
Retracted Article: Epoxy coating with embedded self-healing networks formed by nanogel particles
Abstract
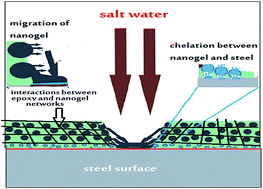
The paper describes the use of a nanogel for the preparation of epoxy-based, self-healing organic coatings for steel. The aim of the research was to use the nanogel as a nano sealant material against salt and humidity and to fill the micro- and nano-cracks which occurred during the curing of the epoxy primer and to apply it as a self-healing organic coating for steel. It is proposed that the self-healing behaviour of epoxy networks composed of nanogel particles occurs from the interconnection between nanogels and epoxy networks. This interconnection can reform rapidly under stress by forming a new structural rearrangement that preserves the mechanical damage to create high strength, self-healing materials. In this case, nanogels with different particle shapes and sizes based on crosslinked N-isopropylacrylamide (NIPAm) copolymer with acrylic acid (AA), 2-acrylamido-2-methylpropane sulfonic acid (AMPS) or 2-hydroxyethyl methacrylate (HEMA) were prepared using two different surfactant free techniques. The morphologies, particle shapes and sizes of nanogels were evaluated at different pH and ionic strengths of aqueous solution. The mechanical properties, salt spray resistance of the epoxy nanogel blend as organic coatings for steel were evaluated. The self-healing mechanism of the prepared nanogel for epoxy organic coating is proposed and was confirmed using scanning electron microscopy.
 Please wait while we load your content...
Please wait while we load your content...

