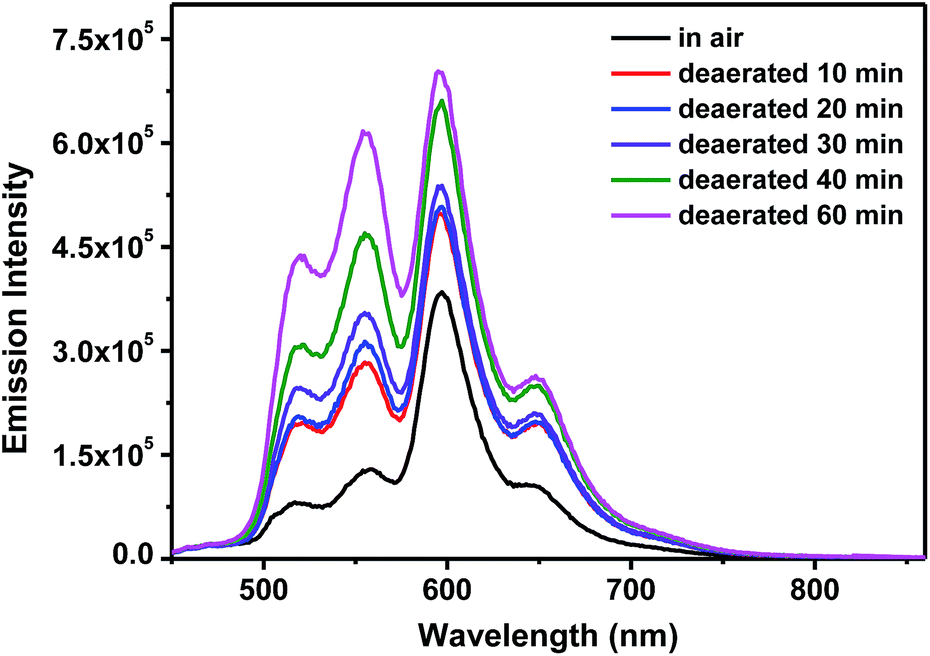 | ||
| Fig. 4 Emission spectra of 5 in toluene under different deaeratation conditions. λex = 436 nm, A436nm = 0.08 in a 1 cm cuvette. | ||
Because the mer-isomers of complexes 4 and 5 both exhibit structured emission at ca. 595 and 650 (sh) nm, and the emission lifetimes are similar to the shorter-lived emission from the fac-isomers, it is reasonable to doubt whether the short-lived emission arises from the trace amount of mer-isomers mixed in the fac-isomers. However, this possibility can be excluded by the absence of the unique 1H-NMR peaks at 6.0–6.2 ppm and the peaks above 9.0 ppm corresponding to the mer-isomers (see the 1H-NMR spectra for the mer-isomers of 4 and 5 in ESI Fig. S1†) and the absence of the corresponding spot to the mer-isomers during the TLC analysis. Even if there are undetectable mer-isomers present, their emission quantum yields are one order of magnitude lower than those for the fac-isomers (∼4–5% for the mer-isomers vs. ∼40% for the fac-isomers). Considering the undetectable amount, we believe that it is impossible for the short-lived emission in the fac-isomers of complexes 3–5 being arising from the undetectable amount of the mer-isomers.
Comparison of the emission of the cationic complex 1 and its neutral counterpart 4 reveals that the neutral complex exhibits a more structured blue-shifted emission with biexponential decays. These differences reflect the different natures of the emitting states in these two complexes. Except for the presence of 3π,π* enriched high-energy emission bands in complex 4, the two low-energy emission bands in 4 are slightly red-shifted with respect to those of 1. This can be rationalized by the extended electron delocalization on the BTF-ppy ligand in 4 versus the bipyridine ligand in 1 (see NTOs in Table 3), which reduces the energy of the emitting 3MLCT states and gives rise to the slightly red-shifted emission bands and a shorter lifetime according to the energy gap law.
At 77 K, the emission spectra of 3–5 are noticeably different from those at room temperature, with the disappearance of the high-energy emission band at 520 nm. Because this band and the 550 nm band have the similar lifetime and the separation between these two bands is approximately 1100 cm−1, it is reasonable to consider this band as a vibronic structure of the 550 nm band. At low temperature, this high-energy vibronic state can't be reached, resulting in the disappearance of this band at 77 K. The similar phenomenon has been reported in literature for another neutral Ir(III) complex.60
Transient absorption
Nanosecond transient absorption (TA) of complexes 1–5 in toluene was investigated to further understand their triplet excited-state characteristics. The spectra for 1–5 at zero delay after excitation are displayed in Fig. 5. The time-resolved TA spectra of 1–5 are shown in ESI Figs. S17–S21.† The triplet excited-state lifetimes, extinction coefficients, and quantum yields are summarized in Table 1.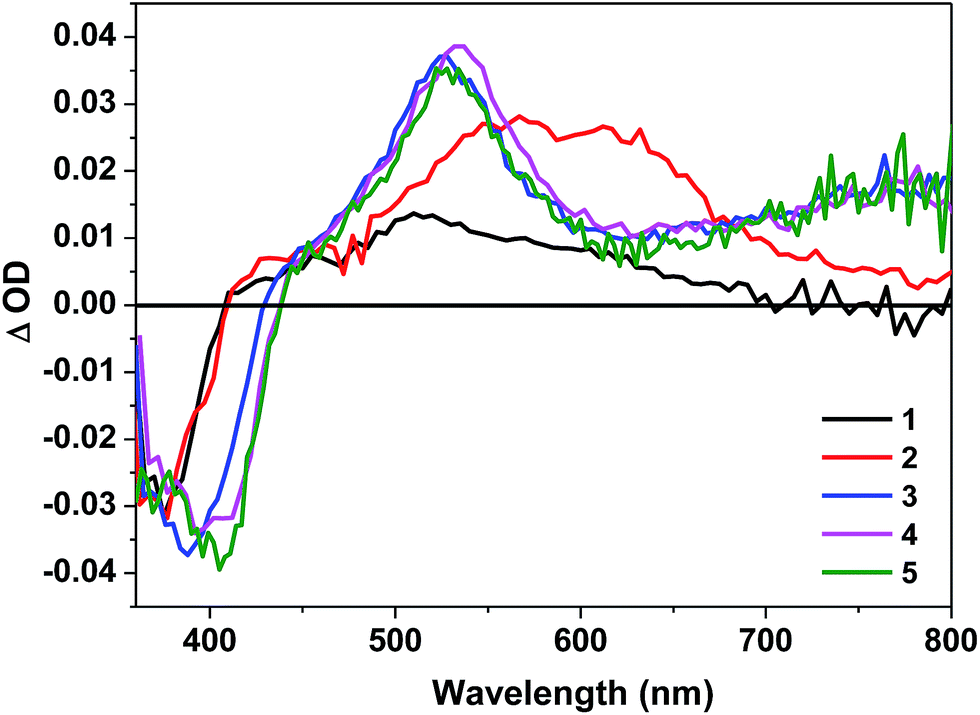 | ||
| Fig. 5 Nanosecond transient difference absorption spectra of 1–5 in toluene at zero time decay (λex = 355 nm, A355 = 0.4 in a 1 cm cuvette). | ||
The TA spectra of all complexes feature a bleaching band in the blue spectral region corresponding to their respective 1π,π* absorption band. Complexes 1 and 2 show moderate and broad TA band spreading the wavelength range of 400–700 nm for 1 and 400–800 nm for 2, and the TA of 2 is red-shifted in comparison to that of 1 due to the more extended π-conjugation in the C^N ligand. The triplet excited-state lifetimes deduced from the decay of the TA is ∼60 ns for 1, suggesting the triplet excited-state absorption is likely from a short-lived 3CT state. In contrast, the TA lifetime of 2 is several microseconds, which is in line with the lifetime deduced from the emission. Therefore, the observed TA for 2 is tentatively attributed to the 3MLCT/3LLCT state.
The positive TA region of 3–5 features two parts: a stronger absorption band in the wavelength range of 450–610 nm and a weaker but broader band in the range of 610–800 nm. The shape of the TA spectra, the ΔODs at the TA band maxima, and the triplet lifetimes of 3–5 are all similar, indicating that the TA likely arises from the similar structural component, i.e. the BTF-ppy ligand. Considering the aforementioned feature, the long lifetimes (tens of microseconds) deduced from the decays of TA (see Table 1), the similarity and difference of the TA spectra with that of 1-L (ESI Fig. S26†), and the similar lifetimes to those obtained from the decay of the long-lived emitting species, we conclude that the observed TA of 3–5 originates from the BTF-ppy ligand centered 3π,π* state, likely mixed with some 3MLCT character in view of the broad TA band in the near-IR region that is lacking in the TA spectrum of the corresponding ligand 1-L.
It should be noted that although the emission of the fac-isomers of 3–5 exhibit biexponential decay, the TA signals decay monoexponentially, with the TA lifetimes being consistent with the longer emission lifetimes. In addition, the TA decay kinetics monitored at various visible and near-IR wavelengths remain the same monoexponential decay character (see the similar TA lifetimes monitored at different wavelengths in ESI Table S8†). The disparity between the emission and TA kinetics for 3–5 is uncommon but not unprecedented because the simultaneous population of two triplet excited states (such as the 3π,π* state and the 3MLCT state) but with only one state emitting and the other state showing transient absorption has been reported for some Ru(II)62 and Pt(II)63,64 complexes. For complexes 3–5, it appears that both states emit, but only the high-lying state exhibits transient absorption. The different emission and TA kinetics was also found in complex 2.
Comparison of the TA features of 4 to those of 1 reveals that replacing the bipyridine ligand with the 2-phenylpyridine ligand results in a broadening of the TA spectrum of 4, and switching the TA excited state from 3CT in 1 to 3π,π*/3MLCT in 4.
Reverse saturable absorption
Complexes 1–5 all possess stronger excited-state absorption than that of the ground state at 532 nm noticing the positive absorption bands in the visible spectral region of their TA spectra. Meanwhile, the triplet-state lifetimes are significantly longer than the ns laser pulse width (4.1 ns). Therefore, it is expected that these complexes would exhibit reverse saturable absorption (RSA) for ns laser pulses at 532 nm. To verify this assumption, nonlinear transmission experiment was carried out for complexes 1–5 in CH2Cl2 solutions with a linear transmission of 90% in a 2 mm cuvette using the 532 nm ns laser pulses and the results are shown in Fig. 6. The transmissions of all of the complexes decrease remarkably with increased incident energy, which clearly demonstrates a strong RSA from these complexes. The strength of the RSA follows this trend: 5 < 4 < 1 ≈ 3 < 2. It appears that the increased number of BTF-ppy ligand reduces the RSA of the neutral complexes while the increased π-conjugation of the ppy ligand in the cationic complexes improves the RSA. However, the neutral complex 4 exhibits a weaker RSA at 532 nm than its cationic counterpart 1.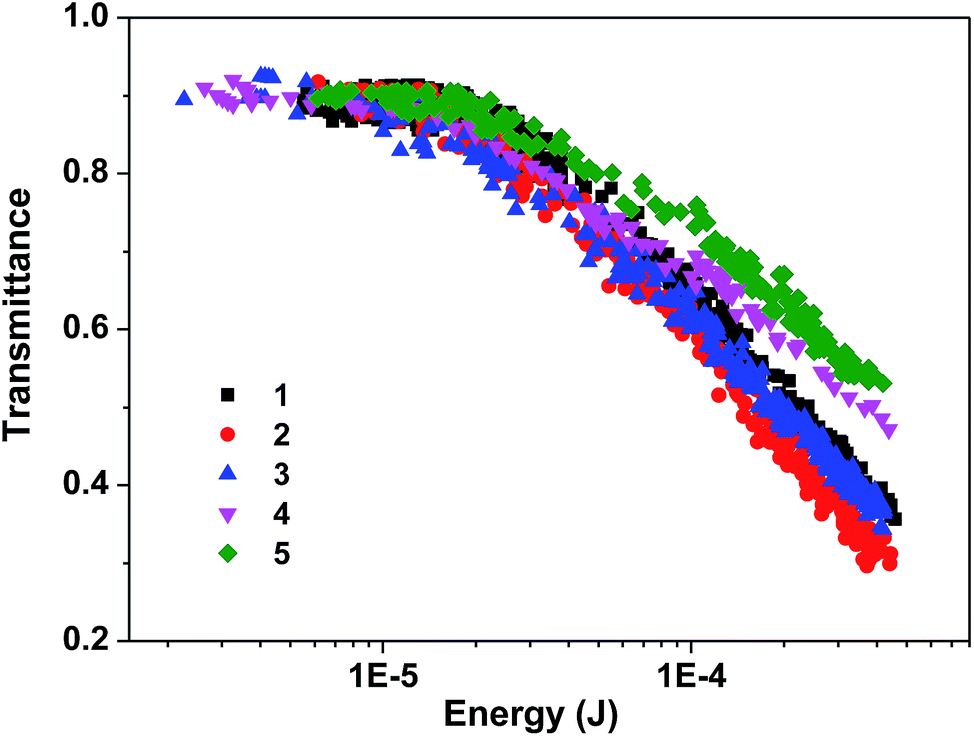 | ||
| Fig. 6 Nonlinear transmission of 1–5 at the linear transmission of 90% in CH2Cl2 solution in a 2 mm cuvette for 532 nm 4.1 ns laser. The radius of the beam waist at the focal point was approximately 96 μm. | ||
The ratios of the excited-state absorption cross section to that of the ground state (σex/σ0) at 532 nm were evaluated to rationalize the RSA trend of these complexes. According to the ground-state molar extinction coefficients at 532 nm and at the wavelength of the TA band maximum, the optical density changes (ΔOD) at 532 nm and at the TA band maximum, the molar extinction coefficient at the TA band maximum (εT1–Tn), and the method described by our group previously,65 the excited-state absorption cross sections (σex) for 1–5 at 532 nm were estimated and the results are shown in Table 4. The trend of the σex/σ0 ratio roughly matches the observed RSA trend. It is clear that the two cationic complexes 1 and 2 have the similar ground-state absorption cross section at 532 nm, but their triplet excited-state absorption cross section is drastically different at 532 nm. Complex 2 that has the triple bond to connect the BTF substituent to the phenylpyridine ligand exhibits much stronger excited-state absorption and thus a stronger RSA. For the three neutral complexes 3–5, both the ground-state absorption and excited-state absorption cross sections at 532 nm increase when the number of BTF-ppy ligand increases, the σex/σ0 ratio remains quite similar from complex 3 to complex 5. However, the triplet quantum yield decreases from complex 3 to complex 5. The combined parameter ΦTσex/σ0 corresponds to the observed RSA trend for 3–5.
| Complex | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| σ0/10−18 cm2 | 0.63 | 0.65 | 0.31 | 0.75 | 0.80 |
| σex/10−18 cm2 | 298 | 394 | 204 | 413 | 530 |
| σex/σ0 | 473 | 606 | 658 | 551 | 663 |
Conclusions
The synthesis, photophysics, and reverse saturable absorption of five iridium(III) complexes with 7-(benzothiazol-2-yl)-9,9-di(2-ethylhexyl)-9H-fluoren-2-yl (BTF) pendant attached to the 2-phenylpyridine (ppy) ligand was reported. The effects of the extension of π-conjugation in the ligand were studied by the comparison between complexes 1 and 2, and the effects of the number of the BTF-ppy ligand were evaluated by comparison among complexes 3, 4, and 5. Assessment of the effects of cationic complex vs. neutral complex was done by comparison of complex 1 with complex 4. The results show that the band maxima of the electronic absorption, emission, and ns transient absorption spectra are all red-shifted by extending the π-conjugation of the C^N ligand or increasing the number of BTF-ppy ligand, and the lowest singlet and triplet excited states of the neutral complex 4 are lowered in comparison to those of the cationic complex 1 (as reflected by the red-shifted electronic absorption and the low-energy emission spectra for 4). The emission of the cationic complexes 1 and 2 are attributed to the 3MLCT/3LLCT state; while for complexes 3–5 the lowest-energy emitting states appear to be the 3π,π*/3MLCT excited states, with another high-energy long-lived emitting state. All complexes possess moderate to strong TA in the visible spectral region, with the TA predominantly emanates from the 3CT states for 1 and 2, and from 3π,π*/3MLCT states for 3–5. The neutral complex 4 exhibits a much broader, stronger, and longer-lived TA in comparison to the cationic complex 1. Reverse saturable absorption of these complexes at 532 nm were evaluated for nanosecond laser pulses. The results demonstrate that these complexes all exhibit strong exhibit strong RSA for ns laser pulses at 532 nm, with a trend of 5 < 4 < 1 ≈ 3 < 2. It appears that the increased number of BTF-ppy ligand reduces the RSA of the neutral complexes while the increased π-conjugation of the ppy ligand in the cationic complexes improves the RSA. However, the neutral complex 4 exhibits a weaker RSA at 532 nm than its cationic counterpart 1. The trend is primarily determined by the ratio of the excited-state absorption cross section to that of the ground state (σex/σ0) at 532 nm, but the triplet quantum yield also plays a role in complexes 3–5.Acknowledgements
The synthesis and photophysical studies of this work was supported by the Army Research Laboratory (W911NF-10-2-0055) to W. Sun. The DFT calculations were supported by the National Science Foundation (NSF DMR-1411086 and CNS-1229316) to W. Sun and S. Kilina. In the frame of computational method development, S. Kilina acknowledges support from the NDSU Advance FORWARD program sponsored by NSF HRD-0811239 joined and ND EPSCoR through the NSF grant EPS-0814442 and financial support of the Alfred P. Sloan Research Fellowship BR2014-073. For computational resources and administrative support, we thank the Center for Computationally Assisted Science and Technology (CCAST) at North Dakota State University.References
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry: A Comprehensive Text, Wiley, New York, 1980 Search PubMed
.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed
- C. Wu, H.-F. Chen, K.-T. Wong and M. E. Thompson, J. Am. Chem. Soc., 2010, 132, 3133–3139 CrossRef CAS PubMed
- D. Bruce and M. M. Richter, Anal. Chem., 2002, 74, 1340–1342 CrossRef CAS PubMed
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed
- M. S. Lowry, W. R. Hudson, R. A. Pascal Jr and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed
- D. Tordera, A. M. Bünzli, A. Pertegás, J. M. Junquera-Hernández, E. C. Constable, J. A. Zampese, C. E. Housecroft, E. Ortí and H. J. Bolink, Chem.–Eur. J., 2013, 19, 8597–8609 CrossRef CAS PubMed
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206–4215 RSC
- E. Margapoti, V. Shukla, A. Valore, A. Sharma, C. Dragonetti, C. C. Kitts, D. Roberto, M. Murgia, R. Ugo and M. Muccini, J. Phys. Chem. C, 2009, 113, 12517–12522 CAS
- E. Margapoti, M. Muccini, A. Sharma, A. Colombo, C. Dragonetti, D. Roberto and A. Valore, Dalton Trans., 2012, 41, 9227–9231 RSC
- K. K.-W. Lo, P.-K. Lee and J. S.-Y. Lau, Organometallics, 2008, 27, 2998–3006 CrossRef CAS
- K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069–12083 RSC
- L. Huynh, Z. Wang, J. Yang, V. Stoeva, A. Lough, I. Manners and M. A. Winnik, Chem. Mater., 2005, 17, 4765–4773 CrossRef CAS
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS
- A. G. Condie, J. C. Gonzáles-Gómez and J. R. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed
- J. B. Edel, A. J. deMello and J. C. deMello, Chem. Commun., 2002, 38, 1954–1955 RSC
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed
- K. Dedeian, P. I. Djurovich, F. O. Garces, G. Carlson and R. J. Watts, Inorg. Chem., 1991, 30, 1685–1687 CrossRef CAS
- A. B. Tamayo, S. Garon, T. Sajoto, P. I. Djurovich, I. M. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 8723–8732 CrossRef CAS PubMed
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS
- R. J. Watts and J. van Houten, J. Am. Chem. Soc., 1974, 96, 4334–4335 CrossRef CAS
- Q. Yan, K. Yue, C. Yu and D. Zhao, Macromolecules, 2010, 43, 8479–8487 CrossRef CAS
- Q. Yan, Y. Fan and D. Zhao, Macromolecules, 2012, 45, 133–141 CrossRef CAS
- K.-Y. Kim, R. T. Farley and K. S. Schanze, J. Phys. Chem. B, 2006, 110, 17302–17304 CrossRef CAS
- Y. Li, N. Dandu, R. Liu, L. Hu, S. Kilina and W. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6556–6570 CAS
- Y. Li, N. Dandu, R. Liu, Z. Li, S. Kilina and W. Sun, J. Phys. Chem. C, 2014, 118, 6372–6384 CAS
- Y. Li, N. Dandu, R. Liu, S. Kilina and W. Sun, Dalton Trans., 2014, 43, 1724–1735 RSC
- H. Zhao, P. V. Simpson, A. Barlow, G. J. Moxey, M. Morshedi, N. Roy, R. Philip, C. Zhang, M. P. Cifuentes and M. G. Humphrey, Chem.–Eur. J., 2015, 21, 11843–11854 CrossRef CAS PubMed
- Z. Li, E. Badaeva, A. Ugrinov, S. Kilina and W. Sun, Inorg. Chem., 2013, 52, 7578–7592 CrossRef CAS PubMed
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef
- P. A. Firey, W. E. Ford, J. R. Sounik, M. E. Kenney and M. A. Rodgers, J. Am. Chem. Soc., 1988, 110, 7626–7630 CrossRef CAS
- F. Guo, W. Sun, Y. Liu and K. S. Schanze, Inorg. Chem., 2005, 44, 4055–4065 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford, CT, USA, 2009 Search PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS
- C. Van Caillie and R. D. Amos, Chem. Phys. Lett., 2000, 317, 159–164 CrossRef CAS
- Q. Zhao, S. Liu, M. Shi, C. Wang, M. Yu, L. Li, F. Li, T. Yi and C. Huang, Inorg. Chem., 2006, 45, 6152–6160 CrossRef CAS PubMed
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS
- M. Maestri, V. Balzani, C. Deuschel-Cornioley and A. von Zelewsky, Adv. Photochem., 1992, 17, 1–68 CAS
- F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464–3471 CrossRef CAS
- L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495–8504 CrossRef CAS PubMed
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed
- A. R. McDonald, M. Lutz, L. S. von Chrzanowski, G. P. M. van Klink, A. L. Spek and G. van Koten, Inorg. Chem., 2008, 47, 6681–6691 CrossRef CAS PubMed
- M. C. DeRosa, D. J. Hodgson, G. D. Enright, B. Dawson, C. E. B. Evans and R. J. Crutchley, J. Am. Chem. Soc., 2004, 126, 7619–7626 CrossRef CAS PubMed
- Q. Yan, K. Yue, C. Yu and D. Zhao, Macromolecules, 2010, 43, 8479–8487 CrossRef CAS
- R. J. Watts and D. Missimer, J. Am. Chem. Soc., 1978, 100, 5350–5357 CrossRef CAS
- E. Y.-T. Li, T.-Y. Jiang, Y. Chib and P.-T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Inorg. Chem., 2015, 54, 6582–6593 CrossRef CAS PubMed
- S. Ladouceur, L. Donato, M. Romain, B. P. Mudraboyina, M. B. Johansen, J. A. Wisner and E. Zysman-Colman, Dalton Trans., 2013, 42, 8838–8847 RSC
- Y.-S. Yeh, Y.-M. Cheng, P.-T. Chou, G.-H. Lee, C.-H. Yang, Y. Chi, C.-F. Shu and C.-H. Wang, ChemPhysChem, 2006, 7, 2294–2297 CrossRef CAS PubMed
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC
- R. Liu, Y. Li, Y. Li, H. Zhu and W. Sun, J. Phys. Chem. A, 2010, 114, 12639–12645 CrossRef CAS PubMed
- R. Liu, N. Dandu, Y. Li, S. Kilina and W. Sun, Dalton Trans., 2013, 42, 4398–4409 RSC
- Y. Li, R. Liu, E. Badaeva, S. Kilina and W. Sun, J. Phys. Chem. C, 2013, 117, 5908–5918 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: The 1H NMR spectra for 3–5. The electronic absorption and emission spectra in different solvents, time-resolved transient absorption spectra in toluene for 1–5, 1-L and 2-L. The normalized emission spectra of 3–5 in toluene at different excitation wavelengths. The normalized excitation spectra of 5 in toluene when monitored at different emission wavelengths. The calculated emission state diagram and NTOs for 1–5. See DOI: 10.1039/c5ra20084a |
| ‡ These two authors contribute equally to this project. |
| This journal is © The Royal Society of Chemistry 2016 |