
Polypyrrole derivatives for electrochromic applications
Pinar Camurlu*
Akdeniz University, Department of Chemistry, Antalya, 07058, Turkey. E-mail: pcamurlu@akdeniz.edu.tr; Fax: +90 242 2278911; Tel: +90 242 3102308
First published on 30th October 2014
Abstract
Electrochromic materials display a reversible color change upon electrochemical cycling. In the last two decades there has been increasing interest in the electrochromic properties of polypyrroles. In this article, we highlight and provide insight into the electrochromic properties of polypyrroles in terms of their structure–property relationships without the intention of providing a complete chronological order.
1. Introduction
Chromism, which is described as the reversible change of a material's color resulting from a electrochemical stimulus is called electrochromism. An electrochromic material undergoes a reversible optical change (transmittance and/or reflectance) because of its electrochemically induced oxidation and reduction.1,2 Coloration could take place between a transparent and a colored state, or between two colored states or between multiple steps (multichromic). Depending on the coloration type, these materials can be classified as anodically coloring (coloration upon oxidation) or cathodically coloring (coloration upon reduction). Starting from the first discovery3 this phenomenon has been profoundly investigated by many researchers and it has developed into a huge family, which can be subdivided as metal oxides,4,5 molecular dyes6,7 and conducting polymers.8,9 Electrochromism has attracted significant attention for its potential applications, such as smart mirrors and windows, displays, active optical filters, data storage and camouflage material for military purposes.10 However, the commercialization of these materials depends on their switching times, optical contrast, coloration efficiency and long term stability.Electrochromic contrast, which is often measured as the percent transmittance change (Δ%T) of the electrochromic material at a specific wavelength, is an essential parameter for characterizing the electrochromic materials.11 For applications such as smart windows, in which the difference between the bleached and colored states is expected to be the highest, the electrochromic contrast is generally recorded on a wide range. For these applications, a bleached state with a transmission of at least 80% within the visible window is necessary. To convey more information on the perception of transmittance to the human eye, measurements on the relative luminance change during an electrochromic switch are also considered to be useful.12 Basically, switching time (response time) is defined as the time required to switch between the two extreme redox states of the electrochromic material. It is generally followed by a square wave potential step method coupled with optical spectroscopy. Depending on the conductivity and solvation of the electrochromic material, ion diffusion within the film, the switching time of an electrochromic material could be in the order of several minutes to milliseconds. In addition, the magnitude of the applied potential, thickness and morphology of the film are known to cause effects on the switching characteristics. Coloration efficiency (CE), which basically defines the change in optical density per injected charge, is a fundamental parameter for investigating the power requirements of electrochromic materials. CE at a given wavelength (λ) can be calculated using the following equation:12
| CE (λ) = ΔOD(λ)/(Q/A) = log[Tb/Tc]/(Q/A) |
ΔOD(λ) is the change in the optical density at λmax, Q the injected charge, A the electrode area and Tb and Tc are the bleached and colored transmittance values, respectively. The increase in CE generally indicates the more effective use of the injected charge. The stability of electrochromic materials is generally reported as the number of redox cycles that a material stand without significant loss in the performance, which is related with the irreversible oxidation or reduction at extreme potentials, side reactions with water or oxygen and heat release in the system during switches. On the other hand, Optical memory, indicates the persistence of the color under open circuit conditions, which is of fundamental importance for electrochromic devices.
Since the discovery of the electrochromic effect by Deb,3 a tremendous amount of studies were performed on metal oxides, such as WO3, NiO, MoO3, TiO2, Ta2O5, Nb2O5, and mixed metal oxides.4 The electrochromic response of these materials is based on the reversible modulation of the oxidation states of the transition metal, which depends on the injection of positive ions (H+, Li+, Na+ and K+) and electrons into multivalent transition metal oxide.5 Among many metal oxides, tungsten oxide has been the most extensively studied due to its high coloration efficiency (typically greater than 50 cm2 C−1) and stability.4 The material is known for its cathodic coloration from colorless to blue, which could be prepared by techniques such as sputtering, thermal evaporation and the sol–gel method. In recent years, particular efforts have been devoted on nanostructured WO3 with various surface morphologies such as nanoparticles, nanorods and nanowires. In general, nanostructured WO3 exhibits promising switching times (below 5 s) and moderate coloration efficiency around 35 to 65 cm2 C−1.11 Interestingly, having large active surface area and extensive grain boundaries, the electrochemically prepared nano-WO3 revealed improved electrochromic performance, in which an optical contrast of 88.51% and an excellent coloration efficiency of 137 cm2 C−1 were achieved.13 Recently, composite materials based on conducting polymers and metal oxide nanostructures have been actively studied. Among these, the composites of polyaniline and WO3 revealed promising features showing color variation from dark blue at negative potentials to violet-green at positive potentials.10
Almost 40 years ago, polyacetylene, which is the simplest form of a conjugated polymer, was shown to reveal near metallic electrical conductivity upon chemical doping.14 This breakthrough caused an explosion of an interdisciplinary research area, and the family of conducting polymers has tremendously grown upon chemical and/or electrochemical polymerization of aromatic molecules such as pyrrole, thiophene, aniline, furan, carbazole. Although the original intention was to pursue their ability to conduct electricity, these materials were shown to exhibit far more to offer in various applications. In fact, the scientist who triggered dawning of this new era were awarded with 2000 Nobel Prize in Chemistry “for the discovery and development of conductive polymers”.
Among conducting polymers, polypyrroles (PPy) is by far the most extensively studied polymer because of their low oxidation potential, water solubility and the low cost of its monomer (pyrrole).15 Polypyrrole derivatives, in general, possess exclusive properties, such as moderate environmental stability, good redox properties and enhanced compatibility in aqueous systems, which pave the way for their utilization in numerous applications including batteries, supercapacitors, electrochemical sensors, mechanical actuators, electromagnetic interference shielding, drug delivery systems and optoelectronic devices.
For the synthesis of conducting polymers electrochemical polymerization is a frequently used technique because it requires a small amount of monomer and it provides an effective platform that allows the investigation of in situ growing process of the polymer and further analysis by electrochemical and spectroscopic techniques. The technique is rather simple and it offers the direct grafting of the intrinsically conducting polymer onto an electrode surface without the need for a subsequent doping process.16 Despite the fact that polypyrrole was one of the first to be electrochemically synthesized, there is an ongoing controversy about its polymerization mechanism. Due to the complexity of the film formation process and the lack of effective approaches for investigating the reaction kinetics, the reaction path is not crystal clear. Nevertheless, the mechanism is considered to entail the formation of a radical cation, which is followed by either radical-cation/radical-cation coupling or the reaction of a radical-cation with a neutral monomer. The mechanism (Fig. 1) described by Diaz et al.,17,18 which was later supported by the theoretical studies of Waltman et al.,19,20 is the most frequently referred mechanism in literature.21 According to Diaz's approach following the first electrochemical step (E), which consists of the oxidation of the monomer into a radical cation, the coupling of the two radicals cations results in the formation of a dihydro dimer-cation. Consecutively, the chemical step (C) occurs, which leads to dimer formation followed by the loss of two protons and re-aromatization. Because of the extended conjugation over two rings, having lower oxidation potential than the monomer, the dimer readily oxidizes to form the radical cation (E) and undergoes coupling with a monomeric radical. Electropolymerization is proposed to proceed through to a general E(CE)n mechanism, in which consecutive electrochemical and chemical steps takes place until the oligomers become insoluble and precipitate onto the electrode surface.22 However, further studies have shown the presence of many other competitive multistep reactions, and the susceptible nature of the radical cation intermediates regarding the nucleophilicity of the polymerization medium.23 Typically, one electron is removed from the polymeric backbone for every three to four monomer units, which results in the intrinsic electrical conductivity and provides a delocalized p-electron band structure. In the oxidized state, the polymer is charge balanced with anions, termed also as 'dopants', which are incorporated into the film to maintain electrical neutrality.
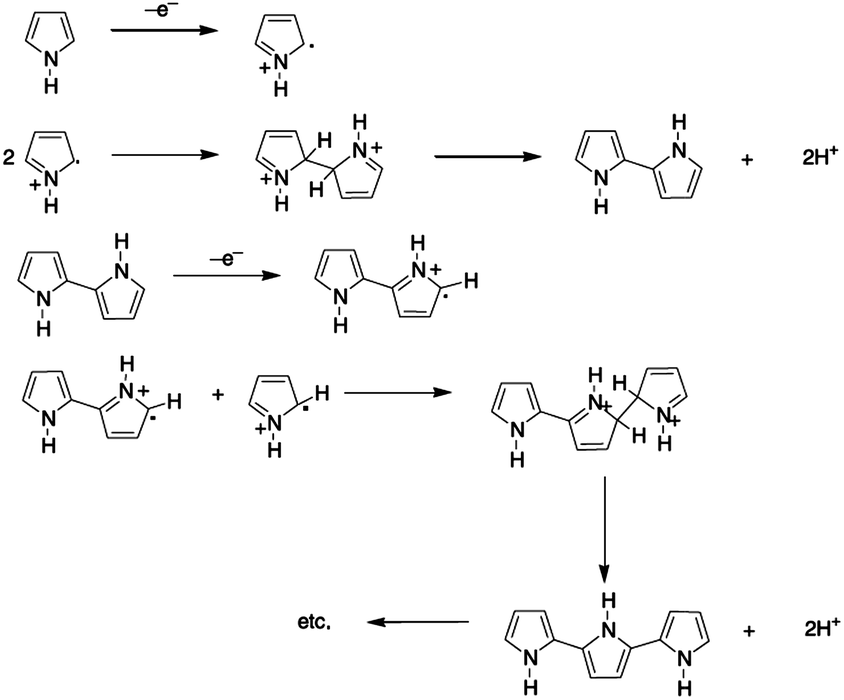 | ||
| Fig. 1 Proposed mechanism for electropolymerization of pyrrole by Diaz et al. | ||
2. Band structure and electrochromism of polypyrrole
Among the conducting polymers, polypyrrole was the first to be investigated for its optoelectronic properties, whose electronic structure could be reversibly changed with doping.18 Fig. 2a represents the energy diagram of PPy in neutral form. Having two-fold coordination, all conjugated polymers are susceptible to structural distortion. Upon oxidation (removal of π-electrons from the valence band) of neutral PPy, the local relaxation of the benzoid structure towards quinoid-like structure occurs, which creates radical cations (polarons).24–26 The formation of polarons induces two new energy levels (Fig. 2b) that are symmetrically positioned within the bandgap. Thus, two new electronic transitions at longer wavelengths emerge. Further oxidation results in the formation of bipolarons, which are the charge carriers of the coupled cations (dication). With an empty lower energy state, bipolarons are signified by the broad, low energy absorptions due to the transitions from the top of the valence band (Fig. 2c). As the polymer is oxidized further, the bipolaronic energy state overlaps and forms intermediate band structures as given in Fig. 2d. Thus, the optical state (and the color) of the polymer is altered by the formation of new electronic states (p-type doping), which results in the development of the lower energy absorptions. Fig. 3 shows the spectral variations of PPy upon doping.27 Potentially all the conducting polymers are electrochromic, which is based on the creation and destruction of charge carriers (polarons and bipolarons).28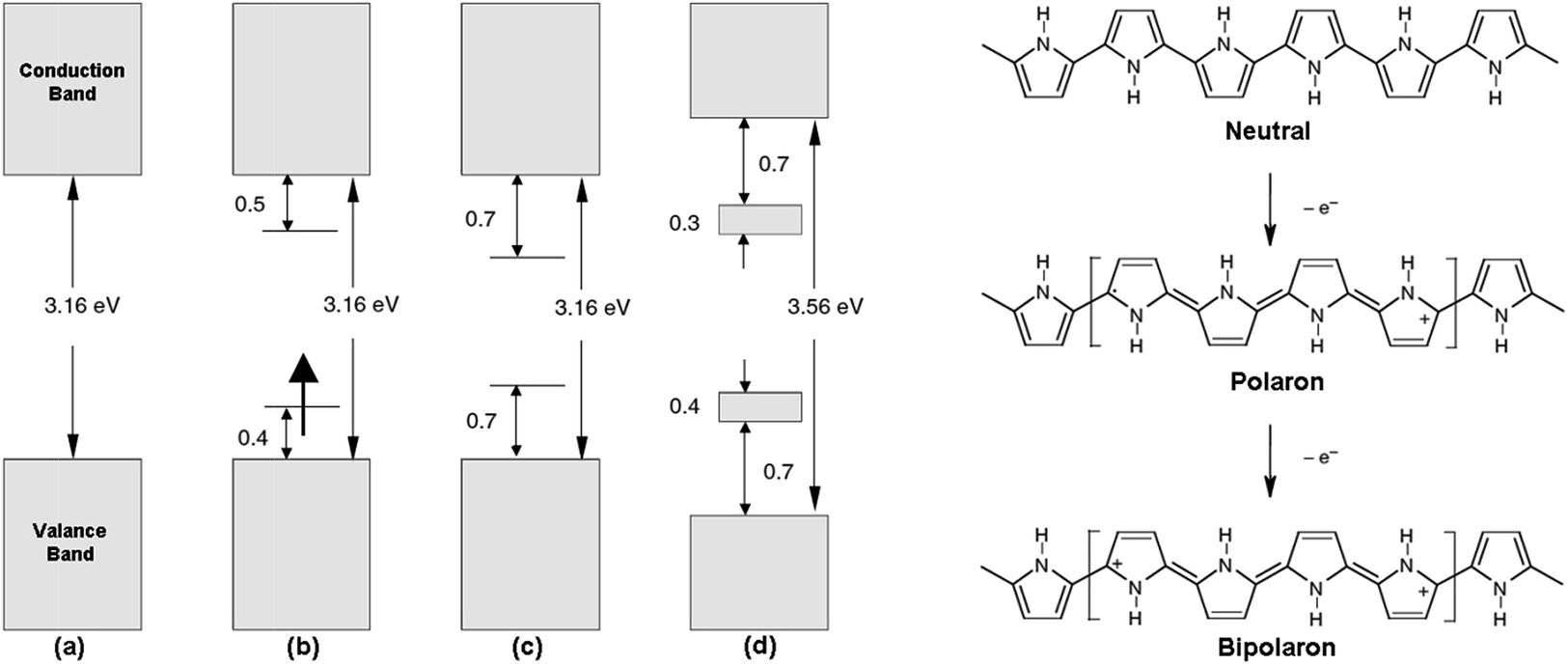 | ||
| Fig. 2 Electronic energy diagrams and structures for (a) neutral PPy, (b) polaron, (c) bipolaron, and (d) fully doped PPy. | ||
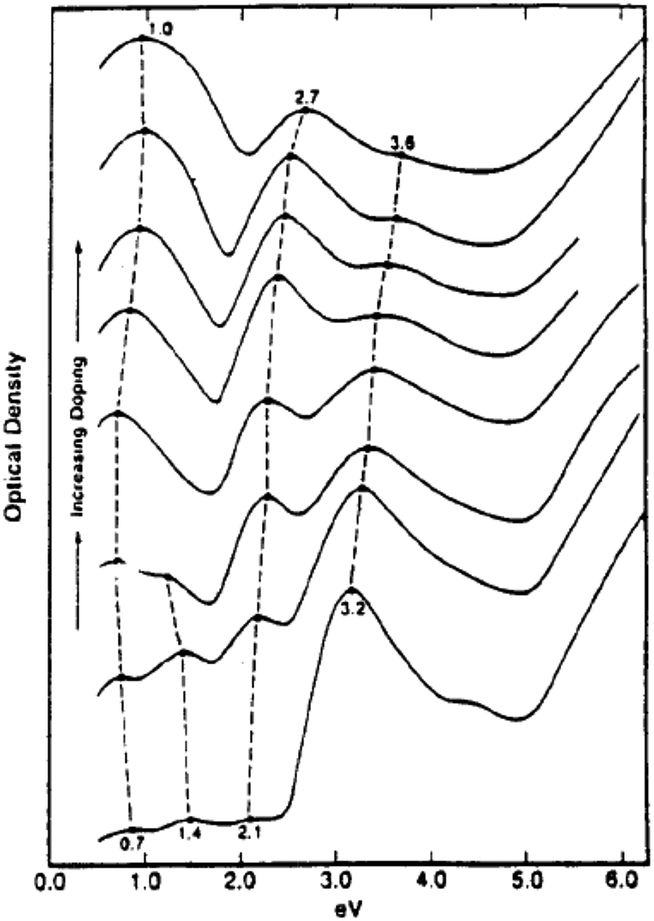 | ||
| Fig. 3 Optical absorption spectra of ClO4-doped polypyrrole as a function of dopant concentration. The dopant level increases from the bottom curve (almost neutral polypyrrole) to the top curve (33 mol% doping level). Reprinted with permission from ref. 27 Copyright 1983 AIP Publishing LLC. | ||
To date, immense efforts have been devoted to comprehend the underlying factors that contribute to the bandgap of these polymers. The energies related to the bond alternation (EΔr), the mean deviation from planarity (Eθ), the aromatic resonance energy (ERes), the inductive and mesomeric electronic effects of substituents (ESub) and the interchain interactions (EInt) have shown to play part on the bandgap of conjugated polymers,29 which is expressed to be; Eg = EΔr + Eθ + ERes + ESub + Eint. Thus, by diverting the electronic nature of the polymer backbone and controlling the interchain interactions, the bandgap and ultimately the coloration of all the conducting polymers could be modified. Polymers with Eg greater than 3.0 eV are generally termed as anodically coloring because they are colorless at the neutral state, while absorbing (colored) in the visible region in the oxidized state.30 On the other hand, those with Eg less than 1.5 eV serve as cathodically coloring materials, which are colored in the neutral state. Polymers with intermediate gaps show distinct color changes throughout the visible region, such as polythiophene (Eg = 2.0 eV, red to blue). The band gap could be categorized as both an optical and electronic bandgap. The former bandgap refers to the energy difference between the valance band and the conduction band, which is determined by spectroscopic techniques. Rather than demanding approaches such as a Tauc plot. The optical bandgap of the electrochromic polymers is readily determined from the low-energy absorption edge of the electronic absorption spectrum. On the other hand, the electronic bandgap is measured by electrochemical methods, which is generally higher than the optical bandgap due to the presence of an electrode–film interface charge barrier. Following the determination of the onset of oxidation and reduction potentials of the polymer and comparing these values with a reference compound, the HOMO and LUMO energy levels and subsequently, the electrochemical bandgap of the polymer are calculated.22,29
The optical bandgap of PPy is reported to be ∼2.7 eV,18 whereas its electronic band gap is calculated as 3.15 eV.24 The polymer appears yellow-green and blue-gray in its neutral and oxidized forms, respectively (Fig. 4). The polymer has the tendency to degrade upon repetitive switching and only the extremely thin films of the polymer could be completely de-doped. Nevertheless, polypyrrole derivatives with improved electrochromic properties are available with a tailor-made functionalization of the repeat units, because the coloration in the conducting polymers mainly depends on the bandgap and its band structure.
 | ||
| Fig. 4 Electrochromism in polypyrrole. | ||
An ideal electrochromic material is expected to have high optical contrast between its extreme states, a short response time and high stability. In conducting polymers, the rates of color change mainly rely on the rate at which the doping–dedoping process occurs. Because the redox switching of the conducting polymers is based on the transfer of ions, the rate of color change mainly depends on both the morphology of the polymer and polymer–electrode interactions. To understanding the effect of dopant ions, Girotto et al. added indigo carmine, an inherently electrochromic, redox active material having dianionic character, during the electrochemical polymerization of pyrrole in the presence of dodecylsulfate.31 The resultant polymer revealed a higher coloration efficiency (137 cm2 C−1) and optical contrast (37%) compared to polypyrrole/dodecylsulfate at 700 nm. Both dodecylsulfate and indigo carmine doped polypyrroles displayed pale yellow (in the neutral state) and dark blue color (in the oxidized state). Furthermore, the effect of utilization of azo (Remazol Black B) and anthraquinone (Dianix Red) dyes was investigated.32 The former and the latter dye doped polypyrroles revealed the electrochromic efficiency of 175 and 100 cm2 C−1, respectively. Moreover, studies showed that polypyrroles synthesized in the presence of Remazol Black B and Dianix Red dyes revealed shorter switching times, which was considered to be because of the enhanced mass and electronic transport with the formation of “nano-channels”. Recently, similar type of studies were conducted, in which the electrosynthesis and the spectroelectrochemical characterization of eriochrome cyanine R doped polypyrrole was performed.33 The dye doped polymer revealed enhanced optoelectronic properties relative to pristine PPy in terms of its optical contrast (27%) and switching time (less than 1.5 s). Yamada et al. investigated the electrochromic properties of PPy on Au nano-brush electrodes, in order to enhance the switching time and life time of PPy.34 Such approach was deliberately chosen (Fig. 5) to improve the mass supply from the bulk of the solution and to compensate the adverse effect of volume change of the copolymer (deterioration of the adhesion between the polymer film and electrode) during switching. PPy films immobilized on Au nano-brush electrodes showed higher coloration efficiency and switching stability when compared to the PPy films on the Au planar electrode.
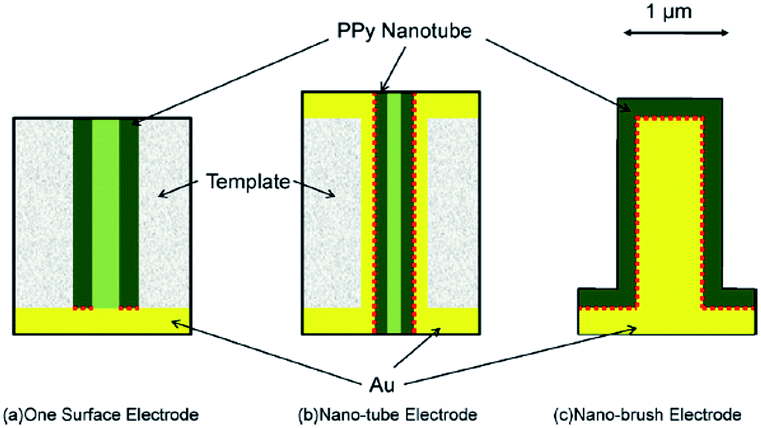 | ||
| Fig. 5 Schematic diagrams for differences in the electrode configurations and the effective contacting area between the Au electrode and polypyrrole nano-tubes. Dotted lines indicate the effective contact surface. (a) One surface electrode; (b) nano-tube electrode; (c) nano-brush electrode. Reprinted with permission from ref. 34 Copyright 2009 Elsevier. | ||
Hybrid and composite materials represent an important class of materials, which are expected to combine the merits of its constituents such as flexibility, short response time, stability. In 1988 electrochromic composite films of polypyrrole and phosphotungstate were prepared by the electropolymerization of pyrrole in aqueous solutions, which contain the sodium salt of phosphotungstate. Because PPy and phosphotungstate switch at different potentials, the composite film switched from yellow to red and finally to blue upon oxidation, showing the typical coloration behavior of both PPy and phosphotungstate.35 In addition, another organic–inorganic hybrid material, nickel oxide/polypyrrole (NiO/PPy) thin film showed enhanced switching time (0.601 s for coloration and 0.395 s for bleaching) and coloration efficiency (358 cm2 C−1) compared to pristine PPy and NiO.36 Most importantly, NiO/PPy presented a dramatically improved electrochemical stability (from about 500 switches for PPy to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 switches for NiO/PPy) in contrast to PPy. Recently, Takagi et al. formulated a nanohybrid film with PPy, which was photoelectrochemically synthesized from the pyrrole monomer, which was attached to TiO2 surface.37 The response time of the nanohybrid film was determined to be significantly slow than that of the PPy coated onto a typical FTO electrode. By reducing the polymer thickness and using the fast electron transport property of the TiO2, the nanohybrid film revealed the fastest reduction response time of 0.4 ms. Interestingly, PPy-poly(ethylene-co-vinyl alcohol) (EVOH) composite films, which were prepared by electrochemical polymerization of pyrrole on lithium perchlorate containing EVOH matrices, changed color reversibly from yellow to brown with markedly improved durability against the repeated application of potentials.38 In another study, the stability of PPy was enhanced by introducing a triblock copolymer, poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol), into the oxidant solution during a vapor phase polymerization process.39 The resultant PPy demonstrated 20-fold increase in electrical switching stability compared to pristine PPy. In an another effort to achieve improved properties,40 carbon nanotube (CNTs) functionalized poly(N-methylpyrrole) was electrochemically synthesized in a hydrophobic ionic liquid and a single layer electrochromic device was constructed with a SnO2:F counter electrode. The coloration of the device was from dark brown to pale yellow with an unstable transient green color. The device exhibited high cycle life and optical transmission (at 550 nm) of 69% and 44% in the bleached and color states, respectively.
000 switches for NiO/PPy) in contrast to PPy. Recently, Takagi et al. formulated a nanohybrid film with PPy, which was photoelectrochemically synthesized from the pyrrole monomer, which was attached to TiO2 surface.37 The response time of the nanohybrid film was determined to be significantly slow than that of the PPy coated onto a typical FTO electrode. By reducing the polymer thickness and using the fast electron transport property of the TiO2, the nanohybrid film revealed the fastest reduction response time of 0.4 ms. Interestingly, PPy-poly(ethylene-co-vinyl alcohol) (EVOH) composite films, which were prepared by electrochemical polymerization of pyrrole on lithium perchlorate containing EVOH matrices, changed color reversibly from yellow to brown with markedly improved durability against the repeated application of potentials.38 In another study, the stability of PPy was enhanced by introducing a triblock copolymer, poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol), into the oxidant solution during a vapor phase polymerization process.39 The resultant PPy demonstrated 20-fold increase in electrical switching stability compared to pristine PPy. In an another effort to achieve improved properties,40 carbon nanotube (CNTs) functionalized poly(N-methylpyrrole) was electrochemically synthesized in a hydrophobic ionic liquid and a single layer electrochromic device was constructed with a SnO2:F counter electrode. The coloration of the device was from dark brown to pale yellow with an unstable transient green color. The device exhibited high cycle life and optical transmission (at 550 nm) of 69% and 44% in the bleached and color states, respectively.
3. Tuning of electrochromic properties of PPy
3.1. Copolymerization
Controlling the bandgap of the polymer through structural modification, copolymerization, blending and lamination are the techniques, which are applied to tune the color of the conducting polymers.41 Copolymerization is an easy, facile approach to unite the distinct properties of the comonomers, which might lead to an exciting combination of the properties possessed by the corresponding homopolymers. In a work of Ak et al.,42 an enhancement of the electrochromic properties of PPy was achieved by the electrochemical copolymerization of pyrrole with octa(thiophenephenyl)silsesquioxane (OPS). The copolymer (OPS–PPy) revealed multichromic behavior showing color change from yellow, red, gren-gray, blue. OPS–PPy switched more rapidly (0.4 s) compared to PPy (1.1 s) and revealed a noticeable increase in the optical contrast (Fig. 6) (from 17% to 30% at 730 nm). Such an improvement was attributed to the loose packing of the PPy chains because of the presence of OPS units, leading to more accessible doping sites and the facile ion movement during the redox switching. In a recent work,43 the copolymerization of pyrrole with 3,4-ethylenedioxythiophene (EDOT) was performed in boron trifluoride diethyl etherate. The maximum absorption wavelength of π–π* transition was blue-shifted (from 462 nm to 556 nm) with the increase in the EDOT content of the feed. Resultant copolymers exhibited bandgaps between 1.71 to 1.64 eV. Moreover, copolymers based on pyrrole and EDOT were synthesized in aqueous micellar solution with various feed ratios of pyrrole/EDOT.44 The copolymers synthesized at the feed ratios (pyrrole/EDOT) of 1/5 and 1/15, showed light brown to light blue and dark blue to light blue coloration upon doping, respectively. Compared to PPy, the stabilities of the copolymers were presumably improved because of the incorporation of EDOT units. Wen et al.45 produced a pyrrole containing copolymer, which was achieved by the electropolymerization of pyrrole and 2,2′-dithiodianiline in propylene carbonate. The UV-visible spectrum of the copolymer showed three optical transitions at λmax 390, 540, and 800 nm for different applied potentials.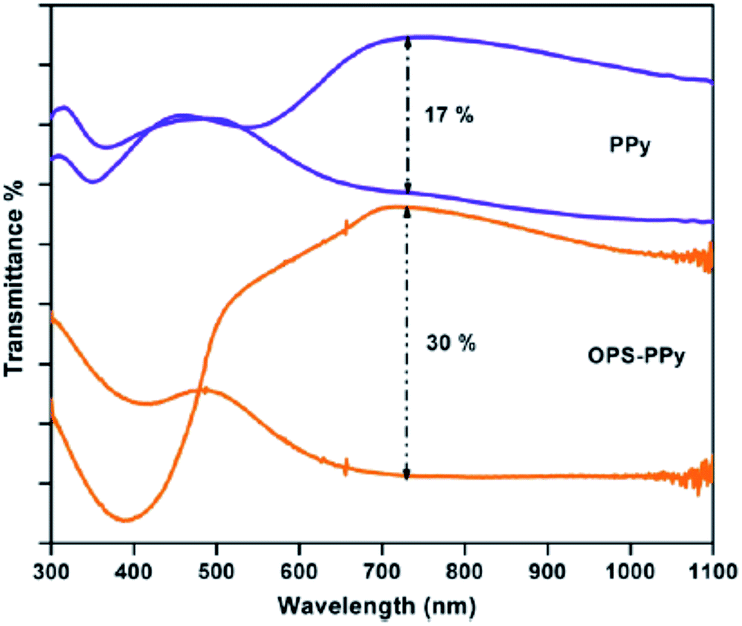 | ||
| Fig. 6 Transmittance spectra of PPy and OPS–PPy in two extreme (oxidized and neutral) states. Reprinted with permission from ref. 42 Copyright 2008 Elsevier. | ||
3.2. Structural modification
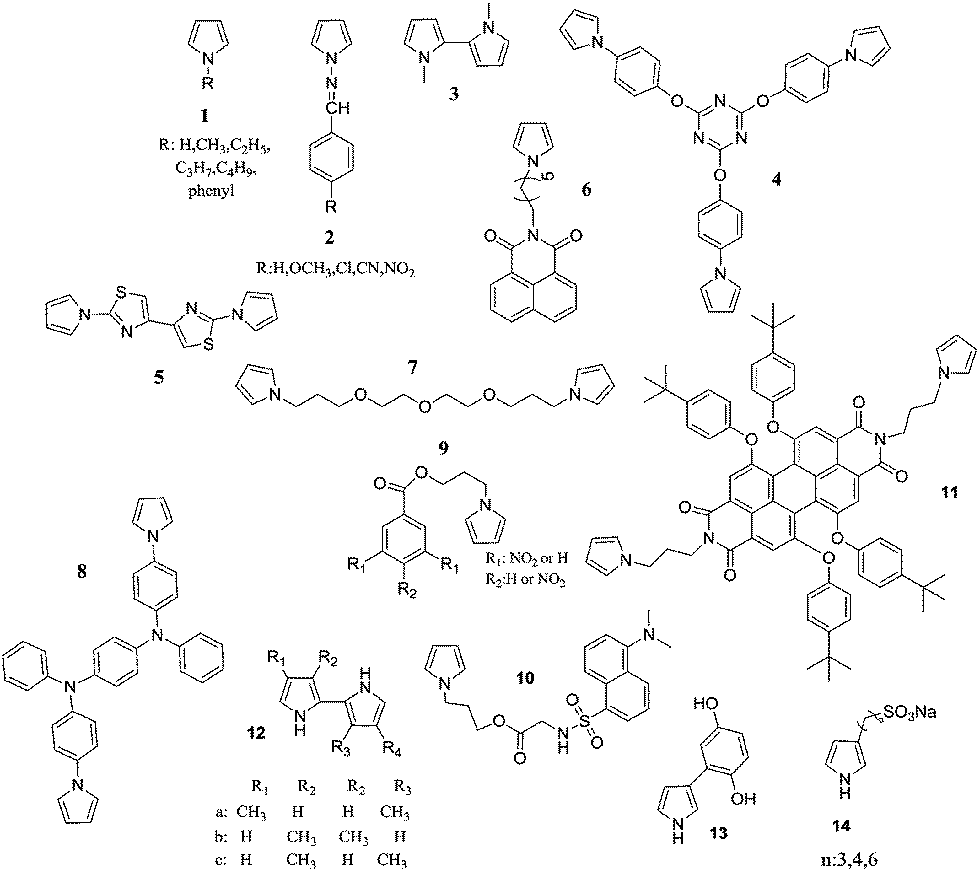
Another polypyrrole derivative, which was synthesized by the electrochemical polymerization of a star shaped, triazine containing monomer (4), displayed red and turquoise colors in the fully oxidized and reduced states, respectively.50 The optical bandgap of the polymer was found to be 2.97 eV. The polymer switched colors in 1.1 s and had an optical contrast of 20% (measured at 700 nm). The bithiazole containing pyrrole (5) derivative was electrochemically polymerized both in the absence and presence of EDOT.51 The resultant polymers exhibited optical bandgaps ranging from 2.60 to 1.75 eV (depending on synthesis conditions). The multichromic copolymers revealed short switching times and useful optical contrast of 0.6 s and 54%, respectively. Soluble, n-dopable, florescent, electrochromic polypyrrole derivatives were synthesized by both the chemical and electrochemical polymerization of 6.52 The optical bandgap of poly(6) was calculated to be 2.99 eV, 2.37 eV. The polymer exhibited a switching time of 1.63 s and an optical contrast of 33.37%. An etheric member of N-linked polybispyrroles (poly(7)), on the other hand, appeared transparent yellow in the neutral state, light pink in the intermediate state, and blue in the oxidized state.53 Poly(8) that contains a triphenylamine derivative presented color variations form brown to blue in 0.6 s (95% of the full change) with an optical contrast of 52% (at 617 nm).54 Recently, two nitrobenzoyl pyrrole derivatives (9) were synthesized and subjected to electrochemical/chemical polymerization. The resultant polymers displayed color changes from pale green to dark grey upon oxidation.55 Similarly, a dansyl substituent pyrrole derivative (10) was electropolymerized and a greenish-yellow to greyish-green coloring, green light emitting polymer was achieved.56 In another inspiring study, the electrochemical deposition of a pyrrole-1-yl substituted perylene diimide (11) was achieved.57 Poly(11) turned from red (λmax: 573 nm) to emerald (λmax: 788 nm) upon oxidation. For poly(11) the time required for reaching 90% of the ultimate absorbance was calculated as 160 s. Such a slow response was attributed to the low conductivity of the polymer, which was considered to be because of the large separation of perylene diimide, as well as the insulating nature of the polypyrrole backbone in the operating potential range. In 2002, synthesis and electropolymerization of pyrrol-1-yl substituted phthalocyanines was achieved. The resultant displayed charge–discharge behavior, which was accompanied by a reversible electrochromic color change.58
In addition to their important role in modification of the electrical and optical properties of the polymers, substitution through 3- and 4-positions of thiophene (as of pyrrole) is expected to prevent the undesirable α–β couplings during polymerization, which are known to decrease effective conjugation length and the solubility of polythiophenes. In general, substitution through 3- and 4-positions allows the polymerization to proceed through 2- and 5-positions, which yields a polymer with fewer structural defects.
1990s witnessed the tremendous improvement in the electrochromic properties of polythiophene derivatives through the breathtaking growth of the poly(3,4-alkylenedioxythiophene) (PXDOT) family. PXDOT derivatives have developed as easily oxidizable, low bandgap materials with good stability.68 Birth of PXDOT derivatives was achieved by the pioneer work of Jonas et al.,69 who were the first to anodically polymerize EDOT. The resultant cathodically coloring polymer had a bandgap of 1.6 eV and displayed coloration from almost transparent (with a sky-blue tint) to dark blue. PEDOT exhibited a bandgap i.e., 1.1 eV lower than that of PPy and 0.5 eV lower than that of polythiophene. The decline in the bandgap of the polymer was attributed to the tendency of the oxygen atoms to donate electron density, which results in an increase in highest occupied molecular orbital (HOMO) of the π-system and leads to a decline in the bandgap. Later on, intense studies were devoted to expand the range of substituted 3,4-alkylenedioxy thiophenes having different ring sizes and substituents. The response time of both PEDOT and 7-membered PProDOT69,70 was recorded as 2.2 s, whereas for the 8-membered PBuDOT it was 1.3 s.71 Moreover, optical contrast (Δ%T) of the polymers increased with the increase in size of the alkylenedioxy ring from 44% for PEDOT to 54% for PProDOT to 63% for PBuDOT. Among these, ProDOT has become the center of attention owing to its facile functionalization at the central carbon of the propylene bridge.
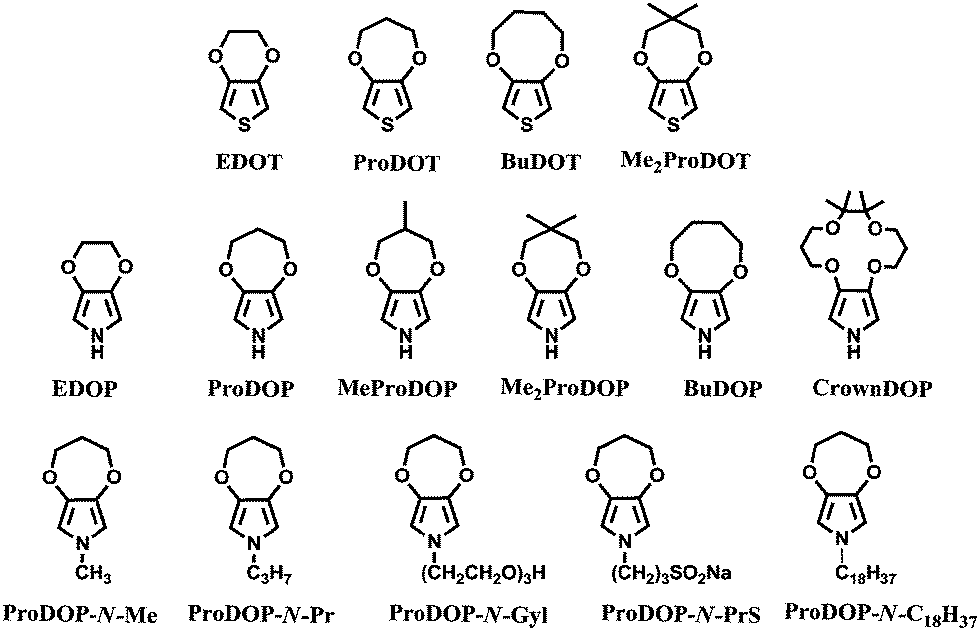
In particular, the dimethyl substituted derivative Me2PProDOT shows a highly transmissive doped state with 78% optical contrast and a switching time of 0.3 s (reported at 95% of full contrast).72
Following the breathtaking growth of the PXDOT family, a new class of easily oxidized electroactive conjugated polymer was born through the attachment of alkylenedioxy bridge to the 3- and 4-positions of pyrrole, namely poly(3,4-alkylenedioxypyrrole)s (PXDOPs).73 The pioneer member, PEDOP revealed a bandgap of 2.0 eV with an optical contrast of 59%.74 Contrary to its cathodically coloring thiophene analogue (PEDOT), PEDOP displays red color in neutral state and sky blue-transparent in the oxidized state. The scheme above reflects some of the members of this unique family, which includes various dialkyl, alkylene, N-hydro, and N-alkyl derivatives. It was shown that the optoelectronic properties of PXDOPs can be modified by changing the size, the composition of the alkylene bridge and N-substitution (Table 1). Contrary to its thiophene analogs, increasing the ring size from 6-membred to 7- and 8-membred ring in pyrrole derivatives did not gradually increase the optical contrast.
| Monomer | Eg [eV] | Colors | λmax [nm] |
|---|---|---|---|
| a Eg: optical bandgap (eV); λmax: wavelength of maximum absorption; n: neutral; i: intermediate; d: doped. | |||
| EDOP | 2.0 | Red(n) sky blue(d) | 537 |
| ProDOP | 2.2 | Orange(n) brown(i) gray-blue(d) | 522 |
| MeProDOP | 2.2 | Orange(n) red-brown(i) light blue(d) | 530 |
| Me2ProDOP | 2.2 | Orange(n) red-brown(i) light blue(d) | 534 |
| BuDOP | 2.2 | Orange(n) blue-gray(d) | 533 |
| CrownDOP | 2.4 | Yellow(n) transparent gray(d) | 490 |
| ProDOP-N-Me | 3.0 | Deep purple(n) dark green(i) blue(d) | 476 |
| ProDOP-N-Gly | 3.4 | Colorless(n) pink(i) tan(i) gray-blue(d) | 460 |
| ProDOP-N-PrS | 2.9 | Colorless(n) pink(i) gray(i) blue-gray(d) | 460 |
| PProDOP-N-C18H37 | 2.96 | Colorless(n) faint pink(i) slightly grey(d) | 350 |
For instance, as in the case of poly[3,4-(propylenedioxy)pyrrole] (PProDOP), MePProDOP and Me2PProDOP, functionalization of the alkyl bridge resulted in the formation of multichromic polymers with bandgaps of 2.2 eV.75 These polymers switched from orange to red-brown (or orange-brown) and finally to a light blue when fully neutralized, partially oxidized, fully oxidized, respectively. Me2PProDOP, exhibiting an optical contrast of 76% (at 534 nm) retained 90% of its electroactivity after undergoing 40000 potential switches. On the other hand, PProDOP-N-Pr revealed higher bandgap compared to its N-hydro derivative (PProDOP) because of the distortion of the polymer backbone, giving rise to a decline in the effective conjugation and lowering of the valance band.76 Moreover, the utilization of sterically demanding substituents, as in the case of PProDOP-N-Gly and PProDOP-N-PrS, increased the bandgap up to 3.4 eV and lead to full visible light transmissivity in the neutral state (Fig. 7). In general, N-alkylated PXDOP derivatives have shown to have large bandgaps high conductivity, and small half-wave potentials, which made them strong candidates as anodically coloring materials in electrochromic devices. One such example was provided in a 2002 study,77 in which a complementary controlled dual type electrochromic device was constructed (Fig. 8), having PProDOP–NPrS as the anodically and Me2PProDOT as the cathodically coloring polymer. It was shown that the utilization of PProDOP–NPrS not only provided a transmissivity window throughout the entire visible spectrum but also enhanced the optical contrast of the device from 56% to 68% (measured at 580 nm). Moreover, the device was found to retain 86% of its optical response after 20000 double potential steps. In another study, the films of PProDOP were paired with PEDOT and PProDOP in a stacked polymer arrangement to achieve dual-polymer electrochromic films.78 By coupling of these polymers logically and by keeping each layer under separate potentiostatic control, a wide range of colors was achieved.
 | ||
| Fig. 7 Absorbance spectroelectrochemistry for thin films of (a) PProDOP-N-Me switched between −0.5 and 0.7 V, (b) PProDOP-N-Pr switched between −0.4 and 0.6 V, (c) PProDOP-N-Gly switched between −0.2 and 0.7 V, and (d) PProDOP-N-PrS switched between −0.4 and 0.5 V. Reprinted with permission from ref. 76 Copyright 2003 American Chemical Society. | ||
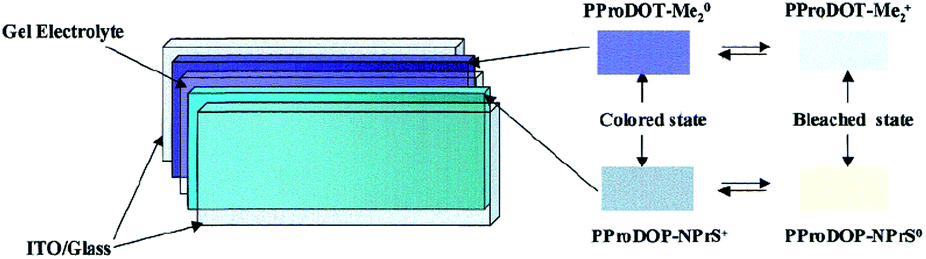 | ||
| Fig. 8 The general scheme for construction of a dual polymer electrochromic device. ITO: indium tin oxide. Reprinted with permission from ref. 77 Copyright 2002 American Chemical Society. | ||
Furthermore, a solution-processable, minimally color-changing dioxypyrrole-based polymer (PProDOP-N-C18H37) with a high level of electroactivity was achieved.79 The neutral polymer (Eg = 2.96 eV) exhibited its maximum absorption in the UV region with an onset at 420 nm and appeared to be colorless (Fig. 9). At the intermediate state, the polymer's transparency decreased and it displayed a faint pink hue because of the formation of polaron bands centered at around 525 nm. Upon further oxidation, the polymer showed bipolaronic transitions in near infrared region with a very slight grey hue. During repetitive redox cycling, the percent electroactivity of the polymer dropped only 10% in the first 35000 cycles. Moreover, dual polymer absorptive/transmissive devices were constructed with poly((2,2-bis(2-ethylhexyloxymethyl)-propylene-1,3-dioxy)-3,4-thiophene-2,5-diyl) (ECP-Magenta) and PProDOP-N-C18H37, in which the latter function as the charge balancing, low coloration efficiency, counter electrode. The ability of the polymer to exhibit minimal color change during a switch resulted in a high contrast and maximum transmittance of the electrochromic device in the bleached state. Later on, the very same electrochromic polymers were utilized in the manufacturing of organic photovoltaic-powered electrochromic displays.80 Using roll coating methods and printable electrolytes, fully printable and laminated devices on flexible substrates were constructed. The devices of various sizes were constructed, having optical contrast of 58% at a visible wavelength of 550 nm and switching times of <10 s. With this pioneer study the fabrication of a self-powered OPV/ECD module was established.
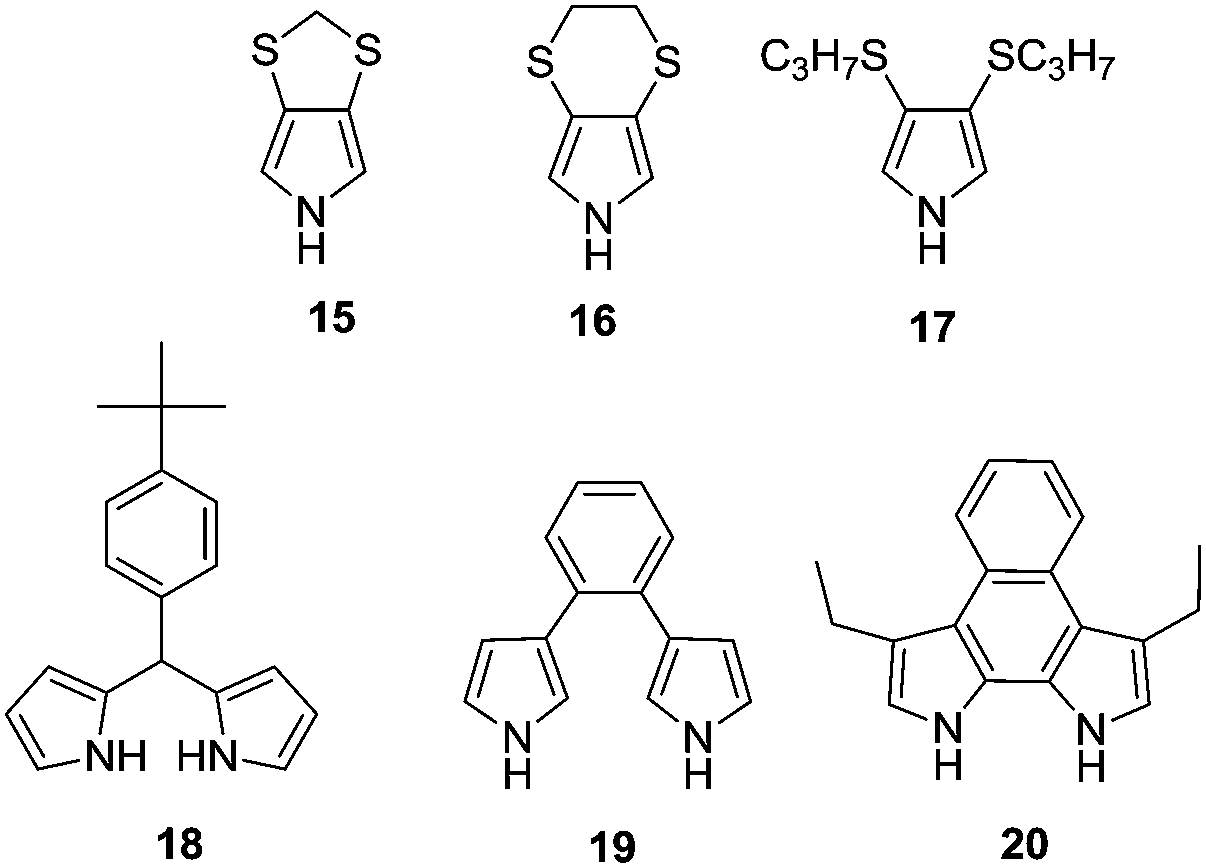
 | ||
| Fig. 9 UV/Vis-NIR transmittance spectra for the neutral (A), intermediate (B), and oxidised (C) states of PProDOP-N-C18H37 of increasing thickness, spray-cast onto ITO glass. Images show the films of 70 nm (D) and 343 nm (E) thickness at the voltages indicated.79 | ||
To further elucidate the effect of 3,4-disubstitution on polypyrrole derivatives; poly(3,4-methylenedithiopyrrole) (poly(15)), poly(3,4-ethylenedithiopyrrole) (poly(16)) and poly(3,4-bis(propylthio)pyrrole) (poly(17)) were also synthesized81 whose bandgaps were determined to be 1.8, 2.1 and 2.3 eV, respectively. These bandgaps are lower than PPy and comparable to that of PXDOPs as previously discussed. The relative increase in the bandgaps was considered to be because of the variation in the biaryl torsional angles, which is stimulated by the size of the alkylthio substituents. Interestingly, these polymers showed significantly high in situ conductivities, which are comparable to PPy. A moderate bandgap polymer (Eg = 2.39 eV) was synthesized upon the electrochemical polymerization of dipyrromethane derivative (18).82 The polymer revealed a color change from yellow to blue upon oxidation with a switching time of 1.2 s. Upon the electrochemical copolymerization of 18 with EDOT, a new polymer with lower bandgap (1.7 eV) was achieved.83 In another study, a β-linked dipyrrole monomer (19) was developed and electrochemically polymerized.84 It was proposed that the oxidation of the monomer leads to an intramolecular cyclization followed by polymerization to obtain an α–α coupled polymer. The polymer displayed yellow to black coloration upon oxidation. Following this study, a new structure (20) was particularly designed via annulation of the phenyl ring to offer enforced planarity between the adjacent pyrroles and to provide unequivocal reactivity at the pyrrole α-positions because of the blocking of the β-positions by alkyl substituents.85 This multichromic polymer goes through color variations from pale-yellow, to red, to green, and then to blue upon oxidation (Fig. 10). Such behavior is attributed to the formation of neutral, polaron, bipolaron and transverse bipolaron states. The coloration efficiency and switching time of the polymer were determined to be 6 to 7 s and 98 cm2 C−1 at 700 nm, respectively.
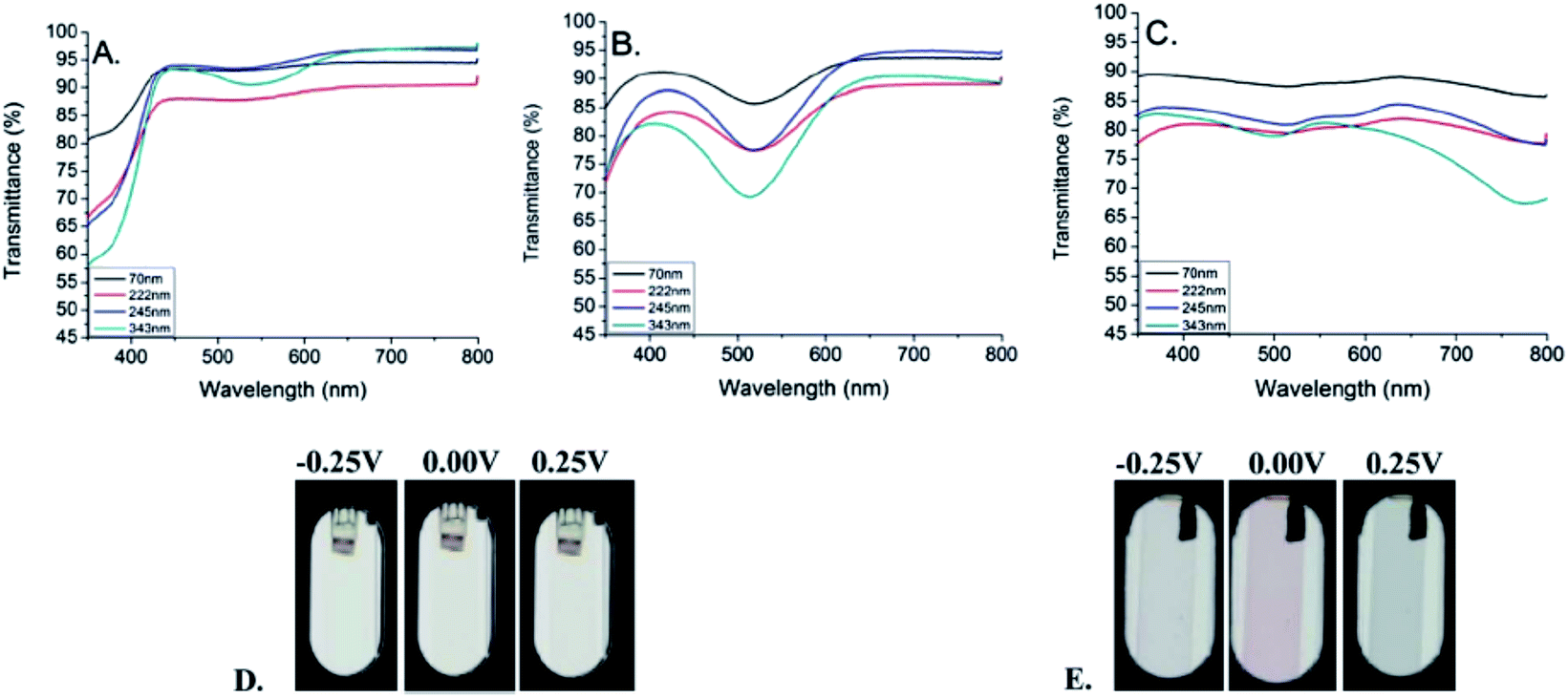
 | ||
| Fig. 10 Results of spectroelectrochemical studies of poly(20) film deposited on an ITO electrode. Arrows denote the changes in the spectra associated with a decrease in the applied potential. Reprinted with permission from ref. 85 Copyright 2010 American Chemical Society. | ||
As an alternative approach, Trofimov et al. synthesized a conducting polymer with a repeat unit of 2-(2-thienyl)-1H-pyrrole (28a) in order to combine the electrochromic properties of polythiophene and PPy.90 The polymer (poly(28a)) displayed orange to black coloration with an optical contrast of 19% (at 450 nm). The polymer had a bandgap of 1.6 eV, which is significantly lower than two parent polymers. The bleaching process coloration efficiency and switching time of the polymer was calculated as 233 cm2 C−1 and 1.3 s (90% of the total absorbance span), respectively. In order to address the effect of substituents, the pyrrole unit was functionalized with ethyl (28b) and n-propyl (28c) substituents and subjected to electrochemical polymerization.91 The resultant polymers were multichromic and showed five different colors ranging from dark orange, orange-yellowish, brown, blue to blue-grayish. For both polymers, switching time was in the range of a few seconds with a coloration efficiency of 107–108 cm2 C−1 during the bleaching process. In order to shed light on the interactions between selenophene and pyrrole derivatives, another inspiring study was performed in 2009, where a series of 2-(selenophen-2-yl)pyrrole derivatives were synthesized and electrochemically polymerized.92 Poly(29a) revealed λmax at 445 nm and it displayed color variation from orange to black upon oxidation as in the case of its thiophene analogue poly(28a). On the other hand, poly(29b) (λmax = 488 nm) and poly(29c) (λmax = 459 nm) displayed deep-orange and greyish-blue at their extreme states. The optical contrasts of these polymers were in the range of 13% and 26% in the visible region.
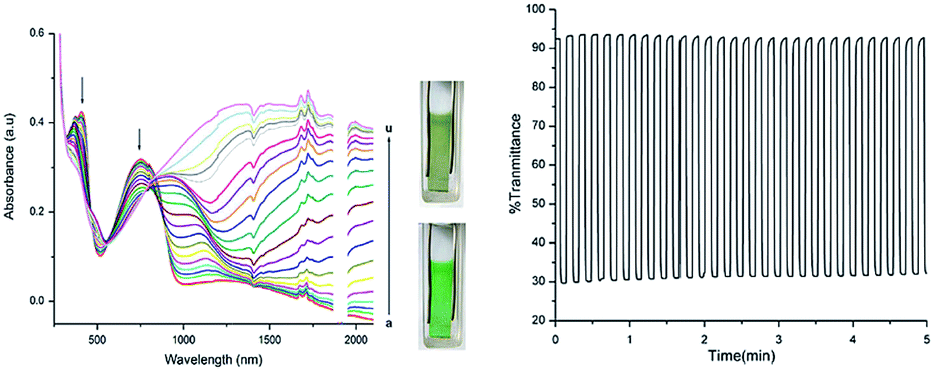
In recent years, an exciting class of conducting polymer, poly(2,5-dithienyl-1-substituted-pyrrole) (PSNS) poly(30) derivatives have been thoroughly investigated for their electrochromic properties.93–126 Because of their terarylenic structures, all the monomers and their corresponding polymers (Fig. 11) exhibited low oxidation potentials. In general all homopolymers (Eg = 1.9 to 3.1 eV) displayed yellow to blue coloration with switching times of 1.3 to 2 s. On the other hand, polymers synthesized by the chemical polymerization of benzyl,105 phenyl,104 4-fluorophenyl,96 petafluorophenyl,95 4-nitrophenyl,97 benzenamine98 substituted derivatives were soluble in common organic solvents. In a recent study clickable, azide containing polymers based on 1-(2-azido-ethyl)-2,5-dithiophene-2-yl-1H-pyrrole (SNS–N3) were synthesized. This polymer was shown to offer a multipurpose platform for simple, effective post-functionalization of PSNS under mild conditions.93 Later on, both p- and n-dopable naphthalimide containing PSNS derivatives were synthesized.120 Other than chemical tailoring by the pyrrole unit, coloring tuning in PSNS derivatives was also accomplished via copolymerization. Copolymers of some of the PSNS derivatives with EDOT revealed an impressive set of multichromic polymers with enhanced electrochromic properties. For instance, the copolymer synthesized by the potentiodynamic electrolysis of 1-benzyl-2,5-di(thiophen-2-yl)-1H-pyrrole and EDOT revealed enhanced optic contrast, switching time compared to parent polymer and multichromism throughout the entire visible region, displaying claret red, yellow, green, and blue colors upon the variation of the applied potential.127 Towards multicolored electrochromic polymers, the selectivity of the electrochemical polymerization was utilized to synthesize compositionally different copolymers of 4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl)benzenamine and EDOT by varying the applied potential at a constant monomer feed ratio. Fig. 12a represents the absorption spectrum of eight different copolymers (in the neutral state), which were synthesized at eight different potentials. As seen, there is a progressive increase in the wavelength of maximum absorption between 431 to 538 nm with the increase in polymerization potential. Fig. 12b reflects the colors of these polymers at various oxidation states.98 In a later recent study, 16 different ambipolar, multichromic naphthalimide containing copolymers were synthesized by controlling the electrochemical copolymerization conditions such as feed ratio, the electrochemical polymerization method and potential.120 Fig. 13 represents the colorimetry studies of the polymers, which were synthesized during potentiodynamic cycling.
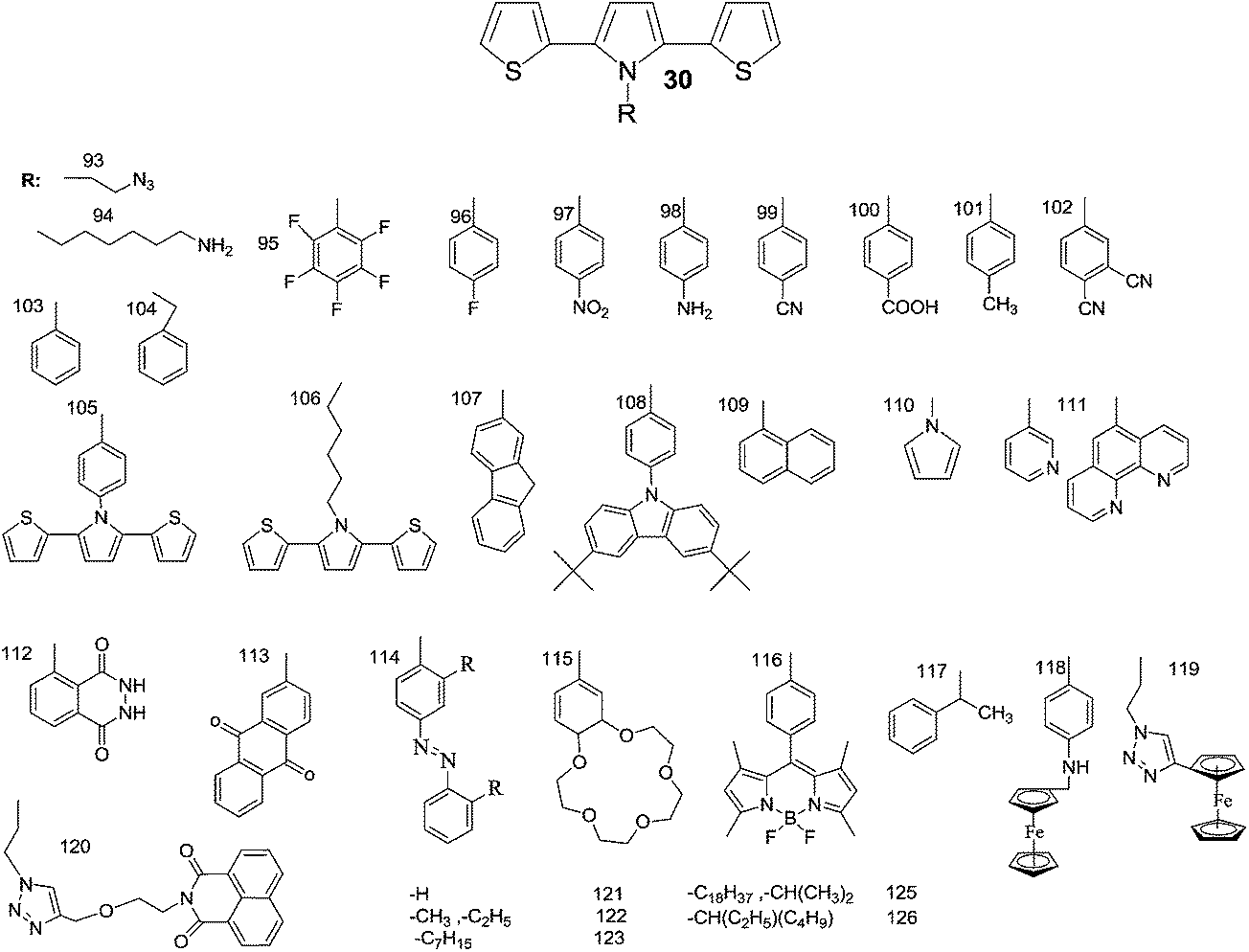 | ||
| Fig. 11 Schematic representation of 2,5-dithienyl-1-substituted-pyrrole derivatives available in literature. (Given numbers relates the structures with the relevant articles). | ||
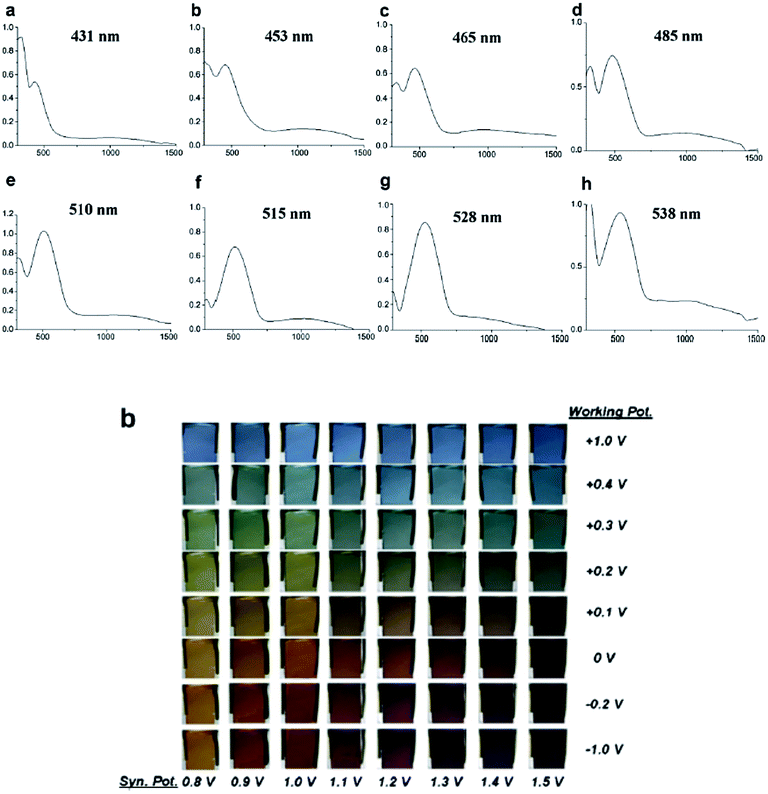 | ||
| Fig. 12 Optical spectra of copolymers of 4-(2,5-di(thiophen-2-yl)-1H-pyrrol-1-yl)benzenamine and EDOT at applied potentials of (a) 0.8, (b) 0.9, (c) 1.0, (d) 1.1, (e) 1.2, (f) 1.3, (g) 1.4, and (h) 1.5 V. Reprinted with permission from ref. 98 Copyright 2008 Elsevier. | ||
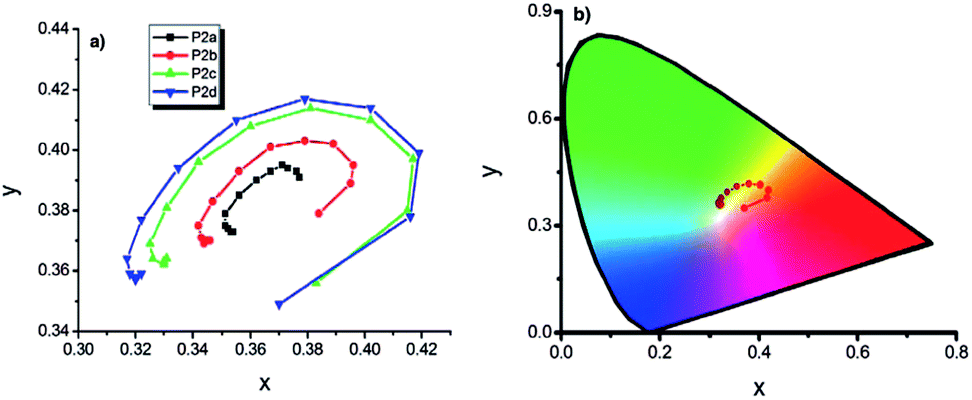 | ||
| Fig. 13 (a) x, y data of P2 type polymers, (b) representation of chromaticity data of P2d on “horse shoe” diagram (copolymers synthesized via potentiodynamic scanning from 0.0 V to 1.0 V, to 1.1 V, to 1.2 V, to 1.3 V in EDOT rich media are abbreviated as P2a, P2b, P2c, P2d respectively). Reprinted with permission from ref. 120 Copyright 2013 The Electrochemical Society. | ||
An interesting class of acceptor group is benzazole derivatives, which are known for their high electron accepting ability through the two electron-withdrawing imine nitrogen atoms. Because of the differences in chemical properties between nitrogen, sulfur and selenium atoms, pyrrole polymers containing benzothiadiazole (31), benzoselenadiazole (32) and benzotriazole (33) were synthesized.130,131 Poly(31), poly(32) and poly(33) exhibited bandgaps of 1.12, 1.08 and 1.6 eV, respectively. Poly(31), poly(32) switched from green to blue at around 1 s with an approximate optical contrast of 35%.130 On the other hand, poly(33) displayed color transition from blue to light blue upon oxidative doping.131 Because of the outstanding electron withdrawing capacity of the benzothiadiazole unit, this polymer was n-type dopable with a light blue appearance. On the other hand, the polymer, which incorporates pyrrole with tert-butylphenyl substituted quinoxaline poly(34) exhibited two distinct absorption maxima (Fig. 14) due to its high energy (λmax = 408 nm) and low energy (λmax = 745 nm) π–π* transitions.132 The color of the polymer changed from saturated green to brownish green at 0.6 s (measured at 408 nm) with a coloration efficiency of 85 cm2 C−1. In order to enhance the electrochromic properties of the copolymer of (34) with bis(3,4-ethylenedioxythiophene), (BiEDOT) was synthesized.133 The polymer revealed multicolor electrochromic properties with distinct colors (purple, gray, light green, blue) and a reasonable switching time (1.2 s) and optical contrast (23%) in the visible range. Another polymer of pyrrole with 2,3-di(thiophen-2-yl)quinoxaline (poly(35)) exhibited a bandgap of 1.0 eV and a green to gray coloration upon doping.134 The polymer possessed 66% optical contrast in the near IR region with a switching time of 1.2 s. Subsequently, the copolymer of (35) with BiEDOT was synthesized.135 The resulting polymer had a λmax at 525 nm (Eg = 1.4 eV) with a purple color in neutral state. The copolymer achieved 10% optical contrast around 2 s in the visible range. Despite their small bandgaps (1.5 eV), vinylene-linked donor–acceptor polypyrroles (poly(36)), revealed only modest optical contrast.136
 | ||
| Fig. 14 Spectroelectrochemistry and electrochromic switching poly(31) film on ITO-coated glass slide. Reprinted with permission from ref. 132 Copyright 2009 Elsevier. | ||
4. Conclusion and outlook
Among many electrochromic materials, conducting polymers have been the center of interest because of their good mechanical properties, non-dependence to the angle of vision, effective tuning of the color through the tailoring of polymer's chemical structure, high coloration efficiency and short switching times. For some time, synthetic challenges of pyrrole chemistry limited the progress of polypyrroles when compared with polythiophenes. However, with the recent synthetic advances, the polymers with building blocks of pyrrole derivatives have matured into an extensive family, having a low oxidation potential and relative stability and conductivity. Extensive studies on the modification of polypyrrole's structure through N-substitution, 3,4-substitution and utilization of donor acceptor approach paved the way for the control of critical issues. The utilization and modification of alkylenedioxypyrrole building blocks affords a new family of anodically coloring polymers with a low oxidation potential.Unfortunately, despite the presence of a vast variety of electrochromic polymers (especially for polythiophene derivatives) not many of them are of practical utility. This is mainly due to unresolved technical issues of electrochemical polymerization (which is probably not the best technique for commercialization) or the absence of stable polymers, which are compatible with modern printing techniques such as ink-jet printing or roll to roll processes. Further efforts should now be headed toward designing polymers that are solution-processable and have at least one accessible highly transmissive state with long-term environmental stability. Very recently, the stability of conducting polymer films was improved by employing barrier foils capable of protecting the films from atmospheric oxygen and/or UV irradiation.137 Despite the compromise in optical contrast and switching the ability of the polymers, the application of these barriers drastically lowered the photochemical decomposition rate of the polymer films. Especially for large-area devices the issues such as long response time, poor stability and high cost might be overcome through utilization of nanostructures.138 Having large specific surface areas, ultrathin nanowires or nanotubes are expected to reveal both a fast and stable electrochromic response. One such study was conducted with PEDOT nanotubes where the responses of less than 10 ms was achieved because of the presence of diffusion distances as short as 10–20 nm.139 Despite the low optic contrast, this case study underlined the importance of structural control for achieving the requirements of display technologies. Moreover, the issues with electrode-polymer interactions, electrode adhesion, electrolyte–polymer interactions, and the formulation of robust manufacturing processes and methodologies of effective encapsulation are yet to be addressed. The utilization of gallium doped ZnO (GZO), antimony doped tin oxide (ATO), PEDOT:PSS, carbon nanotubes or graphene coated glass were shown to provide liable alternatives for the typical optically transparent electrode, ITO. Because of its high bending radius, chemical inertness, large surface area and low surface resistivity single-walled carbon nanotube (SWCNT) films have recently become a featured candidate.140 Unfortunately, not having coordinative interactions and hydrogen bonding, some of the devices constructed with SWCNT were shown to suffer from long term delamination for which the issue was later resolved by tailoring of polymer's structure.141 Moreover, to improve the mass transport and to compensate the adverse effect of volume change during electrochemical switching (which causes the deterioration of the adhesion between the polymer film and electrode) the utilization of nano-structured electrodes should be considered. Continuing the research especially on stability, electrode–polymer interactions would further enhance the development of the field. With this being done, the polymer electrochromics will be in our daily life as displays, e-papers, rear-view mirrors, smart windows etc. which are only limited to the imagination of researchers.
Notes and references
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2 CrossRef CAS PubMed.
- R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147 RSC.
- S. K. Deb, Appl. Opt., Suppl., 1969, 3, 192 Search PubMed.
- D. T. Gillaspie, R. C. Tenent and A. C. Dillon, J. Mater. Chem., 2010, 20, 9585 RSC.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2000, 60, 201 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC.
- R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971 CrossRef CAS.
- A. L. Dyer and J. R. Reynolds, Electrochromism of Conjugated Conducting Polymers, in Handbook of Conducting Polymers Third Edition Conjugated polymers theory, synthesis, properties, and characterization, ed. T. A. Skotheim and J. R. Reynolds, CRC Press Taylor & Francis Group, 2007 Search PubMed.
- P. Çamurlu and L. Toppare, in Electrochromism in Conducting Polymers Chromic Materials, Phenomena and Their Technological Applications, ed. P. R. Somani, Applied Science Innovations Private Ltd., Maharasthra, 2010 Search PubMed.
- P. Monk, R. Mortimer and D. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, 2007 Search PubMed.
- V. K. Thakur, G. Ding, J. Ma, P. S. Lee and X. Lu, Adv. Mater., 2012, 24, 4071 CrossRef CAS PubMed.
- A. A. Argun, P.-H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401 CrossRef CAS.
- D. S. Dalavi, R. S. Devan, R. A. Patil, R. S. Patil, Y.-R. Ma, S. B. Sadale, I. Y. Kim, J.-H. Kim and P. S. Patil, J. Mater. Chem. C, 2013, 1, 3722 RSC.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, F. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098 CrossRef CAS.
- L.-X. Wang, X.-G. Li and Y.-L. Yang, React. Funct. Polym., 2001, 47, 125 CrossRef CAS.
- L. B. Groenendaal, G. Zotti, P. H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855 CrossRef CAS.
- B. L. Funt and A. F. Diaz, Organic Electrochemistry: an Introduction and a Guide, Marcel Dekker, New York, 1991, pp. 1337 Search PubMed.
- E. M. Genies, G. Bidan and A. F. Diaz, J. Electroanal. Chem., 1983, 149, 101 CrossRef CAS.
- R. J. Waltman and J. Bargon, Can. J. Chem., 1985, 64, 76 CrossRef.
- R. J. Waltman and J. Bargon, Tetrahedron, 1984, 40, 3963 CrossRef CAS.
- S. Sadki, P. Schottland, N. Brodie and G. Sabouraud, Chem. Soc. Rev., 2000, 29, 283 RSC.
- J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS.
- M. Zhou and J. Heinze, J. Phys. Chem. B, 1999, 103, 8443 CrossRef CAS.
- S. H. Cho, K. T. Song and J. Y. Lee Recent Advances in Polypyrrole, in Handbook of Conducting Polymers Third Edition Conjugated polymers theory, synthesis, properties,and characterization, ed. T. A. Skotheim and J. R. Reynolds, CRC Press Taylor & Francis Group, 2007 Search PubMed.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309 CrossRef CAS.
- A. O. Patil, A. J. Heeger and F. Wudl, Chem. Rev., 1988, 88, 183 CrossRef CAS.
- K. Yakushi, L. J. Lauchlan, T. C. Clarke and G. B. Street, J. Chem. Phys., 1983, 79, 4774 CrossRef CAS PubMed.
- S. Panero, S. Passerini and B. Scrosati, Mol. Cryst. Liq. Cryst., 1993, 229, 97 CrossRef CAS.
- J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed.
- P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2002, 77, 117 CrossRef.
- E. M. Girotto and M. A. De Paoli, Adv. Mater., 1998, 10, 790 CrossRef CAS.
- J. Ferreira, M. J. L. Santos, R. Matos, O. P. Ferreira, A. F. Rubira and E. M. Girotto, J. Electroanal. Chem., 2006, 591, 27 CrossRef CAS PubMed.
- F. Tavoli and N. Alizadeh, J. Electroanal. Chem., 2014, 720–721, 128 CrossRef CAS PubMed.
- K. Yamada, K. Seya and G. Kimura, Synth. Met., 2009, 159, 188 CrossRef CAS PubMed.
- T. Shimidzu, A. Ohtani, M. Aiba and K. Honda, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 3941 RSC.
- A. C. Sonavane, A. I. Inamda, D. S. Dalavi, H. P. Deshmukh and P. S. Patil, Electrochim. Acta, 2010, 55, 2344 CrossRef CAS PubMed.
- S. Takagi, S. Makuta, A. Veamatahau, Y. Otsuka and Y. Tachibana, J. Mater. Chem., 2012, 22, 22181 RSC.
- M. Shibata, K.-I. Kawashita, R. Yosomiya, H. An and Y. Haga, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35, 1207 CrossRef.
- R. Brooke, D. Evans, P. Hojati-Talemi, P. Murphy and M. Fabretto, Eur. Polym. J., 2014, 51, 28 CrossRef CAS PubMed.
- S. Ahmad and S. Singh, Electrochem. Commun., 2008, 10, 895 CrossRef CAS PubMed.
- I. D. Brotherstona, D. S. K. Mudigondaa, J. M. Osborna, J. Belka, J. Chena, D. C. Lovedaya, J. L. Boehmea, J. P. Ferrarisa and D. L. Meeker, Electrochim. Acta, 1999, 44, 2993 CrossRef.
- M. Ak, B. Gacal, B. Kiskan, Y. Yagci and L. Toppare, Polymer, 2008, 49, 2202 CrossRef CAS PubMed.
- Z. Zhao-yang, T. Yi-jie, X. Xiao-qian, Z. Yong-jiang, C. Hai-feng and Z. Wen-wei, J. Appl. Polym. Sci., 2013, 129, 1506 CrossRef.
- T. Yi-jie, C. Hai-feng, Z. Wen-wei, Z. Zhao-yang and L. Dong-qing, Synth. Met., 2012, 162, 728 CrossRef PubMed.
- T. C. Wen, C. H. Yang, Y. C. Chen and W. C. Lin, J. Electrochem. Soc., 2003, 150, D123 CrossRef CAS PubMed.
- A. F. Diaz, J. Castillo, K. K. Kanazawa and J. A. Logan, J. Electroanal. Chem., 1982, 133, 233 CrossRef CAS.
- Y. Murakami and T. Yamamoto, Polym. J., 1999, 31, 476 CrossRef CAS.
- W. A. Gazotti, M.-A. De Paoli, G. Casalbore-Miceli, A. Geri and G. Zotti, J. Appl. Electrochem., 1999, 29, 753 CrossRef CAS.
- M.-A. De Paoli, G. Casalbore-Miceli, E. M. Girotto and W. A. Gazotti, Electrochim. Acta, 1999, 44, 2983 CrossRef.
- M. Ak, M. S. Ak and L. Toppare, Macromol. Chem. Phys., 2006, 207, 1351 CrossRef CAS.
- P. Camurlu and C. Gültekin, Smart Mater. Struct., 2012, 21, 025019 CrossRef.
- P. Camurlu, E. Eren and C. Gültekin, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4847 CrossRef CAS.
- O. Mert, A. S. Demir and A. Cihaner, RSC Adv., 2013, 3, 2035 RSC.
- M. Ouyang, G. Wang and C. Zhang, Electrochim. Acta, 2011, 56, 4645 CrossRef CAS PubMed.
- E. C. S. Coelho, V. B. Nascimento, A. S. Ribeiro and M. Navarro, Electrochim. Acta, 2014, 123, 441 CrossRef CAS PubMed.
- A. K. A. Almeida, J. M. M. Dias, A. J. C. Silva, D. P. Santos, M. Navarro, J. Tonholo, M. O. F. Goulart and A. S. Ribeiro, Electrochim. Acta, 2014, 122, 50 CrossRef CAS PubMed.
- W. Choi, H. C. Ko, B. Moon and H. Lee, J. Electrochem. Soc., 2004, 151, E80–E83 CrossRef CAS PubMed.
- N. Trombach, O. Hild, D. Schlettwein and D. Wöhrle, J. Mater. Chem., 2002, 12, 879 RSC.
- H. Masuda, S. Tanaka and K. Kaeriyama, J. Chem. Soc., Chem. Commun., 1989, 725 RSC.
- K. Kaeiyama, M. Sato and K. Hamada, Makromol. Chem., Rapid Commun., 1989, 171 CrossRef.
- D. Delabouglise, J. Roncali, M. Lemaire and F. Garnier, J. Chem. Soc., Chem. Commun., 1989, 475 RSC.
- K. Kaeriyama, S. Tanaka, M.-A. Sato and K. Hamada, Synth. Met., 1989, 28, C611 CrossRef CAS.
- M. Salmon, A. F. Diaz, A. J. Logan, M. Krounbi and B. Bargon, Mol. Cryst. Liq. Cryst., 1982, 83, 265 CrossRef.
- T. Jarosz, A. Brzeczek, K. Walczak, M. Lapkowski and W. Domagala, Electrochim. Acta, 2014, 137, 595 CrossRef CAS PubMed.
- T. Benincori, E. Brenna and F. Sannicolo, Chem. Mater., 2000, 12, 1480 CrossRef CAS.
- A. B. Kon, J. S. Foos and T. L. Rose, Chem. Mater., 1992, 4, 416 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve, E. W. Meijer and H. Wynberg, Chem. Mater., 1989, 1, 650 CrossRef CAS.
- F. Jonas and L. Schrader, Synth. Met., 1991, 831, 41 Search PubMed.
- D. M. Welsh, A. Kumar, E. W. Meijer and J. R. Reynolds, Adv. Mater., 1999, 11, 1379 CrossRef CAS.
- D. M. Welsh, A. Kumar, M. C. Morvant and J. R. Reynolds, Synth. Met., 1999, 102, 967 CrossRef CAS.
- A. Kumar, D. M. Welsh, M. C. Morvant, F. Piroux, K. A. Abboud and J. R. Reynolds, Chem. Mater., 1998, 10, 896 CrossRef CAS.
- C. L. Gaupp, D. M. Welsh, R. D. Rauh and J. R. Reynolds, Chem. Mater., 2002, 14, 3964 CrossRef CAS.
- R. M. Walczak and J. R. Reynolds, Adv. Mater., 2006, 18, 1121 CrossRef CAS.
- C. L. Gaupp, K. Zong, P. Schottland, B. C. Thompson, C. A. Thomas and J. R. Reynolds, Macromolecules, 2000, 33, 1132 CrossRef CAS.
- P. Schottland, K. Zong, C. L. Gaupp, B. L. Thompson, C. A. Thomas, I. Giurgiu, R. Hickman, K. A. Abboud and J. R. Reynolds, Macromolecules, 2000, 33, 7051 CrossRef CAS.
- G. Sonmez, I. Schwendeman, P. Schottland, K. W. Zong and J. R. Reynolds, Macromolecules, 2003, 36, 639 CrossRef.
- I. Schwendeman, R. Hickman, G. Sonmez, P. Schottland, K. Zong, D. M. Welsh and J. R. Reynolds, Chem. Mater., 2002, 14, 3118 CrossRef CAS.
- E. Unur, J.-H. Jung, R. J. Mortimer and J. R. Reynolds, Chem. Mater., 2008, 20, 2328 CrossRef CAS.
- E. P. Knott, M. R. Craig, D. Y. Liu, J. E. Babiarz, A. L. Dyer and J. R. Reynolds, J. Mater. Chem., 2012, 22, 4953 RSC.
- J. Jensen, H. F. Dam, J. R. Reynolds, A. L. Dyer and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 536 CrossRef CAS.
- H. Li, C. Lambert and R. Stahl, Macromolecules, 2006, 39, 2049 CrossRef CAS.
- M. Ak, V. Gancheva, L. Terlemezyan, C. Tanyeli and L. Toppare, Eur. Polym. J., 2008, 44, 2567 CrossRef CAS PubMed.
- M. Ak, H. Cetişli and L. Toppare, Colloid Polym. Sci., 2013, 291, 767 CAS.
- J. M. Nadeau and T. M. Swager, Tetrahedron, 2004, 60, 7141 CrossRef CAS PubMed.
- V. V. Roznyatovskiy, N. V. Roznyatovskaya, H. Weyrauch, K. Pinkwart, J. Tübke and J. L. Sessler, J. Org. Chem., 2010, 75, 8355 CrossRef CAS PubMed.
- G. A. Sotzing, J. R. Reynolds, A. R. Katritzky, J. Soloducho, S. Belyakov and R. Musgrave, Macromolecules, 1996, 29, 1679 CrossRef CAS.
- B. Tsuie, J. L. Reddinger, G. A. Sotzing, J. Soloducho, A. R. Katritzky and J. R. Reynolds, J. Mater. Chem., 1999, 9, 2189 RSC.
- G. A. Sotzing, J. L. Reddinger, A. R. Katritzky, J. Soloducho, R. Musgrave, J. R. Reynolds and P. J. Steel, Chem. Mater., 1997, 9, 1578 CrossRef CAS.
- J. L. Reddinger, G. A. Sotzing and J. R. Reynolds, Chem. Commun., 1996, 1777 RSC.
- C. Pozo-Gonzalo, J. A. Pomposo, J. A. Alduncin, M. Salsamendi, A. I. Mikhaleva, L. B. Krivdin and B. A. Trofimov, Electrochim. Acta, 2007, 52, 4784 CrossRef PubMed.
- C. Pozo-Gonzalo, M. Salsamendi, J. A. Pomposo, H. J. Grande, E. Y. Schmidt, Y. Y. Rusakov and B. A. Trofimov, Macromolecules, 2008, 41, 6886 CrossRef CAS.
- B. A. Trofimov, E. Y. Schmidt, A. I. Mikhaleva, C. Pozo-Gonzalo, J. A. Pomposo, M. Salsamendi, N. I. Protzuk, N. V. Zorina, A. V. Afonin, A. V. Vashchenko, E. P. Levanova and G. G. Levkovskaya, Chem.–Eur. J., 2009, 15, 6435 CrossRef CAS PubMed.
- P. Camurlu and N. Karagoren, React. Funct. Polym., 2013, 73, 847 CrossRef CAS PubMed.
- S. Tarkuc, M. Ak, E. Onurhan and L. Toppare, J. Macromol. Sci., Part A: Pure Appl.Chem., 2008, 45, 164 CrossRef CAS.
- E. Sahin, E. Sahmetlioglu, I. M. Akhmedov, C. Tanyeli and L. Toppare, Org. Electron., 2006, 7, 351 CrossRef CAS PubMed.
- A. Arslan, O. Turkaslan, C. Tanyeli, I. M. Akhmedov and L. Toppare, Mater. Chem. Phys., 2007, 104, 410 CrossRef CAS PubMed.
- S. Varis, M. Ak, C. Tanyeli, I. M. Akhmedov and L. Toppare, Eur. Polym. J., 2006, 42, 2352 CrossRef CAS PubMed.
- E. Yildiz, P. Camurlu, C. Tanyeli, I. M. Akhmedov and L. Toppare, J. Electroanal. Chem., 2008, 612, 247 CrossRef CAS PubMed.
- Y. H. Kim, J. Hwang, J. I. Son and Y. Shim, Synth. Met., 2010, 160, 413 CrossRef CAS PubMed.
- N. A. Lengkeek, J. M. Harrowfield and G. A. Koutsantonis, Synth. Met., 2010, 160, 72 CrossRef CAS PubMed.
- B. Yigitsoy, S. Varis, C. Tanyeli, I. M. Akhmedov and L. Toppare, Thin Solid Films, 2007, 515, 3898 CrossRef CAS PubMed.
- A. Yavuz, B. Bezgin, L. Aras and A. M. Onal, J. Electroanal. Chem., 2010, 639, 116 CrossRef CAS PubMed.
- S. Tarkuc, E. Sahmetlioglu, C. Tanyeli, I. M. Akhmedov and L. Toppare, Electrochim. Acta, 2006, 51, 5412 CrossRef CAS PubMed.
- S. Tarkuc, E. Sahmetlioglu, C. Tanyeli, I. M. Akhmedov and L. Toppare, Sens. Actuators, B, 2007, 121, 622 CrossRef CAS PubMed.
- S. Kiralp, P. Camurlu, G. Gunbas, C. Tanyeli, I. M. Akhmedov and L. Toppare, J. Appl. Polym. Sci., 2009, 112, 1082 CrossRef CAS.
- M. Ak, E. Sahmetlioglu and L. Toppare, J. Electroanal. Chem., 2008, 621, 55 CrossRef CAS PubMed.
- A. Cihaner and F. Algı, Electrochim. Acta, 2008, 53, 2574 CrossRef CAS PubMed.
- S. Koyuncu, C. Zafer, E. Sefer, F. B. Koyuncu, S. Demic, I. Kaya, E. Ozdemir and S. Icli, Synth. Met., 2009, 159, 2013 CrossRef CAS PubMed.
- A. Cihaner and F. Algı, J. Electroanal. Chem., 2008, 614, 101 CrossRef CAS PubMed.
- A. Cihaner, O. Mert and A. S. Demir, Electrochim. Acta, 2009, 54, 1333 CrossRef CAS PubMed.
- J. Hwang, I. J. Son and Y. Shim, Sol. Energy Mater. Sol. Cells, 2010, 94, 1286 CrossRef CAS PubMed.
- D. Asil, A. Cihaner, F. Algı and A. M. Önal, Electroanalysis, 2010, 22, 1 CrossRef.
- G. Wang, X. Fu, J. Huang, L. Wu and Q. Du, Electrochim. Acta, 2010, 55, 6933 CrossRef CAS PubMed.
- A. Cihaner and F. Algı, Electrochim. Acta, 2009, 54, 1702 CrossRef CAS PubMed.
- F. Algı and A. Cihaner, Tetrahedron Lett., 2008, 49, 3530 CrossRef PubMed.
- A. Cihaner and F. Algı, Electrochim. Acta, 2008, 54, 786 CrossRef CAS PubMed.
- U. H. Yıldız, E. Sahin, I. M. Akhmedov, C. Tanyeli and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2215 CrossRef.
- P. Camurlu, Z. Bicil, C. Gültekin and N. Karagoren, Electrochim. Acta, 2012, 63, 245 CrossRef CAS PubMed.
- Z. Bicil, P. Camurlu, B. Yucel and B. Becer, J. Polym. Res., 2013, 20, 228 CrossRef.
- P. Camurlu and N. Karagoren, J. Electrochem. Soc., 2013, 160, H560 CrossRef CAS PubMed.
- G. G. McLeod, G. B. Mahboubian-Jones, R. A. Pethrick, S. D. Watson, N. D. Truong, J. C. Galin and J. Francois, Polymer, 1986, 27, 455 CrossRef CAS.
- A. Berlin, W. Wernet and G. Wegner, Macromol. Chem. Phys., 1987, 188, 2963 CrossRef CAS.
- J. P. Ferraris, R. G. Andrus and D. Hrncir, J. Chem. Soc., Chem. Commun., 1989, 18, 1318 RSC.
- J. P. Ferraris and T. R. Hanlon, Polymer, 1989, 30, 1319 CrossRef CAS.
- P. Camurlu, C. Gültekin and Z. Bicil, Electrochim. Acta, 2012, 61, 50 CrossRef CAS PubMed.
- B. C. Thompson, K. A. Abboud, J. R. Reynolds, K. Nakatani and P. Audeber, New J. Chem., 2 0 05, 29, 1128 Search PubMed.
- P. Camurlu, S. Tarkuc, E. Sahmetlioglu, I. M. Akhmedov, C. Tanyeli and L. Toppare, Sol. Energy Mater. Sol. Cells, 2008, 92, 154 CrossRef CAS PubMed.
- C. A. Thomas, K. Zong, K. A. Abboud, P. J. Steel and J. R. Reynolds, J. Am. Chem. Soc., 2004, 126, 16440 CrossRef CAS PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- D. Baran, G. Oktem, S. Celebi and L. Toppare, Macromol. Chem. Phys., 2011, 212, 799 CrossRef CAS.
- D. Baran, A. Balan, B. M. Esteban, H. Neugebauer, N. S. Sariciftci and L. Toppare, Macromol. Chem. Phys., 2010, 211, 2602 CrossRef CAS.
- S. Celebi, A. Balan, B. Epik, D. Baran and L. Toppare, Org. Electron., 2009, 10, 631 CrossRef CAS PubMed.
- S. Celebi, D. Baran, A. Balan and L. Toppare, Electrochim. Acta, 2010, 55, 2373 CrossRef CAS PubMed.
- A. T. Taskin, A. Balan, B. Epik, E. Yildiz, Y. A. Udum and L. Toppare, Electrochim. Acta, 2009, 54, 5449 CrossRef CAS PubMed.
- A. T. Taskin, A. Balan, Y. A. Udum and L. Toppare, Smart Mater. Struct., 2010, 19, 065005 CrossRef.
- A. Abbotto, E. H. Calderon, N. Manfredi, C. M. Mari, C. Marinzi and R. Ruffo, Synth. Met., 2011, 161, 763 CrossRef CAS PubMed.
- J. Jensen, M. V. Madsen and F. C. Krebs, J. Mater. Chem. C, 2013, 1, 4826 RSC.
- J. Wang, X. W. Sun and Z. Jiao, Materials, 2010, 3, 5029–5053 CrossRef CAS PubMed.
- C. I. Cho, W. J. Kwon, S.-J. Choi, P. Kim, S.-A. Park, J. Kim, S. J. Son, R. Xiao, S.-H. Kim and S. B. Lee, Adv. Mater., 2005, 17, 171 CrossRef.
- M. Nikolou, A. L. Dyer, T. T. Steckler, E. P. Donoghue, Z. Wu, N. C. Heston, A. G. Rinzler, D. B. Tanner and J. Reynolds, Chem. Mater., 2009, 21, 5539 CrossRef CAS.
- S. V. Vasilyeva, E. Unur, R. M. Walczak, E. P. Donoghue, A. G. Rinzler and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2009, 1, 2288 CAS.
| This journal is © The Royal Society of Chemistry 2014 |