
Pyridine–triazole ligands for copper-catalyzed aerobic alcohol oxidation†
Abstract
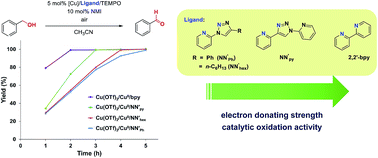
A series of Cu(NN′)2(OTf)2 complexes containing pyridine–triazole ligands [OTf = OSO2CF3; NN′ = NN′Ph (1), NN′hex (2), NN′py (3)] with different substituents at the triazole N4 position or 2,2′-bipyridine (bpy; 4) have been synthesized. Crystal structures of 1 and 3 reveal a trans-isomer with strong preference for regular-type triazole coordination (for 3) whereas the Cu–bipyridine complex 4 is more stable in a cis-form. Cyclic voltammetry of 1–4 suggest that the electron-donating strength follows the trend: bpy > NN′py > NN′hex ∼ NN′Ph. The catalyst systems consisting of 5 mol% Cu(OTf)2/NN′/TEMPO (TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxy) in the presence of 2 × 2.0 cm2 Cu0 sheets as a reducing agent and 10 mol% N-methylimidazole (NMI) exhibit good activities for aerobic oxidation of benzyl alcohol to benzaldehyde. Catalytic studies have shown that the activities were higher with more electron-rich N-based ligands. Furthermore, oxidation of aliphatic alcohols such as 1-hexanol and 2-methyl-1-pentanol using the Cu catalyst system with the NN′py ligand at room temperature afforded the corresponding aldehydes in >99% and 46% yields, respectively after 24 h.
 Please wait while we load your content...
Please wait while we load your content...

