
Tunable near white light photoluminescence of lanthanide ion (Dy3+, Eu3+ and Tb3+) doped DNA lattices†
Sreekantha Reddy Dugasani a,
Byeongho Parkbc,
Bramaramba Gnapareddya,
Sreedhara Reddy Pamanjid,
Sanghyun Yooa,
Keun Woo Leea,
Seok Leeb,
Seong Chan Junc,
Jae Hun Kimb,
Chulki Kim*b and
Sung Ha Park*a
a,
Byeongho Parkbc,
Bramaramba Gnapareddya,
Sreedhara Reddy Pamanjid,
Sanghyun Yooa,
Keun Woo Leea,
Seok Leeb,
Seong Chan Junc,
Jae Hun Kimb,
Chulki Kim*b and
Sung Ha Park*a
aDepartment of Physics and Sungkyunkwan Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon 440-746, Korea. E-mail: sunghapark@skku.edu
bSensor System Research Center, Korea Institute of Science and Technology (KIST), Seoul 136-791, Korea. E-mail: chulki.kim@kist.re.kr
cSchool of Mechanical Engineering, Yonsei University, Seoul 120-749, Korea
dDepartment of Physics, Sri Venkateswara University, Tirupati 517502, India
First published on 17th June 2015
Abstract
For more than two decades, structural DNA nanotechnology has been investigated, yet researchers still have not clearly determined the functional changes and the applicability of DNA structures resulting from the introduction of a variety of ions. Lanthanide ions, such as Dy3+, Eu3+ and Tb3+, are interesting rare earth ions that have unique characteristics applicable to photonics. Here, we have constructed lanthanide ion doped double-crossover DNA lattices, a new class of functional DNA lattices, grown on a silica substrate. Deformation-free lattices were fabricated on a given substrate, and dopant ions were introduced to study their photoluminescence characteristics. The photoluminescence of the lanthanide ion-doped DNA lattices exhibited broad emission spectra in the visible region and a tendency of near white light emission composed of various colours. The intensity of the distinct spectral lines produced by the photoluminescence increased as the doping concentration of the ions reached the critical point, and the intensity then decreased with a further increase in the ions. Photoluminescence quenching was also observed when the excitation wavelength increased. These phenomena are the result of energy transfer between the DNA and the dopant ions. Finally, we make use of chromaticity diagrams to identify the colour coordinates of the luminescence produced by the lanthanide ion-doped DNA lattices, and this information may be useful to construct efficient bio-photonic devices or sensors in the future.
The motivation behind fabricating structural lanthanide ion (Ln3+)-doped DNA (Ln3+-DNA) lattices is to control and improve the physical properties of the DNA molecules, particularly for applications in the field of optoelectronics. Herein, we have selected the excellent luminescence in the visible region for Ln3+, such as dysprosium ions (Dy3+), europium ions (Eu3+), and terbium ions (Tb3+). Artificially designed DNA nanostructure is one of the most promising biomaterials because selective bindings of Ln3+ into the DNA molecules allows for the transfer of functionality, which makes it possible to utilize such molecules in a variety of applications in science and engineering. In addition, Ln3+ can be used to transform DNA molecules into luminescent probes. DNA is an efficient host of functional materials1 in the sense that programmable DNA nanostructures can be used as templates to align various materials, making Ln3+-DNA lattices as one of good candidates to produce bio light emitting diodes, biophotonics and biosensors. DNA is an important bio-macromolecule2 that offers specific binding sites for a wide range of functional molecules and metal ions.2–12 Although DNA luminescence can be enhanced by attaching fluorescent dyes, the size of these dyes is much larger than Ln3+, and their attachment into DNA molecules often requires modifying specific sites to produce ligands in the DNA sequences. In addition, fluorescent dyes produce an inherent toxicity and also exhibit aggregation and relatively less binding to DNA at a low resolution as compared to Ln3+.
Previous reports have focused on enhancing the luminescence of selective cations attached to single- and double-stranded DNA in a solution phase. Tb3+ enhances the luminescence of single-strand DNA to detect single base pair mismatches in duplex sequences,13 and the fluorescence sensitivity of Eu3+-doped protein and DNA molecules14 has also been previously reported. Tb3+ cations enhance the fluorescence as a consequence of an interaction with DNA because Tb3+ has the appropriate electronic transition to serve as an acceptor for long distance energy transfer (ET) from near ultraviolet absorbing nucleic acid bases.15 When DNA was used as an electron blocking layer in organic light emitting diodes, improved electroluminescent characteristics have been observed in both green and blue emissions.16 Such devices adopting DNA molecules as an electron blocking layer exhibit an appreciable luminescence when compared to conventional organic light emitting diodes without the DNA layer. Altogether, the dynamic admixtures of artificially designed and sequence programmable DNA nanostructures with Ln3+ will provide important luminescence applications with unique characteristics in terms of sensitivity, precision, and signal response. Here, we report on a simple but efficient fabrication method for Ln3+-DNA lattices to enhance the luminescence of DNA lattices. Ln3+-DNA lattices may serves as DNA detectors by controlling the ion doping concentration of Ln3+ as well.
In the present study, we utilize two-dimensional double-crossover (DX) DNA nanostructures17,18 consisting of two repeating DX tiles: DX1 and DX2. A unit DX tile is organized into two DX junctions where two parallel duplexes are tied up. The two types of DX tiles – each with a width of 4 nm and length of 12 nm – that were used to construct the DX DNA lattices on the given substrate are shown in Fig. 1a. Each tile consisted of four strands, and the complementary sticky end pairs are shown as S# and S#′ in the sequence drawings (Fig. 1a). The DX lattices were fabricated on a silica substrate via substrate-assisted growth (SAG)19–24 (for more details, see ESI†). The DNA strands are dissolved with a certain amount of Ln3+ in a common physiological 1× TAE/Mg2+ buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA (pH 8.0), and 12.5 mM magnesium acetate). These samples were stable for hybridization at up to a certain critical concentration, which is referred to as an optimum concentration Co. The DX lattices with Ln3+ concentration higher than Co are not properly hybridized to form crystalline structures due to the presence of excess Ln3+ introduced before annealing in the test tube. Instead they form amorphous and aggregated structures without a periodic crystalline form (data not shown). To avoid them, we adopted a post-annealing procedure where Ln3+ was added after hybridization in order to maintain deformation-free DX lattices, even at a concentration slightly higher than Co. The experimental data indicated that, by coincidence, all tested ions – Dy3+, Eu3+, and Tb3+ – had the almost same value of 1 mM for Co. The schematics of the DX lattices and the possible representative Ln3+ coordination sites in the DNA molecules are shown in Fig. 1b and c, respectively. The yellow circles in the schematics (Fig. 1c) represent all the possible coordination sites of the Ln3+ in the DNA molecules.
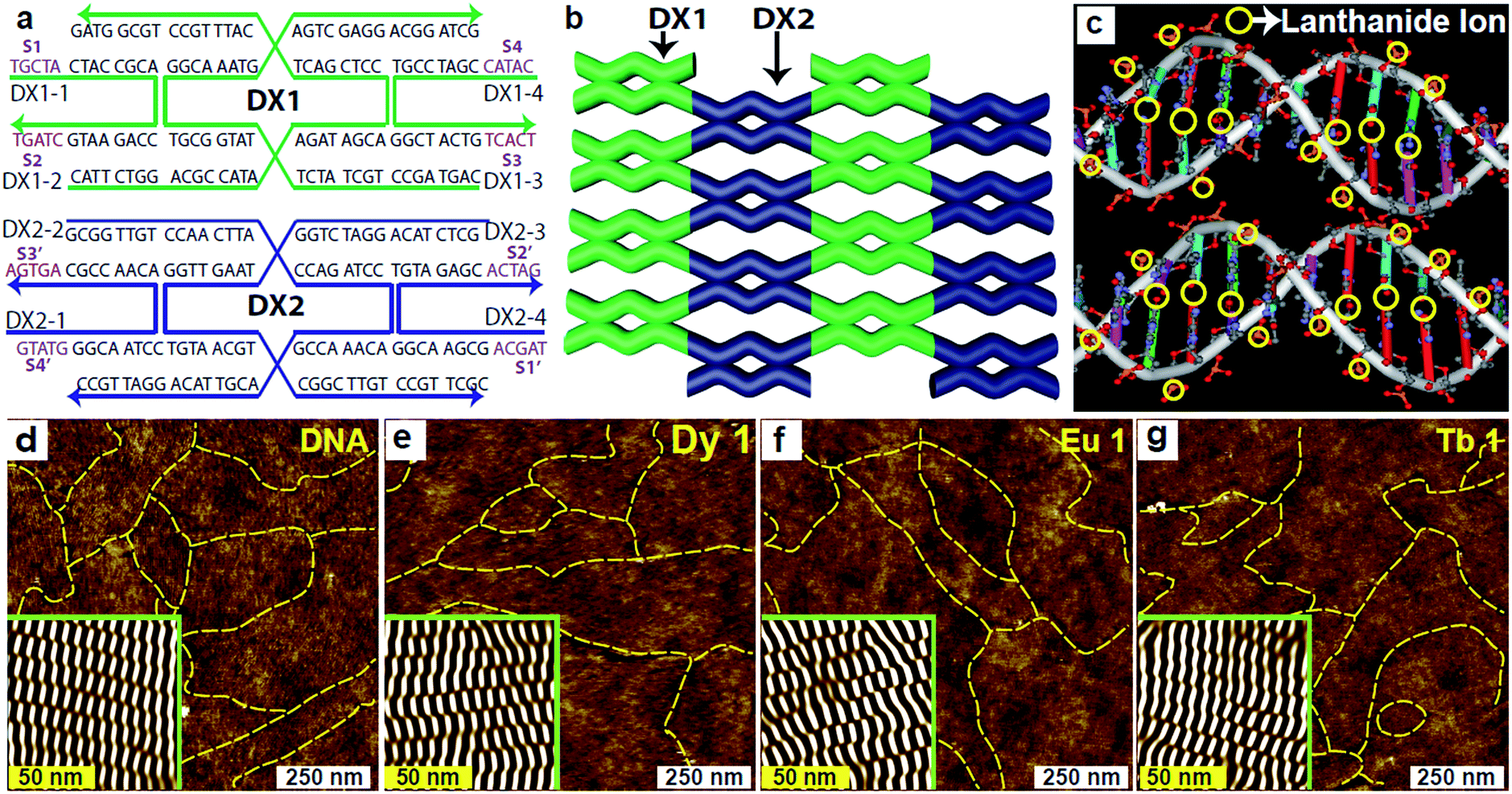 | ||
| Fig. 1 Schematic diagrams of DX sequence map, DX lattice and Ln3+ coordination sites in DNA. (a) DNA sequences of two double-crossover (DX) tiles. Each tile consists of four strands, and each strand arrow points to the 3′ end. The complementary sticky-end pairs are shown as S# and S#′ (violet). (b) An illustration of a DX lattice. (c) Schematic diagram of the representative Ln3+ coordination sites into the DX duplex. Characteristic AFM images of (d–g) pristine, dysprosium ion (1 mM) doped, europium ion (1 mM) doped, and terbium ion (1 mM) doped DNA lattices, labelled as DNA, Dy 1, Eu 1, and Tb 1, respectively. The insets in the AFM images are noise-filtered images reconstructed via fast Fourier transform showing clear periodicities in the DX lattices. The yellow dotted lines in the AFM images reveal the lattice boundaries. | ||
For consistency, other experimental parameters remained fixed during the annealing process, that is, the annealing time to control the thermal energy remained at 24 hours, the substrate was set to 5 × 5 mm2 to determine the total electrostatic interaction between the substrate and DNA molecules, and the sample volume in a test tube was set to 250 μL. On the other hand, the concentration of Ln3+ was adjusted as a control parameter, with distinct concentrations of 0, 0.5, 1, 2, and 4 mM of Dy3+, Eu3+, and Tb3+ into DX lattices. The different concentrations are indicated as Dy 0, Dy 0.5, Dy 1, Dy 2, and Dy 4 for the Dy3+-DNA lattices; Eu 0, Eu 0.5, Eu 1, Eu 2, and Eu 4 for the Eu3+-DNA; and Tb 0, Tb 0.5, Tb 1, Tb 2, and Tb 4 for the Tb3+-DNA. We conducted atomic force microscopy (AFM) to verify that the Ln3+-DNA lattices had formed well on the given substrate. Fig. 1d–g show AFM images of both pristine DNA and of Ln3+-DNA lattices with a critical concentration of each Dy 1, Eu 1, and Tb 1 with deformation-free DX structures with the respective Ln3+ coordination. The insets in the AFM images present noise-filtered images reconstructed via fast Fourier transformation that show clear periodicities in the DX lattices. The yellow dotted lines in all of the AFM images indicate the DX lattice boundaries.
The excitation spectra were captured at fixed emission wavelengths (λem) 466, 538, and 560 nm for pristine DNA; at 464, 558, and 608 nm for Dy 1; at 526, 539, and 586 nm for Eu 1; and at 539, 574, and 593 nm for Tb 1, as shown in Fig. 2a–d. These excitation spectra clearly reveal intense peaks in the range of 350–450 nm, which indicates a proper range of excitation wavelengths (λex) between 350 and 450 nm. The considerable ET can be explained by using the energy level (EL) diagrams shown in Fig. 2e–h. The photoluminescence requires the appropriate excitation energy to excite an electron to a singlet excited state (S1) from the singlet ground state (S0) upon absorbing the photon energy. A preferable approach for relaxation is an internal conversion (IC) in which relaxation from a higher vibrational energy level of an excited state occurs toward the zero level of that excited state. Such a mechanism is referred to as nonradiative energy transfer (NRET).
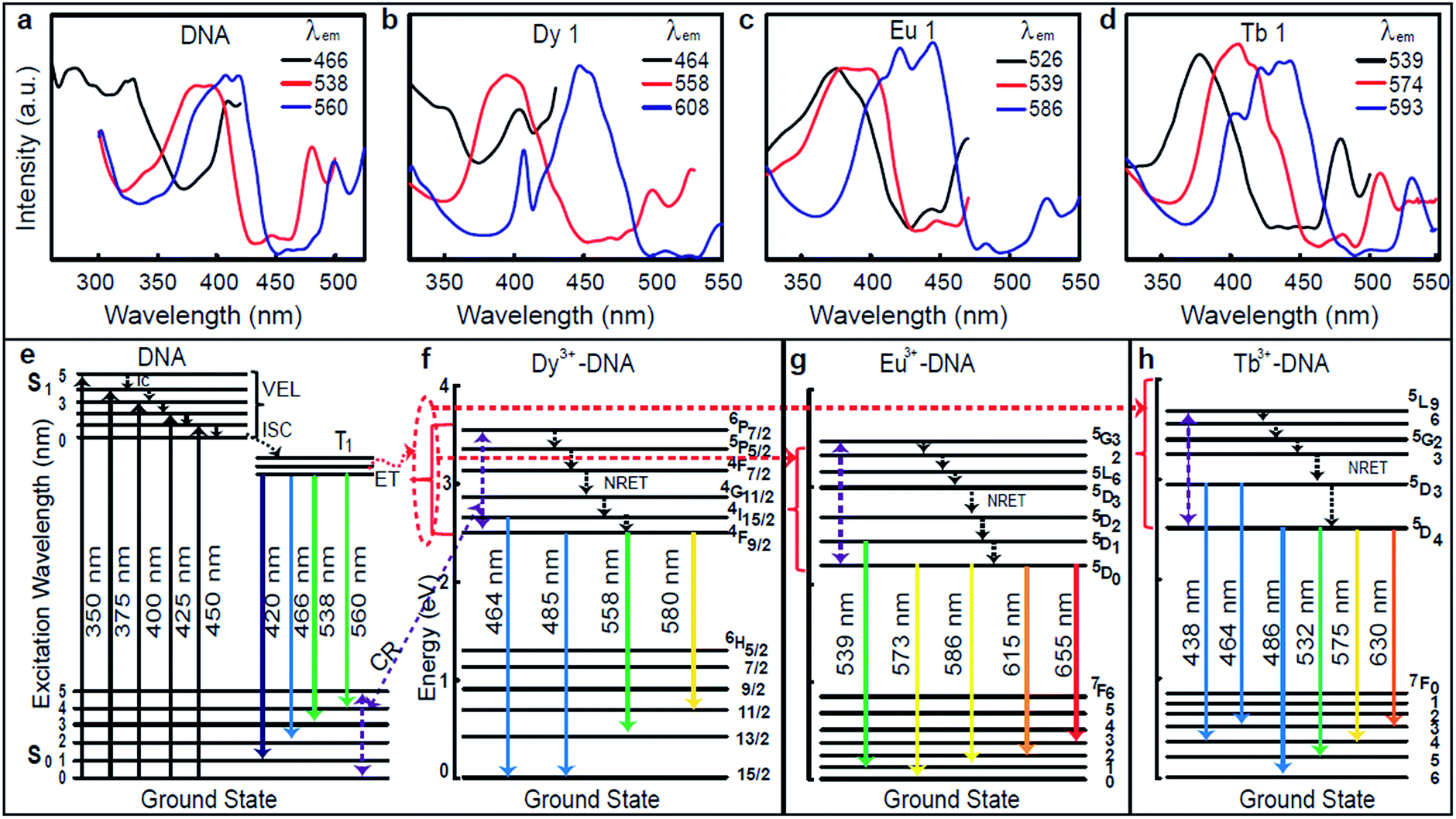 | ||
| Fig. 2 Excitation spectra and representative energy level diagram of pristine DNA and Ln3+-DNA lattices. Excitation spectra of (a–d) pristine, dysprosium ion (1 mM) doped, europium ion (1 mM) doped, and terbium ion (1 mM) doped DNA lattices at various emission wavelengths. (e–h) The representative energy level diagrams for pristine, dysprosium ion (1 mM) doped, europium ion (1 mM) doped, and terbium ion (1 mM) doped DNA lattices show the various excitation (↑) and conceivable emission (↓) wavelength transitions. Here the red dotted regions indicate the energy transfer (ET) from DNA molecules to Ln3+, and the violet dotted regions indicate the energy transfer from Ln3+ to DNA through cross relaxation (CR). (Abbreviation note: IC = internal conversion, VEL = vibrational energy levels, ISC = inter-system crossing, ET = energy transfer, CR = cross relaxation, NRET = nonradiative energy transfer.) | ||
The inter-system crossing (ISC) is a type of NRET that occurs when an electron relaxes to the triplet excited state (T1) from the singlet excited state. Photoluminescence then arises from the release of a photon upon relaxation of the electron from the triplet excited state. The EL diagrams shown in Fig. 2e–h depict the possible emission process for the pristine DNA and Dy3+, Eu3+, and Tb3+-DX lattices with indications of the various excitation (↑) and conceivable emission (↓) wavelength transitions. DNA molecules are well known to exhibit absorption in the range of 250–280 nm, thus ET occurs through an IC process within the excited singlet state, then from the excited singlet state to the triplet state through the ISC process, and finally to emissive states.13 At up to Co for each Ln3+, the ET from the excited states of the DNA to the emissive 4I15/2 and 4F9/2 states of the bound Dy3+, to the emissive 5D1 and 5D0 states of bound Eu3+, and to the emissive 5D3 and 5D4 states of bound Tb3+ are expected to take place from the lowest triplet excited state in Ln3+-DNA lattices. Above Co for each ion, the ET might occur from Ln3+ to the DNA through cross relaxation (CR) (violet dotted lines in Fig. 2e–h) due to the excess Ln3+ binding nonspecifically to DNA molecules. Such an ET results in the enhancement of the luminescence in Ln3+-DNA lattices.
The photoluminescence spectra of the Ln3+-DNA lattices as a function of Ln3+ concentration with a λex of 350 nm at room temperature are shown in Fig. 3. The corresponding columns indicate the photoluminescence spectra of the same samples under various λex's of 375, 400, 425, and 450 nm. These emission spectra present distinct emission peaks and intensities. The results obviously differentiated the Ln3+ doping levels in the DNA molecules. With various λex and concentrations of Dy3+ in the Dy3+-DNA lattices, the photoluminescence spectra exhibit emissions from the DNA singlet excited state ET to the Dy3+ in the visible spectral regions correspond to the 4I15/2 excited state to 6H15/2 ground state, and 4F9/2 excited state to the 6H15/2 ground state as well as to the 6H13/2,11/2 states. The ET pathway of the emission spectral lines is shown in Fig. 2e and f. Here, the red dotted circle and the violet dotted line indicated the ETs from DNA to Dy3+ (≤Co of Dy3+) and from Dy3+ to DNA (>Co of Dy3+) in the Dy3+-DNA lattices, respectively. The characteristic spectral lines of Dy3+ were previously identified at 485 and 580 nm in different host.25 The blue lights at 464 and 485 nm were observed due to the magnetic dipoles allowing 4I15/2 → 6H15/2, and 4F9/2 → 6H15/2 transitions, respectively. A relatively light doping of Dy3+ into DNA lattices, compared to Co, exhibits broad green spectral line at 558 nm. This green luminescence has been associated with the presence of specific ion bindings into DNA which allowed 4F9/2 → 6H13/2 transitions at 558 nm. Interestingly, 4F9/2 → 6H11/2 spectral transition at ∼580 nm (yellow) was strongly sensitive to the ion concentrations which might be due to the CR between DNA and Dy3+.
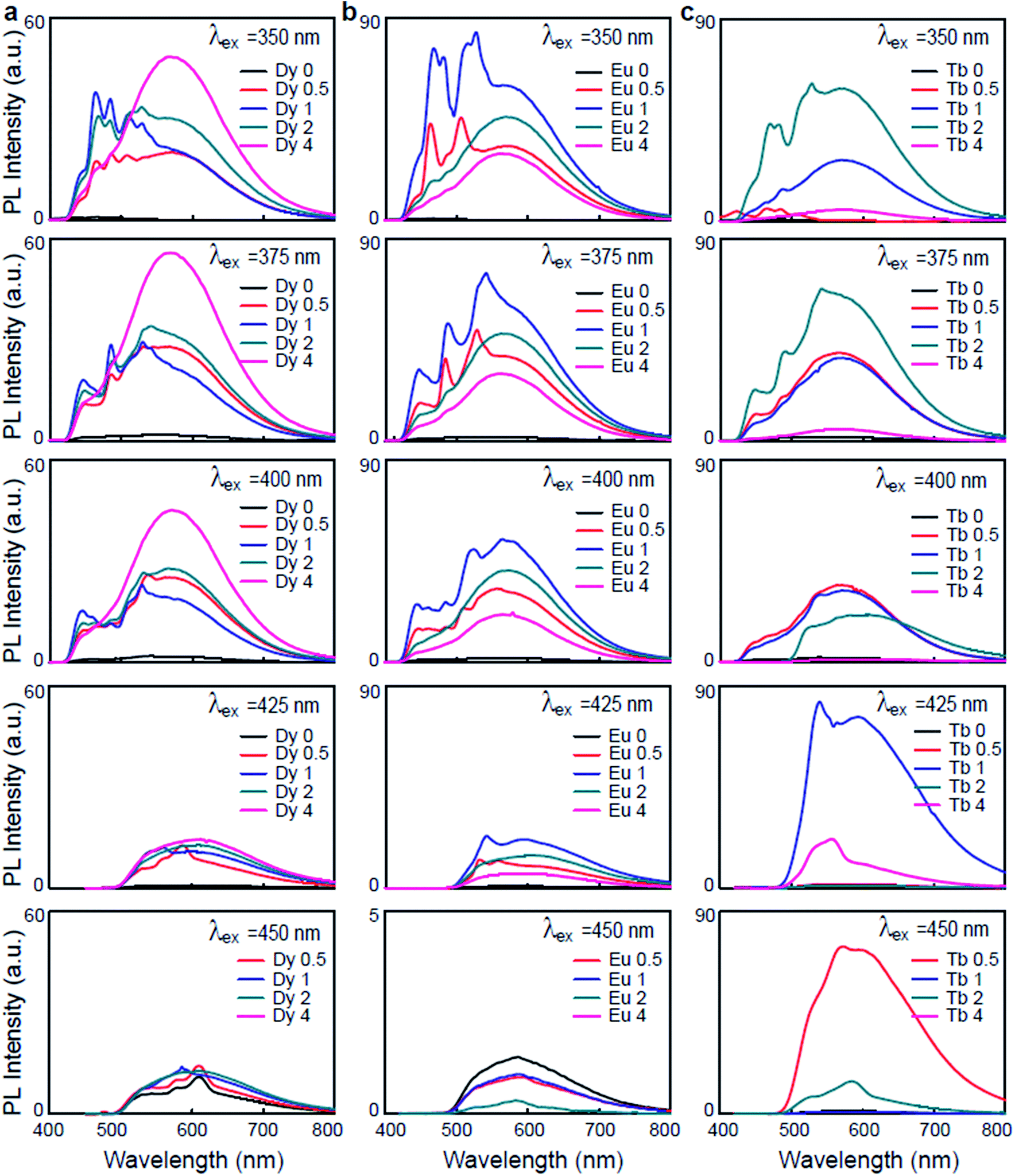 | ||
| Fig. 3 Emission spectra of Ln3+-DNA lattices under various excitation wavelengths. The emission spectra of (a) dysprosium ion doped, (b) europium ion doped, and (c) terbium ion doped DNA lattices with various ion concentrations of 0, 0.5, 1, 2, and 4 mM under excitation wavelengths of 350, 375, 400, 425, and 450 nm. | ||
Fig. 3b shows the variation in the photoluminescence spectra of the Eu3+-DNA lattices as the ion concentration increases. The emission spectra consist of several emission lines originating from the transitions of the emissive state 5D1 to the ground state 7F1 and of 5D0 to the ground state 7FJ (J = 0, 1, 2, 3) in the 4f6 configuration of Eu3+. Fig. 2e and g show the ET pathway of the emission spectral lines, and here, the red dotted line indicates the ET from DNA to Eu3+ (≤Co of Eu3+), and the violet dotted line indicates the ET from Eu3+ to DNA (>Co of Eu3+) in Eu3+-DNA lattices through CR. The emission intensities increased with the increase of the ion concentration up to Co 1 mM of Eu3+, and then decreased after the further increase of the ion concentration. The characteristic Eu3+ peaks were observed at 573, 586, and 615 nm, which are similar as those that were previously reported.26 At lower concentrations of Eu3+ into the DNA lattices, a green emission band at 539 nm was exhibited, which allowed the emission transition of 5D1 → 7F1. At higher concentrations, the relative intensities of 5D0 → 7F0, 5D0 → 7F1, 5D0 → 7F2, and 5D0 → 7F3 transitions were quite sensitive to the concentration of Eu3+ in a manner similar as that for Dy3+. The photoluminescence quenching in the Eu3+-DNA lattices might depend upon λex and Eu3+ concentration. With the given excitation of light, Eu3+ can interact with neighbouring ions through various magnetic dipole–dipole, electric dipole–dipole, electric dipole–quadrupole, and electric quadrupole–quadrupole interactions.
Fig. 3c shows the photoluminescence spectra of the Tb3+-DNA lattices as a function of Tb3+ concentration. At λex of 350 nm, the spectral intensities of the emissions increased as the ion concentration increased up to 2 mM, and then decreased. The blue and green emissions (lower) and yellow and red emissions (higher) were dominant at lower and higher concentration of Tb3+ into DNA lattices relative to Co. Under various λex and Tb3+ concentration, the Tb3+-DNA lattices showed emissions from the DNA singlet excited state ET to the Tb3+ in the visible spectral regions, which corresponded to the 5D3 excited state to the 7F4,3 ground states; and from 5D4 excited state to the 7F6 ground state and the 7F5,4,3 levels. As for Dy3+ and Eu3+, the ET pathway of the emission spectral lines is shown in Fig. 2e and h. The red dotted line indicates the ET from DNA to Tb3+ (≤Co of Tb3+), and violet dotted line indicates the ET from Tb3+ to DNA (>Co of Tb3+) in the Tb3+-DNA lattices. The characteristic emission peaks for Tb3+ could be observed at 438, 464, 486, 532, 575, 630 nm, which match quite well with those reported in previous studies.26 The emission transitions that were allowed were 5D3 → 7F4 (blue) at 438 nm, 5D3 → 7F3 (blue) at 464 nm, 5D4 → 7F6 (blue) at 486 nm, 5D4 → 7F5 (green) at 532 nm, 5D4 → 7F4 (yellow) at 575 nm, and 5D4 → 7F3 (orange) at 630 nm. The lightly-doped Tb3+-DNA lattices exhibited a high intensity with sharp blue and broad green spectral lines at 486 and 532 nm. The intensity gradually decreased by reducing the given excitation energy of the Ln3+-DNA lattices, which could be due to the fact that the energy was not sufficient for ET from donor to acceptor states.
The variations in the photoluminescence intensities and the spectral line positions were a result of the different transitions in the Ln3+-DNA lattices manifesting that Ln3+ ions were well coordinated into DNA. At Ln3+ concentrations higher than Co, more crucial description was often needed due to inhomogeneous and nonspecific bindings that occurred as well. These undesired bindings to the DNA lattices led to slight distortions in the DNA lattice structures, and consequently decreased the intensity of the luminescence. The intensities of the blue spectral lines were found to heavily rely upon the critical Co of Ln3+. They gradually increased at up to Co, which was 1 mM for Dy3+ and Eu3+ and 2 mM of Tb3+, and a decrease in intensity followed as the ion concentrations further increased, which indicated high sensitivity of the luminescence to the concentration of the Ln3+ dopant and determined the controllable optical properties of the DNA lattices. In addition, a significant blue luminescence resulted from the dominant blue transition in the DNA lattices when the Ln3+ were optimally coordinated with homogeneous and specific bindings at Co. Furthermore, the luminescence in the green to orange spectral region was found to be stronger for lightly doped DNA lattices than for highly doped ones. This means that further quenching of the luminescence had to be taken into account as a result of the differences in the ion concentration. When the excitation spectra were monitored in the fixed blue to orange emission spectral region, the emissions were observed to be preferentially excited through higher excited states of the 4fn configuration, i.e., 4f9 of Dy3+, 4f6 of Eu3+ and 4f8 of Tb3+.
Overall, the photoluminescence spectra produced broad emissions which might be due to the extremely thin thickness (∼0.6 nm) of the Ln3+-DNA lattices. The known diameter of the DNA duplex is of ∼1.9 nm, but the DNA duplex on a substrate has a diameter of 1.2 ± 0.2 nm in a buffer solution and 0.6 ± 0.2 nm in a dry state due to the presence of electrostatic attraction between the DNA molecules and the substrate.27,28 Our observations indicate that the Ln3+-DNA lattices have a significant luminescence in a wide range of wavelengths where the Dy3+ yellow lines merge with broad blue-green emissions, Eu3+ red lines merge with broad green-orange emissions, and Tb3+ green lines merge with broad blue-yellow emissions. The ET has an advantage in that it improves Ln3+ emissions produced as a result of the transfer from an excited state of the donor bases to the emissive state of the Ln3+ acceptors. The efficiency of the ET can be controlled by adjusting the Ln3+ concentration, the excitation energy, and the proper coordination sites of the ions, i.e., the base pairs and the phosphate backbones.13,15,29 Interestingly, divalent metal ion bindings to DNA have been previously described by a number of researchers. Eichhorn et al.30 suggested that the metal ion binding sites in the DNA molecule were phosphate groups and electron donor atoms on heterocyclic bases. If zinc ion (Zn2+) doped DNA duplexes were prepared through a freeze-dry method, the chemical configuration of the Zn2+ revealed covalent bonding of Zn2+ with nitrogen atoms in the base-pairs due to the extremely dry condition.8 Similarly Lee et al. reported intercalation of the Zn2+ between the base-pairs in order to study the cooperative conformational change in the DNA duplexes.31
To better understand the origin of the photoluminescence via Ln3+-DNA lattices, we analyzed the resulting photoluminescence spectra through Gaussian and Lorentzian functions. The typical Gaussian fittings of the UV-visible photoluminescence spectra exhibits various peaks located at 471, 527, 580, 620, 635, 651, and 717 nm for Dy 4; 566, 572, 616, 630, 655, and 710 for Eu 2; 438, 464, 486, 532, 575, and 630 nm for Tb 1, as shown in Fig. 4a–c. Fig. 4d–f show the position of the intense emission peaks, as well as the height, area, and full width at half maximum (FWHM), as a function of the Ln3+ concentration under an λex of 400 nm. Appreciable red shift is observed in the peak position as the ion concentration increases, which is a strong evidence of Ln3+ coordination into the DNA. The peak height and area of the Dy3+-DNA lattices increase as the ion concentration increases, and the FWHM values increase to up to 1 mM and then decrease. For the Eu3+-DNA and Tb3+-DNA lattices, the peak height, area, and FWHM values increase as the ion concentration increases up to 1 mM of Eu3+ and 2 mM of Tb3+, and then decrease as the concentration increases further. The bending tendency of the FWHM values and the associated critical doping level of the Ln3+-DNA lattices can be explained as follows: the Ln3+ prefer to intercalate between the base-pairs and to bind with the phosphate backbone up to Co, forming long chains that serve as luminescent arrays inside the duplex without disturbing the original DNA structures much. An excess amount of Ln3+ (Ln3+ concentration >Co) into the DNA causes them to bind to improper and indiscriminate sites, which in turn creates stress on the helical properties of the DNA.
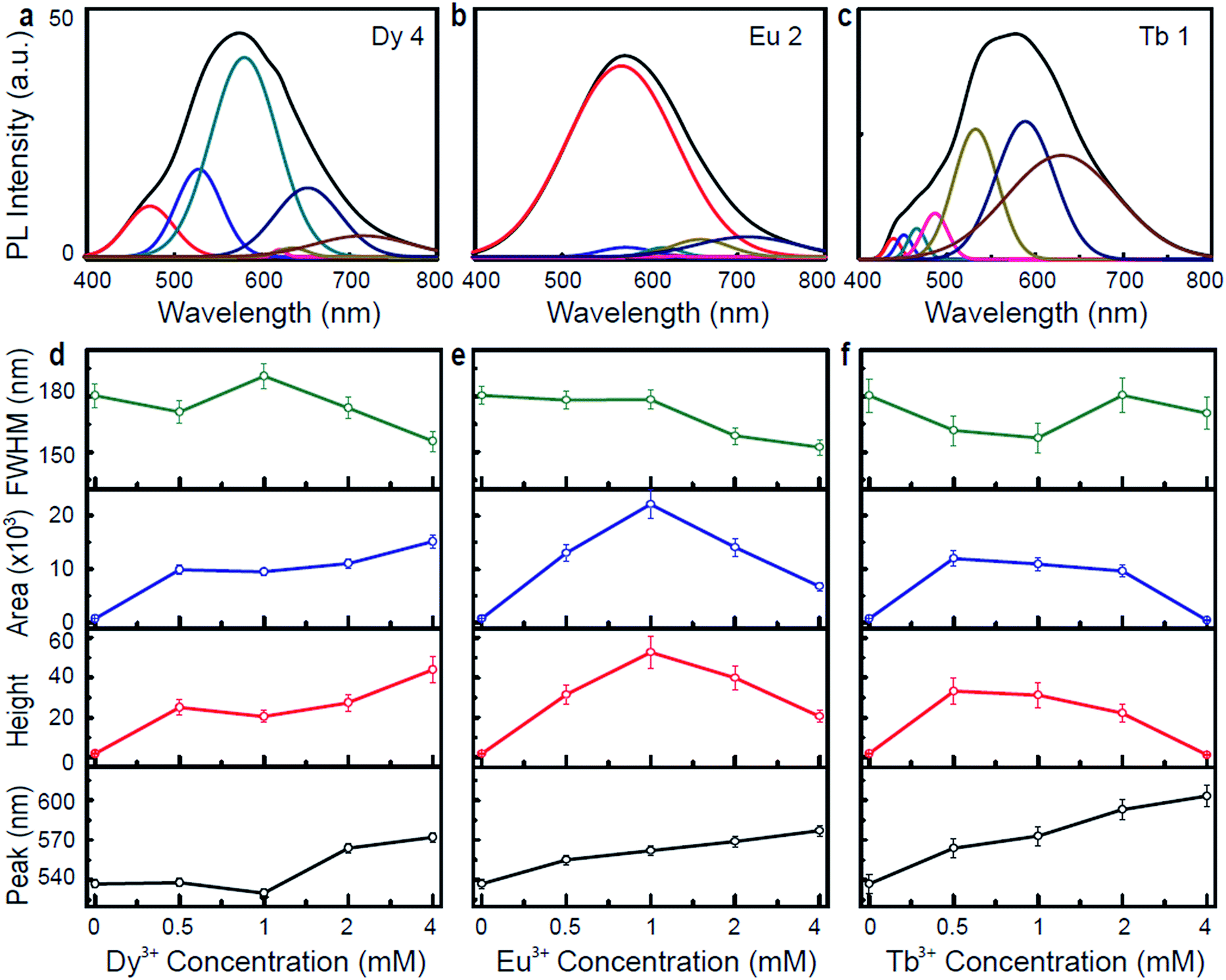 | ||
| Fig. 4 Typical Gaussian fitting and emission analysis of Ln3+-DNA lattices. Gaussian fitting of (a) dysprosium ion (4 mM) doped, (b) europium ion (2 mM) doped, and (c) terbium ion (1 mM) doped DNA lattices under specific excitation wavelengths of 400 nm. Representation of intense emission peak position, height, area, and full width at half maximum (FWHM) as function of the Ln3+ concentration of (d) dysprosium ion doped, (e) europium ion doped, and (f) terbium ion doped DNA lattices. These values are extracted from the emission spectra under excitation wavelengths of 400 nm using Gaussian and Lorentzian functions. | ||
In general, the trichromatic system of colorimetry recommended by the Commission Internationale de l'éclairage (CIE) (1931) is used to specify the colour of light. The CIE standards provide two coordinates along the x and y directions that are related to the emission wavelength and the energy distribution of the light. Fig. 5a–c show the colour coordinate chromaticity diagrams of Dy3+-DNA, Eu3+-DNA, and Tb3+-DNA lattices as a function of the Ln3+ concentrations under typical λex, such as 375, 350, and 350 nm, respectively. Fig. 5d–f show the colour coordinates in the CIE 1931 chromaticity diagram of the Ln3+-DNA lattices as a function of λex at a critical concentration of each ion into DNA. The diagram indicates colour changes from blue to orange as the ion concentration varies. Interestingly, the emission quite close to the white light can be possible by obtaining the appropriate mixture of visible transitions of the Ln3+.
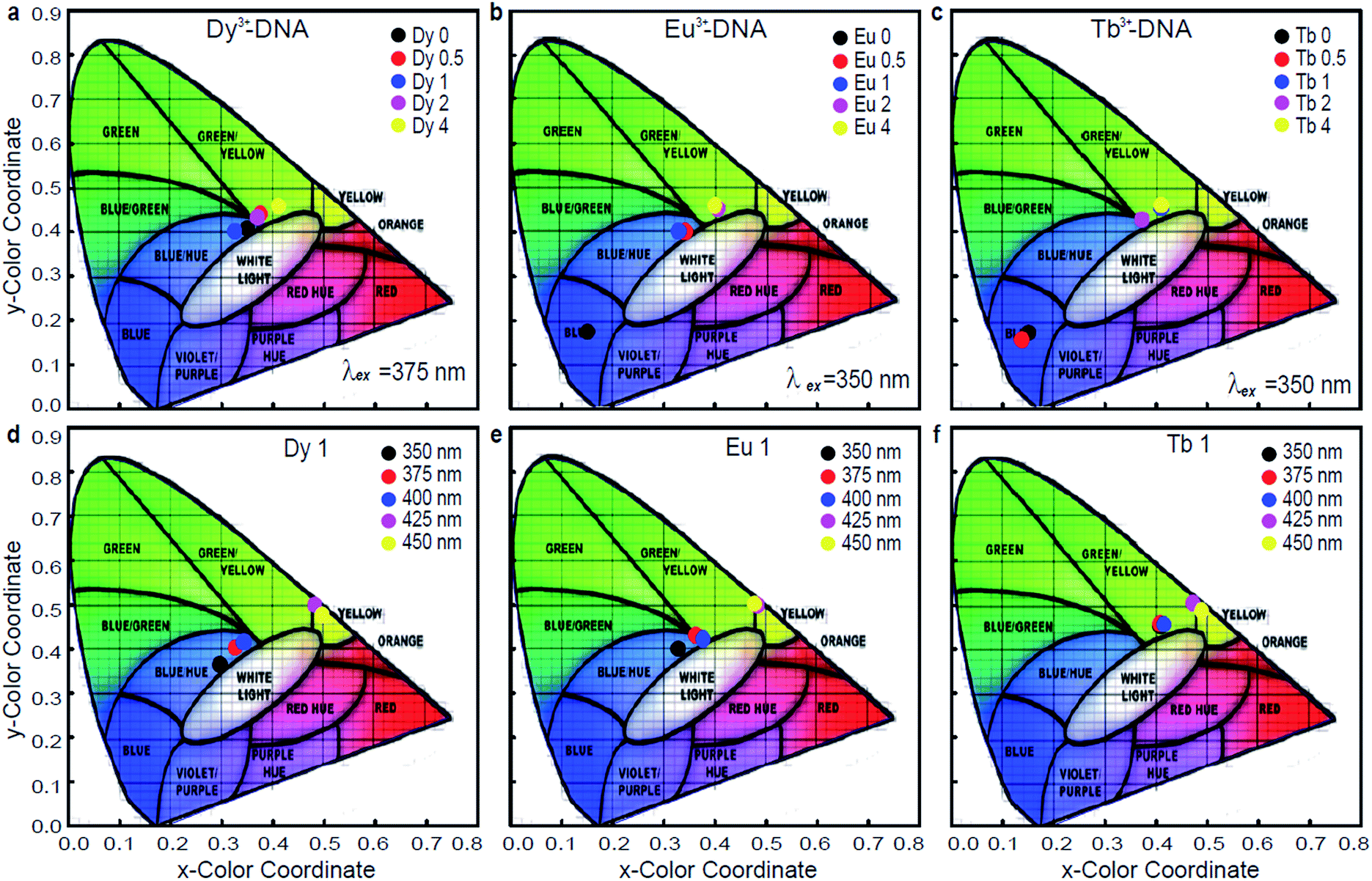 | ||
| Fig. 5 Possible colour coordinates controlled by various Ln3+ concentration and excitation wavelength. Chromaticity diagram of (a) dysprosium ion doped, (b) europium ion doped, and (c) terbium ion doped DNA lattices with various Ln3+ concentrations under particular excitation wavelengths such as 375, 350, and 350 nm, respectively. The chromaticity coordinates on the CIE 1931 chromaticity diagram of (d) dysprosium ion doped, (e) europium ion doped, and (f) terbium ion doped DNA lattices with critical concentrations of each ion under various excitation wavelengths. | ||
The activation of ET phenomena through variations in Ln3+ concentration permits an excellent control of emission chromaticity across the CIE 1931 diagram, e.g., (x, y) colour coordinates from (0.154, 0.178) to (0.423, 0.462), (0.401, 0.464), and (0.411, 0.465) can be obtained for Dy3+-DNA, Eu3+-DNA, and Tb3+ DNA, respectively, at a fixed λex of 350 nm. These variations in colour coordinates for the Ln3+-DNA lattices are mainly a result of the different intensities of blue, green, yellow, and orange spectral lines. The ET between the host DNA molecules and the Ln3+ cations originate from the Ln3+-DNA emitting center. The ability to tune the colour coordinates by controlling the Ln3+ concentration in the DNA nanostructures can be useful for practical applications due to the simple fabrication process. Even though the colour coordinates are also slightly controllable by adjusting λex, the colour efficiency and yield might be reduced because the emission intensities were much suppressed at longer λex than at shorter ones, as shown in Fig. 3.
Conclusions
In conclusion, we studied the photoluminescence of the ultra thin Ln3+-DNA lattices as a function of both λex and Ln3+ concentration. By adding Ln3+ into DNA, the luminescence colour of the spectral lines varies from blue to orange. At a particular concentration of each Ln3+, the intensity of the photoluminescence is enhanced due to the homogeneous and specific bindings to DNA via intercalation between base-pairs and binding on phosphate backbones. The coordination of Ln3+ into DNA has originated as an alternative to overcome some of the limitations of the traditional fluorescent labels, such as organic dyes or quantum dots. The ultimate advantage of adopting DNA lattices combined with Ln3+ ions is the ability to control the spectrum of the luminescence with Ln3+ concentrations in precision. The insight gained from this study opens the door to searching for new functional DNA nanostructures for practical applications towards the fabrication of bio-photonic devices and biosensors with better sensitivity, efficiency, and photo-stability.Acknowledgements
This research was supported by the Basic Science Research Program (NRF-2013R1A1A2061731) for S.R.D. and by the Nano Material Technology Development Program (NRF-2012M3A7B4049801 & NRF-2012M3A7B4049804) of the National Research Foundation of Korea (NRF) for S.H.P. & C.K., through funding from the Ministry of Education, Science and Technology (MEST) and the Ministry of Science, ICT & Future Planning (MSIP) in Korea.Notes and references
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–224 RSC.
- A. Condon, Nat. Rev. Genet., 2006, 7, 565–575 CrossRef CAS PubMed.
- B. A. Armitage, Top. Curr. Chem., 2005, 253, 55–76 CAS.
- H. Ihmels and D. Otto, Top. Curr. Chem., 2005, 258, 161–201 CrossRef CAS.
- S. R. Dugasani, N. H. Lee, J. Lee, B. Kim, S. Hwang, K. W. Lee, W. N. Kang and S. H. Park, Sci. Rep., 2013, 3, 1819 CrossRef PubMed.
- S. J. Kim, J. Jung, K. W. Lee, D. Yoon, T. S. Jung, S. R. Dugasani, S. H. Park and H. J. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 10715–10720 CAS.
- A. Rakitin, P. Aich, C. Papadopoulos, Y. Kobzar, A. S. Vedeneev, J. S. Lee and J. M. Xu, Phys. Rev. Lett., 2001, 86, 3670–3673 CrossRef CAS.
- A. Omerzu, B. Anzelak, I. Turel, J. Strancar, A. Potocnik, D. Arcon, I. Arcon, D. Mihailvic and H. Matsui, Phys. Rev. Lett., 2010, 104, 156804 CrossRef.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS PubMed.
- S. Pal, Z. Deng, B. Ding, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 2700–2704 CrossRef CAS PubMed.
- K. Keren, R. S. Berman, E. Bushstab, U. Sivan and E. Braun, Science, 2003, 302, 1380–1382 CrossRef CAS PubMed.
- H. T. Maune, S. P. Han, R. D. Barich, M. Bockrath, W. A. Goddard and P. W. K. Rothermund, Nat. Nanotechnol., 2010, 5, 61–66 CrossRef CAS PubMed.
- K. L. F. Patty and T. Claudia, J. Am. Chem. Soc., 1999, 121, 1–7 CrossRef.
- A. K. Saha, K. Kross, E. D. Kloszewski, D. A. Upson, T. L. Toner, R. A. Snow, C. D. V. Black and V. C. Desai, J. Am. Chem. Soc., 1993, 115, 11032–11033 CrossRef CAS.
- M. D. Topal and J. R. Fresco, Biochemistry, 1980, 19, 55315537 CrossRef.
- J. A. Hagen, W. Li, A. J. Steckl and J. G. Grote, Appl. Phys. Lett., 2006, 88, 171109 CrossRef PubMed.
- T. J. Fu and N. C. Seeman, Biochemistry, 1993, 32, 3211–3220 CrossRef CAS.
- E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539–544 CrossRef CAS PubMed.
- S. Hamada and S. Murata, Angew. Chem., Int. Ed., 2009, 48, 6820–6823 CrossRef CAS PubMed.
- X. Sun, S. H. Ko, C. Zhang, A. E. Ribbe and C. Mao, J. Am. Chem. Soc., 2009, 131, 13248–13249 CrossRef CAS PubMed.
- J. Lee, S. Kim, J. Kim, C. W. Lee, Y. Roh and S. H. Park, Angew. Chem., Int. Ed., 2011, 50, 9145–9149 CrossRef CAS PubMed.
- A. Kulkarni, B. Kim, S. R. Dugasani, P. Joshirao, J. Kim, C. Vyas, V. Manchanda, T. Kim and S. H. Park, Sci. Rep., 2013, 3, 2062 Search PubMed.
- J. Lee, S. Hamada, S. Hwang, R. Amin, J. Son, S. R. Dugasani, S. Murata and S. H. Park, Sci. Rep., 2013, 3, 2115 Search PubMed.
- S. R. Dugasani, J. Kim, B. Kim, P. Joshirao, B. Gnapareddy, C. Vyas, T. Kim, S. H. Park and V. Manchanda, ACS Appl. Mater. Interfaces, 2014, 6, 2974–2979 CAS.
- M. R. N. Soares, M. J. Soares, A. J. S. Fernandes, L. Rino, F. M. Costa and T. Monteiro, J. Mater. Chem., 2011, 21, 15262–15265 RSC.
- Z. Liu, M. F. Wu, S. H. Wang, F. K. Zheng, G. E. Wang, J. Chen, Y. Xiao, A. Q. Wu, G. C. Guo and J. S. Huang, J. Mater. Chem. C, 2013, 1, 4634–4639 RSC.
- S. H. Park, M. W. Prior, T. H. LaBean and G. Finkelstein, Appl. Phys. Lett., 2006, 89, 033901 CrossRef PubMed.
- M. Antognozzi, M. D. Szczelkun, A. N. Round and M. J. Miles, Single Mol., 2002, 3, 105–110 CrossRef CAS.
- D. Gersanovski, P. Colson, C. Houssier and E. Fredericq, Biochim. Biophys. Acta, Gene Struct. Expression, 1985, 824, 313–323 CrossRef CAS.
- G. L. Eichhorn, Advances in Inorganic Biochemistry, ed. G. L. Eichhorn and L. G. Marzilli, Elsevier, New York, 1982, vol. 3, p. 2 Search PubMed.
- J. S. Lee, L. J. P. Latimer and R. S. A. Reid, Biochem. Cell Biol., 1993, 71, 162–168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07360j |
| This journal is © The Royal Society of Chemistry 2015 |